

Graphical Abstract
CD/CS-SCU-NPs were prepared by ionic cross-linking method. Orthogonal design was used to optimize the formulation. The optimum formulation was tested in vitro and in vivo.
Keywords: Carbamazepine, Blood-brain barrier, Nanoparticles, Nasal drug delivery, Pharmacokinetics, Chitosan
Abstract
Epilepsy is considered as a common and diverse set of chronic neurological disorders and its symptoms can be controlled by antiepileptic drugs (AEDs). The presence of p-glycoprotein and multi-drug resistance transporters in the blood-brain barrier could prevent the entry of AEDs into the brain, causing drug resistant epilepsy. To overcome this problem, we propose using carboxymethyl chitosan nanoparticles as a carrier to deliver carbamazepine (CBZ) intra-nasally with the purpose to bypass the blood-brain barrier thus to enhance the brain drug concentration and the treatment efficacy. Results so far indicate that the developed CBZ-NPs have small particle size (218.76 ± 2.41 nm) with high drug loading (around 35%) and high entrapment efficiency (around 80%). The in vitro release profiles of CBZ from the NPs are in accordance with the Korsmeyer-peppas model. The in vivo results show that both encapsulation of CBZ in nanoparticles and the nasal route determined the enhancement of the drug bioavailability and brain targeting characteristics.
1. Introduction
Epilepsy has been defined as a recurrent and unpredictable set of chronic neurological disorders in normal brain [1]. The main symptoms are recurrent and unprovoked seizures or a single seizure together with brain alterations which enhance the chance of future seizures. Epileptic seizures are triggered by abnormal, excessive or hypersynchronous neuronal activity in the brain [2]. About 50 million people all over the world are diagnosed to have epilepsy, and almost 90% of the people with epilepsy are found in developing countries [3]. Epilepsy generally cannot be cured but its symptoms can be controlled using antiepileptic drugs (AEDs). However, even with those drugs, many patients with epilepsy still could not have the seizures under control [3]. For epilepsy treatment, the drug should reach the central nervous system in the brain. However, the central nervous system is well protected by the blood-brain barrier (BBB) which maintains its homeostasis and protects the internal environment of brain from toxicities outside. It also restricts entry of many drugs for treatment of brain disease [4]. One theory on the causes of drug resistance in epilepsy is the transporter hypothesis [5]. Experimental and clinical evidence supporting the transporter hypothesis indicated that increased expression of efflux transporters at the BBB in the focal tissue limits the penetration of AEDs to the focus.
A lot of strategies have been investigated to overcome the BBB to deliver therapeutics to the CNS. With the development in nanotechnology and nanoscience, novel approaches have been employed to deliver drug into the brain. In brief, nanoparticles (NPs) are colloidal particles with the particle size between 1 and 1000 nm [6]. NPs are normally prepared by encapsulating the drug into a vesicle or by dispersing the drug within a matrix and the product will have significant advantages for drug delivery, such as better bioavailability, systemic stability, higher drug loading, longer blood circulation time and selective distribution in the organs/tissues with longer half-life [6]. In recent years, nanoparticulate drug delivery systems have been widely studied for brain targeting [[7], [8]].
To bypass the BBB and deliver drugs to the brain, several methods have also been used such as osmotic and chemical opening of the BBB, intracerebral injection or implantation, intravenous (IV) injection, and intranasal delivery [9], etc. Most of the polymer-based brain drug delivery systems containing large molecules are administered systemically, in most cases via IV injection in order to avoid the first pass effect in gastrointestinal tract [10]. Recently, intranasal drug administration has been widely studied as an alternative to IV administration since it could directly deliver drugs into the brain by bypassing the BBB. For instance, a liposomal formulation following intranasal administration has been prepared to deliver rivastigmine into the brain [11]. Delivery of proteins to the brain by cationic liposomes via intranasal route has also been studied [12]. As a result, intranasal administration route, as a non-invasive drug administration route, could be a new option for treatment of central nervous system diseases.
Generally speaking, nasal delivery system is an administration route to deliver drugs from the nasal cavity to the blood circulation system or to specific targets which cannot be reached by conventional administration routes. In particular, recent studies showed that nasal delivery is superior to other routes of administration in bypassing blood-brain barrier to target brain tissues effectively [13]. Thus intranasal drug delivery system could be better received by patients and might improve the drug transport to the brain. However, there is limitation for nasal delivery, as the mucociliary clearance limits the time allowed for drug absorption in the nasal cavity. Thus mucoadhesive materials, such as starch and chitosan have been employed to overcome such limitation [[14], [15]].
Chitosan, a linear polysaccharide extracted from chitin, is a material that has been shown to be mucoadhesive [16]. With organic and inorganic acids, free amino groups resultant from the deacetylation process of chitin can form positively charged chitosan salts. Chitosan is a good option in nasal delivery as it binds to the nasal mucosal membrane with an increased retention time. Moreover, chitosan is also an excipient that could improve the dissolution rate of hydrophobic drugs and is a good absorption enhancer [17]. Nevertheless, chitosan still possess limitations according to its own structure: its solubility could only be attained in acidic aqueous solution below pH 6.5 [18]. Improvement could be obtained by chemical modification, such as quaternization or introducing hydrophilic or carboxyalkyl groups to chitosan. Compared with other water-soluble chitosan derivatives, carboxymethyl chitosan (CMC) has been widely studied because it is easy to synthesize, it has ampholytic property with ample possible applications [19].
Carbamazepine (CBZ) is used for clinical treatment for seizure disorders, trigeminal neuralgia and most recently manic depressive illness [20]. The mechanism of CBZ treatment is to stabilize the inactivated state of sodium channels, causing fewer of these channels to open and brain cells will become less excitable. CBZ has also been shown to potentiate GABA receptors made up of α1, β2 and γ2 subunits [21]. However, CBZ has an unfavorable function called autoinduction, meaning it could induce the expression of the hepatic microsomal enzyme system CYP3A4, which unfortunately also metabolizes CBZ itself [22]. Once the drug is administrated orally, the concentration of CBZ in plasma is predictable and follows a baseline clearance/half-life values that have been established for the specific patients. However, as the concentration of CBZ increases in the liver, the CYP3A4 activity also increases, resulting in a sharp rise in drug clearance, leading to shorter half-life. Due to its narrow therapeutic range (4–12 µg/ml), frequent fluctuation of plasma concentration could precipitate many side effects.
In summary, to overcome the BBB and have better treatment efficacy for antiepileptic drugs, this study focused on the application of carboxymethyl chitosan nanoparticles for nasal drug administration of CBZ. As the current antiepileptic drugs are all used by oral administration, the nasal delivery could be an alternative strategy to achieve a better brain drug penetration while reducing systemic drug exposure for the antiepileptic drugs.
2. Materials and methods
2.1. Chemical and reagents
Carbamazepine USP-grade was purchased from Sigma–Aldrich; medium molecular weight Chitosan was supplied by Aldrich Chemical Company; Sodium hydroxide (1M) was obtained from Merck Chemicals Co., Ltd (Germany); Phosphate buffer (pH7.4) was purchased from Vivantis Technologies Sdn Bhd (USA). All other chemical solvents and reagents used were of analytical grade.
2.2. Animals
Adult male C57BL mice aged between 6 and 7 weeks and weighing 20–30 g were supplied by local certified animal facility (Comparative Medicine, Center for Life Science, National University of Singapore). Mice were housed under controlled environment with free access to tap water and standard rodent diet. All the experiments involving animals and their care were approved by the Institutional Animal Care and Use Committee (IACUC), National University of Singapore with the protocol number of R13-4467(A2)14.
2.3. Synthesis of CMC
Carboxymethyl chitosan (CMC) was prepared following the method reported previously [23]: chitosan (10 g) and sodium hydroxide (15 g) were suspended in isopropanol (100 ml) to swell and alkalize at room temperature for 1 h. The monochloro-acetic acid (10 g) was dissolved in 20 ml isopropanol, and added to the reaction mixture drop-wise within 30 min and reacted for 4 h at 55 °C. Then the reaction was stopped by removing the reaction mixture from heat and discarding the isopropanol (Fig. 1). Ethyl alcohol (80%) was added and the solid product was filtered and rinsed with 80%~90% ethyl alcohol to desalt and dewater, and vacuum-dried at 50 °C. The product yield of CMC from the synthetic scheme was calculated by the following formula:
Fig. 1.

Synthetic routes of CMC (1. O-CMC, 2. N-CMC).
2.3.1. The degree of substitution of CMC
The degree of substitution (DS) of CMC was determined by pH titration. The accurately weighed CMC was dissolved in water, and then the pH was adjusted to less than 2 with hydrochloric acid (12 mol/l). The standard sodium hydroxide solution (1 mol/l) was used for titration. The amount of aqueous sodium hydroxide was determined by the second order differential method [19]. The DS can be calculated by the following equation:
where mCMC is the mass of CMC (g), 161 and 58 are the molecular weights of glucosamine (chitosan skeleton unit) and a carboxymethyl group respectively, VNaOH and CNaOH are the respective volume and molarity of the aqueous sodium hydroxide solution for the titration [19].
2.3.2. FT-IR spectroscopy
Fourier transform infrared spectroscopy (FT-IR) was used to verify the chemical structure of the synthesized CMC. The measurements were carried out on a FT-IR (Perkin Elmer, Model 100). Samples were analyzed in the atmospheric environment, and the FT-IR curves of chitosan and CMC were acquired [19]. The synthesized CMC was characterized by using a Perkin Elmer PE2400 Series II CHNS/O analyzer.
2.3.3. NMR spectroscopy
Nuclear magnetic resonance (NMR) analyses were carried out as described previously [24]: 15 mg of the CMC sample was dissolved in 1 ml of deuterated water before analyzed using an AC-400 Bruker spectrometer at 353 K. Elemental analysis data were obtained using a Carlo Erba elemental analyzer. Samples were tested in both 1H NMR and 13C NMR.
2.4. Preparation of CBZ-NPs
Drug-loaded nanoparticles were prepared by a modified method reported previously [25]. Briefly, CBZ was dissolved in one part of acetone; and the CMC was separately dissolved in one part of distilled water. The CBZ solution was added dropwise into the polymer solution with magnetic stirrer. After forming the completely dispersed particles, the dispersions were sonicated using a sonic dismembrator (Fisher Scientific Co., Model 500) for about 30 min at 250 W and the time interval for sonication was 10 min. Then the mixture was stirred with magnetic stirrer for about 4 h to evaporate off the organic solvent. The obtained nanoparticle suspension was filtrated through a 0.45 µm filter membrane to remove the unincorporated CBZ aggregates, and the drug-loaded nanoparticle suspensions were obtained. The dried CBZ-NPs was obtained after freeze-drying and kept under vacuum for 48 h.
In order to compare the pharmacokinetic profiles of CBZ-NPs with CBZ, a CBZ-solution was prepared by dissolving CBZ in water containing 2% HP-β-CD to increase its solubility. The drug concentration of the CBZ-solution is the same as that of the CBZ-NPs.
2.5. Formulation optimization
The CBZ/CMC mass ratio (A), the concentration of CMC (B) and the volume of acetone (C) were selected as the main factors. The orthogonal experiment design table of the three factors and three levels are showed in Table 1. Three batches of CBZ-NPs were prepared using the optimal formulation for verification.
Table 1.
Factors and levels for formulation optimization.
| Factors | A |
B |
C |
|---|---|---|---|
| CBZ/CMC mass ratios (mg/mg) | Concentration of CMC (%) | Volume of acetone (ml) | |
| 1 | 1:1 | 0.1 | 0.5 |
| 2 | 1:2 | 0.2 | 2 |
| 3 | 1:3 | 0.5 | 5 |
2.6. Characterization of CBZ-NPs
2.6.1. Transmission electron microscopic (TEM) studies
Particle morphology was examined by transmission electron microscopy (TEM, JEOL 2010). Samples (the nanoparticulate suspensions) were dropped onto the Formvar-coated copper grids. After complete drying, the samples were stained using 2% w/v tungstophosphoric acid (TPA). Image capture and analysis were done using Digital Micrograph and Soft Imaging Viewer Software.
2.6.2. Size and zeta potential analysis of the CBZ-NPs
Photon correlation spectroscopy (Zetasizer 3000; Malvern Instruments, Malvern UK) was used to measure the particle size and zeta potential of all drug-loaded nanoparticles [26]. Before analysis, all samples were diluted with appropriate distilled water. Z-average particle size, polydispersity index (PDI), and zeta potential were all measured in triplicates at room temperature.
2.6.3. Drug-loading rate studies
The dried CBZ-NPs samples (5 mg) were transferred to a 25 ml volumetric flask. A total volume of 10 ml of methanol was added and the obtained dispersion was sonicated for 15 min to dissolve CBZ. Samples were withdrawn from the undiluted solution using a syringe, then filtered (PTFE 0.45 µm) before detection [25]. The concentration of CBZ in methanol was determined by high performance liquid chromatography (HPLC). The drug-loading rate (DL %) can be calculated by the following equation:
Contents of CBZ in the NPs were determined by HPLC. The HPLC system consisted of a pump (Model LC-20A, Shimadzu, Japan), a reversed-phase C18 column (150 × 4.5 mm, 3 µm) maintained at room temperature, a variable wavelength UV detector (Model SPD-20A, Shimadzu, Japan) at 285 nm. The mobile phase was composed of 70% methanol and 30% water (v/v) and was delivered at a flow rate of 0.4 ml/min. The injection volume was 10 µl introduced by an autosampler and the retention time was 6.5 min. Quantification of the compounds was carried out by measuring the peak areas in relation to those of standards chromatographed under the same condition.
2.6.4. Entrapment efficiency
The entrapment efficiency was determined by centrifugation method as reported previously. 1 ml of CBZ-NPs was centrifuged at 10000 rpm for 15 min (4 °C) and the clear supernatant solutions were obtained. After dilution by 10-flod methanol, concentrations of CBZ in the supernatant (free drug) were determined by HPLC. The entrapment efficiency (EE %) could be calculated by the following formula:
where Cfree was the CBZ concentration in the supernatant after centrifugation while Ctotal was the initial amount of drug added during the preparation of CBZ-NPs.
2.7. In vitro drug release
The release of CBZ from CBZ-NPs was evaluated using dialysis technique [26]. Dialysis bag with a molecular weight cut off 350 Da (Sigma-Aldrich Co.) was soaked in diffusion medium (PBS pH7.4). 2 ml of CBZ-NPs suspension was placed in each dialysis bag (n = 3), then sealed at both ends with medicell clips (Spectrum, USA), and placed at the bottom of dissolution vessels containing 150 ml PBS pH 7.4. The study was carried out in a USP dissolution apparatus (Pharma Test, Germany), at 37 ± 0.5 °C using an agitation speed of 300 rpm. Aliquots of 1 ml samples were collected at predetermined time points (0.25, 0.5, 1, 1.5, 2, 4, 6, 8, 12 and 24 h), then added with the same volume of fresh release medium each time to maintain a constant volume. The samples were filtered with filter cartridge (PTFE 0.45 µm) before assayed by HPLC for CBZ at 285 nm.
2.8. Pharmacokinetics study
Thirty-six healthy male mice weighing 20~30g were randomly divided into two groups, the CBZ-NPs and CBZ-SL with dose of 2 mg/Kg. The drug solution used for administration in the experiment was 1 mg/ml. For intranasal administration, 25µl per nostril samples were administrated intra-nasally by slow infusion over ~1 min through a polyethylene tubing which was attached to a microsyringe as described previously [27]. The polyethylene tubing was inserted approximately 0.7 cm into the nostril. For both groups, blood samples of 0.3 ml were withdrawn by cardiac puncture at 0.25, 0.5, 1, 2, 3, 4 h after administration [28]. Sample at one time point was collected from an individual mouse. Then plasma samples were stored at −20 °C immediately after collection until analysis. Following the completion of blood collection, the mice were sacrificed and the skull of the mice was opened after separation of the head, and the whole brain was collected. The brain samples were rinsed with phosphate buffer and stored immediately at −80 °C until analysis. In order to determine the concentration of CBZ in the brain, the whole brain was weighed and homogenized after mixing in 1:1 (w/v) ratio with distilled water.
CBZ was extracted from plasma and brain samples by the liquid–liquid extraction (LLE) method. Briefly, an exact 100 µl sample of plasma was well mixed using vortex with 280 µl acetonitrile and 20 µl phenacetin working solution (200 µg/ml in acetonitrile) as internal standard [27]. Then the mixture was centrifuged at 13000 rpm for 10 min. A total of 50 µl of the supernatant of the sample was injected into the HPLC. Similar to the processing of plasma samples, 100 µl of the brain-distilled water homogenate was added to 280 µl acetonitrile and 20 µl phenacetin working solution (200 µg/ml in acetonitrile) as internal standard, vortexed and then centrifuged at 13000 rpm for 10 min. Finally, 50 µl of the supernatant was injected into the HPLC for analysis.
Samples of CBZ in plasma and brain tissues were assayed using a modified method of HPLC after extraction. The HPLC system (Shimadzu LC-20AD, Japan) consisted of a C18 analytical column (4.6 mm × 150 mm, 3.5 µm) maintained at room temperature and a Shimadzu SPD- 20A-UV detector with wavelength set at 285 nm. The mobile phase was made up of methanol: water (6:4) and was delivered at a flow rate of 0.4 ml/min. The injection volume was 50 µl.
Pharmacokinetic parameters were estimated using model independent methods. The parameters used for evaluation were the maximum plasma or brain tissue concentration (Cmax) and the corresponding time (Tmax) which were recorded directly from the individual plasma or brain concentrations against time profiles. The area under the plasma or brain concentration–time curve (AUC0–∞) and the mean residence time (MRT) were calculated.
3. Results and discussion
3.1. Synthesis and characterization of CMC
3.1.1. Yield of product and degree of substitution
CMC was prepared by etherification method using CS and monocholoroacetic acid in a NaOH-isopropanol system. The main factor which influenced the preparation of CMC was the mass ratio of CS, NaOH and ClCH2COOH [19], while for the reaction time and temperature, there was no significant impact on the preparation process. When the mass ratio of NaOH: ClCH2COOH was less than 1:1, it was difficult to start the reaction, meaning the reaction only started under strong alkaline condition. Moreover, with the increase of CS: ClCH2COOH mass ratio, the degree of substitution increased and the yield of product decreased indicating that the carboxymethyl reaction would start first on the -OH group rather than the -NH2 group. According to the results stated above, when the mass ratio of the CS: NaOH: ClCH2COOH was 2: 3: 2, the yield of product from the synthetic scheme was 84.6%.
The changes of pH value in response to the addition of sodium hydroxide were shown in Fig. 2. There are two sharp rises of pH values in the curve: the first one was at the volume of 13 ml of sodium hydroxide and the second one was at the volume of 19 ml. Thus according to the formula, CMC presented a substitution degree of 0.364.
Fig. 2.
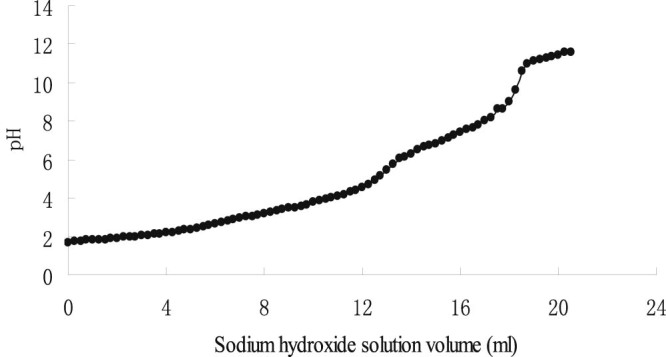
Estimation of degree of substitution of CMC by titration with sodium hydroxide.
3.1.2. FT-IR spectroscopy
In the spectra of FT-IR spectroscopy (Fig. 3) for chitosan, distinctive absorption bands appear at 3433 cm−1 (O-H stretch), 2923-2920 cm−1 (C-H stretch), 1656 cm−1 (acetylated amino group), 1383 cm−1 (C-O stretch of primary alcoholic group(-CH2-OH)), 1155 cm−1 (asymmetric stretching of the C-O-C bridge), 1200-897 cm−1 (the free primary amino group (-NH2) at the C2 position) are characteristic of its saccharine structure. Compared with chitosan, two new bands at 1598 cm−1 and 1415 cm−1 were observed in the spectrum of the CMC, indicating the existence of two functional groups -COOH and -CH2- in the carboxymethylated derivatives. It was also noted that the peaks at 1383 and 1155 cm−1 were disappeared and the range of 1200~897 cm−1 were decreased. The results confirmed that carboxymethylation on both the amino and hydroxyl groups but mainly occurred at hydroxyl groups of chitosan. These findings were in agreement with the results of a previous study [19].
Fig. 3.

FT-IR spectroscopy of (A) chitosan and (B)CMC.
3.1.3. 1H NMR
The proton NMR spectrum of CMC in D2O was shown in Fig. 4. The resonance of H–1D (4.8 ppm), H–1A (4.622 ppm), 3 acetyl-protons (2.051 ppm), H3–6 protons (3.5–3.9 ppm), H–2D proton (3.305 ppm) are the basic assignments of chitosan resonance which can be found in the 1H NMR spectrum of chitosan described previously [19]. The protons resonances of the 3- and 6-substituted carboxymethyl (–O–CH2COOD) of CMC can be observed between 4.05 and 4.55 ppm. The result proved that the amino groups were partly carboxymethylated along with the hydroxyl groups, indicating that our product complies with the previously reported parameters of CMC [[19], [23]].
Fig. 4.

1H NMR spectrum of CMC.
3.1.4. 13C NMR
The 13C NMR spectrum of CMC was shown in Fig. 5. In the spectrum, we can see obvious signals at 168.409 and 171.133 ppm which are signals for -*CH2COOH substituted on the respective -OH and -NH. There are also three chemical shifts at 70.9, 69.500 and 52.756 ppm which assigned to -*CH2COOH groups substituted on O-6, O-3 and N-2 positions as described previously [[19], [23]].
Fig. 5.

13C NMR spectrum of CMC.
3.2. Formulation optimization and verification
The aim of formulation optimization was to obtain smaller particle size and higher drug loading.
The results of three factors and three levels orthogonal experiment design are shown in Table 2. K is the average of results in each level under each factor (e.g., the average of the “1” under A in Table 2 to give K1, and average of the “2” under A to give K2 etc.). R is the difference between the highest and lowest values of K in each factor. The importance of each factor was determined by the R value. The bigger of R value under the factor, the more important would be this factor. Thus to the particle size, the importance of the factors is in the order of A > B > C. Since the smaller particle size is preferred, the best formulation is A3B2C3. To drug content, the importance of effect is A > C > B. Since the higher drug content is preferred, the best formulation is A2B2C3. In the evaluation of particle sizes and drug content, B2 and C3 are confirmed to the consistently preferred factors. In assessing the factor A, for particle size, the difference between A2 and A3 (229.6 vs 218.5 nm) is only 5% while for drug loading the difference is about 15% (34.85% vs 29.35%). As a result, considering the higher drug loading in A2, the optimum formulation was identified to be A2B2C3.
Table 2.
Results of orthogonal tests.
| Run | A |
B |
C |
Size (nm) |
Drug loading (%) |
|---|---|---|---|---|---|
| CBZ/CMC mass ratios (mg/mg) | Concentration of CMC (%) | Volume of acetone (ml) | |||
| 1 | 1 | 1 | 1 | 239.7 | 20.84 |
| 2 | 1 | 2 | 2 | 217.9 | 37.35 |
| 3 | 1 | 3 | 3 | 253.3 | 26.71 |
| 4 | 2 | 1 | 2 | 238.6 | 35.69 |
| 5 | 2 | 2 | 1 | 231.7 | 30.34 |
| 6 | 2 | 3 | 3 | 218.4 | 38.53 |
| 7 | 3 | 1 | 3 | 195.2 | 33.92 |
| 8 | 3 | 2 | 1 | 223.5 | 28.37 |
| 9 | 3 | 3 | 2 | 236.8 | 25.77 |
| K1 | 240.0 | 224.5 | 231.6 | Size | |
| K2 | 229.6 | 224.4 | 231.1 | ||
| K3 | 218.5 | 236.2 | 222.3 | ||
| R | 21.5 | 11.8 | 9.3 | ||
| K1 | 28.30 | 30.15 | 26.52 | Drug loading | |
| K2 | 34.85 | 32.02 | 22.94 | ||
| K3 | 29.35 | 30.34 | 33.05 | ||
| R | 6.55 | 1.87 | 10.11 | ||
3.3. Characterization of CBZ-NPs
3.3.1. Physical properties of CBZ-NPs
The CBZ-NPs suspension was a semi-transparent solution with particle diameter around 220 nm. The product after freeze drying appeared to be a white powder. Fig. 6 shows the transmission electron microscopic (TEM) photomicrographs. It is evident that the spherical particles were of uniform size.
Fig. 6.

Microphotograph of CBZ-NPs by Transmission Electron Microscope (TEM).
3.3.2. CBZ-NPs size, drug loading rate and entrapment efficiency of the optimum formulation
With the optimized formulation generated from the orthogonal design experiments described above, the following procedure was adopted to prepare the CBZ-NPs: CBZ (10 mg) was dissolved in 5 ml of acetone, and CMC (20 mg) was dissolved in 10 ml of water. The CBZ solution was dropped into CMC solution with magnetic stirrer. After forming complete particle dispersion, the dispersion was sonicated for about 30 min. Then the mixture was stirred with magnetic stirrer for about 4h to evaporate the organic solvent. The obtained nanoparticle suspension was filtrated through a 0.45 µm filter membrane to remove the unincorporated CBZ aggregates, and the drug-loaded nanoparticle suspension was then obtained. The properties of the three batches CBZ-NPs prepared using the optimization formulation are showed in Table 3. The results indicate that the absolute zeta potential values of the CBZ-NPs are all over 30, suggesting these nanoparticles to have good physical stability (i.e., phase separation) in the solution [[26], [29], [30]].
Table 3.
Physicochemical characterization of three batches CBZ-NPs prepared by optimal experimental condition (n = 3 ± SD).
| No | Particle size (nm) | PDI | Zeta potential (mV) | Drug Loading (%) |
Entrapment Efficiency (%) |
|---|---|---|---|---|---|
| 1 | 216.5 | 0.189 | −34.9 | 33.72 ± 0.33 | 78.48 ± 2.13 |
| 2 | 221.3 | 0.232 | −32.1 | 36.45 ± 0.21 | 81.92 ± 1.66 |
| 3 | 218.5 | 0.276 | −33.3 | 34.96 ± 0.18 | 79.05 ± 1.94 |
3.3.3. In-vitro release studies
The mean cumulative % of CBZ released from CBZ-NPs versus time plot is shown in Fig. 7, which illustrates a biphasic release of the drug from CBZ-NPs. The drug release rate was faster in the first 4h and reached a plateau at 12h. The initial slight burst release can be attributed to CBZ molecules located at or near to the surface of CBZ-NPs. In vitro release kinetics appeared to be in accordance with the Korsmeyer-peppas model (ln Q = 0.3113ln t + 3.6316, r2 = 0.9729, whereas Q is a fraction of drug released at time t; 0.3113 is the release exponent, n; 3.6316 is the ln (release rate constant), k). The n value was less than 0.45, suggesting that CBZ was released from CBZ-NPs through a mechanism of Fickian diffusion that was controlled by concentration gradient, diffusion distance and degree of swelling of the matrix [31].
Fig. 7.
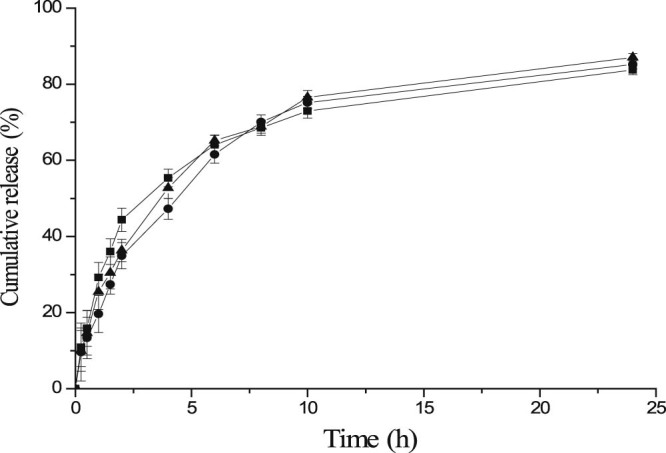
Release profiles of CBZ from the 3 batches of CBZ-NPs prepared under the optimal experimental condition in pH 7.4 PBS media at 37 °C, 300 rpm in water bath (n = 3) (–●–batch 1, –▲– batch 2, –■– batch 3).
3.4. Pharmacokinetics and brain targeting studies
The results obtained from the in vivo tests in mice were shown in Fig. 8 which expressed the CBZ concentration levels in the plasma and brain samples taken from mice after nasal administration of CBZ-NPs and CBZ-SL as a control group. Results showed that the in vivo administration of CBZ-NPs remarkably increased the CBZ concentrations found in the plasma and brain when compared to the concentrations found after administration of CBZ-SL. The two-tailed paired t-test also showed statistically significant differences (P < 0.05) between the CBZ-NPs and the CBZ-SL at each time point. It is believed that drug uptake into brain from the nasal mucosa is via two different pathways. One is the systemic pathway, where some of the drug is absorbed into the systemic circulation and subsequently reaches the brain by crossing the BBB. The other is the olfactory pathway, where drug can travel from the olfactory region in the nasal cavity directly into cerebrospinal fluid (CSF) and brain tissue [32]. The brain concentrations of CBZ were higher than the plasma concentrations after nasal administration using CBZ-NPs demonstrated the existence of a direct pathway from the nasal olfactory area into the brain.
Fig. 8.
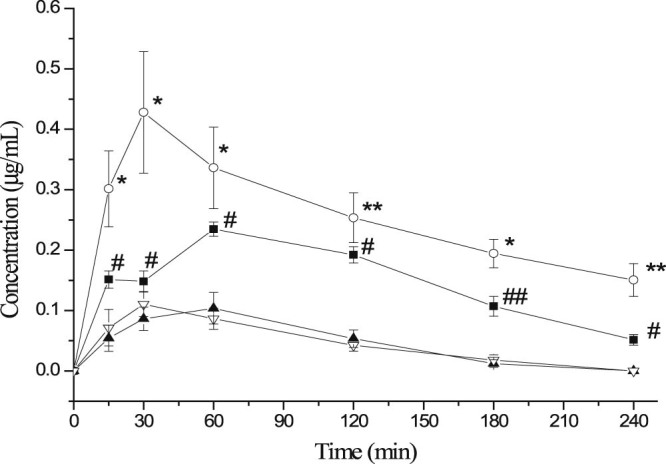
The mean concentration of CBZ (µg/ml) in mice plasma and brain after intranasal administration at dose of 2 mg/kg of CBZ-NPs (—■— plasma, —○—brain, n = 3 ± SD) and CBZ-SL (—▲—Plasma, —▽—Brain, n = 3 ± SD). Paired t-test: (* and #) significantly different from CBZ-SL, P < 0.05, (** and ##) very significantly different from CBZ-SL, P < 0.01.
The pharmacokinetic parameters of CBZ after intranasal administration of CBZ-NPs and CBZ-SL were shown in Table 4. The plasma AUC0–∞ after intranasal administration using CBZ-NPs was about 2.6 times higher than that using CBZ-SL (36.28 versus 13.88 min·µg/ml); and the brain AUC0–∞ was more than 8.1 times higher than that using CBZ-SL (93.01 versus 7.51 min·µg/ml). The low bioavailability of CBZ-SL in plasma and brain could be due to the low water solubility of the drug that determined by both a low dissolution rate and probably the formation of a saturated drug solution on the surface of mucosa.
Table 4.
Pharmacokinetic parameters for CBZ-NPs and CBZ-SL after intranasal administration (n = 3).
| Parameters | CBZ-NPs |
CBZ-SL |
||
|---|---|---|---|---|
| Plasma | Brain tissue | Plasma | Brain tissue | |
| Tmax(min) | 50.2 ± 11.48 | 41.4 ± 13.32 | 32.1 ± 4.64 | 36.0 ± 8.12 |
| Cmax(µg/ml) | 0.23 ± 0.10 | 0.50 ± 0.13 | 0.15 ± 0.02 | 0.11 ± 0.04 |
| AUC0–∞(min·µg/ml) | 36.28 ± 3.17 | 93.01 ± 2.35 | 13.88 ± 1.07 | 7.51 ± 0.65 |
| MRT(min) | 89.4 ± 10.1 | 138.6 ± 11.5 | 41.4 ± 9.1 | 53.4 ± 4.4 |
A considerably high CBZ concentration in the brain was reached 41.4 min after administration of CBZ-NPs (Cmax about 0.50 µg/ml; Tmax 41.4 min) compared to the one in the plasma (Cmax about 0.23 µg/ml; Tmax 50.2 min). CBZ concentrations in the brain were always remained significantly higher (P < 0.05) compared to the one in plasma during 240 min. These results demonstrated that by using the mucoadhesive material in the formulation, the contact time of the formulation in the nasal mucosa was increased which results in a better brain targeting characteristic. Significantly higher AUC0–∞ and Cmax in brain samples for CBZ-NPs compared to CBZ-SL were attributed to the addition of CMC in the formulation [33]. These findings were in good agreement with those reported previously [[29], [30]]. The results of in vivo studies showed that nanoparticulate system based on CMC was able to promote a rapid drug absorption through the nasal mucosa and to remarkably improve the bioavailability of the drug.
In conclusion, a direct transport pathway of CBZ to brain tissue from the nasal cavity has been confirmed. The encapsulation of CBZ in nanoparticles and the nasal route could determine the enhancement of the drug bioavailability and brain targeting characteristics: the high CBZ absorption through nasal mucosa was due to the administration of drug loaded carboxymethyl chitosan nanoparticles, which could adhere on the mucosal surface thus improve the dissolution of the drug in the aqueous environment of the mucosa. It has been demonstrated that the bioadhesive nanoparticles have a significant effect on the mucosal uptake of drugs [34].
4. Conclusion
In this study, a carboxymethyl chitosan nanoparticle of CBZ (CBZ-NPs) was developed and the formulation was administered intranasally with the objective to enhance its delivery to the brain. The CBZ brain to plasma exposure in AUC was 150% when the CBZ-NPs were administered intranasally. This showed that CBZ was preferentially delivered to the brain when the CBZ-NPs were administered intranasally. The results have demonstrated that it was possible to prepare a nasal formulation of CBZ which resulted in a fast and pronounced absorption, with a potential for clinical application in acute situations. The result is promising, though the delivery of CBZ through the intranasal route is limited by the volume / amount of drug being administered.
Many AEDs are potent drugs but unfortunately these substances are often associated with unwanted side effects. If it was possible to administer CBZ nasally, dose and side-effects could be reduced. This pathway could prove to be a useful route for AEDs which do not normally pass the blood–brain barrier in sufficient amounts. In summary, a direct transport pathway of CBZ to brain tissue from the nasal cavity has been confirmed. Compared with the conventional oral and intravenous drug formulations, intranasal drug formulation could provide more direct delivery of AEDs to the brain, leading to higher treatment efficacy.
Conflicts of interest
The authors declare that there are no conflicts of interest.
Acknowledgement
The study was supported by the Academic Research Fund, Faculty of Science, National University of Singapore, R148-000-180-112.
Footnotes
Peer review under responsibility of Shenyang Pharmaceutical University.
References
- 1.Fisher R.S., Van Emde Boas W., Blume W. Epileptic seizures and epilepsy: definitions by the International Leqgue Against Epilepsy (ILAE) and the International Bureau for Epilepsy (IBE) Epilepsia. 2005;46:470–472. doi: 10.1111/j.0013-9580.2005.66104.x. [DOI] [PubMed] [Google Scholar]
- 2.Bromfield E.B., Cavazos J.E., Sirven J.I. American Epilepsy Society; West Hartford (CT): 2006. An introduction to epilepsy. [PubMed] [Google Scholar]
- 3.Epilepsy fact sheets. 2017. http://www.who.int/mediacentre/factsheets/fs999/en/ World Health Organization; Available from:
- 4.Daneman R., Zhou L., Agalliu D. The mouse blood-brain barrier transcriptome: a new resource for understanding the development and function of brain endothelial cells. PLoS ONE. 2010;5:e13741. doi: 10.1371/journal.pone.0013741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Löscher W., Klitgaard H., Twyman R.E. New avenues for anti-epileptic drug discovery and development. Nat Rev Drug Discov. 2013;12:757–776. doi: 10.1038/nrd4126. [DOI] [PubMed] [Google Scholar]
- 6.Wu W., He Q.G., Jiang C.Z. Magnetic iron oxide nanoparticles: synthesis and surface functionalization strategies. Nanoscale Res Lett. 2008;3:397–415. doi: 10.1007/s11671-008-9174-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kreuter J. Nanoparticulate systems for brain delivery of drugs. Adv Drug Deliv Rev. 2001;47:65–81. doi: 10.1016/s0169-409x(00)00122-8. [DOI] [PubMed] [Google Scholar]
- 8.Lockman P.R., Mumper R.J., Khan M.A. Nanoparticle technology for drug delivery ascross the blood brain barrier. Drug Dev Ind Pharm. 2002;28:1–13. doi: 10.1081/ddc-120001481. [DOI] [PubMed] [Google Scholar]
- 9.Costantino H.R., Illum L., Brandt G. Intranasal delivery: physicochemical and therapeutic aspects. Int J Pharm. 2007;337:1–24. doi: 10.1016/j.ijpharm.2007.03.025. [DOI] [PubMed] [Google Scholar]
- 10.Sarin H. Recent progress towards development of effective systemic chemotherapy for the treatment of malignant brain tumors. J Transl Med. 2009;7:77. doi: 10.1186/1479-5876-7-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Arumugam K., Subramanian G.S., Mallayasamy S.R. A study of rivastigmine liposomes for delivery into the brain through intranasal route. Acta Pharm. 2008;58:287–297. doi: 10.2478/v10007-008-0014-3. [DOI] [PubMed] [Google Scholar]
- 12.Migliore M.M., Vyas T.K., Campbell R.B. Brain delivery of proteins by the intranasal route of administration: a comparison of cationic liposomes versus aqueous solution formulations. J Pharm Sci. 2009;99(4):1745–1761. doi: 10.1002/jps.21939. [DOI] [PubMed] [Google Scholar]
- 13.Serralheiroa A., Alvesb G., Fortuna A. Intranasal administration of carbamazepine to mice: a direct delivery pathway for brain targeting. Eur J Pharm Sci. 2014;60:32–39. doi: 10.1016/j.ejps.2014.04.019. [DOI] [PubMed] [Google Scholar]
- 14.Jain A.K., Khar R.K., Ahmed F.J. Effective insulin delivery using starch nanoparticles as a potential trans-nasal mucoadhesive carrier. Eur J Pharmaceut Biopharmaceut. 2008;69:426–435. doi: 10.1016/j.ejpb.2007.12.001. [DOI] [PubMed] [Google Scholar]
- 15.Jiang L.M., Gao L., Wang X.Q. The application of mucoadhesive polymers in nasal drug delivery. Drug Dev Ind Pharm. 2010;36:323–336. doi: 10.1080/03639040903170750. [DOI] [PubMed] [Google Scholar]
- 16.Aranaz I., Mengibar M., Harris R. Functional characterization of chitin and chitosan. Curr Chem Biol. 2009;3:203–230. [Google Scholar]
- 17.Alonso M.J., Sanchéz A. The potential of chitosan in ocular drug delivery. J Pharm Pharmacol. 2003;55:1451–1463. doi: 10.1211/0022357022476. [DOI] [PubMed] [Google Scholar]
- 18.Tang H., Zhang P., Kieft T.L. Antibacterial action of a novel functionalized chitosan-arginine against gram-negative bacteria. Acta Biomater. 2010;6:2562–2571. doi: 10.1016/j.actbio.2010.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mourya V.K., Inamdara N.N., Tiwari A. Carboxymethyl chitosan and its applications. Adv Mat Lett. 2010;1:11–33. [Google Scholar]
- 20.Kramlinger K.G., Phillips K.A., Post R.M. Rash complicating carbamazepine treatment. J Clin Psychopharmacol. 1994;14:408–413. [PubMed] [Google Scholar]
- 21.Granger P., Biton B., Faure C. Modulation of the gamma-aminobutyric acid type-A receptor by the antiepileptic drugs cambamazepine and phenytoin. Mol Pharmacol. 1995;47:1189–1196. [PubMed] [Google Scholar]
- 22.Tolou-Ghamari Z., Zare M., Habibabadi J.M. A quick review of carbamazepine pharmacokinetics in epilepsy from 1953 to 2012. J Res Med Sci. 2013;18:S18–S85. [PMC free article] [PubMed] [Google Scholar]
- 23.Zhong C., Zhao J., Huang M. Preparation of N, O-carboxymethyl chitosan by the approach of two-step alkalization-reaction conditions influencing the degree of substitution. Fine Chem. 2004;21:338–341. [Google Scholar]
- 24.Vaghania S.S., Patelb M.M., Satish C.S. Synthesis and characterization of pH-sensitive hydrogel composed of carboxymethyl chitosan for colon targeted deivery of ornidazole. Carbohydr Res. 2012;347:76–82. doi: 10.1016/j.carres.2011.04.048. [DOI] [PubMed] [Google Scholar]
- 25.Gavini E., Hegge A.B., Rassu G. Nasal administration of Carbamazepine using chitosan microspheres: in vitro/in vivo studies. Int J Pharm. 2006;307:9–15. doi: 10.1016/j.ijpharm.2005.09.013. [DOI] [PubMed] [Google Scholar]
- 26.Nair R., Kumar A.C.K., Priya V.K. Formulation and evaluation of chitosan solid lipid nanoparticles of carbamazepine. Lipids Health Dis. 2012;13:72. doi: 10.1186/1476-511X-11-72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Barakat N.S., Omar S.A., Ahmed A.A.E. Carbamazepine uptake into rat brain following intra-olfactory transport. J Pharm Pharmacol. 2006;58:63–72. doi: 10.1211/jpp.58.1.0008. [DOI] [PubMed] [Google Scholar]
- 28.Nishimura A., Honda N., Sugioka N. Evaluation of carbamazepine pharmacokinetic Profiles in mice with kainic acid-induced acute seizures. Biol Pharm Bull. 2008;31:2302–2308. doi: 10.1248/bpb.31.2302. [DOI] [PubMed] [Google Scholar]
- 29.Jogani V.V., Shah P.J., Mishra P. Intranasal mucoadhesive microemulsion of tacrine to improve brain targeting. Alzheimer Dis Assoc Disord. 2008;22:116–124. doi: 10.1097/WAD.0b013e318157205b. [DOI] [PubMed] [Google Scholar]
- 30.Patel R.B., Patel M.R., Bhatt K.K. Microemulsion-based drug delivery system for transnasal delivery of Carbamazepine: preliminary brain-targeting study. Drug Deliv. 2015;21:1–7. doi: 10.3109/10717544.2014.908980. [DOI] [PubMed] [Google Scholar]
- 31.Siepmann J., Siepmann F. Mathematical modeling of drug delivery. Int J Pharm. 2008;364:328–343. doi: 10.1016/j.ijpharm.2008.09.004. [DOI] [PubMed] [Google Scholar]
- 32.Illum L. Transport of drugs from the nasal cavity to the central nervous system. Eur J Pharm Sci. 2000;11:1–18. doi: 10.1016/s0928-0987(00)00087-7. [DOI] [PubMed] [Google Scholar]
- 33.Van der Lubben I.M., Verhoef J.C., Borchard G. Chitosan and its derivatives in mucosal drug and vaccine delivery. Eur J Pharm Sci. 2001;14(3):201–207. doi: 10.1016/s0928-0987(01)00172-5. [DOI] [PubMed] [Google Scholar]
- 34.Alpar H.O., Somavarapu S., Atuah K.N. Biodegradable mucoadhesive particulates for nasal and pulmonary antigen and DNA delivery. Adv Drug Deliv Rev. 2005;57:411–430. doi: 10.1016/j.addr.2004.09.004. [DOI] [PubMed] [Google Scholar]