

Graphical Abstract
Keywords: β-cyclodextrin, Chitosan, Nanoparticle drug delivery system, Ketoprofen
Abstract
β-cyclodextrin (CD) grafted N-maleoyl chitosan (CD-g-NMCS) with two different degrees of substitution (DS) of N-maleoyl (DS = 21.2% and 30.5%) were synthesized from maleic anhydride and chitosan bearing pendant cyclodextrin (CD-g-CS). CD-g-NMCS based nanoparticles were prepared via an ionic gelation method together with chitosan and CD-g-CS nanoparticles. The size and zeta potential of prepared CD-g-NMCS nanoparticles were 179.2~274.0 nm and 36.2~42.4 mV, respectively. In vitro stability test indicated that CD-g-NMCS nanoparticles were more stable in phosphate-buffered saline compared with chitosan nanoparticles. Moreover, a poorly water-soluble drug, ketoprofen (KTP), was selected as a model drug to study the obtained nanoparticle's potentials as drug delivery carriers. The drug loading efficiency of CD-g-NMCS20 nanoparticles were 14.8% for KTP. MTT assay showed that KTP loaded CD-g-NMCS nanoparticles were safe drug carriers. Notably, in vitro drug release studies showed that KTP was released in a sustained-release manner for the nanoparticles. The pharmacokinetic of drug loaded CD-g-NMCS20 nanoparticles were evaluated in rats after intravenous administration. The results of studies revealed that, compared with free KTP, KTP loaded CD-g-NMCS20 nanoparticles exhibited a significant increase in AUC0→24h and mean residence time by 6.6-fold and 2.9-fold, respectively. Therefore, CD-g-NMCS nanoparticles could be used as a novel promising nanoparticle-based drug delivery system for sustained release of poorly water-soluble drugs. The carboxylic acid groups of the CD-g-NMCS molecule provide convenient sites for further structural modifications including introduction of tissue- or disease- specific targeting groups.
1. Introduction
In recent years, with the fast advance of modern nanotechnology [1], nanoparticles (NP) based drug delivery system made of biodegradable and biocompatible materials have emerged as a novel platform for drug delivery [2]. Such systems have many advantages, which could not only solubilize drugs, protect drugs from degradation, and control the drug release rate to prolong the drug's effective therapeutic duration, but also could achieve targeted drug delivery to specific sites [3]. The research in our group had been focused on nanoparticle-based drug delivery system (NDDS) based on chitosan and cyclodextrins for poorly water-soluble drugs [4], [5], [6].
Chitosan (CS) is a natural cationic polysaccharide polymer composed of N-acetylglucosamine and glucosamine residues which has been widely used for preparation of NDDS due to its attractive properties such as biocompatibility, biodegradability, tendency for formation of nanoparticles, and antimicrobial activity [7]. Chitosan based nanoparticles were reported for delivery of poorly water-soluble drug [8] and water-soluble protein [9] as well. In addition, they were also widely explored in gene delivery system recently [10], [11]. Different from chitosan, β-cyclodextrin (β-CD) has a hydrophilic outer surface and a lipophilic central cavity to accommodate a variety of lipophilic drugs [12], resulting in increased solubility of the incorporated drug, enhanced permeation for macromolecular drugs [13], and inhibition of certain protease activities [14].
With combined advantages of β-CD and chitosan, β-CD grafted with chitosan (CD-g-CS) was synthesized and used in many fields [15]. However, the cationic nature of chitosan requires acidic media to maintain its solubility, while in neutral or slight basic condition such as physiological condition of pH 7.4, it is poorly soluble, which limits its practical use. Hence, structural modification to introduce a hydrophilic functional group and improve its solubility is often necessary and crucial for aimed applications [16]. For this reason, many chitosan derivatives including dicarboxymethyl and quaternized chitosan, N-maleoyl chitosan, carboxymethyl chitosan and N-sulfofuryl chitosan with a solubility range of 3–10 mg/ml have been developed [16].
Herein we reported cyclodextrin grafted N-maleoyl chitosan (CD-g-NMCS) nanoparticles as drug-delivery system for poorly water-soluble drugs. CD-g-NMCS polymers with two different degree of substitution (DS) were synthesized and confirmed by 1H NMR, FT-IR and XRD. CD-g-CS and CD-g-NMCS nanoparticles were prepared via ionic gelation method using sodium tripolyphosphate (TPP) as cross-linking agent. The cytotoxicity of CS nanoparticles and CD-g-NMCS nanoparticles were examined. Finally, Ketoprofen (KTP), a potent non-steroidal anti-inflammatory drug commonly used for the treatment of acute and chronic rheumatoid arthritis, was selected as the model drug for encapsulation and sustained drug-release studies. The short half-life, poor solubility in water, low bioavailability and side effects in the gastrointestinal tract [17] make KTP particularly suitable for formulation of a controlled-release dosage form [18]. Pharmacokinetic parameters of these KTP loaded nanoparticles were investigated in rats.
2. Materials and methods
2.1. Materials
Chitosan (Mw = 110 kDa, deacetylation degree >90%) was purchased from Zhejiang Aoxing Biotechnology Co., Ltd., Zhejiang, China. β-cyclodextrin (β-CD) was purchased from Shanghai Kayon Biological Technology Co., Ltd., Shanghai, China. p-Toluenesulfonyl chloride was purchased from Ling-feng Chemical Reagent Co., Ltd., Shanghai, China. Maleic anhydride, sodium tripolyphosphate (TPP) and mannitol were purchased from Sinopharm Chemical Reagent Co., Ltd., Shanghai, China. Ketoprofen was purchased from Shanghai Aidie Chemical Reagent Co. Ltd., Shanghai, China. All other reagents used were commercially available and were of analytical grade.
2.2. Synthesis of cyclodextrin grafted N-maleoyl chitosan
2.2.1. Synthesis of mono-6-deoxy-6-(p-toluenesulfonyl)-β-cyclodextrin
Mono-6-deoxy-6-(p-toluenesulfonyl)-β-cyclodextrin (6-OTs-β-CD) was synthesized as described by the previously reported method [19]. Briefly, β-CD (30.0 g) was suspended in 250 ml of water, and NaOH (3.24 g) in 10 ml of water was added dropwise over 6 min. The suspension became homogeneous and slightly yellow before the addition was complete. p-Toluenesulfonyl chloride (5.04 g) in 15 ml of acetonitrile was added dropwise over 8 min, causing immediate formation of a white precipitate. After 2 h of stirring at 23 °C, the precipitate was removed by filtration and the filtrate was refrigerated overnight at 4 °C. The resulting white precipitate was recovered by filtration. After drying for 12 h, the white solid was obtained.
2.2.2. Synthesis of β-cyclodextrin grafted with chitosan (CD-g-CS)
CD-g-CS was synthesized following procedure in our previous work [20]. Briefly, chitosan (1.0 g) was dissolved in 1% (v/v) acetic acid (pH 4, 80 ml). The solution of 6-OTs- β-CD (1.0–5.0 g) in 40 ml N, N-dimethylformamide (DMF) was added into the chitosan solution. The reaction mixture was refluxed at 100 °C for 16 h and dialyzed with deionized water for 3 days. The solution was then lyophilized to give a cotton like powder of CD-g-CS. The CD-g-CS with different degrees of substitution of β-CD could be obtained by adjusting mass ratio of 6-OTs-β-CD to chitosan. The substitution degree of CD-g-CS was calculated using phenol- sulfuric acid method.
2.2.3. Synthesis of cyclodextrin grafted N-maleoyl chitosan (CD-g-NMCS)
CD-g-CS (1.0 g) was dissolved in 240 ml 2% acetic acid (v/v) at room temperature, and 5 ml of the maleic anhydride (0.17–1.0 g,) solution in 5 ml acetone was added dropwise with vigorous stirring. After 5 h of stirring at 25 °C, the reaction pH was adjusted to 7.0–8.0 by the dropwise addition of NaOH solution (1.0 M), causing immediate formation of a white precipitate. The resulting precipitate was filtered, washed with acetone, and desiccated to give CD-g-NMCS. The different maleoyl substitute degrees of CD-g-NMCS could be obtained by adjusting mass ratio of maleic anhydride to CD-g-CS.
2.3. Characterization of CD-g-NMCS
The Fourier transform infrared (FT-IR) spectrum was recorded on Nicolet 5700 instrument (Nicolet Instrument, Thermo Company, USA). Samples were prepared as KBr pellet and scanned against a blank KBr pellet background at wavenumber range 4000–400 cm−1 with resolution of 4.0 cm−1.
The 1H NMR spectra of the samples was carried out on a Bruker DPX300 spectrometer (Bruker, Germany). Chitosan and CD-g-CS were dissolved in a mixed solvent of CD3COOD and D2O, CD-g-NMCS was dissolved in D2O. DSCOOH was determined by 1H NMR.
The X-ray diffraction (XRD) patterns of the sheet sample was recorded on an X-ray diffractometer (D/Max2500VB2+/Pc, Rigaku, Japan) with area detector operating at a voltage of 40 kV and a current of 50 mA using CuKα radiation source (λ = 0.154 nm). The scanning rate was 5°/min and the scanning scope of 2θ was from 10° to 80° at room temperature.
2.4. Solubility test
The solubility of chitosan, CS-g-CD, CD-g-NMCS20 (DSCOOH = 21.2%) and CD-g-NMCS30 (DSCOOH = 30.5%) in aqueous solution was determined by measuring their optical transmittances. A weighed amount of materials was placed in large screw-cap tubes (50 ml) containing ultrapure water. The solution pH was adjusted to 4 with 1.0 mol/l HCl and left overnight at a final concentration of 4 mg/ml. Then, the pH was readjusted with 0.1 mol/l NaOH to pH gradients ranging from 4 to 9. The absorbance of solutions was measured at 420 nm using a UV-visible spectrometer (Jasco V-530).
2.5. Determination of molecular weight
The viscosity average molecular weight was determined by measuring the relative viscosity with an Ubbelohbe viscometer (Sigma-Aldrich, USA). The solvent system used was 1% (v/v) CH3COOH. The viscosity average molecular weight (Mν) was calculated from the intrinsic viscosity η using the Mark-Houwink equation (η = K Mνα). The k and α values were 16.8 × 10−3 and 0.81, respectively.
2.6. Cell viability assay of CD-g-NMCS polymer
Cell viability was evaluated by evaluation of the viability of human lung adenocarcinoma epithelial (A549) cell line by MTT method. A549 cells were cultured in Dulbecco's modified Eagle's medium (DMEM) in 5% CO2. The cells were seeded in 96-well plates with 1 × 105 cells per well and incubated for 24 h. Then, the old growth medium was replaced with 200 µl of fresh medium containing various amounts of chitosan, CD-g-CS, CD-g-NMCS20 or CD-g-NMCS30 polymer (0.5, 1.0, 2.0, 4.0, 8.0, 10.0 mg/ml) and cells were incubated for 24, 48 and 72 h, respectively. After incubation, culture medium and 20 µl of MTT solution was used to replace the mixture in each well. After further incubation for 4 h in incubator, the culture medium in each well was replaced by 100 µl of isopropanol to dissolve the insoluble formazan-containing crystals. The optical density was read on a microplate reader at 570 nm.
2.7. Preparation of CD-g-NMCS nanoparticles
Chitosan, CD-g-CS and CD-g-NMCS nanoparticles were prepared using the mild ionic gelation method according to the procedure previously developed [20]. Briefly, TPP (0.125–1.0 mg/ml) was added dropwise to chitosan, CD-g-CS or CD-g-NMCS solution (0.5–2.0 mg/ml, pH 4.0–6.0) with magnetic stirring at 800 rpm and continued stirring for 30 min at room temperature to obtain blank nanoparticles.
For the preparation of drug-loaded CD-g-NMCS nanoparticles, the KTP solution with various concentrations was added slowly to CD-g-NMCS (DSCOOH = 0, 21.2% and 30.5%, pH 5.0) solution by magnetic stirring (800 rpm) for 30 min at room temperature. Then, TPP solution was added dropwise to the mixture with mild stirring (800 rpm) for another 30 min. The weight ratio of CD-g-NMCS to TPP used throughout this study was 10:1, which was obtained from the results of several trials. The resulting suspension was subjected to particle size analysis. Particles were washed and ultracentrifuged. Dry nanoparticles were obtained after lyophilized with 3% mannitol as lyophilized protection agent.
2.8. Characterization of CD-g-NMCS nanoparticles
The particle size, zeta potential and size distribution of nanoparticles were measured by dynamic light scattering (DLS, NanoZS4700 nanoseries, Malvern Instruments, UK). For transmission electron microscopy (TEM, JEM- 2010JEOL, Japan), a 10 µl of polymer micelle solution of 10 mg/ml was carefully dropped onto clean copper grids, then dried at room temperature for 30 min.
2.9. Cell viability assay of KTP laoded CD-g-NMCS nanoparticles
A549 cells were cultured in DMEM in 5% CO2. The cells were seeded in 96-well plates with 1 × 105 cells per well and incubated for 24 h. Then, the old growth medium was replaced with 200 µl of fresh medium containing free KTP solution, KTP loaded CS nanoparticles or KTP loaded CD-g-NMCS nanoparticles (0.0005, 0.001, 0.005, 0.01, 0.05 and 0.1 mg/ml) and cells were incubated for 24 h, respectively. After incubation, culture medium and 20 µl of MTT solution was used to replace the mixture in each well. After further incubation for 4 h in incubator, the culture medium in each well was replaced by 100 µl of isopropanol to dissolve the insoluble formazan-containing crystals. The optical density was read on a microplate reader at 490 nm.
2.10. Stability study
The chitosan, CD-g-CS and CD-g-NMCS nanoparticles were freshly prepared, isolated and resuspended at 0.1% (w/v) in pH 6.8 phosphate-buffered saline (PBS). The nanoparticles were incubated at 37 °C under agitation (100 rpm), and samples were collected at time points of 0, 0.5, 1, 2, 4, 6, 12, 24, 48 and 72 h. The size distribution of the nanoparticles was measured by DLS. Each experiment was performed in triplicate.
2.11. Determination of KTP by HPLC
KTP was determined using a modified reverse-phase HPLC system (Agilent Technologies, Santa Clara, CA, USA). Chromatographic separation was conducted on a Diamonsil® C18 column (4.6 × 250 mm, 5 µm, Dikma, Beijing, Peoples' Republic of China). The ternary mobile phase consisted of methanol/0.01 M KH2PO4 buffer (70/30, v/v) and adjusted to pH 3.0 with phosphoric acid, which was delivered at an isocratic flow rate of 1 ml/min at 40 °C. The detection wavelength was set at 260 nm. The injection volume was 20 µl, and the retention time of KTP was 7.9 min.
2.12. Determination of entrapment efficiency and loading efficiency
The drug content in nanoparticles was calculated from the difference between the total amount of drug added in the nanoparticles and the amount of unentrapped drug in the aqueous medium. The KTP content was measured by UV-visible spectrophotometry at 260 nm. A blank sample was made from nanoparticles without loaded KTP but treated similarly as the KTP-loaded nanoparticles. Each sample was measured in triplicate. The entrapment efficiency (EE) and loading efficiency (LE) were calculated as:
2.13. In vitro release studies
In vitro release profiles of KTP from drug-loaded nanoparticles were investigated for 24 h in the PBS. 2 mg KTP-loaded nanoparticles and 5 ml release medium were put into a dialysis tube (MWCO: 14,000 Da). The dialysis tube was placed in 30 ml release medium at 37 °C and stirred continuously at 100 rpm. At specific time intervals, 5 ml release medium were withdrawn and replaced with 5 ml fresh release medium. The concentration of the released KTP was determined by UV spectrophotometer at 260 nm. The analysis was performed in triplicate for each sample.
2.14. Animal testing
Nine female SD rats (200 ± 10 g) were randomly divided into three groups, Group A: free KTP treated group; Group B: KTP loaded CS nanoparticles treated group; Group C: KTP loaded CD-g-NMCS nanoparticles treated group. The animals were injected in tail vein with a dose of 10 mg KTP/kg. The blood samples (0.3 ml) were drawn from fundus oculi at a designated time intervals. Blood samples were centrifuged to obtain plasma (50 µl). 1 ml of acetonitrile was added into the plasma and vortexed to precipitate protein. The mixture was centrifuged and 0.9 ml of upper was transferred and dried by nitrogen gas. The residue was dissolved with 200 µl mobile phase and the KTP concentration in plasma was assayed by HPLC.
2.15. Statistical analysis
Multiple group comparisons were conducted using one-way analysis of variance (ANOVA). All data analysis was executed using the IBM SPSS Statistics 17.0. All data were presented as a mean value with its standard deviation indicated (mean ± SD). P-values less than 0.05 were considered to be statistically significant.
3. Results and discussion
3.1. Synthesis and characterization of CD-g-CS and CD-g-NMCS
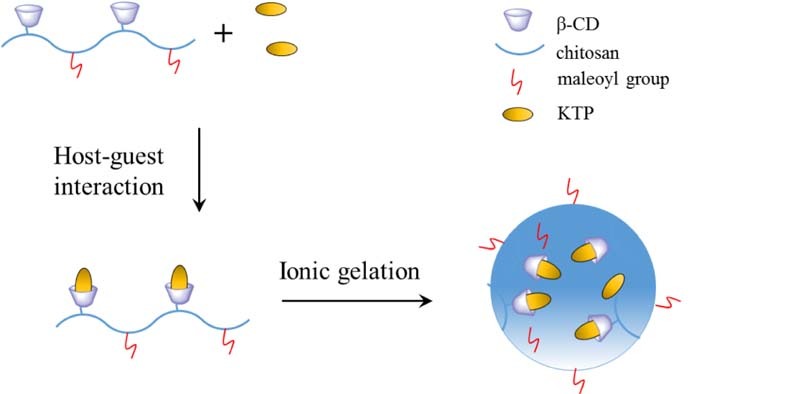
As shown in Fig. 1, chitosan was first reacted with 6-OTs-β-CD in the presence of acetic acid to functionalize partial amino groups of the chitosan with β-CD and obtained CD-g-CS [20]. Then CD-g-CS further reacted with maleic anhydride and functionalized the unreacted amino groups of CD-g-CS to afford CD-g-NMCS. The obtained chitosan derivatives were characterized by FT-IR (Fig. 2), 1H NMR spectroscopy (Fig. 3), and XRD spectrum (Fig. 4).
Fig. 1.

Synthetic pathway of CD-g-NMCS polymer.
Fig. 2.
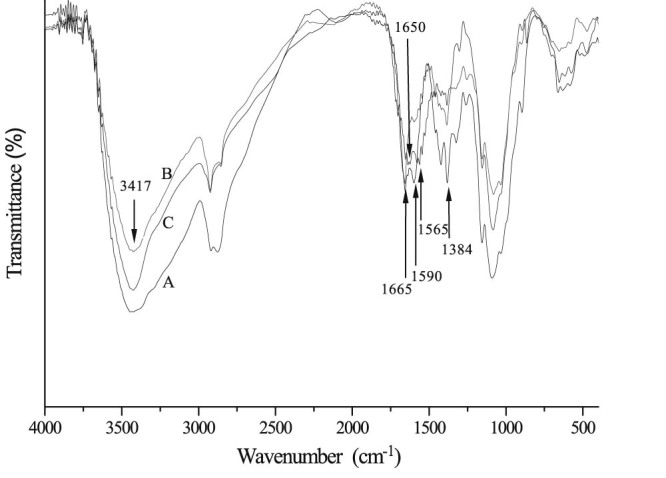
FT-IR spectra of CS (A), CD-g-CS (B) and CD-g-NMCS (C).
Fig. 3.

1H NMR spectra of CS (A), CD-g-CS (B) and CD-g-NMCS (C).
Fig. 4.
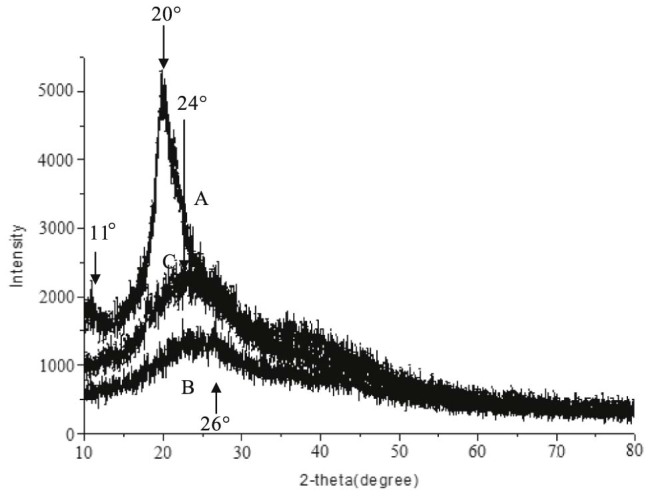
XRD spectra of CS (A), CD-g-CS (B) and CD-g-NMCS (C).
The FT-IR spectrum of chitosan (Fig. 2A) showed a broad -OH stretch absorption band between 3500 and 3100 cm−1 and the aliphatic C-H stretch between 2990 and 2850 cm−1. The peak at 1384 cm−1 represented the -C-O stretch of primary alcoholic group (-CH2-OH). The FT-IR spectrum of CD-g-CS (Fig. 2B) showed both patterns of chitosan and β-cyclodextrin. The dominant peaks at wavenumbers 3417 cm−1 and 1384 cm−1 were due to -OH stretching and -C-O stretching of chitosan backbone, respectively. Further, the characteristic peak of the β-pyranyl vibration of chitosan at 894.9 cm−1 and the characteristic peak of the α-pyranyl vibration of β-CD at 942.8 cm−1 both appeared. The peak at 890 cm−1 was the characteristic bands of α-(1, 4) glucopyranose in β-CD. Fig. 2C showed the FT-IR spectrum of CD-g-NMCS. Peaks appeared at 1565 cm−1 and 1650 cm−1 represented the stretching vibration of C-N from acyl group (-NH-CO-) and the stretching vibration of C = C from maleoyl group, respectively. Furthermore, the peak at 1665 cm−1, which represented the free primary amino group (-NH2), was decreased because of the substitution of maleoyl group. These results demonstrated the successful synthesis of CD-g-NMCS.
As shown in Fig. 3A, the 1H NMR assignments of chitosan were as follows: δ 3.08 (H2); 3.65–3.82 (H1, H3, H4, H5, H6). Compared with the 1H NMR of chitosan, the 1H NMR spectrum of CD-g-CS (Fig. 3B) showed multiple signals at 5.0–3.0 ppm, which were due to the H2' -H6' protons of CD and H3-H6 of chitosan. The singlet at 1.99 ppm was due to the H2 proton of the glucosamine (Fig. 3B). Fig. 3C showed the 1H NMR spectrum of CD-g-NMCS. The multiplet proton signals at 5.0–3.0 ppm corresponded to the protons of CD, and the proton signal at 6.3 was due to the ethylenic bond proton in maleoyl. These results indicated the successful synthesis of CD-g-NMCS.
X-ray diffraction spectra of chitosan exhibited two reflections at 2θ values of 11° and 20° (Fig. 4A), which was assigned to crystal form I and crystal forms II, respectively, by Samuels [21]. Compared with chitosan, XRD spectra of CD-g-CS (Fig. 4B) and CD-g-NMCS (Fig. 4C) showed broad single peak pattern at 2θ values of 26° and 24°, respectively, indicating low crystallinity and considerably more amorphous than chitosan. We hypothesize that the decrease in the crystallinity of CD-g-CS and CD-g-NMCS is due to deformation of the strong hydrogen bonds in the chitosan backbone as the amino groups are functionalized by 6-OTs-β-CD or maleic anhydride.
3.2. Solubility study
Fig. 5 exhibited the transmittance of CD-g-NMCS in water with various pH compared with chitosan and CD-g-CS at 5 mg/ml. It was found out that the water solubility of chitosan, CD-g-CS and CD-g-NMCS decreased with the increase of pH. Chitosan, CD-g-CS, CD-g-NMCS20 and CD-g-NMCS30 showed excellent water solubility at low pH due to protonation of amino groups. At the pH range of 5.0–8.0, the CD-g-NMCS20 and CD-g-NMCS30 showed better water solubility than chitosan and CD-g-CS, which indicated that maleoyl conjugation enhances the solubility of chitosan.
Fig. 5.

The influence of pH on water solubility of CS, CS-g-CD and CD-g-NMCS.
Data were expressed as mean ± SD (n = 3).
3.3. Molecular weight determination
Compared with chitosan, the apparent viscosity average molecular weight of CD-g-CS, CD-g-NMCS20 and CD-g-NMCS30 increased (Table 1). The results clearly reflected the sequential increase of molecular weight due to the graft of CD and maleoyl groups, and further increase of the molecular weight through increase of maleoyl substitution.
Table 1.
Viscosity average molecular weight of CS, CD-g-CS and CD-g-NMCS.
| Sample | η | (Da) |
|---|---|---|
| CS | 203.6 | 1.10 × 105 |
| CD-g-CS | 230.2 | 1.28 × 105 |
| CD-g-NMC20 | 234.6 | 1.31 × 105 |
| CD-g-NMC30 | 237.5 | 1.33 × 105 |
Note: η: intrinsic viscosity; : viscosity average molecular weight.
3.4. Cell toxicity assays of CD-g-NMCS polymer
The in vitro toxicity of CD-g-CS, CD-g-NMCS20 and CD-g-NMCS30 was studied by the MTT assay using A549 cell as the model cell line. The results were shown in Fig. 6, CD-g-NMCS, CD-g-CS, and CS showed almost no cytotoxicity in 24, 48 and 72 h.
Fig. 6.
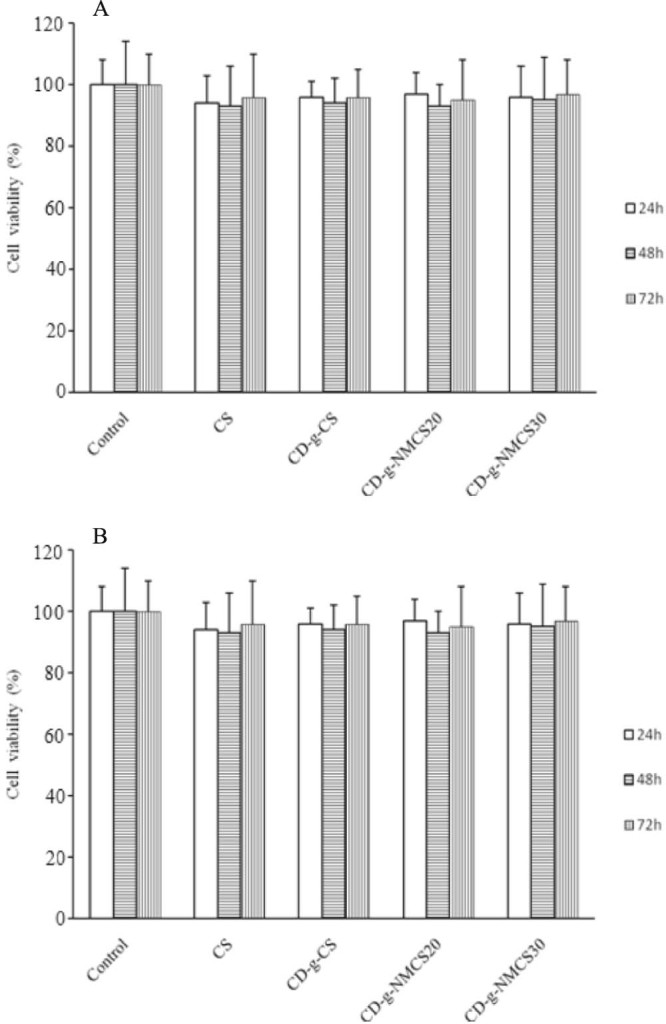
Cell viability assay with (A) CS, CD-g-CS and CD-g-NMCS polymer (4 mg/ml), (B) various concentrations of CD-g-NMCS polymer. Data were expressed as mean ± SD (n = 3).
We also conducted a dose-dependent MTT assay by co-incubation A549 cells with various concentrations (0.1–10 mg/ml) of CD-g-NMCS20 for 24, 48 and 72 h (Fig. 6B). CD-g-NMCS showed no cytotoxicity up to 10 mg/ml for 72 h. The results show that CD-g-NMCS is a safe and biocompatible material for use as a potential drug carrier.
3.5. Preparation and characterization of CD-g-NMCS nanoparticles
These nanoparticles formation with 1.0 mg/ml CD-g-NMCS and 0.5 mg/ml TPP (CD-g-NMCS/TPP = 5/1, v/v) were examined at various pH values (from 4.0 to 6.0). Results showed that CD-g-NMCS could form nanoparticles between pH 4.5 and pH 5.5 (Table 2). Besides, various weight ratios of CD-g-CS to TPP were also studied for nanoparticle formation, since the inter and intra molecular linkages between TPP and positively charged amino groups of CD-g-NMCS are known to be responsible for the gelation process and the right weight ratio of CD-g-NMCS to TPP is critical for the success of nanoparticle formation. Results showed that the formation of nanoparticles was only possible for a certain weight ratio range of CD-g-CS to TPP from 10/1 to 15/1 (Table 2).
Table 2.
Physicochemical properties of CD-g-NMCS nanoparticles.
| pH | CD-g-NMCS (mg/ml) |
CD-g-NMCS/TPP (w/w) |
Morphology | Size (nm) |
Zeta potential (mV) |
PI |
|---|---|---|---|---|---|---|
| 4.0 | 1.0 | 10/1 | Unformed | N.D. | — | — |
| 4.5 | 1.0 | 10/1 | Spherical | 179.2 ± 16.1 | 42.4 ± 3.1 | 0.33 |
| 5.0 | 0.5 | 5/1 | Precipitation | N.D. | — | — |
| 5.0 | 1.0 | 10/1 | Spherical | 197.7 ± 14.2 | 38.1 ± 2.5 | 0.18 |
| 5.0 | 1.0 | 20/1 | Precipitation | N.D. | — | — |
| 5.0 | 1.0 | 5/1 | Precipitation | N.D. | — | — |
| 5.0 | 1.5 | 15/1 | Spherical | 274.0 ± 9.8 | 40.9 ± 1.6 | 0.40 |
| 5.0 | 2.0 | 20/1 | Unformed | N.D. | — | — |
| 5.5 | 1.0 | 10/1 | Spherical | 225.0 ± 18.9 | 36.2 ± 1.9 | 0.16 |
| 6.0 | 1.0 | 10/1 | Precipitation | N.D. | — | — |
Note: DSCOOH of CD-g-NMCS is 21.2%; N.D.: no data; PI: polydispersity index. Data were expressed as mean ± SD (n = 3).
The properties of KTP loaded nanoparticles were summarized in Table 3. The drug loading efficiency of CD-g-NMCS20 and CD-g-NMCS30 nanoparticles were 14.8 and 15.0%, respectively. KTP loaded CD-g-CS and CD-g-NMCS nanoparticles all displayed high entrapment efficiency, which were all over 60%. This was probably due to the interactions consisting of a mixture of host-guest complexation with β-CD, hydrogen bonding with β-CD polymer, physical adsorption into the pores and an ionic interaction between CD-g-NMCS and KTP.
Table 3.
Physicochemical properties of KTP loaded CD-g-NMCS nanoparticles.
| Nanoparticles | KTP (mg) |
EE (%) |
LE (%) |
Size (nm) |
Zeta potential (mV) |
PI |
|---|---|---|---|---|---|---|
| CS | 4.0 | 55.4 ± 2.7 | 16.2 ± 1.4 | 237.6 ± 25.1 | 28.1 ± 2.9 | 0.28 |
| CD-g-CS | 0.5 | 23.2 ± 1.4 | 8.8 ± 1.7 | 183.2 ± 2.1 | 33.4 ± 2.0 | 0.27 |
| 1.0 | 24.4 ± 0.4 | 10.2 ± 1.1 | 217.3 ± 4.1 | 40.2 ± 3.8 | 0.21 | |
| 2.0 | 33.8 ± 1.4 | 12.5 ± 2.3 | 219.0 ± 9.2 | 32.3 ± 3.4 | 0.25 | |
| 4.0 | 60.7 ± 4.2 | 14.0 ± 2.1 | 211.3 ± 17.0 | 35.4 ± 1.5 | 0.26 | |
| 8.0 | 50.7 ± 3.0 | 12.7 ± 1.5 | 233.5 ± 5.3 | 39.1 ± 2.8 | 0.18 | |
| CD-g-NMCS20 | 4.0 | 62.6 ± 2.8 | 14.8 ± 1.9 | 306.9 ± 47.5 | 21.5 ± 2.5 | 0.21 |
| CD-g-NMCS30 | 4.0 | 65.4 ± 3.1 | 15.0 ± 2.2 | 327.0 ± 55.1 | 21.7 ± 1.9 | 0.25 |
Note: EE: entrapment efficiency; LE: loading efficiency; PI: polydispersity index. Data were expressed as mean ± SD (n = 3).
The morphology of the CD-g-NMCS nanoparticles was investigated by TEM. As shown in Fig. 7, nanoparticles had spherical morphology and maintained in non-aggregation state.
Fig. 7.

TEM image of CD-g-NMCS nanoparticles.
3.6. Cell viability assay of KTP loaded CD-g-NMCS nanoparticles
The in vitro toxicity of free KTP, KTP loaded CS nanoparticles and KTP loaded CD-g-NMCS nanoparticles was studied by the MTT assay in A549 cells. The results were shown in Fig. 8, free KTP, KTP loaded CS nanoparticles and KTP /CD-g-NMCS nanoparticles did not show significant cytotoxicity in 24 h, which indicated that nanoparticles can be non-toxic and tissue-compatible drug carriers.
Fig. 8.

In vitro cytotoxicity of free KTP, KTP loaded CS nanoparticles and KTP loaded CD-g-NMCS nanoparticles in A549 cells for 24 h. Data were expressed as mean ± SD (n = 3).
3.7. Stability study
The stability study was performed in pH 6.8 PBS buffer, a simulated biological fluid. The time-dependent size change of nanoparticles in PBS buffer reflected their stability as shown in Fig. 9. Generally, the particle size changes fall into three stages: an instantaneous stage in which particle size reduces significantly in the first several hours, followed by an aging stage in which the particle size remained nearly unchanged, and then the swelling/aggregation stage. Swelling was possibly due to the inflow of water into the nanoparticles by osmosis and it would lead to the polymer matrix fracture. Aggregation might be due to collision and adhesion of nanoparticles during extended storage.
Fig. 9.
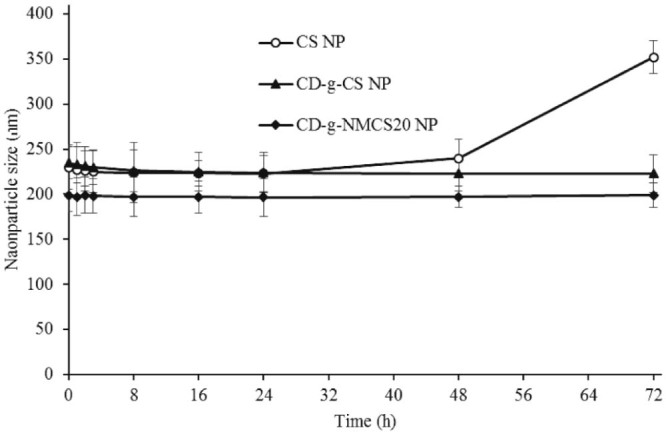
Size change of CD-g-NMCS nanoparticles in pH 6.8 PBS. Data were expressed as mean ± SD (n = 3).
In Fig. 9, the CD-g-CS and CD-g-NMCS nanoparticles remained stable after incubation in PBS for up to 3 d, while the control chitosan nanoparticles showed quick particle swelling upon incubation in PBS for over 24 h. The increased stability of CD-g-CS and CD-g-NMCS nanoparticles was likely due to a more compact inner structure resulting from increased intramolecular interactions between the grafted cyclodextrin and the chitosan main chain. Therefore, the introduction of β-CD to chitosan resulted in improved stability of chitosan in simulated biological fluids, a preferred property for drug delivery.
3.8. In vitro drug release
The in vitro drug release profiles of KTP from loaded CD-g-NMCS nanoparticles with different DSCOOH (0, 21.2% and 30.5%) were investigated. The results were shown in Fig. 10. All the curves showed the same trend. The KTP release rate from nanoparticles was initially very fast within the first two hours and then decreased significantly over hours. The burst release was likely from KTP molecules adsorbed onto the surface of nanoparticles, and the slow release curve reflected the diffusion-controlled release of absorbed KTP molecules deep inside nanoparticles. Among nanoparticles investigated, drug-release rate from CS was the fastest, likely due to its instability. Moreover, drug release from CD-g-NMCS20 nanoparticles was slower than CD-g-NMCS30 nanoparticles.
Fig. 10.
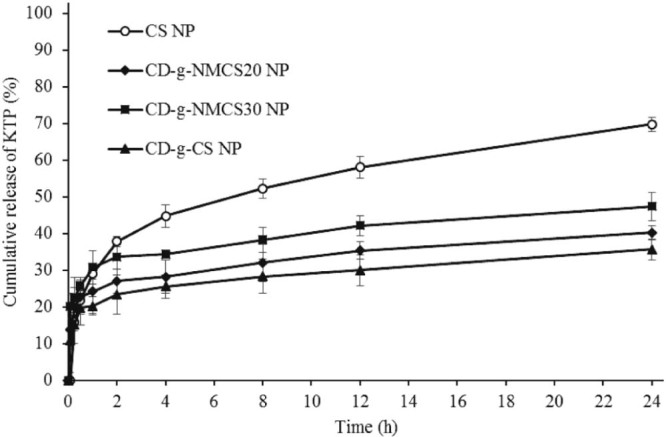
The in vitro release of KTP from drug loaded nanoparticles in pH 6.8 PBS. Data were expressed as mean ± SD (n = 3).
3.9. Animal testing
The plasma concentrations of KTP in rats were quantified by the assay up to 24 h (Fig. 11). In all cases, the blood plasma concentration of KTP was the highest immediately after injection (5 min). The KTP concentration was dropped to hardly detectable level in free drug group, while the drug loaded nanoparticle groups had relative high KTP concentration. This result indicated the sustained release effect of nanoparticle structure. The pharmacokinetic parameters of free KTP, KTP loaded CS nanoparticles or KTP loaded CD-g-NMCS nanoparticles in rats after intravenous administration were shown in Table 4. KTP loaded CD-g-NMCS nanoparticles showed an extended half-life of the elimination phase t1/2β (686 min) compared with free KTP (412 min). Moreover, KTP loaded CD-g-NMCS nanoparticles exhibited a significant increase in AUC0→24h and mean residence time by 6.6-fold and 2.9-fold, respectively. This result stood for prolonged drug duration in vivo. Compared with KTP loaded CS nanoparticles, β-CD also had a positive effect on higher AUC0–24h and MRT because it could form inclusion complex with drug thus slowing down drug release rate and improving drug solubility, leading to the continuous drug content in blood with slow elimination. Pharmacokinetic results showed that KTP loaded CD-g-NMCS nanoparticles released in a sustained manner to maintain KTP plasma concentration.
Fig. 11.
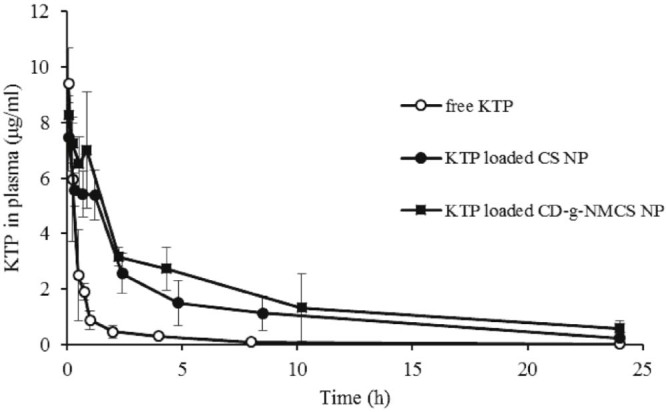
Plasma concentration-time curve of a single dose of free KTP, KTP loaded CS nanoparticles or KTP loaded CD-g-NMCS nanoparticles after intravenous administration (10 mg/kg) in rats. Data were expressed as mean ± SD (n = 3).
Table 4.
Pharmacokinetic parameters for KTP in rats after intravenous administration (10 mg/kg).
| Parameters | Free KTP | KTP loaded CS NP |
KTP loaded CD-g-NMCS NP |
|---|---|---|---|
| t1/2α (min) | 14.70 ± 0.37 | 60.65 ± 11.08 | 68.57 ± 9.03 |
| t1/2β (min) | 412.6 ± 183.2 | 486.3 ± 155.5 | 686.2 ± 134.2* |
| AUC0-24h(µg ⋅ min/ml) | 397.7 ± 25.3 | 1894.3 ± 305.1 | 2632.7 ± 233.0* |
| AUC0-∞ (µg ⋅ min/ml) | 431.9 ± 35.3 | 1945.7 ± 237.2 | 3122.2 ± 129.0 |
| MRT (min) | 281.0 ± 108.9 | 531.7 ± 121.7 | 816.3 ± 113.0* |
| CL (ml/kg ⋅ min) | 23.2 ± 1.9 | 5.2 ± 0.7 | 3.2 ± 0.1 |
Note: t1/2α: a rapid distribution half-life; t1/2β: elimination half-life; AUC: the area under the concentration-time curve; MRT: mean residence time; CL: clearance. Data were expressed as mean ± SD (n = 3). *P < 0.05 versus KTP loaded chitosan nanoparticles.
4. Conclusions
In this study, CD-g-NMCS polymers with different DSCOOH were synthesized and characterized by IR, 1H NMR and XRD. The water solubility, average molecular weight, and cytotoxicity of the CD-g-NMCS polymers were examined. Compared with chitosan or CD-g-CS, the solubility of CD-g-NMCS was improved. CD-g-NMCS nanoparticles were then successfully obtained by ionic gelation method to study their potentials as a new drug carrier using poorly water-soluble drug KTP as a model drug. CD-g-NMCS nanoparticles were proved to be safe and stable nanocarrier in PBS. Moreover, in vitro release study indicated that KTP was released from the nanoparticles in a sustained-release manner, which was affected by DSCOOH. The higher AUC0→24h value indicated that this nanocarrier had a sustained drug-release profile. Overall, we demonstrated that the CD-g-NMCS nanocarrier has potentials as a promising biodegradable delivery system for sustained release of poorly water-soluble drugs. Moreover, the carboxylic acid groups of the CD-g-NMCS molecule provide convenient sites for further structural modifications including introduction of tissue- or disease- specific targeting groups, thus providing a good platform for targeted therapy.
Conflicts of interest
The authors declare that there are no conflicts of interest.
Acknowledgement
This work was supported by the National Science Foundation of China (No.21577037) and Shanghai Committee of Science and Technology (No.17ZR1406600). This work was also sponsored by Science and Technology Commission of Shanghai Municipality (STCSM, contract No.10DZ2220500) and Shanghai Committee of Science and Technology (grant No.11DZ2260600).
Footnotes
Peer review under responsibility of Shenyang Pharmaceutical University.
References
- 1.Meyer M., Persson O. Nanotechnology-interdisciplinarity, patterns of collaboration and differences in application. Scientometrics. 1998;42:195–205. [Google Scholar]
- 2.Gref R., Minamitake Y., Peracchia M.T. Biodegradable long-circulating polymeric nanospheres. Science. 1994;263:1600–1603. doi: 10.1126/science.8128245. [DOI] [PubMed] [Google Scholar]
- 3.Elzoghby A.O., Samy W.M., Elgindy N.A. Albumin-based nanoparticles as potential controlled release drug delivery systems. J Control Release. 2012;157:168–182. doi: 10.1016/j.jconrel.2011.07.031. [DOI] [PubMed] [Google Scholar]
- 4.Chen Y.Z., Huang Y.K., Chen Y. Novel nanoparticles composed of chitosan and β-cyclodextrin derivatives as potential insoluble drug carrier. Chin Chem Lett. 2015;26:909–913. [Google Scholar]
- 5.Fu D.J., Jin Y., Xie M.Q. Preparation and characterization of mPEG grafted chitosan micelles as 5-fluorouracil carriers for effective anti-tumor activity. Chin Chem Lett. 2014;25:1435–1440. [Google Scholar]
- 6.Ye Y.J., Wang Y., Lou K.Y. The preparation, characterization, and pharmacokinetic studies of chitosan nanoparticles loaded with paclitaxel/dimethyl-β-cyclodextrin inclusion complexes. Int J Nanomedicine. 2015;10:4309–4319. doi: 10.2147/IJN.S83508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shao Y., Li L., Gu X. Evaluation of chitosan–anionic polymers based tablets for extended-release of highly water-soluble drugs. Asian J Pharm Sci. 2015;10:24–30. [Google Scholar]
- 8.Jameela S.R., Latha P.G., Subramoniam A. Antitumour activity of mitoxantrone-loaded chitosan microspheres against Ehrlich ascites carcinoma. J Pharm Pharmacol. 1996;48:685–688. doi: 10.1111/j.2042-7158.1996.tb03951.x. [DOI] [PubMed] [Google Scholar]
- 9.Gan Q., Wang T. Chitosan nanoparticle as protein delivery carrier – systematic examination of fabrication conditions for efficient loading and release. Colloid Surf B Biointerfaces. 2007;59:24–34. doi: 10.1016/j.colsurfb.2007.04.009. [DOI] [PubMed] [Google Scholar]
- 10.Mao H.Q., Roy K., Troung-Le V.L. Chitosan-DNA nanoparticles as gene carriers: synthesis, characterization and transfection efficiency. J Control Release. 2001;70:399–421. doi: 10.1016/s0168-3659(00)00361-8. [DOI] [PubMed] [Google Scholar]
- 11.Alex S.M., Rekha M.R., Sharma C.P. Spermine grafted galactosylated chitosan for improved nanoparticle mediated gene delivery. Int J Pharm. 2011;410:125–137. doi: 10.1016/j.ijpharm.2011.02.067. [DOI] [PubMed] [Google Scholar]
- 12.Loftsson T., Duchêne D. Cyclodextrins and their pharmaceutical applications. Int J Pharm. 2007;329:1–11. doi: 10.1016/j.ijpharm.2006.10.044. [DOI] [PubMed] [Google Scholar]
- 13.Yang Y., Gao J., Ma X. Inclusion complex of tamibarotene with hydroxypropyl-β-cyclodextrin: preparation, characterization, in-vitro and in-vivo evaluation. Asian J Pharm Sci. 2016;12:187–192. doi: 10.1016/j.ajps.2016.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Matsubara K., Ando Y., Irie T. Protection afforded by maltosyl-beta-cyclodextrin against alpha-chymotrypsin-catalyzed hydrolysis of a luteinizing hormone-releasing hormone agonist, buserelin acetate. Pharm Res. 1997;14:1401–1405. doi: 10.1023/a:1012120705408. [DOI] [PubMed] [Google Scholar]
- 15.Manakker F.V.D., Vermonden T., Nostrum C.F.V. Cyclodextrin-based polymeric materials: synthesis, properties, and pharmaceutical/biomedical applications. Biomacromolecules. 2009;10:3157–3175. doi: 10.1021/bm901065f. [DOI] [PubMed] [Google Scholar]
- 16.Holme K.R., Perlin A.S. Chitosan N-sulfate. A water-soluble polyelectrolyte. Carbohydr Res. 1997;302:7–12. doi: 10.1016/s0008-6215(97)00117-1. [DOI] [PubMed] [Google Scholar]
- 17.Maestrelli F., González-Rodríguez M.L., Rabasco A.M. Effect of preparation technique on the properties of liposomes encapsulating ketoprofen–cyclodextrin complexes aimed for transdermal delivery. Int J Pharm. 2006;312:53–60. doi: 10.1016/j.ijpharm.2005.12.047. [DOI] [PubMed] [Google Scholar]
- 18.Sheng J.J., Kasim N.A., Chandrasekharan R. Solubilization and dissolution of insoluble weak acid, ketoprofen: effects of pH combined with surfactant. Eur J Pharm Sci. 2006;29:306–314. doi: 10.1016/j.ejps.2006.06.006. [DOI] [PubMed] [Google Scholar]
- 19.Petter R.C., Salek J.S., Sikorski C.T. Cooperative binding by aggregated mono-6-(alkylamino)-beta-cyclodextrins. J Am Chem Soc. 1989;112:3860–3868. [Google Scholar]
- 20.Yuan Z.T., Ye Y.J., Gao F. Chitosan-graft-β-cyclodextrin nanoparticles as a carrier for controlled drug release. Int J Pharm. 2013;446:191–198. doi: 10.1016/j.ijpharm.2013.02.024. [DOI] [PubMed] [Google Scholar]
- 21.Samuels R.J. Solid state characterization of the structure of chitosan films. J Polym Sci. 1981;19:1081–1105. [Google Scholar]