

Abstract
For efficient mucosal vaccine delivery, nanoparticulate antigens are better taken by microfold cells in the nasal associated lymphoid tissue and also dendritic cells. Nanoparticles based on polymers such as chitosan (CHT) and its water soluble derivative, trimethylchitosan (TMC), could be successfully used as carrier/adjuvant for this purpose. Sodium alginate, a negatively charged biopolymer, could modify the immunostimulatory properties of CHT and TMC NPs and increase their stability. Sodium alginate (ALG)-coated chitosan (CHT) and trimethylchitosan (TMC) nanoparticles (NPs) loaded with inactivated PR8 influenza virus were successfully prepared by direct coating of the virus with CHT or TMC polymers to evaluate their immunoadjuvant potential after nasal immunization. After nasal immunizations in BALB/c mice, PR8-CHT formulation elicited higher IgG2a and IgG1 antibody titers compared with PR8-TMC. ALG coating of this formulation (PR8-CHT-ALG) significantly decreased the antibody titers and a less immune response was induced than PR8-TMC-ALG formulation. PR8-TMC-ALG formulation showed significantly higher IgG2a/IgG1 ratio, as criteria for Th1-type immune response, compared with PR8-CHT-ALG and PR8 virus alone. Altogether, the PR8-TMC-ALG formulation could be considered as an efficient intranasal antigen delivery system for nasal vaccines.
Keywords: Chitosan, Trimethyl chitosan, Alginate, PR8 influenza virus, Nasal immunization
Graphical abstract
1. Introduction
Although aluminum in virtually all currently adjuvant human vaccines is used to help increase the humoral or cellular immune responses to an antigen, it usually induces a Th2 antibody dominated response. Accordingly, aluminum-based vaccines are not suitable for intracellular pathogens, chronic infections or cancers. Therefore, enormous efforts have been done to develop new adjuvants that are able to induce both humoral and cellular immune responses.
Intramuscular (Im) or subcutaneous (SC) are the most commonly administration routes of currently available vaccines that elicit robust and strong systemic immune responses. Since, these vaccines create pain upon injection and are not able to elicit mucosal immune responses, a desirable non-invasive immunization is necessary [1], [2], [3], [4], [5], [6], [7]. Among the non-invasive routes, nasal administration is of especial interest due to induction of mucosal immunity [8], [9]. Unfortunately, there are some drawbacks associated with nasal administration that should be considered in order to elicit an acceptable mucosal and systemic response. For example, the mucociliary clearance, the tolerogenic nature of mucosal epitheliums and the large size of antigen are some vital factors that lead to the decrease of the residence time of antigen and its uptake through nasal epithelium and eventually complicate a robust immune response. A common way to tackle these problems is to incorporate antigens into the mucoadhesive polymeric NPs [10]. This strategy prolongs the residence time of antigens and also protects against enzymes [11]. Additionally, nanoparticulated antigens are more taken by dendritic cells (DCs) and could cross the epithelial barrier more easily through a famous kind of cells which are called microfold cells in the nasal associated lymphoid tissue (NALT) [12], [13].
Chitosan (CHT) is a biodegradable, biocompatible and safe cationic mucoadhesive polymer that is made by treating the chitin shells of shrimp which numerous studies have shown its potential as an enhancer polymer for nasal vaccine delivery across mucosal surfaces [14], [15], [16], [17]. CHT can increase the permeability of the mucosal barrier possibly through disruption of intracellular tight junctions. Since, CHT has a limited aqueous solubility at alkaline or neutral pH, a derivative of CHT, N, N, N-trimethyl chitosan (TMC) has been developed as a mucoadhesive polymer for nasal vaccination to overcome the problem, which has an improved solubility over a wide range of pH [18], [19]. Sodium alginate (ALG) is also a negatively charged polymer that could be simply coated around the positively charged CHT or TMC [20]. ALG could modify the immunostimulatory properties of NPs, increase their stability [21], [22] as well as prevent a burst release of loaded antigens [23].
Human influenza A/Puerto Rico/8//1934 (H1N1) virus (PR8) was the most common cause of human influenza at 2009. That is the subtype of influenza A virus with a negative charge and could be easily associated with positively charged polymers such as CHT or TMC [24], [25].
In this study, we investigate whether PR8-loaded TMC or CHT NPs, with or without ALG coat, are suitable antigen carrier systems to enhance in vivo transmucosal antigen delivery.
2. Materials and methods
2.1. Materials
IgG2a and IgG1 secondary antibodies were obtained from Zymed Inc. (USA). Coating and detection mAb antibodies for IFN-γ and IL-4 as well as streptavidin-HRP were obtained from Mabtech (Sweden). Concanavalin A and ALG (low molecular weight) were purchased from Sigma Alderich (USA). RPMI1640 culture medium, penicillin–streptomycin solution and fetal calf serum (FCS) were purchased from Sigma Aldrich (USA). CHT was purchased from Fluka (USA). TMC was synthesized and characterized from above mentioned chitosan as reported in our previous studies [1].
BALB/c mice and PR8 antigen were obtained from the Pasteur Institute of Iran. Experiments were performed in accordance with the guidelines and regulations for the care and use of animals implemented by the ethics committee of Mashhad University of Medical Sciences (Approval number: IR.MUMS.REC.1392.23). Also the animal studies were performed based on the European Community guidelines as accepted principles for the use of experimental animals.
2.2. Preparation of PR8-CHT-ALG and PR8-TMC-ALG NPs
NPs were prepared with different weight ratio of various components including PR8 antigen, CHT or TMC polymers and ALG coating for finding the best results including the smallest particle size and polydispersity index (PDI) as well as the highest surface charge (zeta potential). The 1:4:6 (w/w/w) ratios of the PR8:CHT/TMC:ALG showed the best mentioned physicochemical properties and used for further studies. First, the PR8-CHT and PR8-TMC NPs were prepared by adding PR8 solution to CHT or TMC polymers (dispersed in phosphate buffer (PB), pH 6) at the ratio of 1:4 (w/w) and gently vortex-mixed for about 5 s [1], [9], [26]. Next, the formulations containing (1:4)/6 (w/w) ratios of PR8-CHT (or TMC)/ALG were also prepared. All of the obtained formulations were prepared in PB and stored at 2–8 °C. The final PR8 concentration in the formulations was set as 15 µg/dose/mouse for in vivo administration.
2.3. Characterization of NPs
Dynamic light scattering analysis (NANO-Zetasizer, Malvern, UK) was used to determine the zeta potential, mean particle size and PDI of NPs. Also, to evaluate the stability of NPs, each 5 d, the zeta potential, mean particle size and PDI of different formulations (in PB buffer, pH 6) were evaluated at 4 °C for 30 d
2.4. In vivo vaccination protocol
The in vivo evaluation of immunoadjuvant potential of prepared NPs was investigated in female BALB/c mice. Seven groups were immunized intranasally (In) with: 1. PBS solution as a negative control, 2 and 3. PR8 antigen (15 µg/mouse, given intramuscularly (Im) or intranasally, 4 and 5. PR8-CHT and PR8-TMC NPs (15 µg PR8 antigen + 64 µg CHT or TMC/mouse) and 6 and 7. PR8-CHT-ALG and PR8-TMC-ALG NPs (15 µg PR8 antigen + 64 µg CHT or TMC + 96 µg ALG/mouse). Six mice in each group were injected three times in 2-week intervals with these NPs. For nasal immunization, an intraperitoneal injection of xylazine and ketamin (10 and 100 µg/g body weight, respectively) were used to anesthetize the mice. Finally, a total volume of 5 µl of each formulation was administered into the two separated nostrils [1]. For Im immunization, a total volume of 100 µl of each formulation was administered.
2.5. Antibody isotype assay
Ten days after the last booster injection, the mice blood samples were obtained by heart puncture and retro-orbital bleeding. The blood was allowed to coagulate at 4 °C and then the serum was collected by centrifugation for 10 min at 14 000 rpm. The serum samples were kept at −20 °C [27]. The sera of vaccinated BALB/c mice were used to titrate both of IgG2a and IgG1 antibodies using ELISA technique [28]. Briefly, 96-well plates were coated with 0.5 µg/50 µl PR8 antigen in bicarbonate buffer (pH 9.6) and incubated overnight at 4 °C. After washing the plates, they were blocked with 300 µl of 2.5% BSA in PBS-tween per well for 1 h at 37 °C. Different dilutions of serum were added to the plates for 75 min at 37 °C. After washing with PBS–tween solution, plates were treated with IgG1 and IgG2a secondary antibodies based on the manufacturer's instructions (Zymed Inc., USA). Optical density was measured using a microplate reader (StatFax® 4200 microplate reader, NEOGEN® Corporation, USA) at 450 nm and a reference wavelength of 630 nm.
2.6. Statistical analysis
GraphPad Prism version 6 was used to perform the statistical analysis. Also, two-way analysis of variance (ANOVA) and Tukey's multiple comparison test were used to analyze the obtained data. Data were showed as mean ± standard deviation (SD).
3. Results and discussion
3.1. Characterization of NPs
In the present study, NPs were prepared by a simple incubation method in which the different components were gently mixed to each other [1]. As a result, the coating of NPs with ALG significantly increased the particle size to more than 100 nm that suggested the presence of an ALG coating layer. Also, ALG has a negative charge, thus resulting in significant decrease in zeta potential for the ALG-coated NPs as compared with non-coated NPs. The characteristic features of obtained NPs were summarized in Table 1. In our previous study, The TMC and CHT NPs loaded with hepatitis B surface antigen (HB) showed more positively charged of 14.6 and 13.9 mV, respectively [1].
Table 1.
Zeta potential, particle size, PDI of NPs.
| Formulations | Ratio | Z-average mean | Intensity mean | Volume mean | Number mean | PDI | Zeta potentials |
|---|---|---|---|---|---|---|---|
| diameter (nm) | diameter (nm) | diameter (nm) | diameter (nm) | (mV) | |||
| PR8 antigen | – | 284.3 | 284.3 | 284.3 | 284.3 | 0.41 | −9.3 |
| PR8-CHT | 1:4 | 380.6 | 633.9 | 977.2 | 234.7 | 0.43 | + 5.9 |
| PR8-TMC | 1:4 | 318.2 | 607.9 | 1205.8 | 194.8 | 0.39 | + 7.0 |
| PR8-CHT-ALG | 1:4:6 | 471.1 | 439.8 | 371.9 | 239.5 | 0.66 | −32.8 |
| PR8-TMC-ALG | 1:4:6 | 453.2 | 247.2 | 248.5 | 268.9 | 0.56 | −29.6 |
Additionally, Fig. 1 shows that these NPs have great stability and no significant differences were found in particle size, PDI as well as zeta potential during the period of 30 d [20]. However, the negative results of PR8-CHT-ALG formulation could be attributed to the agglomeration of these NPs during the preparation process. Totally, the prepared NPs with this simple and scalable method showed suitable physicochemical properties, stability and could be used in vivo for their immunoadjuvant potential.
Fig. 1.

Stability of prepared NPs in PB buffer (pH 6). The particle size (A), PDI (B) as well as zeta potential (C) of NPs were measured, each 5 d, during the period of 30 d in 4 °C. Data represented as mean ± SD (n = 3).
3.2. Antibody response
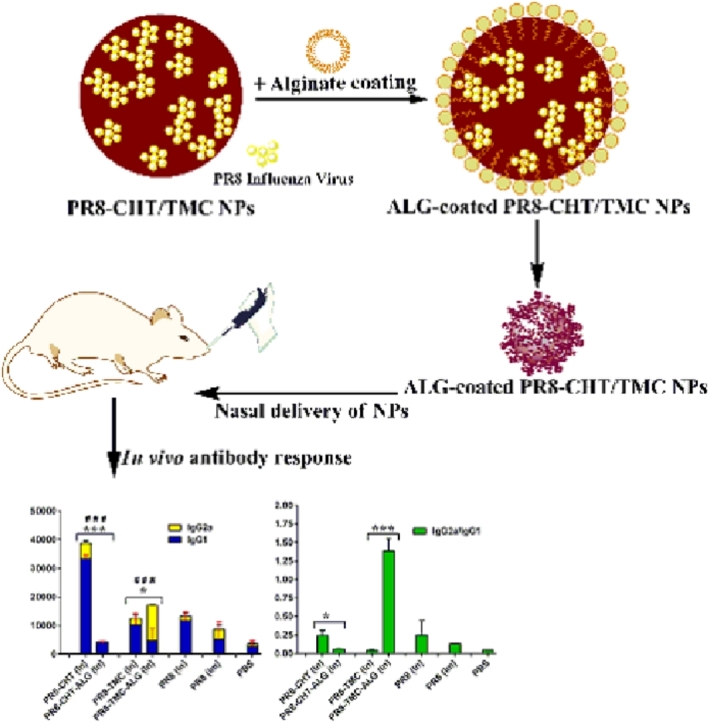
The adjuvant potential of prepared NPs was investigated by the immunization of BALB/c mice [1]. Animals were immunized with different formulations, three times with two weeks intervals. 10 d after the last immunizations, the sera from the freshly killed mice were collected. The sera of vaccinated BALB/c mice were used to titrate IgG1 and IgG2a antibodies using ELISA technique. In this study, Fig. 2A and B shows that CHT or TMC-administrated PR8 virus, as efficient immunoadjuvants, could significantly increase immunological protection against PR8 whole virus after In administration. In our previous study, TMC and CHT NPs loaded with hepatitis B surface antigen (HB) showed similar potential as mucosal immunoadjuvants for HB [1]. Xie et al. showed that H pylori-CHT particles managed to create an significantly higher immunological protection in 60% mice compared with H pylori antigen alone (P < 0.05).
Fig. 2.

The level of anti-PR8 specific IgG1 and IgG2a (A); and the ratio of IgG2a/IgG1 antibody titers (B) of BALB/c mice immunized by In and Im routes, 10d after the last booster injection with different formulations. The assays were performed using an ELISA method in triplicate at different dilutions of serum samples. Significant differences between the IgG1 titers of ALG-coated NPs (In) and none-coated ones was marked as *(P < 0.05) and ***(P < 0.001). Significant difference of IgG2a titers between ALG-coated NPs (In) and none-coated ones was labeled with ###(P < 0.001). Data represented as mean ± SD (n = 6).
Additionally, the amounts of IgG2a and IgG1 secretions are usually considered in favor of Th1 cellular immune response and Th2 humoral immune response, respectively [29]. Our results showed that the prepared NPs could create a mixed Th1/Th2 response [30]. For example, Fig. 1A shows that significant higher IgG1 (P < 0.001) and IgG2a (P < 0.01) antibody titers in sera of mice immunized with PR8-CHT NPs might be considered as an indicator of a mixed Th1/Th2 profile. In contrast, the ALG-coated PR8-TMC NPs (PR8-TMC-ALG) generated a significantly higher IgG2a antibody titer than the PR8-CHT-ALG ones (P < 0.001) that indicate a Th2 profile for these NPs. According to our results, the ALG coating could increase the IgG2a antibody titer, however, the negative results on PR8-CHT-ALG NPs (P < 0.001) could be attributed to the agglomeration of this NP during the preparation process and subsequently lower uptake of this formulation by M cells and APCs. Ajdary et al. showed that the bacille calmette-guerin (BCG) encapsulated in ALG microspheres induced equal or better Th1 immune responses than standard BCG vaccination by oral administration [31].
It also has been shown that influenza antigens are potent inducers of IgG2a antibody responses in mice and results in a Th1-type immune response that is generally considered as a critical factor for producing a variety of prophylactic and therapeutic vaccines, especially those are to prevent or treat viral infections and intracellular pathogens [32], [33]. Therefore, it is beneficial to elicit a higher ratio of IgG2a/IgG1 antibody titer for immunization against influenza virus. The ratio of IgG2a/IgG1 antibody titer is usually considered as an indicator of Th profile [34]. When the IgG2a/IgG1 ratio is higher, it is in favor of Th1 immune response [35]. Fig. 2B shows that the IgG2a/IgG1 ratio was significantly higher in intranasally administered PR8-TMC-ALG NPs than those induced with other formulations (P < 0.001). This notable achievement showed that TMC-ALG adjuvanted PR8 vaccines are potent inducer of Th1 immune responses after In delivery.
4. Conclusion
It is concluded that the CHT and TMC NPs are efficient immunoadjuvants for immunization against PR8 whole influenza virus. This effect was proved after immunization by the In route. Generally, the PR8-TMC NPs induced less immune responses compared with PR8-CHT NPs after In administration. However, the ALG-coated NPs could generate superior immune response to the non-coated ones, especially in the PR8-TMC-ALG NPs.
Conflicts of interest
The authors declare that there is no conflicts of interest.
Acknowledgments
The data presented are part of Amir-Hossein Sabbaghi Pharm.D. thesis (Grant number: 911042) supported by Vice Chancellor for Research, Mashhad University of Medical Sciences.
References
- 1.Tafaghodi M, Saluja V, Kersten GF. Hepatitis B surface antigen nanoparticles coated with chitosan and trimethyl chitosan: impact of formulation on physicochemical and immunological characteristics. Vaccine. 2012;30:5341–5348. doi: 10.1016/j.vaccine.2012.06.035. [DOI] [PubMed] [Google Scholar]
- 2.Amorij JP, Hinrichs WL, Frijlink HW, Wilschut JC, Huckriede A. Needle-free influenza vaccination. Lancet Infect Dis. 2010;10:699–711. doi: 10.1016/S1473-3099(10)70157-2. [DOI] [PubMed] [Google Scholar]
- 3.Saluja V, Amorij JP, van Roosmalen ML. Intranasal delivery of influenza subunit vaccine formulated with GEM particles as an adjuvant. AAPS J. 2010;12:109–116. doi: 10.1208/s12248-009-9168-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tafaghodi M, Rastegar S. Preparation and in vivo study of dry powder microspheres for nasal immunization. J Drug Target. 2010;18:235–242. doi: 10.3109/10611860903434035. [DOI] [PubMed] [Google Scholar]
- 5.Amin M, Jaafari MR, Tafaghodi M. Impact of chitosan coating of anionic liposomes on clearance rate, mucosal and systemic immune responses following nasal administration in rabbits. Coll Surf B Biointerfaces. 2009;74:225–229. doi: 10.1016/j.colsurfb.2009.07.024. [DOI] [PubMed] [Google Scholar]
- 6.Hirschberg HJ, van de Wijdeven GG, Kraan H, Amorij JP, Kersten GF. Bioneedles as alternative delivery system for hepatitis B vaccine. J Control Release. 2010;147:211–217. doi: 10.1016/j.jconrel.2010.06.028. [DOI] [PubMed] [Google Scholar]
- 7.Tafaghodi M, Jaafari M, Sajadi Tabassi S. Nasal immunization studies by cationic, fusogenic and cationic-fusogenic liposomes encapsulated with tetanus toxoid. Curr Drug Deliv. 2008;5:108–113. doi: 10.2174/156720108783954833. [DOI] [PubMed] [Google Scholar]
- 8.Slütter B, Hagenaars N, Jiskoot W. Rational design of nasal vaccines. J Drug Target. 2008;16:1–17. doi: 10.1080/10611860701637966. [DOI] [PubMed] [Google Scholar]
- 9.Hagenaars N, Mastrobattista E, Verheul RJ. Physicochemical and immunological characterization of N, N, N-trimethyl chitosan-coated whole inactivated influenza virus vaccine for intranasal administration. Pharm Res. 2009;26:1353–1364. doi: 10.1007/s11095-009-9845-y. [DOI] [PubMed] [Google Scholar]
- 10.Köping-Höggård M, Sánchez A, Alonso MJ. Nanoparticles as carriers for nasal vaccine delivery. Expert Rev Vaccines. 2005;4:185–196. doi: 10.1586/14760584.4.2.185. [DOI] [PubMed] [Google Scholar]
- 11.Almeida A, Alpar H. Antigen delivery systems: immunological and technological issues. Harwood Academic Publishers; Amsterdam: 1997. Mucosal immunisation with antigen-containing microparticles; pp. 207–226. [Google Scholar]
- 12.Verheul RJ, Amidi M, van der Wal S. Synthesis, characterization and in vitro biological properties of O-methyl free N, N, N-trimethylated chitosan. Biomaterials. 2008;29:3642–3649. doi: 10.1016/j.biomaterials.2008.05.026. [DOI] [PubMed] [Google Scholar]
- 13.Mangal S, Pawar D, Garg NK. Pharmaceutical and immunological evaluation of mucoadhesive nanoparticles based delivery system(s) administered intranasally. Vaccine. 2011;29:4953–4962. doi: 10.1016/j.vaccine.2011.04.112. [DOI] [PubMed] [Google Scholar]
- 14.Günbeyaz M, Faraji A, Özkul A, Puralı N, Şenel S. Chitosan based delivery systems for mucosal immunization against bovine herpesvirus 1 (BHV-1) Eur J Pharm Sci. 2010;41:531–545. doi: 10.1016/j.ejps.2010.08.011. [DOI] [PubMed] [Google Scholar]
- 15.Illum L, Jabbal-Gill I, Hinchcliffe M, Fisher A, Davis S. Chitosan as a novel nasal delivery system for vaccines. Adv Drug Deliv Rev. 2001;51:81–96. doi: 10.1016/s0169-409x(01)00171-5. [DOI] [PubMed] [Google Scholar]
- 16.van der Lubben IM, Kersten G, Fretz MM. Chitosan microparticles for mucosal vaccination against diphtheria: oral and nasal efficacy studies in mice. Vaccine. 2003;21:1400–1408. doi: 10.1016/s0264-410x(02)00686-2. [DOI] [PubMed] [Google Scholar]
- 17.Dodane V, Khan MA, Merwin JR. Effect of chitosan on epithelial permeability and structure. Int J Pharm. 1999;182:21–32. doi: 10.1016/s0378-5173(99)00030-7. [DOI] [PubMed] [Google Scholar]
- 18.Amidi M, Mastrobattista E, Jiskoot W, Hennink WE. Chitosan-based delivery systems for protein therapeutics and antigens. Adv Drug Deliv Rev. 2010;62:59–82. doi: 10.1016/j.addr.2009.11.009. [DOI] [PubMed] [Google Scholar]
- 19.Boonyo W, Junginger HE, Waranuch N, Polnok A, Pitaksuteepong T. Chitosan and trimethyl chitosan chloride (TMC) as adjuvants for inducing immune responses to ovalbumin in mice following nasal administration. J Control Release. 2007;121:168–175. doi: 10.1016/j.jconrel.2007.05.025. [DOI] [PubMed] [Google Scholar]
- 20.Tafaghodi M, Kersten G, Jiskoot W. Nano-adjuvanted polio vaccine: Preparation and characterization of chitosan and trimethylchitosan (TMC) nanoparticles loaded with inactivated polio virus and coated with sodium alginate. Nanomed J. 2014;1:220–228. [Google Scholar]
- 21.Démoulins T, Bassi I, Thomann-Harwood L. Alginate-coated chitosan nanogel capacity to modulate the effect of TLR ligands on blood dendritic cells. Nanomed Nanotechnol Biol Med. 2013;9:806–817. doi: 10.1016/j.nano.2013.01.002. [DOI] [PubMed] [Google Scholar]
- 22.Oliveira CR, Rezende CM, Silva MR. A new strategy based on SmRho protein loaded chitosan nanoparticles as a candidate oral vaccine against schistosomiasis. PLoS Negl Trop Dis. 2012;6:e1894. doi: 10.1371/journal.pntd.0001894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Borges O, Borchard G, Verhoef JC, de Sousa A, Junginger HE. Preparation of coated nanoparticles for a new mucosal vaccine delivery system. Int J Pharm. 2005;299:155–166. doi: 10.1016/j.ijpharm.2005.04.037. [DOI] [PubMed] [Google Scholar]
- 24.Johnson A, Chen L-M, Winne E. Identification of influenza A/PR/8/34 donor viruses imparting high hemagglutinin yields to candidate vaccine viruses in eggs. PLoS One. 2015;10 doi: 10.1371/journal.pone.0128982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu H, Bungener L, ter Veer W. Preclinical evaluation of the saponin derivative GPI-0100 as an immunostimulating and dose-sparing adjuvant for pandemic influenza vaccines. Vaccine. 2011;29:2037–2043. doi: 10.1016/j.vaccine.2011.01.012. [DOI] [PubMed] [Google Scholar]
- 26.Hagenaars N, Mania M, de Jong P. Role of trimethylated chitosan (TMC) in nasal residence time, local distribution and toxicity of an intranasal influenza vaccine. J Control Release. 2010;144:17–24. doi: 10.1016/j.jconrel.2010.01.027. [DOI] [PubMed] [Google Scholar]
- 27.Mosafer J, Teymouri M, Abnous K, Tafaghodi M, Ramezani M. Study and evaluation of nucleolin-targeted delivery of magnetic PLGA-PEG nanospheres loaded with doxorubicin to C6 glioma cells compared with low nucleolin-expressing L929 cells. Mater Sci Eng C. 2017;72:123–133. doi: 10.1016/j.msec.2016.11.053. [DOI] [PubMed] [Google Scholar]
- 28.Badiee A, Jaafari MR, Khamesipour A. Leishmania major: immune response in BALB/c mice immunized with stress-inducible protein 1 encapsulated in liposomes. Exp Parasitol. 2007;115:127–134. doi: 10.1016/j.exppara.2006.07.002. [DOI] [PubMed] [Google Scholar]
- 29.Sayın B, Somavarapu S, Li X. Mono-N-carboxymethyl chitosan (MCC) and N-trimethyl chitosan (TMC) nanoparticles for non-invasive vaccine delivery. Int J Pharm. 2008;363:139–148. doi: 10.1016/j.ijpharm.2008.06.029. [DOI] [PubMed] [Google Scholar]
- 30.Chadwick S, Kriegel C, Amiji M. Nanotechnology solutions for mucosal immunization. Adv Drug Deliv Rev. 2010;62:394–407. doi: 10.1016/j.addr.2009.11.012. [DOI] [PubMed] [Google Scholar]
- 31.Ajdary S, Dobakhti F, Taghikhani M. Oral administration of BCG encapsulated in alginate microspheres induces strong Th1 response in BALB/c mice. Vaccine. 2007;25:4595–4601. doi: 10.1016/j.vaccine.2007.03.039. [DOI] [PubMed] [Google Scholar]
- 32.Coulie PG, van Snick J. Enhancement of IgG anti-carrier responses by IgG2 anti-hapten antibodies in mice. Eur J Immunol. 1985;15:793–798. doi: 10.1002/eji.1830150810. [DOI] [PubMed] [Google Scholar]
- 33.Coutelier J-P, Van der Logt J, Heessen F, Warnier G, Van Snick J. IgG2a restriction of murine antibodies elicited by viral infections. J Exp Med. 1987;165:64–69. doi: 10.1084/jem.165.1.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Maruggi G, Chiarot E, Giovani C. Immunogenicity and protective efficacy induced by self-amplifying mRNA vaccines encoding bacterial antigens. Vaccine. 2017;35:361–368. doi: 10.1016/j.vaccine.2016.11.040. [DOI] [PubMed] [Google Scholar]
- 35.Tao W, Fu T, He Z. Evaluation of immunostimulatory effects of N-(2-hydroxy) propyl-3-trimethylammonium chitosan chloride for improving live attenuated hepatitis a virus vaccine efficacy. Viral Immunol. 2017;30:120–126. doi: 10.1089/vim.2016.0099. [DOI] [PubMed] [Google Scholar]