

Abstract
A simple and rapid UPLC–MS/MS method to simultaneously determine gemcitabine and its L-carnitine ester derivative (2'-deoxy-2', 2'-difluoro-N-((4-amino-4-oxobutanoyl) oxy)-4-(trimethyl amm-onio) butanoate-cytidine, JDR) in rat plasma was developed and validated. The conventional plasma sample preparation method of nucleoside analogues is solid-phase extraction (SPE) which is time-consuming and cost-expensive. In this study, gradient elution with small particles size solid phase was applied to effectively separate gemcitabine and JDR, and protein precipitation pretreatment was adopted to remove plasma protein and extract the analytes with high recovery(>81%). Method validation was performed as per the FDA guidelines, and the standard curves were found to be linear in the range of 5–4000 ng/ml for JDR and 4–4000 ng/ml for gemcitabine, respectively. The lower limit of quantitation (LLOQ) of gemcitabine and JDR was 4 and 5 ng/ml, respectively. The intra-day and inter-day precision and accuracy results were within the acceptable limits. Finally, the developed method was successfully applied to investigate the pharmacokinetic studies of JDR and gemcitabine after oral administration to rats.
Keywords: Gemcitabine, L-carnitine, Prodrug, Pharmacokinetics, UPLC/MS/MS
Abbreviations: JDR, 2'-deoxy-2', 2'-difluoro-N-((4-amino-4-oxobutanoyl)oxy)-4-(trimethyl amm-onio) butanoate-cytidine; SPE, solid-phase extraction; LLOQ, lower limit of quantification; PK, pharmacokinetic; OCTN2, organic cation/carnitine transporters 2; ESI, electrospray ionization; THU, Tetrahydrouridine; IS, internal standard; QC, quality control
1. Introduction
Gemcitabine is a water-soluble pyrimidine nucleoside analogue with significant anticancer activity for several types of cancers, such as pancreatic adenocarcinoma, bladder cancers and breast cancers [1], [2]. Unfortunately, due to high hydrophilicity and poor membrane permeability, it is difficult for gemcitabine to enter the blood circulation after oral administration. Therefore, gemcitabine is administered by intravenous injection in clinic due to its low oral bioavailability [3]. Although many efforts have been made to design advanced intravenous drug delivery system, oral route is still the most preferred route due to its patient compliance, therapeutic efficacy and convenience [4].
Prodrug strategies based on various gastrointestinal nutrient transporters have been utilized to overcome undesirable and pharmacokinetic properties of drug, since these transporters play an important role in the oral absorption of nutrients and therapeutic drugs. It has been reported that valdidanosine and valdecitabine could be transported across intestinal epithelium by oligopeptide transporter 1 [5], [6], [7]. In order to further improve the oral bioavailability of nucleoside drugs, some new attempts are required. For example, prednisolone–carnitine conjugate was studied for nasal delivery mediated by organic cation/carnitine transporters 2 (OCTN2) [8]. This high-affinity carnitine transporter is highly expressed in kidney, trachea and intestine and is a promising target protein because it can transport organic cations and nutrients, such as carnitine and ergothioneine [9], [10], [11], [12], [13]. OCTN2 is also crucial for the β-oxidation and energy metabolism of fatty acids, and responsible for a primary systemic carnitine deficit. Following this idea, we synthesized the L-carnitine ester derivative of gemcitabine as shown in Fig. 1, namely JDR, to enhance the oral bioavailability of gemcitabine. To compare oral pharmacokinetics of gemcitabine and JDR, it was necessary to develop a sensitive method for simultaneous quantification of both drugs.
Fig. 1.

Structure of JDR and gemcitabine. (A) gemcitabine, (B) JDR.
Various quantitative methods have been developed for the determination of gemcitabine and its derivatives. Wickremsinhe et al. developed a method for the determination of LY2334737 (gemcitabine prodrug), gemcitabine and its metabolite (dFdU) by LC–MS/MS and column switching chromatography [14]. Bowen et al. had validated a method for the analysis of gemcitabine and dFdU using solid phase extraction (SPE) [15]. However, these methods have some notable limitations, such as time-consuming and cost-expensive features of SPE sample preparation, unconventional column switching chromatography.
There are several challenges in the quantification of gemcitabine and its analogues. The similar chemical structure requires highly selective methods for their quantification. What is more, it is difficult to be simultaneously retained in RP system for the compounds with different polarity. Last, the current available quantification methods have been developed based on SPE of a large-volume sample and long analytical time. However, a simple and efficient protein precipitation method with short analytical time was rarely used in nucleoside analogue pretreatment due to low extraction recovery and matrix effect. Therefore, developing a simple and reliable method to simultaneously determine gemcitabine and its analogue is critical to the pre-clinical study of gemcitabine prodrugs.
In the present study, a rapid, sensitive UPLC–MS/MS method was developed to simultaneously determine JDR and gemcitabine. Compared with the previous assay methods of plasma volume of 100 µl and single run time of 5 min, this method provided shorter analysis time (3 min), reduced volume requirements (50 µl) and simplified plasma sample pretreatment. The lower limit of quantification (LLOQ) of gemcitabine was 4 ng/ml, which was sensitive enough to detect relatively low concentration of gemcitabine in rat plasma. This method was successfully applied to characterize the pharmacokinetic profiles of JDR and gemcitabine after a single dose of oral administration.
2. Materials and methods
2.1. Chemicals and reagents
Gemcitabine (98.8% purity) was purchased from Nanjing Chemlin Chemical Industry Co., Ltd (Jiangsu, Nanjing, PR China). L-carnitine was obtained from Kaiyuan Hengtai Chemical Co., Ltd (Liaoning, Shenyang, P R China). IS (99% purity, didanosine) was supplied by JiaXing I sen Chemical Co., Ltd (Zhejiang, Jiaxing, PR China). JDR (97.2% purity) was synthesized in Shenyang Pharmaceutical University (Shenyang, China). Ammonium acetate (HPLC grade) was obtained from Tianjin Kemiou Chemical Reagent Co. (Tianjin, China). Ultra pure water was prepared by EASYPURE®II RF/UV system (Boston, MA, USA). Tetrahydrouridine (THU), the cytidine deaminase inhibitor, was purchased from J&K Scientific (HPLC grade). HPLC-grade methanol was purchased from Fisher Scientific (Fairlawn, NJ, USA). All other chemicals were of analytical grade.
2.2. Synthesis of JDR
2.2.1. Synthesis of benzyl carnitine
L-carnitine (0.322 g, 2 mmol) was dissolved in 10 ml DMF and heated to 140 °C. Benzyl bromide (0.342 g, 2 mmol) was added dropwise, and the reaction was allowed to reflux 2 h. The solvent was concentrated under reduced pressure and the residue purified by recrystallization from acetonitrile. The product was concentrated under reduced pressure, and dried under high vacuum at room temperature until the weight was constant. Yield: 85% of a white powder. MS (ESI): m/z = 253.1 [M + H+].
2.2.2. Synthesis of 5-(benzyloxy)-3-(3-carboxypropanoyloxy)-N, N, N-trimethyl-5-oxo pentan-1- aminium
Succinic acid (0.2 g, 2 mmol) was dissolved in 10 ml 1, 4-dioxane and heated to reflux. Sulfoxide chloride (0.238 g, 2 mmol) was added dropwise, and the reaction was allowed to reflux 4 h. The solvent was concentrated under reduced pressure and the residue was dissolved in 10 ml DMF. Benzyl carnitine (0.504 g, 2 mmol) and triethylamine (0.202 g, 2 mmol) were then filled in the reaction flask and heated to 60 °C overnight. The solvent was evaporated, and the residue was purified by column chromatography on a silica gel, eluting with methanol in dichloromethane (gradient 5–10%). Yield: 53% of a yellow powder. MS (ESI): m/z = 353.2 [M + H+].
2.2.3. Synthesis of JDR
5-(benzyloxy)-3-(3-carboxypropanoyloxy)-N, N, N-trimethyl-5-oxopentan-1-aminium (0.35 g, 1 mmol) and gemcitabine (0.263 g, 1 mmol) were filled in the reaction flask. After complete melting, DCC (0.412 g, 2 mmol) and DMAP (0.012 g, 0.2 mmol) were added in the flask under room temperature overnight. The solvent was evaporated, and the residue was purified by column chromatography on a silica gel, eluting with methanol in dichloromethane (gradient 5–10 %). The pure intermediate was dissolved in methanol, and then Pd/C (10 %) was added. The mixture was stirred under H2 at room temperature for 15 minutes and filtered. The solvent was evaporated, and product was collected in a vacuum. Yield: 67% of a white powder. MS (ESI): m/z = 507.2 [M + H+]. The purity determined by HPLC was 98.2%. 1H NMR (400 MHz, DMSO-d6): δ 10.86 (d, J = 286.0 Hz, 2H), 8.31 (d, J = 7.6 Hz, 1H), 7.20 (d, J = 7.5 Hz, 1H), 6.48 (s, 1H), 6.15 (t, J = 7.1 Hz, 1H), 5.86 (dd, J = 13.9, 4.8 Hz, 2H), 5.39 (s, 1H), 5.25 (s, 1H), 4.28–4.03 (m, 8H), 3.86 (d, J = 8.6 Hz, 1H), 3.76 (s, 1H), 3.61 (ddt, J = 16.3, 12.4, 7.8 Hz, 10H), 3.36 (d, J = 19.8 Hz, 10H), 3.08 (s, 9H), 2.75 (s, 2H), 2.56 (dd, J = 11.9, 4.5 Hz, 4H).
2.3. Instrumentation
An ACQUITY triple-quadrupole tandem mass spectrometer equipped with an electrospray ionization (ESI) interface (Waters Corp., Milford, MA, USA) was used. BEH C18 column (50 mm × 2.1 mm, 1.7 µm, Waters) was used for chromatographic separation. Data acquisition was performed by MassLynx 4.1 software with QuanLynx program (Waters Corp., Milford, MA, USA).
2.4. UPLC–MS/MS condition
The mobile phase was composed of methanol (A) and water (B) (containing 2 mM ammonium acetate) at a flow rate of 0.2 ml/min. A gradient elution was performed as described below, 0 ~0.2 min, 5% A; 0.21 ~1.2 min, 70% A; 1.21 ~3 min, 5% A. The column and auto-sampler temperatures were set at 40 °C and 10 °C, respectively. The injected volume was 5 µl.
For three compounds, the positive ion mode of electrospray ionization (ESI) was chosen for JDR, gemcitabine and negative ion mode for IS. Quantification was achieved by multiple reactions monitoring (MRM) mode. Conditions of the ESI source were optimized as follows: the capillary was set at 4.0 kV, desolvation gas temperature was 350 °C, and cone voltage was 40, 30 and 30V for JDR, gemcitabine and IS, Collision Energies (CE) was 25, 15 and 20 eV for JDR, gemcitabine and IS. The most sensitive ion transitions were selected for the monitoring of m/z 507.0→162.1 for JDR, m/z 264.0→112.0 for gemcitabine, and m/z 235.1→134.9 for IS.
2.5. Sample preparation
An aliquot of 50 µl rat plasma sample in 1.5 ml Eppendorf tube was added with 25 µl IS solution (1500 ng/ml) and 225 µl of methanol. Then the samples were vortex-mixed for 3 min. The sample was centrifuged at 13,000 × g for 5 min. An aliquot of 5 µl of the supernatant was injected for the LC-MS/MS analysis.
2.6. Preparation of standard and quality control (QC) samples
Standard stock solutions of JDR, gemcitabine and IS were prepared by dissolving the accurately weighed reference compounds in water at the concentrations of 8 µg/ml, 4 µg/ml and 150 µg/ml, respectively. A series of working standard solutions of JDR, gemcitabine and IS were prepared by diluting standard stock solution with methanol at appropriate concentrations. All solutions were stored in a 4 °C freezer and brought to room temperature before use.
Calibration samples and QC samples were prepared according to sample preparation item. Methanol (75 µl) was replaced by 25 µl JDR and 50 µl gemcitabine standard solutions in THU-pretreated rat plasma. The final concentrations of blood sample were 5, 15, 125, 500, 3200, and 4000 ng/ml for JDR, 4, 12, 125, 500, 3200 and 4000 ng/ml for gemcitabine and 750 ng/ml for IS. QC samples were obtained in the same manner with three levels of 15, 125 and 3200 ng/ml for JDR and 12, 125 and 3200 ng/ml for gemcitabine.
2.7. Method validation
The selectivity of this method was evaluated by comparing chromatograms of six different batches of blank rat plasma with the corresponding spiked rat plasma at LLOQ for interferences between analytes, IS and endogenous substances. Linearity was measured by liner regression with a weighted (1/x2) least-squares analysis. Intra- and inter-day precision (the relative standard deviation, RSD %) and accuracy (the relative error, RE %) were verified by analysis of three levels of QC samples (n = 6) on 3 different days. The matrix effect on the ionization of the analytes was tested by comparing the peak areas between post extraction sample and standard solutions at different concentrations. Recovery was calculated by comparing the mean peak areas of a pre- and post- extraction spiked sample at low, middle and high QC concentrations. The stability was assessed by determining QC samples (n = 3) in three freeze/thaw cycles (−80 to 20 °C), long-term sample storage (-80 °C for 2 months) and short-term sample storage (20 °C for1 h). The stability of extracted samples in the autosampler was also assessed at 10 °C for 8 h.
2.8. Pharmacokinetic study in rats
Twelve Wistar male rats weighting 180 to 220 g were supplied by the Animal center of the Shenyang Pharmaceutical University (Shenyang, China). All protocols for animal experiments were approved by Shenyang Pharmaceutical University Animal Care and Use Committee. The validated analytical method was then applied to the pharmacokinetic study after oral administration of JDR (equivalent to 50 mg/kg gemcitabine) or gemcitabine (50 mg/kg) to rats. The rats were randomly divided into two groups and fasted for 12 h until the administration of the drug. Serial blood samples were obtained at 0.083, 0.25, 0.5, 1, 2, 3, 4, 6, 8, 10, 12, 24, 48 h for JDR and 0.25, 0.5, 2, 4, 8, 12, 24 h for gemcitabine, respectively. Blood samples were collected into THU-pretreated heparinized tubes (10 mg/ml) and immediately centrifuged at 3000 × g for 15 min and supernatant (plasma) was stored at −80 °C prior to analysis.
2.9. Statistical analysis
Descriptive statistics were showed as mean ± standard deviation. The pharmacokinetic parameters (including Cmax, Tmax, AUC, T1/2) of gemcitabine in control group and prodrug group were estimated using a one-tailed Student t test at the P < 0.05 level.
3. Results and discussions
3.1. Optimization of chromatographic and mass conditions
UPLC–MS/MS operation parameters were optimized for the determination of analytes. The standard solutions of analytes were infused with the mobile phase into the mass spectrometer with ESI as the ionization source. The response of analytes to ESI were evaluated by recording the full-scan mass spectrum in both positive and negative ionization modes, and they had a good mass spectrum response in positive mode for gemcitabine and JDR, and negative mode for IS. Therefore, the most sensitive molecular ion transitions were with ESI for the MRM determination of m/z 264.0→112.0 for gemcitabine, m/z 507.0→162.1 for JDR and m/z 235.1→134.9 for IS as shown in Fig. 2. Other main mass spectrometry parameters, such as capillary voltage, cone voltage, collision energy and desolvation temperature were optimized to obtain the optimal response of analytes.
Fig. 2.
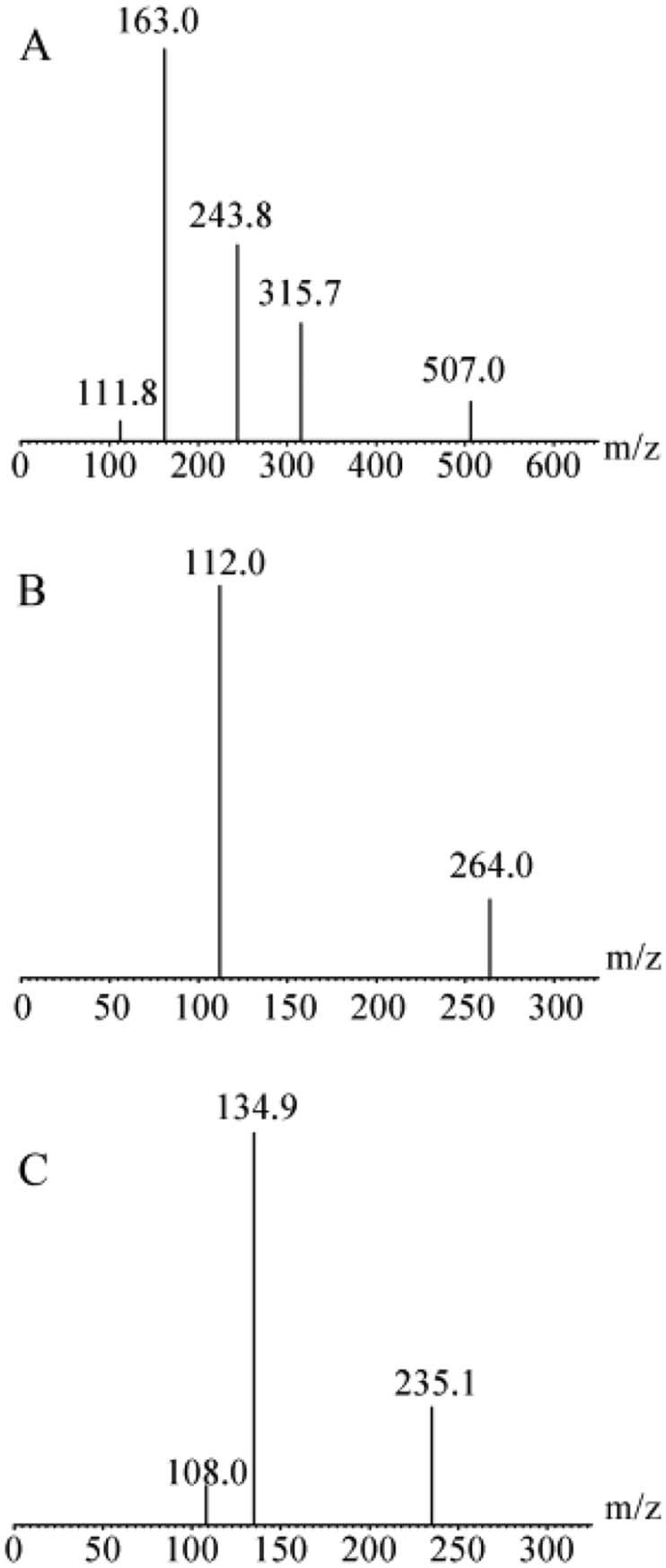
Product ion mass spectra of [M + H]+ ions of prodrug and gemcitabine. (A) JDR, (B) gemcitabine and (C) [M-H]- ions didanosine.
The separation and ionization of analytes are affected by compositions of mobile phase. Therefore, the selection of mobile phase is important for improving peak shape, sensitivity and short run time. Acetonitrile and methanol were both tested as organic modifier of mobile phase. A lower MS response was caused by addition of acetonitrile. Therefore, methanol was chosen as the organic modifier. It was known that the additives of mobile phases can affect the LC resolution and MS response of analytes. In the present work, the role of added formic acid and ammonium formate at various concentrations in the mobile phases were investigated. The results showed that the additive of ammonium formate improved the ionization of analytes. Particularly, the addition of 2 mM ammonium acetate not only enhanced the MS response of analytes but also improved the peak shapes of analytes. As a result, the addition of 2 mM ammonium acetate in the mobile phase was used in this study. The use of small particles of stationary phase allowed UPLC to strengthen the limits of both peak capacity and speed of analysis without compromising resolution. In the present work, the column and gradient method were optimized. Several of reversed-phase columns, namely phenomenon Ultracarb C18 (50 mm × 4.6 mm, 2.1 µm), Waters SunFire C18 (50 mm × 4.6 mm, 2.1 µm) and Waters BEH C18 (50 mm × 2.1 mm, 2.1 µm) were studied. Compared with the other two columns, the sensitivity for analytes increased significantly with the use of BEH C18 column. To avoid undesirable matrix effect, gradient elution was used to provide a better MS response and peak shape. Various different gradient conditions were investigated and optimized. The peaks were efficiently separated from the interference with a signal-to-noise ratio of more than 10 for all analytes of interest.
3.2. Sample preparation
In order to inhibit the transformation of gemcitabine during sample preparation, the inhibitor were screened at three different concentrations (2, 5 and 10 mg/ml in rat plasma) under room temperature. THU at 10 mg/ml was the most effective inhibitor for stabilizing gemcitabine in rat plasma.
In recent years, solid-phase extraction (SPE) is often applied in the plasma pretreatment for nucleoside drugs, due to clean sample and high sensitivity. However, high cost and tedious-operation is inevitable. In this study, the simple protein precipitation using methanol or acetonitrile was applied and the optimal organic solvent was methanol, because the MS response of analytes is not good in acetonitrile. More important, the extraction recovery was greater than 80% for all analytes and there were no matrix effects. Because of the adequate sensitivity of optimized UPLC–MS/MS conditions, the plasma volume was reduced to 50 µl, much lower than the previous methods (100 µl of plasma).
Compared with the previous assay methods of the LLOQ values of gemcitabine was 0.25 ng/ml, this method provided higher LLOQ values (4 ng/ml) [15]. In this study, the supernatant of sample dried under nitrogen cannot be completed since prodrug could be hydrolyzed by a small amount of esterase remaining in biological samples. The result showed that the sensitivity, recovery and matrix effect of the method could meet the needs of pharmacokinetic study.
3.3. Method validation
The establishment and validation of this method were performed according to the FDA bioanalytical method validation guidelines.
For specificity, analyses of blank samples of plasma were obtained from six different rats, blank samples spiked with JDR (5 ng/ml), gemcitabine (4 ng/ml), and IS (750 ng/ml) and plasma samples after oral administration of JDR. Fig. 3 represented the chromatogram of LLOQ. Any interference from plasma could not be detected around the analytes or the internal standard.
Fig. 3.

Representative MRM chromatograms of JDR, gemcitabine and didanosine (IS) in rat plasmas. (A1–A3): a blank rat plasma sample; (B1–B3): a blank rat plasma sample spiked with JDR (5 ng/ml), gemcitabine (4 ng/ml) and didanosine (750 ng/ml); (C1–C3): a rat plasma sample following an oral dose of JDR at 96 mg/kg (calculated as gemcitabine) to a Wister rat.
Linear regression was achieved with 1/x2 as weighing factor and standard curve were established. All coefficients (r) were greater than 0.99, which showed a good linearity over the studied concentration range. The LLOQ was 5 ng/ml for JDR and 4 ng/ml for gemcitabine.
The inter-day and intra-day precision and accuracy for all analytes from three QC levels were summarized in Table 1. The precision were fitted and the RSD % were over the range of 0.8% to 3.7% for JDR and 0.9% to 5.3% for gemcitabine, respectively, while the RE% ranged from −9.9% to 4.0%. These results indicated that this method was accurate and reliable.
Table 1.
Inter-run accuracy and precision following a 3-batch validation of JDR and gemcitabine in rat plasma (data were mean ± SD, n = 18).
| Analytes | Nominal conc. (ng/ml) | Intra-day | Inter-day | ||||
|---|---|---|---|---|---|---|---|
| Measured concentration (ng/ml) | Precision (RSD%) | Accuracy (RE%) | Measured concentration (ng/ml) | Precision (RSD%) | Accuracy (RE%) | ||
| JDR | |||||||
| 15.00 | 15.35 ± 1.4 | 9.4 | −1.6 | 15.12 ± 0.5 | 3.4 | −3.1 | |
| 125.00 | 116.13 ± 3.8 | 3.2 | −7.1 | 112.60 ± 4.2 | 3.7 | −9.9 | |
| 3200.00 | 3573.15 ± 61.5 | 1.7 | 11.6 | 3083.75 ± 25.9 | 0.8 | 1.8 | |
| Gemcitabine | |||||||
| 12.00 | 11.26 ± 1.2 | 10.4 | 4.3 | 11.02 ± 0.3 | 2.3 | 2.0 | |
| 125.00 | 124.66 ± 3.9 | 3.1 | −0.3 | 129.96 ± 6.9 | 5.3 | 4.0 | |
| 3200.00 | 2912.59 ± 148.1 | 5.1 | −9.0 | 2931.04 ± 26.2 | 0.9 | −8.4 | |
The recovery and matrix effect of all the QC samples were summarized in Table 2. The mean recovery ranged from 88.5% to 110.8% for JDR, 81.4% to 100.8% for gemcitabine and 102.2% to 111.7% for IS. The matrix effect of plasma for three analytes was more than 90%, indicating that it was negligible.
Table 2.
Extraction efficiency and matrix effect for JDR, gemcitabine in rat plasma (data were mean ± SD, n = 6).
| Extraction efficiency (%) (mean ± SD) | Matrix effect (%) (mean ± SD) | ||||||
|---|---|---|---|---|---|---|---|
| JDR | Gem | IS | JDR | Gem | IS | ||
| Low | 88.5 ± 0.06 | 81.4 ± 0.04 | 102.2 ± 0.03 | Low | 111.3 ± 0.09 | 94.2 ± 0.03 | 99.9 ± 0.01 |
| Mid | 101.4 ± 0.03 | 100.8 ± 0.07 | 106.5 ± 0.04 | Mid | 96.0 ± 0.02 | 102.8 ± 0.01 | 94.1 ± 0.01 |
| High | 110.8 ± 0.08 | 97.3 ± 0.05 | 111.7 ± 0.05 | High | 98.4 ± 0.02 | 96.8 ± 0.03 | 100.2 ± 0.01 |
Table 3 showed the results of the stability under four different conditions. The percent deviations for all analytes were less than 8% from theoretical value. The results showed that pre-preparative sample was stable when stored at room temperature for 1 h, −80 °C for 2 months, three freeze-thaw cycles, and kept in the auto-sampler at 10 °C for 8 h.
Table 3.
Stability of JDR and gemcitabine in rat plasma exposed to various storage conditions (data were mean ± SD, n = 3).
| Concentration(ng/ml) | RSD% | RE% | ||
|---|---|---|---|---|
| Added | Founded | |||
| Bench-top (20 °C for 1 h) | ||||
| JDR | ||||
| Low | 15.00 | 15.46 ± 1.3 | 8.6 | −-0.9 |
| Mid | 125.00 | 113.58 ± 1.3 | 4.3 | −9.1 |
| High | 3200.00 | 3169.38 ± 150.5 | 4.7 | −1.0 |
| Gemcitabine | ||||
| Low | 12.00 | 10.29 ± 1.0 | 10.0 | −4.8 |
| Mid | 125.00 | 134.78 ± 1.6 | 1.2 | 7.8 |
| High | 3200.00 | 2915.15 ± 95.7 | 3.3 | −8.9 |
| Autosampler rack at 10 °C for 8 h | ||||
| JDR | ||||
| Low | 15.00 | 16.78 ± 0.3 | 1.5 | 7.5 |
| Mid | 125.00 | 112.35 ± 5.6 | 5.0 | −10.1 |
| High | 3200.00 | 3129.46 ± 137.9 | 4.4 | −2.2 |
| Gemcitabine | ||||
| Low | 12.00 | 10.90 ± 1.3 | 12.0 | 0.9 |
| Mid | 125.00 | 136.30 ± 1.8 | 1.4 | 9.1 |
| High | 3200.00 | 2996.32 ± 151.9 | 5.1 | −6.4 |
| Three freeze/thaw cycles (-80 to 20 °C) | ||||
| JDR | ||||
| Low | 15.00 | 15.17 ± 1.6 | 10.5 | −2.8 |
| Mid | 125.00 | 109.17 ± 1.4 | 1.2 | −12.7 |
| High | 3200.00 | 3078.27 ± 54.5 | 1.8 | −3.8 |
| Gemcitabine | ||||
| Low | 12.00 | 10.58 ± 0.7 | 7.0 | −2.0 |
| Mid | 125.00 | 134.30 ± 11.6 | 8.7 | 7.4 |
| High | 3200.00 | 2966.93 ± 150.4 | 5.1 | −7.3 |
| Freezing at -80 °C for 2 months | ||||
| JDR | ||||
| Low | 15.00 | 15.29 ± 1.9 | 12.3 | −2.0 |
| Mid | 125.00 | 110.75 ± 2.3 | 2.1 | −11.4 |
| High | 3200.00 | 3092.26 ± 124.3 | 4.0 | −3.4 |
| Gemcitabine | ||||
| Low | 12.00 | 10.57 ± 1.3 | 12.7 | −2.1 |
| Mid | 125.00 | 130.38 ± 10.6 | 8.1 | 4.3 |
| High | 3200.00 | 3195.42 ± 15.4 | 0.5 | −0.1 |
3.4. Application to pharmacokinetic
This validated method was successfully applied to a pharmacokinetic study after oral administration of JDR or gemcitabine to the Wistar rats at a dose of 50 mg/kg (calculated as gemcitabine). The mean plasma concentration-time curves of JDR and gemcitabine were displayed in Fig. 4. The pharmacokinetic parameters of JDR and gemcitabine were calculated using noncompartmental method and were summarized in Table 4. With a short peak time Tmax (JDR) of 1 h, JDR demonstrated a faster absorption rate than gemcitabine. Furthermore, the plasma elimination half-time t1/2 of gemcitabine released from JDR was significantly longer than the parent drug gemcitabine, suggesting that JDR was more stable in plasma than gemcitabine and the bioactivation of JDR to gemcitabine was slowly performed by the esterase in plasma. Finally, the area under the curve of gemcitabine following oral administration of JDR was 16132.95 mg ⋅ h /l, approximate 3-fold increase compared to that of gemcitabine (5031.61 mg ⋅ h/ l). Hence, the pharmacokinetic results indicated that L-carnitine ester derivative of gemcitabine can improve the oral bioavailability of gemcitabine.
Fig. 4.
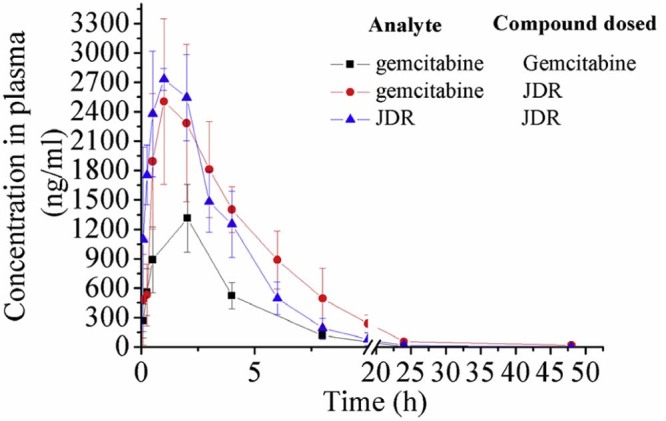
Mean plasma concentration–time profiles of JDR and gemcitabine in Wistar male rats (data were mean ± SD, n = 6). (▲) JDR and (●) gemcitabine, following oral administration of JDR to rats (96 mg/kg, calculated as gemcitabine); (■) gemcitabine, following oral administration of gemcitabine to rats (50 mg/kg).
Table 4.
Pharmacokinetic parameters of JDR and gemcitabine in rat after oral administration JDR and gemcitabine (data were mean ± SD, n = 6).
| Ingredients | Cmax (ng/ml) | Tmax | AUC0-24 (mg ⋅ h /l) | T1/2 (h) |
|---|---|---|---|---|
| JDR-JDR | 4153.18 ± 1927.40 | 1.08 ± 0.9 | 12828.59 ± 2204.5 | 6.31 ± 3.9 |
| JDR-gemcitabine | 2658.04 ± 1212.6** | 1.00 ± 0.0 | 16132.95 ± 5128.0** | 6.36 ± 2.1** |
| Gemcitabine | 1181.17 ± 47.1 | 1.50 ± 0.9 | 5031.61 ± 627.4 | 2.61 ± 1.1 |
P < 0.01 compared with gemcitabine.
4. Conclusions
A simple, highly sensitive and reliable UPLC–MS/MS analytical method that can simultaneously determine JDR and gemcitabine in rat plasma has been developed and validated. The method required relatively simply sample pretreatment and accomplished high sensitivity using as small as 50 µl volume of plasma. The established method provided the LLOQs of 5 ng/ml for JDR and 4 ng/ml for gemcitabine with a single run time of 3 min. An effective method to select cytidine deaminase inhibitors to stabilize gemcitabine in rat plasma was developed. This method was successfully implemented to pharmacokinetic study of JDR and gemcitabine to rats after oral administration.
Conflicts of interest
The authors declare that there is no conflicts of interest.
Acknowledgments
We thank the financial support from the National Natural Science Foundation of China (No. 81173009) and Technology Bureau in Shenyang (No. ZCJJ2013402). We also thank the financial support from Project for New Century Excellent Talents of Ministry of Education (No.NCET-12-1015), Specific Science Foundation of Shenyang Pharmaceutical University (No. ZCJJ2014409) and National Undergraduate Training Program for Innovation and Entrepreneurship (2016).
Footnotes
Peer review under responsibility of Shenyang Pharmaceutical University.
References
- 1.Abbruzzese J.L. New applications of gemcitabine and future directions in the management of pancreatic cancer. Cancer. 2002;95:941–945. doi: 10.1002/cncr.10753. [DOI] [PubMed] [Google Scholar]
- 2.Wolff R.A. Chemotherapy for pancreatic cancer: from metastatic disease to adjuvant therapy. Cancer J. 2007;13:175–184. doi: 10.1097/PPO.0b013e318074e6c3. [DOI] [PubMed] [Google Scholar]
- 3.Bender D.M., Bao J., Dantzig A.H. Synthesis, crystallization, and biological evaluation of an orally active prodrug of gemcitabine. J Med Chem. 2009;52:6958–6961. doi: 10.1021/jm901181h. [DOI] [PubMed] [Google Scholar]
- 4.Yun Y., Cho Y.W., Park K. Nanoparticles for oral delivery: targeted nanoparticles with peptidic ligands for oral protein delivery. Adv Drug Deliv Rev. 2013;65:822–832. doi: 10.1016/j.addr.2012.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang Y., Sun J., Gao Y. An HPLC-MS/MS method for simultaneous determination of decitabine and its valyl prodrug valdecitabine in rat plasma. J Chromatogr B Analyt Technol Biomed Life Sci. 2013;917–918:78–83. doi: 10.1016/j.jchromb.2012.12.040. [DOI] [PubMed] [Google Scholar]
- 6.Yan Z., Sun J., Wang J. Simultaneous determination of didanosine and its amino acid prodrug, valdidanosine by hydrophilic interaction chromatography coupled with electrospray ionization tandem mass spectrometry: application to a pharmacokinetic study in rats. J Chromatogr B Analyt Technol Biomed Life Sci. 2010;878:466–470. doi: 10.1016/j.jchromb.2009.11.041. [DOI] [PubMed] [Google Scholar]
- 7.Zhang Y., Gao Y., Wen X. Current prodrug strategies for improving oral absorption of nucleoside analogues. Asian J Pharm Sci. 2014;9:65–74. [Google Scholar]
- 8.Mo J.X., Shi S.J., Zhang Q. Synthesis, transport and mechanism of a type I prodrug: L-carnitine ester of prednisolone. Mol Pharm. 2011;8:1629–1640. doi: 10.1021/mp100412z. [DOI] [PubMed] [Google Scholar]
- 9.Tamai I., Yabuuchi H., Nezu J. Cloning and characterization of a novel human pH-dependent organic cation transporter, OCTN1. FEBS Lett. 1997;419:107–111. doi: 10.1016/s0014-5793(97)01441-5. [DOI] [PubMed] [Google Scholar]
- 10.Tamai I., Ohashi R., Nezu J. Molecular and functional identification of sodium ion-dependent, high affinity human carnitine transporter OCTN2. J Biol Chem. 1998;273:20378–20382. doi: 10.1074/jbc.273.32.20378. [DOI] [PubMed] [Google Scholar]
- 11.Tamai I., Ohashi R., Nezu J.I. Molecular and functional characterization of organic cation/carnitine transporter family in mice. J Biol Chem. 2000;275:40064–40072. doi: 10.1074/jbc.M005340200. [DOI] [PubMed] [Google Scholar]
- 12.Grundemann D., Harlfinger S., Golz S. Discovery of the ergothioneine transporter. Proc Natl Acad Sci USA. 2005;102:5256–5261. doi: 10.1073/pnas.0408624102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nakamura T., Yoshida K., Yabuuchi H. Functional characterization of ergothioneine transport by rat organic cation/carnitine transporter Octn1 (slc22a4) Biol Pharm Bull. 2008;31:1580–1584. doi: 10.1248/bpb.31.1580. [DOI] [PubMed] [Google Scholar]
- 14.Wickremsinhe E.R., Lee L.B., Schmalz C.A. High sensitive assay employing column switching chromatography to enable simultaneous quantification of an amide prodrug of gemcitabine (LY2334737), gemcitabine, and its metabolite dFdU in human plasma by LC-MS/MS. J Chromatogr B Analyt Technol Biomed Life Sci. 2013;932:117–122. doi: 10.1016/j.jchromb.2013.06.008. [DOI] [PubMed] [Google Scholar]
- 15.Bowen C., Wang S., Licea-Perez H. Development of a sensitive and selective LC-MS/MS method for simultaneous determination of gemcitabine and 2,2-difluoro-2-deoxyuridine in human plasma. J Chromatogr B Analyt Technol Biomed Life Sci. 2009;877:2123–2129. doi: 10.1016/j.jchromb.2009.06.002. [DOI] [PubMed] [Google Scholar]