

Highlights
-
•
HPMC was used to inhibit crystallization both in solid dispersions and tablets.
-
•
Fluid-bed technique was employed to realize the scaling-up of solid dispersions.
-
•
Dissolution results became reliable with the usage of discriminatory media.
-
•
The results of the bioavailability showed a higher AUC0–12 h value for fluid-bed tablets, compared to Nimotop™.
Keywords: Solid dispersion, Nimodipine, HPMC E5, Fluid-bed, Bioavailability
Abstract
Solid dispersion (SD) systems have been extensively used to increase the dissolution and bioavailability of poorly water-soluble drugs. To circumvent the limitations of polyvinylpyrrolidone (PVP) dispersions, HPMC E5 was applied in the formulation process and scaling-up techniques, simultaneously. In this study, SD of nimodipine (NMP) and corresponding tablets were prepared through solvent method and fluid bed granulating one step technique, respectively. Discriminatory dissolution media were used to obtain reliable dissolution results. Meanwhile, the stability study of SDs was investigated with storage under high temperature and humidity conditions. Moreover, the solubility of SDs was measured to explore the effect of carriers. The preparations were characterized by DSC, PXRD, and FTIR. Dramatical improvements in the dissolution rate of NMP were achieved by the ingenious combination of the two polymers. Binary NMP/PVP/HPMC-SDs released steadily, while the dissolution of single NMP/PVP-SDs decreased rapidly in water. The fluid-bed tablets (FB-T) possessed a similar dissolution behavior to the commercial Nimotop™ tablets. The characterization patterns implied that NMP existed in an amorphous state in our SDs. Furthermore, the results of stability tests suggested a better stability of the binary SDs. A special cooperative effect of PVP and HPMC was discovered on dissolution characteristics of NMP SDs and tablets, which could be extended to other drugs henceforth. Finally, the bioavailability of FB-T was evaluated in beagle dogs with Nimotop™ as the reference, and the results showed a higher AUC0–12hvalue for FB-T.
Graphic abstract
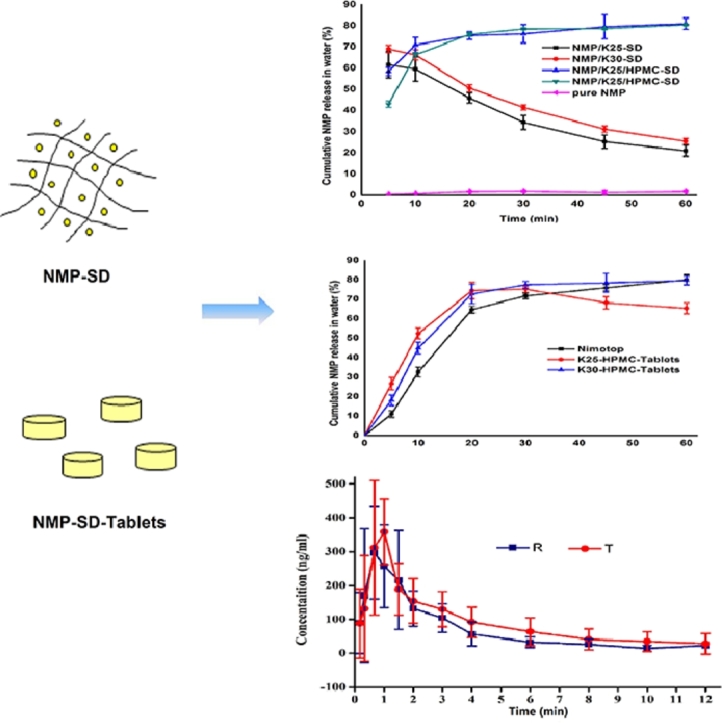
1. Introduction
NMP is known as a dihydropyridine calcium channel blocker applied in the treatment of patients with subarachnoid hemorrhage [1]. As a class II drug in the Biopharmaceutics Classifcation Scheme, NMP has a high permeability, but a poor solubility in water, which leads to a low bioavailability and limited clinical efficacy [2]. For such drugs, drug release is an imperative and limiting step [3]. And improving the release profile of these drugs could make it possible to enhance their bioavailability [4]. Therefore, various advanced approaches such as salt formation, particle size reduction and complexation have been exploited to enhance the dissolution rate [5]. Notably, solid dispersion is considered as a promising strategy to raise dissolution profile of sparingly soluble drugs [6–8].
Most often, amorphous organic polymers such as PVP, polyethylene glycol (PEG) and various cellulose derivatives are capable of forming the amorphous solid solutions [4,9]. Particularly, SD prepared with relative high proportions of PVP tends to perform a higher drug solubility and release rate than those with high proportions of drug [7 10,11]. Meanwhile, the chain length of PVP plays a crucial part in the dissolution rate of the scattered drug from its SD formulation [4,12]. However, PVP exhibits a quick inhibition to the precipitation of the drug from the supersaturated solution, the drug may precipitate out faster than that prepares with HPMC, performing a sharp decrease of the dissolution rate in less than an hour, while the drug with HPMC showed a smooth release for more than an hour [13,14]. Due to its swelling and dissolution properties in aqueous solution, HPMC is of great importance as a water soluble polymer carrier in controlling the release of drug from SD system, which is probably related to the formation of soluble complexes between the water-soluble polymers and insoluble drug [15]. HPMC is commonly used on account of its effects on both crystal growth inhibition and extension of supersaturated conditions [16]. Moreover, Yamashita et al. reported that SD formulations with HPMC have been adopted to improve in vivo drug absorption [17]. Possibly, it is expected that SDs with HPMC could exhibit a better stability.
Unexpectedly, despite all that super advantages, the number of commercial products resulting from SD strategies is relatively low [18]. It is primarily imputed to the manufacturing difficulties, the poor stability problems, and the scale-up problems of the SDs. From a commercial point of view, the most common SD product is tablet, which is the most preferred preparation ascribed to its obvious virtues and high patients compliance [19,20]. Therefore, in order to realize the scale-up of SD, the approach of fluid bed granulation is utilized to formulate NMP tablets, which is an efficient strategy to solve above problems, form solid dispersion, and even achieve the pharmacoeconomics. Also, the granules obtained from this technique exhibit excellent flowability, compressibility and composition uniformity [21–23].
In this article, a series of solid dispersions and tablets with PVPs and HPMC have been developed to select a proper ratio of drug and carrier by the standard of higher drug solubility, release rate and good stability. The first purpose of present work is to investigate if the PVP and HPMC could affect the release rate of NMP with the usage of differentiating media, simultaneously, as Suziki and Sunada reported that both HPMC and PVP exhibited a high compatability with nifedipine [14]. Phosphate buffer (pH 4.5) with 0.05% SDS (37 ± 0.5 °C) was selected as the discriminatory dissolution medium for the formulation screening, which have been proven its excellent discriminating power to evaluate the relationship between in vitro dissolution and in vivo absorption [24,25]. Also, purified water was used as the other powerful discriminatory dissolution medium which could help select better formulations and predict carriers’ potency of inhibiting crystallizing because the commercial Nimotop™ tablets could release well in it [25]. Then, the stability of single PVP-SDs and binary SDs was analyzed in high temperature and high humidity. Based on the results of SDs, NMP tablets were formulated by fluid bed technique to achieve the scale-up. Finally, the behavior of NMP tablets in vivo was compared with Nimotop™ in beagle dogs.
2. Materials and methods
2.1. Materials
NMP was purchased from Zhengzhou Ruikang Pharmaceutical Company (Zhengzhou, Henan, China). The reference commercial brand of nimodipine tablets was Nimotop™ 30 mg tablet (batch No.20150130/22, Bayer AG, Germany). Hydroxypropylmethyl cellulose E5 (HPMC-E5), magnesium stearate, and microcrystalline cellulose (MCC-SH101) were kindly gifted from Anhui Shanhe Pharmaceutical Excipients Co., Ltd. (Huainan, China). Polyvinylpyrrolidone K25 (PVP K25), Polyvinylpyrrolidone K30 (PVP K30) and Kollidon CL (PVPP) were provided by BASF Co., Ltd. (Shanghai, China). Sodium dodecyl sulfate (SDS) was purchased from Tianjin Damao chemical reagent factory (Tianjin, China). All reagents were analytical grade.
2.2. Preparation of NMP-SDs
NMP-SDs were prepared by solvent evaporation with ethanol. When PVP and HPMC were dissolved simultaneously, a 1:1 (v/v) mixture of ethanol and dichloromethane had to be added. The solvent was heated to 40 °C to help dissolve completely by water bath and then was removed by rotary evaporation under vacuum at same temperature for 1 h. Thus, a series of solid dispersions with NMP/PVP and NMP/PVP/HPMC mass ratio were prepared. After complete removal of the solvent, the formulations were dried overnight in a vacuum desiccator at 25 °C. The dry amorphous SDs were passed through a proper mesh sieve breezily to produce a homogeneous entirety.
2.2.1. Optimization of the particle size of the NMP-SDs
The dried amorphous materials were milled by a mortar and passed through 80, 60 and 40 mesh sieves, respectively, to find out a better particle size with a bigger drug release.
2.2.2. Measurement of solubility of NMP and SD formulations
The NMP solubility was determined to investigate the mechanism of the increase in dissolution of SD. An excess of NMP, and SD formulations were added to 30 ml of water and acetate buffer (pH 4.5) containing 0.05% SDS. Then the samples were kept under stirring at 150 rpm in air constant temperature oscillator (37 ± 2 °C) (CHA-S) for 72 h until a fixed concentration of the drug was achieved. Supernatants were collected and filtered through 0.22 µm filters before measuring by HPLC (Hypersil ODS2, 4.6 mm × 150 mm, 5 µm; HITACHI, Japan) at 236 nm. The mobile phase was composed of water and methanol (35/65, v/v). The column temperature was sited at 25 °C, and the flow rate was kept at 1.0 ml/min.
2.2.3. Stability study
The samples were reserved in a desiccator under vacuum at 60 °C, 75% relative humidity (NaCl saturated solution), separately. The dissolution studies on the SDs were managed after 10 d.
2.3. Preparation of NMP-SD tablets by fluid bed method
Granulation experiments were carried out in a top-spray lab Type Multifunctional Fluid Bed granulator (FLZB 1.5, Chinese Mechatronics, Changzhou, China), with total working volume of 4 l. Additives were comprised of MCC and PVPP, which had been pre-sieved through an 80 mesh sieve to get uniform for granulation. Firstly, a solid dispersion solution which should be clear and transparent was formulated by dissolving NMP and PVP in a certain amount of ethanol, which was also utilized as the binder of the particles. Then the binder solution was sprayed at a rate of 8–10 g/min during granulation. The compressor flow rate was 25–50 m3/h, which was adjusted to make certain a good and consistent flow image throughout the whole process [22]. The heating temperature of fluidized bed was 40 °C. Then the granule samples were passed through a 40 mesh sieve to separate unusual lumps. Then the excipients (including HPMC E5, disintegrants and lubricants) were weighed accurately and blended fully. Finally, the powder blends were filled into the dies and compressed into tablets with the usage of a ZP-5 type rotary tablet press (Shanghai, China).
2.4. Dissolution studies of SDs and tablets
The dissolution studies were performed according to dissolution test Apparatus 2 of NMP tablets as described in the BP 2015 edition (RC806D, Tianjin TiandaTianfa Technology Co., Ltd., Tianjin, China). The dissolution tests were implemented at 37 ± 0.5 °C at a rotation speed of 75 rpm. A certain amount of SD (30 mg as NMP) powders were added to 900 ml dissolution apparatus directly, and 5 ml of the samples were withdrawn at 5, 10, 20, 30, 45, 60 min and filtered with 0.22 µm filters. The filtrate was analyzed by using UV spectrometer at 356 nm (model UV-1102, Techcomp Ltd, China).
2.5. Solid-state characterization of NMP crystalline, SDs and fluid-bed granules
2.5.1. Differential scanning calorimetry (DSC)
DSC analyses were performed with a METTLER (Switzerland) calorimeter to obtain the thermograms. Samples 4 mg approximately were weighed and sealed in aluminum crucibles with perforated lids. The samples were heated from 30 °C to 160 °C at a heating rate of 10 °C/min under nitrogen atmosphere with a flow rate of 40 ml/min.
2.5.2. Powder X-ray diffraction (PXRD)
Powder X-ray diffraction (Rigaku Corporation, Tokyo, Japan) was carried out to research the solid state properties. The measurements were performed at room temperature, scanning over the 2θ angles from 3° to 50° at steps of 0.05°. Measurement parameters were set as follows: target, Cu; voltage, 40 kV; current, 30 mA.
2.5.3. Fourier transform infrared spectroscopy (FTIR)
The IR spectra were obtained in the spectral region 4000–400 cm–1 with a resolution of 4 cm–1 on a Fourier-transform infrared spectroscopy (FTIR) (Bruker Corporation, Switzerland) with the KBr disk method.
2.6. Pharmacokinetic study
2.6.1. Administration program
The pharmacokinetic study was approved by the Ethics Committee of Shenyang Pharmaceutical University. The study design involved a single dose of two treatments and two periods. Each dog was given two preparations with a one week washout period between each. Six beagle dogs were used for each treatment group. After fasting overnight, preparations containing 30 mg NMP (Nimotop™, FB-K30 Tablets) were given to the beagle dogs with water. 2 ml of blood samples were obtained at predetermined times (0.17, 0.33, 0.67, 1, 1.5, 2, 3, 4, 6, 8, 10 and 12 h) after administration. Plasma was separated from samples by centrifugation (3500 rpm for 10 min) and stored at −20 °C until analysis within one month.
2.6.2. Plasma sample preparation
Plasma (50 µl) was spiked with 50 µl internal standard solution (lacidipine, 100 ng/ml dissolved in methanol) and 50 µl methanol. The sample was subsequently made alkaline with 50 µl 0.1 mol/l NaOH solution and extracted with 3 ml solvent (n-hexane/diethylether = 1:1, v/v) by vortexing for 3 min. After centrifugation at 3500 rpm for 10 min and the supernatant was transferred to a conical tube. The separated organic phase was then evaporated at 37 °C. The residue was reconstituted with 100 µl mobile phase and vortexed for 3 min. After centrifugation at 12 000 rpm for 10 min, a 5 µl aliquot was injected into the UPLC-MS/MS system.
2.6.3. Analysis conditions
The analysis was carried out on an ACQUITYTM UPLC system (Waters Corp., Milford, MA, USA) with cooling autosampler and column oven. An ACQUITY UPLC™ C18 column (100 mm × 2.1 mm, 1.7 µm; Waters Corp., Milford, MA, USA) was employed for separation with the column temperature maintained at 35 °C. The chromatographic separation was achieved with a mobile phase composed of 0.2% formic acid and acetonitrile (17/83, v/v). The flow rate was set at 0.20 ml/min.
The ESI source was operated in positive ionization mode. The quantification was performed using multiple reaction monitoring (MRM) of the transitions of m/z 419.3→343.3 for nimodipine and m/z 473.3→354.3 for lacidipine (IS), respectively. The linear range was 5–1000 ng/ml with an r (correlation coefficient) value of no less than 0.99.
3. Results and discussion
3.1. In vitro dissolution studies
3.1.1. Dissolution behavior of different SDs under discriminatory dissolution media
The typical PVPs (PVP-K25 and PVP-K30) which have similar abilities of crystallization inhibition were selected. Meanwhile, the capabilities of controlling release and inhibiting crystallization of HPMC-E5 were investigated under the existence of PVP in the SDs.
Five proportions of SDs (passed through a 40 mesh sieve) of drug-PVP K25 = 1:2, 1:3, 1:4, 1:5, 1:6 and drug-PVP K30 = 1:2, 1:3,1:4, 1:5, 1:6 weight ratios were prepared and their dissolutions were measured. According to the dissolution behavior (data were not shown), the in vitro performance of SDs at drug-polymer ratios of drug-PVP K25 = 1:3, drug-PVP K30 = 1:3 were considered favorable under phosphate buffer (pH 4.5) with 0.05% SDS (Fig. 1A). Both the SDs brought the release of NMP to over 90%, and the SDs showed a little decrease after the biggest release.
Fig. 1.

Release rates of (A) PVP-SDs with different sizes and (B) binary SDs, pure NMP in acetate buffer pH 4.5 (0.05% of SDS), (C) SDs and pure NMP in water.
Meanwhile, the favorable particle size was searched by milling the favorable ratios of SDs and passing through an 80, 60 and 40 mesh sieve severally. Obviously, the SDs with lower particle size showed an agglomeration phenomenon and registered a severe decrease to 50% release of NMP, which revealed that the particle size impacted seriously on the solubility of the drug from SDs and a proper particle size was indispensable. Hence, the SDs with passing through a 40 mesh sieve were recommended.
Furthermore, the dissolution behavior of PVP-SDs (40 mesh) in purified water was performed (Fig. 1C). Surprisingly, both the PVP-SDs displayed a rapid decline of release in less than 30 min, which implied the water instability of the SDs.
By understanding the advantages of controlling the release of drug from SD system of HPMC, HPMC E5 was selected to prepare the SDs cooperatively. Also, a series of drug-PVP K25-HPMC E5 = 1:1:1, 1:2:1, 1:3:1, 1:4:1, 1:2:2, 1:1.5:1.5 and drug-PVP K30-HPMC E5 = 1:1:1, 1:2:1, 1:3:1, 1:4:1, 1:2:2, 1:1.5:1.5 weight ratios of SDs (all of 40 mesh) were prepared and their dissolutions were observed. For the viscous speciality of the HPMC E5, all of these SDs were required to be milled to pass through a 40 mesh sieve. On the basis of the measured dissolution behavior (data were not shown), the vitro performance of SDs at drug-polymer ratios of 1:3:1, 1:2:2, 1:1.5:1.5 showed a commendable release, drug–polymer ratios of drug-PVP K25-HPMC E5, drug-PVP K30-HPMC E5 = 1:3:1 were considered favorable under both dissolution medium (Fig. 1B), considering the operability and pharmacoeconomics. The SDs with HPMC E5 displayed a gently release of more than 85% of NMP under phosphate buffer (pH 4.5) with 0.05% SDS in 60 min. Meanwhile, nearly 80% of NMP in the SDs was released under purified water (Fig. 1C), which presented a significant increase compared to NMP raw material, and was similar to Nimotop™ (Fig. 2B). Undoubtedly, with the help of HPMC E5, a huge improvement was developed in the quality of the solid dispersions.
Fig. 2.

Release rates of tablets and commercial Nimotop™ in (A) acetate buffer pH 4.5 (0.05% of SDS) and (B) in water.
3.1.2. Dissolution behavior of NMP fluid bed tablets
According to the dissolution results of SDs, drug-polymer ratios were fixed as 1:3 (drug-PVP) in preparing fluid bed tablets. Both PVP-K25 and PVP-K30 were explored to discover whether they had distinct effects in the tablets’ drug release. The amounts of HPMC E5 were determined by the effects of inhibiting crystallization. Therefore, three concentrations (9%, 6%, 3%, w/w) of HPMC E5 in the tablets were prepared and their dissolutions were characterized to determine the favorable ratio. On the grounds of the dissolution behavior, the favorable concentration of HPMC E5 was 3% and 6% in FB-K25 tablets and FB-K30 ones, respectively (Fig. 2A). Obviously, PVP-K30 tablets manifested a continued rise of the release rate in 60 min, while PVP-K25 tablets revealed a downtrend after 30 min under the purified water (Fig. 2B), which implied the longer chain length, the deferred crystallization. Given the above, FB-K30 tablets with 6% HPMC E5 were the most recommended formulation. Proper amount of HPMC E5 made a prodigious contribution to inhibiting crystallization, especially illuminated in the discriminatory dissolution media of water.
3.2. Solubility of NMP and SDs
To seek the effect of SDs on the solubility of NMP, the solubility of SD formulations was measured, especially the effect of inhibition of reprecipitation by the binary SDs. Fig. 3 shows the results of solubility of the NMP and SDs in water and 0.05% SDS (pH 4.5), respectively.
Fig. 3.

Solubility of NMP and SDs in water and 0.05%SDS (pH 4.5).
As regards the solubility of SDs in water, increases comparing to the crystalline NMP (1.48 µg/ml) were of 370.27% ± 0.25% (5.48 µg/ml), 410.14% ± 0.10% (6.07 µg/ml), 616.89% ± 1.09% (9.13 µg/ml) and 430.41% ± 1.06% (6.37 µg/ml) for 1:3-SD, 1:1.5:1.5-SD, 1:3:1-SD and 1:4:1-SD, respectively. By contrast, the solubility of NMP and SDs in 0.05%SDS (pH 4.5) were 7.91 µg/ml, 27.15 µg/ml (1:3-SD), 36.03 µg/ml (1:1.5:1.5-SD), 36.07 µg/ml (1:3:1-SD) and 32.60 µg/ml (1:4:1-SD). These results clarify that the increases in dissolution of SDs counted on the improvements of solubility directly. Moreover, the solubility of binary SDs displayed a higher value even in same carrier concentration with the single SD, which revealed the better dissolution behavior of them.
3.3. Stability study
As the amorphous material may precipitate out on aging [26], the re-crystallization of the API leads to the decrease of dissolution level. The stability of SD becomes quite an issue in the commercial application.
The stability study of SDs was performed after storing at a desiccator under vacuum at 60 °C, 75% RH for 10 d, separately. Fig. 4 shows the dissolution changes of the two SDs in such conditions. Obviously, the drug release of single SDs dropped markedly both in two conditions, while the rate of dissolution of binary SDs lowered slightly. The dissolution results suggested the better stability of binary SDs and the power of inhibiting crystallization of HPMC.
Fig. 4.

Release rates of SDs in (A) water storing at 60 °C, (B) 0.05%SDS (pH 4.5) storing at 60 °C, (C) water storing at 75% RH, and (D) 0.05%SDS (pH 4.5) storing at 75% RH.
3.4. Characterization of the solid dispersions and tablets
3.4.1. Differential scanning calorimetry (DSC)
Thermoanalytical methods consist of all that could detect a characteristic of the system as a function of temperature. Among them, differential scanning calorimetry (DSC) is one of the most highly recommended method that enables the quantitative detection of all processes in which energy is required or generated [4]. The DSC curves for the drug, carriers and SDs (40 mesh and 80 mesh) were plotted. As NMP polymorphs can be differentiated via DSC [27], the NMP raw material appeared a single melting presentation at 124.1 °C, fixing its identity as pure Mod I (Fig. 5A).
Fig. 5.

DSC curves of (A) NMP, carriers, physical mixtures and (B) the SDs.
To examine the solid-state of the SDs, DSC profiles of the samples were drawn after a period of storage (3 months in a desiccator) in comparison with their respective physical mixtures, which are shown in Fig. 5B. Obviously, all the SDs did not show any crystalline reflections, comparing to physical mixtures with crystallinity of the drug. It revealed that milling did not change the form of the SDs, and the decrease of dissolution of the SDs (80 mesh) was due to the aggregation phenomena which could be observed during the experiments. Also it explained that the improved solubility is the result of the disordered structure of the amorphous halos.
Moreover, DSC patterns of the fluid bed granules were recorded to detect whether the drug presented as an amorphous form in the tablets (Fig. 6). Expectedly, neither the granules sent crystalline events, by contrast, the physical mixtures of the excipients with obvious crystallization peak of the drug.
Fig. 6.

DSC curves of physical mixtures of excipients and fluid bed granules.
3.4.2. PXRD
PXRD was performed to explain crystallization behavior of nimodipine SDs following preservation in a desiccator for 3 months, as well as the raw material, carriers, physical mixtures, which are shown in Fig. 7. As the reflection at 6.6° is exclusive for Mod I, whilst that at 10.4° is characteristic for Mod II [28], it is obvious that the raw material is composed exclusively of the metastable polymorph Mod I (Fig. 7A), which are corresponded to the results of DSC.
Fig. 7.

PXRD patterns of (A) NMP, carriers, physical mixtures and (B) the SDs.
For all of the physical mixtures, the crystalline reflections of the drug could be clearly observed, which reveals that amorphous phase related to the SDs attributes to the preparation process. Also, as can be observed in Fig. 7B, all of the SDs (40 mesh and 80 mesh) showed amorphous characteristics, accounting for the increase in the solubility of NMP in these formulations and in accordance with the DSC results.
As shown in Fig. 8, both the fluid bed granules presented amorphous characteristics, in agreement with the DSC results.
Fig. 8.

PXRD patterns of physical mixtures of excipients and fluid bed granules.
3.4.3. FTIR spectroscopy
FTIR was carried out to confirm whether the changed chemical structure interactions happened in the SDs systems, as shown in Fig. 9. As NMP has an amine group in its molecule, which may form hydrogen bonds with the carbonyl groups of the PVP macromolecules [29] and the hydroxyls of HPMC E5. Fig. 9A showed characteristic peaks of NMP at 3430 cm−1 ((N—H stretching vibration peaks) [30], 1650 cm−1 (C O stretching vibration peaks) [31,32], and 1270 cm−1 (C—N stretching vibration peak) [15]. As shown in Fig. 9B, the characteristic peak of NMP 1270 cm−1 (C—N stretching vibration peak) nearly disappeared from SDs. Importantly, this kind of interaction between drug and carrier is an extra advantage for the SDs, since they may increase the solubility of the drug into the hydrophilic carriers.
Fig. 9.

FTIR spectra of (A) NMP, carriers, physical mixtures and (B) the SDs.
As for fluid bed granules, the phenomena were similar with those of SDs, and different from their physical mixtures of excipients, as shown in Fig. 10.
Fig. 10.

FTIR spectra of physical mixtures of excipients and fluid bed granules.
3.5. Pharmacokinetic study in vivo
UPLC-MS/MS method was applied to a pharmacokinetic study of nimodipine tablets in beagle dogs. As the vitro behavior of FB-K30 tablet was more similar to Nimotop™, the vivo behavior of it was looked forward to having better results. The profile of the mean plasma concentration-time is shown in Fig. 11.
Fig. 11.

Mean NMP plasma profiles from a single dose (30 mg) bioavailability study compared with Nimotop™ (n = 6).
The Cmax of Nimotop™ (R) and FB-K30 tablets (T) was similar (425.2 ± 90.4 and 431.2 ± 81.8 ng/ml). The AUC0–12 h of Nimotop™ and FB-K30 tablets were 780.4 ± 276.0 ng·h/ml and 1064.4 ± 302.4 ng·h/ml, respectively. The AUC0–∞ of FB-K30 tablets was 1464.2 ± 878.6 ng·h/ml, which was higher than that of Nimotop™ (826.3 ± 295.3 ng·h/ml). The AUC0–12 h and AUC0–∞ of NMP after the oral administration of FB-K30 tablets were 1.4 and 1.8 fold higher than those of Nimotop™. Comparing to Nimotop™, FB-T showed a little decrease in Tmax (0.723 h for T, 0.807 h for R), indicating a faster drug absorption of FB-K30 tablets in vivo. The results of enhancement in bioavailability were in agreement with the results reported in previous study, revealing the positive effect of HPMC in vivo drug absorption [17]. Meanwhile, it could be concluded that fluid bed was quite an efficient technology to improve the oral bioavailability of NMP.
4. Conclusions
A solvent evaporation method was applied to screen carriers based on their ability to inhibit moisture-induced solid-state crystallization of nimodipine. Meanwhile, solid dispersions tablets have been successfully prepared by removing the solvent from the drug and the polymers with the usage of a fluid-bed one-step granulating technique.
The dissolution results of the tablets and PVP-HPMC SDs revealed the success of the cooperative effect of PVPs and HPMC E5, since PVP helped improve the release rate and HPMC E5 contributed to inhibiting crystallization and the stability of the SDs. The solubility in the polymer is directly related to the stabilization of amorphous drug against crystallization [33]. Unignorably, the effect of PVP K30 was superior to that of PVP K25, and the difference was significant on the condition of tablets. Also, dissolution data verified the negative effect of small particle size (80 mesh and 60 mesh) on the PVP-SDs for the aggregation effect, and the particle size of 40 mesh was recommended. Moreover, proper milling made little effect on the morphology of SDs.
Last but not least, the dissolution phenomena of the tablets were corresponded to the behavior of SD, which implied that the application of fluid-bed manufacturing realized the scaling up of SD successfully. The pharmacokinetic study showed that FB-T exhibited a higher bioavailability in vivo to Nimotop™. It is also indicated that the fluid-bed granulating technique could possibly be widely used to the tablets of solid dispersions and may get application in the manufacturing and scaling-up of solid dispersion preparations in the future.
Acknowledgments
Conflicts of interest
The authors declare no competing financial interest.
Contributor Information
Kexin Li, Email: kexinbest@163.com.
Zhonggui He, Email: hezhgui_student@aliyun.com.
References
- 1.Cardoso TM, Rodrigues PO, Stulzer HK, Silva MAS, Matos JDR. Physical–chemical characterization and polymorphism determination of two nimodipine samples deriving from distinct laboratories. Drug Dev Ind Pharm. 2005;31(7):631–637. doi: 10.1080/03639040500216212. [DOI] [PubMed] [Google Scholar]
- 2.Fu J, Xu L, Wang X, et al. Nimodipine (NM) tablets with high dissolution containing NM solid dispersions prepared by hot-melt extrusion. Drug Dev Ind Pharm. 2011;37(8):934–944. doi: 10.3109/03639045.2010.550301. [DOI] [PubMed] [Google Scholar]
- 3.Vasconcelos T, Sarmento B, Costa P. Solid dispersions as strategy to improve oral bioavailability of poor water soluble drugs. Cheminform. 2007;39(25):1068–1075. doi: 10.1016/j.drudis.2007.09.005. [DOI] [PubMed] [Google Scholar]
- 4.Leuner C, Dressman J. Improving drug solubility for oral delivery using solid dispersions. Eur J Pharm Biopharm. 2000;50(1):47–60. doi: 10.1016/s0939-6411(00)00076-x. [DOI] [PubMed] [Google Scholar]
- 5.Baird JA, Taylor LS. Evaluation of amorphous solid dispersion properties using thermal analysis techniques. Adv Drug Deliv Rev. 2012;64(5):396–421. doi: 10.1016/j.addr.2011.07.009. [DOI] [PubMed] [Google Scholar]
- 6.Janssens S, Van den Mooter G. Review: physical chemistry of solid dispersions. J Pharm Pharmacol. 2009;61(12):1571–1586. doi: 10.1211/jpp/61.12.0001. [DOI] [PubMed] [Google Scholar]
- 7.Torrado S, Torrado S, Torrado JJ, Cadórniga R. Preparation, dissolution and characterization of albendazole solid dispersions. Int J Pharm. 1996;140(2):247–250. [Google Scholar]
- 8.Van den Mooter G. The use of amorphous solid dispersions: a formulation strategy to overcome poor solubility and dissolution rate. Drug Discov Today. 2012;9(2):e79–e85. doi: 10.1016/j.ddtec.2011.10.002. [DOI] [PubMed] [Google Scholar]
- 9.Newman A, Knipp G, Zografi G. Assessing the performance of amorphous solid dispersions. J Pharm Sci. 2012;101(4):1355–1377. doi: 10.1002/jps.23031. [DOI] [PubMed] [Google Scholar]
- 10.Ghanavati R, Taheri A, Homayouni A. Anomalous dissolution behavior of celecoxib in PVP/Isomalt solid dispersions prepared using spray drier. Mat Sci Eng C. 2017;72:501–511. doi: 10.1016/j.msec.2016.11.042. [DOI] [PubMed] [Google Scholar]
- 11.Riekes MK, Caon T, de Silva J, et al. Enhanced hypotensive effect of nimodipine solid dispersions produced by supercritical CO2 drying. Powder Technol. 2015;278:204–210. [Google Scholar]
- 12.Yagi N, Terashima Y, Kenmotsu H, Sekikawa H, Takada M. Dissolution behavior of probucol from solid dispersion systems of probucol-poly vinylpyrrolidone. Chem Pharm Bull. 1996;44(1):241–244. [Google Scholar]
- 13.Usui F, Maeda K, Kusai A, Nishimura K, Yamamoto K. Inhibitory effects of water-soluble polymers on precipitation of RS-8359. Int J Pharm. 1997;154(1):59–66. [Google Scholar]
- 14.Suzuki H, Sunada H. Influence of water-soluble polymers on the dissolution of nifedipine solid dispersions with combined carriers. Chem Pharm Bull. 1998;46(3):482–487. doi: 10.1248/cpb.46.482. [DOI] [PubMed] [Google Scholar]
- 15.Oh MJ, Shim JB, Yoo H, et al. The dissolution property of raloxifene HCl solid dispersion using hydroxypropyl methylcellulose. Macromol Res. 2012;20(8):835–841. [Google Scholar]
- 16.Gao P, Morozowich W. Development of supersaturatable self-emulsifying drug delivery system formulations for improving the oral absorption of poorly soluble drugs. Expert Opim Drug Del. 2006;3(1):97–110. doi: 10.1517/17425247.3.1.97. [DOI] [PubMed] [Google Scholar]
- 17.Yamashita K, Nakate T, Okimoto K, et al. Establishment of new preparation method for solid dispersion formulation of tacrolimus. Int J Pharm. 2003;267(1):79–91. doi: 10.1016/j.ijpharm.2003.07.010. [DOI] [PubMed] [Google Scholar]
- 18.Vo LN, Park C, Lee BJ. Current trends and future perspectives of solid dispersions containing poorly water-soluble drugs. Eur J Pharm Biopharm. 2013;85(3):799–813. doi: 10.1016/j.ejpb.2013.09.007. [DOI] [PubMed] [Google Scholar]
- 19.Hare C, Bonakdar T, Ghadiri M, Strong J. Impact breakage of pharmaceutical tablets. Int J Pharm. 2018;536(1):e370–e376. doi: 10.1016/j.ijpharm.2017.11.066. [DOI] [PubMed] [Google Scholar]
- 20.Liu C, Desai KGH. Characteristics of rofecoxib-polyethylene glycol 4000 solid dispersions and tablets based on solid dispersions. Pharm Dev Technol. 2008;10(4):467–477. doi: 10.1080/10837450500299701. [DOI] [PubMed] [Google Scholar]
- 21.Jianping Q, Lu Yi, Wei W. Manufacturing solid dosage forms from bulk liquids using the fluid-bed drying technology. Curr Pharm Des. 2015;21(19):2668–2676. doi: 10.2174/1381612821666150416100639. [DOI] [PubMed] [Google Scholar]
- 22.Rajniak P, Stepanek F, Dhanasekharan K, Fan R, Mancinelli C, Chern R T. A combined experimental and computational study of wet granulation in a Wurster fluid bed granulator. Powder Technol. 2009;189(2):190–201. [Google Scholar]
- 23.Sun N, Wei X, Wu B, Chen J, Lu Y, Wu W. Enhanced dissolution of silymarin/polyvinylpyrrolidone solid dispersion pellets prepared by a one-step fluid-bed coating technique. Powder Technol. 2008;182(1):72–80. [Google Scholar]
- 24.Fu Q, Kou L, Gong C, et al. Relationship between dissolution and bioavailability for nimodipine colloidal dispersions: the critical size in improving bioavailability. Int J Pharm. 2012;427(2):358–364. doi: 10.1016/j.ijpharm.2012.02.022. [DOI] [PubMed] [Google Scholar]
- 25.He Z, Chen X, Liu X, Tang X, Zhao L. Development of a dissolution medium for nimodipine tablets based on bioavailability evaluation. Eur J Pharm Sci. 2004;21(4):487–491. doi: 10.1016/j.ejps.2003.11.009. [DOI] [PubMed] [Google Scholar]
- 26.Serajuddin ATM. Solid dispersion of poorly water-soluble drugs: Early promises, subsequent problems, and recent breakthroughs. J Pharm Sci. 1999;88(10):1058–1066. doi: 10.1021/js980403l. [DOI] [PubMed] [Google Scholar]
- 27.Riekes MK, Kuminek G, Rauber GS, de Campos CE, Bortoluzzi AJ, Stulzer HK. HPMC as a potential enhancer of nimodipine biopharmaceutical properties via ball-milled solid dispersions. Carbohydr Polym. 2014;99(1):474–482. doi: 10.1016/j.carbpol.2013.08.046. [DOI] [PubMed] [Google Scholar]
- 28.Docoslis A, Huszarik KL, Papageorgiou GZ, Bikiaris D, Stergiou D, Georgarakis E. Characterization of the distribution, polymorphism, and stability of nimodipine in its solid dispersions in polyethylene glycol by micro-Raman spectroscopy and powder X-ray diffraction. AAPS J. 2007;9(3):361–370. doi: 10.1208/aapsj0903043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Papageorgiou G, Docoslis A, Georgarakis M, Bikiaris D. The effect of physical state on the drug dissolution rate miscibility studies of nimodipine with PVP. J Therm Anal Calorim. 2009;95(3):903–915. [Google Scholar]
- 30.Barmpalexis P, Kachrimanis K, Georgarakis E. Solid dispersions in the development of a nimodipine floating tablet formulation and optimization by artificial neural networks and genetic programming. Eur J Pharm Biopharm. 2011;77(1):122–131. doi: 10.1016/j.ejpb.2010.09.017. [DOI] [PubMed] [Google Scholar]
- 31.Papadimitriou S, Papageorgiou G, Kanaze F, Manolis G, Bikiaris D. Nanoencapsulation of nimodipine in novel biocompatible poly(propylene-co-butylene succinate) aliphatic copolyesters for sustained release. J Nanomater. 2009;2009 [Google Scholar]
- 32.Tang XC, Pikal MJ, Taylor LS. A spectroscopic investigation of hydrogen bond patterns in crystalline and amorphous phases in dihydropyridine calcium channel blockers. Pharm Res. 2002;19(4):477–483. doi: 10.1023/a:1015147729564. [DOI] [PubMed] [Google Scholar]
- 33.Qian F, Huang J, Hussain MA. Drug-polymer solubility and miscibility: stability consideration and practical challenges in amorphous solid dispersion development. J Pharm Sci. 2010;99(7):2941–2947. doi: 10.1002/jps.22074. [DOI] [PubMed] [Google Scholar]