

Graphical Abstract
Keywords: Liquisolid technique, Dissolution enhancement, Poorly water-soluble drugs, Sustained release, pH variation, Photostability
Abstract
Most of the newly developed drug candidates are lipophilic and poorly water-soluble. Enhancing the dissolution and bioavailability of these drugs is a major challenge for the pharmaceutical industry. Liquisolid technique, which is based on the conversion of the drug in liquid state into an apparently dry, non-adherent, free flowing and compressible powder, is a novel and advanced approach to tackle the issue. The objective of this article is to present an overview of liquisolid technique and summarize the progress of its applications in pharmaceutics. Low cost, simple processing and great potentials in industrial production are main advantages of this approach. In addition to the enhancement of dissolution rate of poorly water-soluble drugs, this technique is also a fairly new technique to effectively retard drug release. Furthermore, liquisolid technique has been investigated as a tool to minimize the effect of pH variation on drug release and as a promising alternative to conventional coating for the improvement of drug photostability in solid dosage forms. Overall, liquisolid technique is a newly developed and promising tool for enhancing drug dissolution and sustaining drug release, and its potential applications in pharmaceutics are still being broadened.
1. Introduction
With the advent of combinatorial chemistry and innovative high-throughput screening, knowledge concerning physicochemical properties (i.e., crystal structures and salt formation) as well as biological factors (such as metabolizing enzymes and transporters) of drug candidates has been extensively accumulated [1]. As a result, a vast number of active pharmaceutical ingredients have been produced. However, most of these drugs are very lipophilic and poorly water-soluble [2]. It is reported that about 40% of the newly developed drugs and nearly 60% of the synthesized chemical entities suffer from solubility issues [3], [4]. Therefore, to enhance the solubility and dissolution of these poorly water-soluble drugs and improve their bioavailabilities are a matter of concern for many pharmaceutical scientists. The bioavailability of these Biopharmaceutical Classification System Class II (BCS II) drugs is often limited by their solubility and dissolution rate in the gastrointestinal tract [5], [6].
Many suitable formulation approaches have been developed to increase the solubility of poorly water-soluble drugs. Micronization technique is the most commonly used approach to improve drug solubility due to an increase in surface area, but the agglomeration tendency of micronized hydrophobic drugs makes it less effective to circumvent the solubility problem, especially when the drug is formulated into tablets or encapsulations [7]. Solid dispersion has gained an active research interest for improving drug dissolution in the past few decades, however its commercial application is very limited and only a few products, such as Kaletra® and Gris-PEG® have become commercially available. The reason mainly lies on its poor stability during storage and lack of understanding of its solid-state structure [8]. Formulating soft gelatin capsules is another widely used approach, whereas it is costly and requires sophisticated technologies [9]. Other approaches, such as inclusion complexation [10], microencapsulation [11], and preparation of nanosuspensions [12], self-nanoemulsions [13] and solid lipid nanoparticles [14] have also been studied for dissolution enhancement of poorly water-soluble drugs. But these approaches involve high production cost and entail advanced preparation method and/or sophisticated machinery.
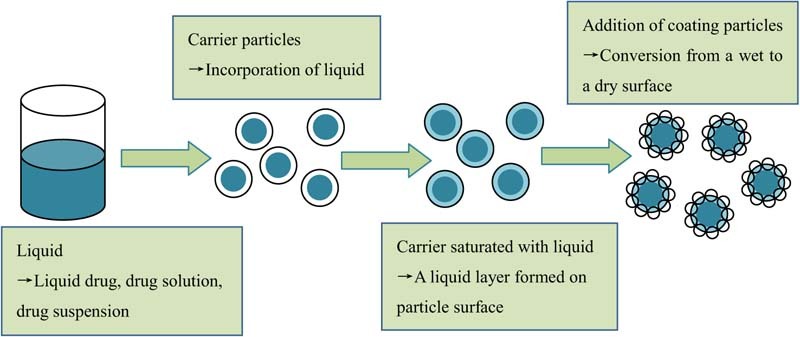
Liquisolid technique, a newly developed and advanced method for dissolution enhancement, can overcome many aforementioned barriers [15], [16], [17]. This technique was first introduced by Spireas et al. and applied to incorporate water-insoluble drugs into rapid release solid dosage forms. The design principle of liquisolid system is to contain liquid medications (i.e., liquid drugs, drug solutions or suspensions) in powdered form and delivery drug in a similar way to soft gelatin capsules containing liquids. Liquisolid technique refers to the conversion of liquid medications into apparently dry, non-adherent, free flowing and compressible powder mixtures by blending the liquid medications with suitable excipients, which are generally termed as carriers and coating materials [18], [19]. The liquid medication is first absorbed into the interior framework of the carrier. Once the interior of the carrier is saturated with liquid medication, a liquid layer is formed on the surface of carrier particles, which is instantly adsorbed by the fine coating materials. Consequently, an apparently dry and free flowing and compressible powder mixture is formed. The mechanism of liquisolid system formation is displayed in Fig. 1. Usually, orally safe, and preferable water-miscible organic solvents with high boiling point, such as propylene glycol and polyethylene glycol (PEG) 400, are used as the liquid vehicles. Carriers refer to porous materials with large specific surface area and high liquid absorption capacity to absorb liquid medication [19]. Various grades of cellulose, starch and lactose can be adopted as carriers. However, only excipients with very fine particle size and highly adsorptive property, such as silica powder, can be used as coating materials [21].
Fig. 1.

Mechanism of liquisolid system formation. Figure adapted from Reference [20].
Even though the drug within liquisolid system is in a solid state, it exists exactly in a completely or partly molecularly dispersed state [22], [23]. Therefore, a liquisolid system may exhibit enhanced dissolution rate due to the increased dissolution area, enhanced aqueous solubility, or improved wetting properties [24]. Apart from dissolution enhancement, liquisolid technique has recently been investigated as a tool to retard drug release [25], [26], [27], to minimize the influence of pH variation on dissolution profile [28], [29], and to improve drug photostability [30]. Finally, it is worth mentioning that liquisolid systems are not associated with stability issues [15], [31], [32], [33]. This article presents an overview of the liquisolid technique and the advance in its applications in pharmaceutics.
2. Theory of liquisolid system
A powder can only retain limited amount of liquid medication while maintaining acceptable flowability and compressibility. Therefore, in order to attain a liquisolid system with acceptable flowable and compressible properties, a mathematical model introduced and validated by Spireas is recommended to calculate the appropriate quantities of carrier and coating material [18], [19], [20]. The model is based on two fundamental properties of a powder, i.e., flowable liquid retention potential ( value) and compressible liquid retention potential ( value). The and values of a powder excipient represent the maximum quantity of liquid vehicle that can be retained in the powder bulk without compromising flowability and compressibility [19]. The value is preferably determined by measuring the angle of slide of the prepared liquid–powder admixture. And the value can be measured by an experiment called pactisity, which is defined as the maximum crushing strength of a tablet with a tablet weight of one gram when compressed at sufficient compression force [19], [21].
The excipients ratio (), which is also known as the carrier/coating ratio, is defined as follows:
| (1) |
Therefore, is the ratio between the weights of carrier () and coating material (). An increase in the value will lead to higher quantities of the carrier and lower amounts of the coating material. As the value is associated with the flowability and compressibility properties, disintegration, and dissolution rate of the liquisolid system, an optimum value of is recommended to be 20 [21], [34]. Another important parameter of the liquisolid system is termed as liquid loading factor (), which is defined as the weight ratio of the liquid medication () and the carrier material () in the liquisolid system.
| (2) |
The liquid loading factor for the production of a liquisolid system with acceptable flowability can be determined by:
| (3) |
Where and values correspond to the flowable liquid retention potential of the carrier and coating material, respectively. Correspondingly, the liquid loading factor to ensure acceptable compressibility of a liquisolid system can be determined by:
| (4) |
Where and values correspond to the compressible liquid retention potential of the carrier and coating material, respectively. Therefore, the optimum liquid loading factor () that produces a liquisolid system with acceptable flowability and compressibility is equal to either or , whichever has the lower value.
As , , , and values are constants for each powder–liquid combination, for a given excipients ratio (), the optimum liquid loading factor () can be calculated according to Equations (3) or (4). Then, according to different drug concentrations, different weights of liquid medication () will be used. Thus, based on the calculated and , the appropriate amount of carrier () and coating material () can be calculated according to Equations (1) and (2), respectively.
3. Advantages and disadvantages of liquisolid technique
3.1. Advantages [16], [30], [35], [36]
Numerous advantages of liquisolid technique have been reported. (i) Huge number of slightly water-soluble, very slightly water-soluble and practically water-insoluble drugs can be formulated into liquisolid systems with enhanced dissolution and bioavailability. (ii) Sustained release formulations with zero order release pattern can be achieved provided that hydrophobic carriers, such as Eudragit® RL and RS, or retarding agents such as hydroxypropyl methylcellulose (HPMC) are used in the liquisolid systems. (iii) This technique has the potential to produce liquisolid tablets or capsules with pH-independent drug release profiles. (iv) It is a promising alternative to conventional coating approach for the improvement of drug photostability in solid dosage forms. (v) The applied excipients are easily available and cost-effective. Besides, the preparation process is simple, which is similar to conventional solid dosage forms (i.e. tablets and capsules). Moreover, the good flowability and compressibility of liquisolid powder make the technique feasible for large-scale production.
3.2. Disadvantages [37], [38]
There are also disadvantages associated with liquisolid technique. (i) The technique is successfully applied for low dose water-insoluble drugs, whereas the incorporation of high dose water-insoluble drugs into liquisolid systems is its main limitation. As these drugs require large quantity of liquid vehicle, therefore, in order to obtain liquisolid powder with good flow and compressible properties, large amounts of carrier and coating material are required. This may increase tablet weight over the limit, which is difficult for patients to swallow. Several strategies have been reported to address the above obstacle. For example, adding some additives (i.e., PVP and PEG 35000) into the liquid medications to increase the viscosity can reduce the quantities of carrier and coating material. Additionally, application of modern carrier and coating materials (such as Fujicalin® and Neusilin®) with large specific surface area (SSA) and high absorption capacity is another efficient way to load high dose water-insoluble drugs. (ii) A high solubility of drug in liquid vehicle is required to prepare liquid solid systems.
4. Formulation design and preparation of liquisolid system
4.1. Formulation design of liquisolid system
4.1.1. Liquid vehicle
Liquid vehicle used in liquisolid systems should be orally safe, inert, not highly viscous, and preferably water-miscible nonvolatile organic solvents, such as propylene glycol, glycerin, PEG 200 and 400, polysorbate 20 and 80, etc [39]. The solubility of drug in nonvolatile solvent has an important effect on tablet weight and dissolution profile. Higher drug solubility in the solvent leads to lower quantities of carrier and coating material, and thus lower tablet weight can be achieved. On the other hand, the higher the drug solubility in the solvent, the greater FM value (the fraction of molecularly dispersed drug) will be, which confers an enhancement of the dissolution rate [16], [40]. The selection of liquid vehicle mainly depends on the aim of study. Namely, a liquid vehicle with high ability to solubilize drug will be selected in the case of dissolution enhancement. While if the aim is to prolong drug release, liquid vehicle with the lowest capacity for solubilizing drug may be chosen [27]. In addition to the drug solubility in liquid vehicle, several other physicochemical parameters such as the polarity, lipophilicity, viscosity, and chemical structure also have significant effects on drug release profiles [21].
Moreover, it is claimed that liquid vehicle can act as a binder in a low concentration, which contributes to the compactness of liquisolid tablets. The reason may lie on the presence of hydroxyl groups in the molecular structure of liquid vehicle which leads to hydrogen bonding between solvents and other excipients in liquisolid formulations [41].
4.1.2. Carriers
Carriers should possess porous surface and high liquid absorption capacity [18]. As carriers allow an incorporation of large amount of liquid medication into the liquisolid structure, the properties of carriers, such as (SSA) and liquid absorption capacity, are of great importance in designing the formulation of liquisolid system. The liquid adsorption capacity mainly depends on the SSA value. Additionally, it is also influenced by the type of coating material and the physicochemical properties of the liquid vehicle, such as polarity, viscosity, and chemical structure [42].
Currently, microcrystalline cellulose (MCC) with SSA of 1.18 m2/g is the most commonly used carrier. Javadzadeh [15] investigated the effect of three grades of MCC (i.e., PH 101, 102, and 200) on the flowability and compressibility as well as the dissolution rate of piroxicam liquisolid tablets. It was observed that liquisolid formulations prepared from MCC PH 101 exhibited better flowability, compressibility, and dissolution profiles compared with those prepared from MCC PH102 and 200. In addition, aging has no significant effect on the hardness and dissolution profiles of the prepared liquisolid tablets. Overall, MCC PH 101 is a suitable carrier to prepare liquisolid systems in terms of flowability, compressibility, and dissolution profile. Apart from MCC, other general carriers, such as lactose (SSA 0.35 m2/g), sorbitol (SSA 0.37 m2/g), and starch (SSA – 0.6 m2/g) have relatively limited applications due to their low SSA values [43]. As a result of the low SSA value of carriers, large amounts of carriers are required for the conversion of liquid medication into apparently dry, free flowing and compressible powder mixture, which will further lead to the increase in tablet weight. In addition to these carriers, Eudragit® RL and RS are also commonly used in the preparation of liquisolid systems with sustained drug release patterns [25].
Recently, several promising carriers with extremely high SSA value and greater liquid absorption capacity are available at the market. For instance, Fujicalin®, a synthetic anhydrous dibasic calcium phosphate, has a SSA value of 40 m2/g and a liquid absorption ability up to 1.2 ml/g [44]. Hentzschel et al. [42] prepared tocopherol acetate liquisolid system using Fujicalin® as a carrier. Their results further confirmed that Fujicalin® is a suitable carrier for preparing liquisolid systems. In addition, Neusilin®, another newly-developed carrier, is an amorphous form of magnesium aluminometasilicate with a SSA value up to 300 m2/g. Neusilin® is commercially available in eleven grades, among which Neusilin® US2 (SSA of 300 m2/g, liquid adsorption capacity up to 3.4 mL/g) is the most commonly used carrier [45]. Vranikova et al. [46] determined the flowable liquid retention potential of Neusilin® US2 for three different nonvolatile solvents, it was observed that 1 gram of Neusilin® US2 could retain up to 1 gram of propylene glycol, 1.16 gram of PEG 400 and 1.48 gram of PEG 200 while maintaining acceptable flowability. Therefore, the large SSA value and high absorption capacity makes Neusilin® US2 an excellent carrier for liquisolid systems. Hentzschel et al. [47] adopted Neusilin® US2 as a carrier to prepare griseofulvin liquisolid system in comparison with Avicel®. The results showed that Neusilin® possessed seven-fold higher liquid adsorption capacity than Avicel®, which allowed a production of liquisolid tablets with lower tablet weights. Furthermore, apart from Fujicalin® and Neusilin®, ordered mesoporous silicates own even larger specific surface (up to 1500 m2/g [48]) and larger pore volume, which enables it to be a promising choice in designing liquisolid formulations. Chen et al. [49] prepared carbamazepine using ordered mesoporous silicates as a carrier. It was clear that ordered mesoporous silicates formed good reservoirs for liquid medication and showed a substantial increase in drug loading capacity.
4.1.3. Coating materials
Coating materials refer to very fine and highly adsorptive materials, such as Aerosil® 200, Neusilin®, and calcium silicate or magnesium aluminometasilicates in a powder form. These materials play a contributory role in covering the wet carrier particles to form an apparently dry, non-adherent, and free flowing powder by adsorbing any excess liquid [50]. It was proved that the replacement of Aerosil® 200 by Neusilin® US2 as a coating material in liquisolid system considerably increased the liquid adsorption capacity and reduced tablet weight [47]. Since Neusilin® can be either a carrier or a coating material, its usage will greatly simplify the preparation procedure of liquisolid formulations [47].
4.1.4. Additives
The disintegration of solid dosage forms obviously influences drug release. Therefore, disintegrants are usually included in liquisolid tablets to allow a fast disintegration. Some commonly used disintegrants in liquisolid system include sodium starch glycolate, croscarmellose sodium, and low substituted hydroxypropyl cellulose [51]. Polyvinylpyrrolidone (PVP) is another promising additive, which has the potential to incorporate high amount of drug into liquisolid systems, and thus reduce the tablet weight [38]. Besides, due to the crystal growth inhibition effect of PVP, liquisolid tablets containing PVP show an improvement of dissolution rate [37]. There is another additive in liquisolid systems – HPMC, which usually acts as a release retarding agent to extend drug release [36].
4.2. General preparation procedures of liquisolid system
Calculated amounts of drug and liquid vehicle are mixed, and then heated or sonicated for completely solubilizing or evenly blending. The following mixing process of the resulted liquid medication with other excipients used in the liquisolid formulation is carried out in three steps as described by Spireas and Bolton [18]. During the first stage, the resulted liquid medication is poured onto calculated quantity of carrier material and blended at an approximate mixing rate of one rotation per second for one minute to facilitate a homogenous distribution of liquid medication throughout the carrier powder. Then, coating material in calculated amount is added and mixed homogenously. In the second stage, the prepared powder mixture is spread as a uniform layer on the surface of a mortar and left standing for 5 min to facilitate a complete absorption of drug medication into the interior framework of carrier and coating materials. In the third stage, disintegrant is added and mixed thoroughly with the above powder mixture, and a final liquisolid system is obtained. The prepared liquisolid system can be further compressed or encapsulated. It has to be mentioned that the mixing speed, mixing time, and standing time can be adapted according to specific case. The preparation procedures of liquisolid system are displayed in Fig. 2.
Fig. 2.

Preparation procedures of liquisolid system. Figure adapted from Reference [52].
5. Applications of liquisolid technique in pharmaceutics
5.1. Liquisolid technique as a tool to enhance drug dissolution
Based on the literatures, liquisolid technique has been widely used to improve the dissolution rate of low dose insoluble drugs, such as prednisolone [21], famotidine [22], valsartan [53], ketoprofen [54], raloxifene hydrochloride [23], clonazepam [24], clofibrate [55], etc. In the case of high dose water insoluble drugs (i.e., carbamazepine), the feasibility of liquisolid technique has also been discussed. Javadzadeh et al. suggested [38] that it is possible to involve liquisolid technique in the incorporation of high dose water-insoluble drugs into liquisolid systems by adding some additives (such as PVP, HPMC and polyethylene glycol 35000), because these additives have the capability to increase the liquid absorption capacity of carrier and coating materials. Hentzschel et al. [42] have shown another potential approach to load high dose of poorly water-soluble drugs into liquisolid systems, namely by using modern carriers (such as Neusilin®) with larger SSA value and higher absorption capacity.
Recently, Pezzini et al. [56] explored the possibility of using this technique to prepare liquisolid pellets for dissolution enhancement of felodipine. It was observed that a liquisolid microenvironment with soft structures and high porosity was formed, which favored the disintegration and dissolution process of felodipine liquisolid pellets. The results indicated that it is feasible to adopt liquisolid pellets as novel drug delivery systems to improve the dissolution rate of poorly water-soluble drugs. A comparative study to corroborate the feasibility of liquisolid technique is performed by Khan et al. [57], in which the liquisolid technique was applied to enhance the dissolution rate of hydrochlorothiazide in comparison with solid dispersion technique. The obtained results showed liquisolid systems enhanced the drug dissolution rate to 95% while it only increased to 88% for solid dispersions. Thus a conclusion could be drawn that the liquisolid technique was more effective than solid dispersion technique in improving the rate and extent of drug release.
Furthermore, the in vivo profiles of liquisolid tablets have been studied by several researchers. For example, Khaled et al. [58] evaluated the in vivo performance of hydrochlorothiazide liquisolid tablets in six male Beagle dogs using two-way crossover design. It was shown that hydrochlorothiazide liquisolid tablets exhibited 15% greater bioavailability than the commercial oral dosage form. Recently, in another study, the clinical evaluation of mosapride citrate liquisolid tablets was performed by Badawy et al. [29] in six healthy male volunteers aged twenty to forty years. A randomized, single dose, two-way crossover open-label design was used for the study. The authors concluded that mosapride citrate liquisolid tablets could increase the oral bioavailability when compared with the commercial counterparts, with significantly improved pharmacokinetic parameters (i.e., Cmax, Tmax, and AUC(0–12)).
Three possible mechanisms of dissolution enhancement for liquisolid systems have been proposed in the literature, namely increased drug surface area, increased drug solubility, and increased wetting properties. Even though the drug is held in a solid dosage form, it is presented either in a solubilized or dispersed state. Therefore, the drug surface area available for dissolution is markedly increased [16], [17], [22]. In addition to the preceding mechanism, the drug solubility could be increased in the aqueous diffusion layer. It is recognized that the relatively small amount of liquid vehicle existed in the liquisolid system may be insufficient to increase the overall drug solubility in the dissolution medium. However, in the microenvironment of diffusion layer between the individual liquisolid primary particle and the dissolution medium, liquid vehicle may act as a co-solvent and diffuses out of the primary particle together with the drug, which might be adequate to increase drug solubility [22], [59], [60]. Moreover, due to the surface activity of liquid vehicles, the interfacial tension between tablet surface and dissolution media can be reduced, which leads to an improved wettability of the hydrophobic drug [31], [60]. Recently, we have improved the dissolution of tadalafil, a poorly water-soluble drug, by employing the liquisolid technique. Meanwhile, the mechanism of enhanced dissolution was also investigated. The results suggested that a reduction of the particle size and crystallinity and an enhancement of the wettability were the main mechanisms for the enhanced dissolution rate of tadalafil [61].
5.2. Liquisolid technique as a tool to sustain drug release
Liquisolid technique is initially designed to enhance the dissolution rate of poorly water-soluble drugs. In the past few years, extensive studies indicated that the liquisolid technique could be utilized as a promising method for preparing sustained release formulations of different drugs [25], [26], [52], [62]. Sustained release formulations are designed to release the drug slowly at a predetermined rate for a certain period of time with high efficacy, high patient compliance, and minimum side effects. One of the main advantages of applying liquisolid technique in prolonging drug release is the possibility to attain a liquisolid system with zero order release kinetics [25], [26], [27]. However, its main limitation lies on the high tablet weight, which is attributed to the high dose of drug used in the sustained release liquisolid formulations (usually higher than that in conventional tablets) [43].The principle behind liquisolid technique to sustain drug release is mainly based on the hypothesis that by involving hydrophobic carriers (i.e. Eudragit® RL and RS) instead of hydrophilic carrier or retarding agents (such as HPMC) in the liquisolid formulations, a prolonged drug release pattern can be achieved [25], [63]. Besides, as the SSA value of the commonly used hydrophobic carriers (such as Eudragit® RL and RS) are usually lower than that of the hydrophilic carriers such as MCC, the amount of coating material (such as silica, a hydrophobic material) that required to convert wetting carrier particles to apparently dry and free flowing powders will be generally higher [25]. This may aid in sustaining drug release. Moreover, it was claimed that by selecting suitable types of liquisolid vehicle, a prolonged drug release pattern could also be obtained [62].
Many attempts have been made to optimize the sustained release liquisolid formulations. Javadzadeh et al. [25] investigated the feasibility of this technique to prolong the release of propranolol hydrochloride. The results showed that liquisolid technique can be adopted as a new tool to prepare sustained release matrices with zero-order release kinetic. The authors pointed out that polysorbate 80 (Tween 80) had an important role in sustaining drug release. Due to the plasticizer effect of Tween 80, the glass transition temperature (Tg) of polymer that applied in the formulation could be reduced. As a result, the polymer chains would coalesce better, which resulted in a fine polymer network with lower porosity and higher tortuosity. During the release process, drug was surrounded and restricted by the fine network, and thus prolonged the drug release. In another innovative study, Nokhodchi et al. [26] evaluated the effect of co-solvent and HPMC on theophylline release. It was concluded that the presence of non-volatile co-solvent was critical for prolonging drug release. The sustained release action of HPMC was amplified and desirable release profile was achieved by changing the type of co-solvent. Similar conclusions were made by Khanfar et al. [64] where venlafaxine hydrochloride liquisolid tablets exhibited greater retardation effect compared with the directly compressed tablets. The type of liquid vehicle was observed to affect drug release significantly. Other important factors included drug concentration in the liquid medication and excipients ratio (). Specifically, drug release from liquisolid tablets could be decreased with the increase of drug concentration. A reduction of drug release was observed in liquisolid tablets with higher R value. This was because the amount of carrier and swelling agents (HPMC) was increased in these formulations, which led to a slow diffusion of drug through the porous carrier and the gel layer formed by HPMC. The authors further concluded that prolonged drug release profiles over a period of twelve hours were obtained from liquisolid tablets containing Tween 80 as a liquid vehicle, Avicel® as a carrier, and HPMC as a retarding agent. Adibkia et al. [52] claimed that the solubility of drug in liquid vehicle had a significant effect on drug release profiles. Additionally, other physicochemical properties such as the formation of micelles, dielectric constant and HLB also affect drug release profiles.
5.3. Liquisolid technique as a tool to minimize the influence of pH variation on drug release
The solubility of weak acids and bases is dependent on the ionization constant (pKa) of the compound and pH of the local environment. Therefore, the dissolution and bioavailability of these drugs are greatly influenced by the pH of gastrointestinal fluids. This further leads to a high degree of inter- and intra-variability in drug bioavailability and therapeutic effects [29], [65]. El-Hammadi et al. [28] first explored the possibility of using liquisolid technique to minimize the influence of pH variation on the release of loratadine. Several liquisolid formulations were prepared using propylene glycol as a liquid vehicle, MCC as a carrier, and silica as a coating material. The dissolution profile of the prepared liquisolid tablets was investigated in three buffered media with pH values of 1.2, 2.5, and 5, respectively. The results indicated that the dissolution rates of liquisolid tablets were significantly higher and less affected by pH variation in comparison with the directly compressed tablets and marketed tablets (Clarityn®). The results suggested that liquisolid technique is a promising tool to minimize the influence of pH variation on the dissolution rate of poorly water-soluble drugs. Similar results were also reported by Chella et al. [33] where an optimized liquisolid formulation was obtained with a significant improvement in dissolution and a less pH-dependent release profile compared to drug alone or its commercial formulation. In another study, Badawy et al. [29] demonstrated the robustness of mosapride citrate (a poorly soluble weak base) liquisolid tablets, which minimize the effect of pH variation on drug release along the gastrointestinal tract with bio-relevant media.
5.4. Liquisolid technique as a promising tool to improve drug photostability in solid dosage forms
A loss of drug potency during the photodegradation process may result in toxic degradation products and causing potential side effects, thus the photostability study is an indispensable part of pre-formulation studies for photosensitive drugs [30], [66]. The principle behind photoprotective action of liquisolid technique is based on the photoprotective property of silicon dioxide (a commonly used coating material in liquisolid system) due to its high refractive index and the capability to diffract light waves of different energies [30].
Khames [30] designed a study to evaluate the possibility of using liquisolid technique as a promising alternative to conventional coating for the improvement of drug photostability. Several liquisolid formulations of amlodipine (a photosensitive drug) were prepared, where Avicel® PH 102 was used as the carrier, nanometer-sized amorphous silicon and titanium dioxide either alone or in combination was used as the coating material. The prepared amlodipine liquisolid formulations were irradiated with visible light, UVA and UVB with different light dose for eight hours. Meanwhile, the conventional film coating tablets and drug alone were tested in the same way for comparison. It was found that all liquisolid formulations showed significant photoprotective effect with a residual drug percentage of 97.37% compared to 73.8% for the drug alone after eight hours of irradiation (P < 0.05). Besides, the photoprotective action of liquisolid tablets was comparable to the conventional film coating tablets (titanium dioxide as the sunscreen, P > 0.05). To be specific, the photoprotective effect of liquisolid tablets was inversely proportional to the excipients ratio (). As a conclusion, liquisolid technique was proved to be a promising alternative to conventional coating for improving drug photostability in solid dosage forms.
6. Conclusion
To enhance the solubility and dissolution of poorly water-soluble drugs is still a matter of concern for pharmaceutical scientists. A review of extensive literatures indicates that the development of liquisolid technique is advancing very fast in the past few years. Liquisolid technique is not only a useful tool to improve the dissolution rate of poorly water-soluble drugs, but also an innovative and excellent method to prepare sustained release tablets with zero order release pattern. Moreover, the technique has exhibited great potential in reducing the effect of pH variation on drug release and improving drug photostability in solid dosage forms. Other potential applications of this technique in pharmaceutics are to be explored in the future. Further studies regarding the development of excellent solvents as well as modern carrier and coating materials for loading high dose drugs are still underway. Currently, much research work still focuses on the formulation development of liquisolid systems and the investigation of in vitro drug release profiles. Future works on the measurement of loading high dose water-insoluble drugs, and in vivo evaluation of liquisolid systems need to be explored and strengthened.
Footnotes
Peer review under responsibility of Shenyang Pharmaceutical University.
Contributor Information
Dongkai Wang, Email: Wangdksy@126.com.
Pingtian Ding, Email: dingpingtian@qq.com.
References
- 1.Lipinski C.A., Lombardo F., Dominy B.W. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 2001;46:3–26. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 2.Stegemann S., Leveiller F., Franchi D. When poor solubility becomes an issue: from early stage to proof of concept. Eur J Pharm Sci. 2007;31:249–261. doi: 10.1016/j.ejps.2007.05.110. [DOI] [PubMed] [Google Scholar]
- 3.Liversidge E.M. Nanocrystals: resolving pharmaceutical formulation issues associated with poorly water soluble compounds. In: Marty J.J., editor. Particles. Marcel Dekker; Orlando: 2002. [Google Scholar]
- 4.Giliyar C., Fikstad D.T., Tyavanagimatt S. Challenges and opportunities in oral delivery of poorly water-soluble drugs. Drug Del Technol. 2006;6:57–63. [Google Scholar]
- 5.Amidon G.L., Lennernas H., Crison J.R. A theoretical basis for a biopharmaceutic drug classification: the correlation of in vitro drug product dissolution and in vivo bioavailability. Pharm Res. 1999;12:413–420. doi: 10.1023/a:1016212804288. [DOI] [PubMed] [Google Scholar]
- 6.Yadav A.V., Shete A.S., Dabke A.P. Formulation and evaluation of orodispersible liquisolid compacts of aceclofenac. Indian J Pharm Educ. 2010;44:227–235. [Google Scholar]
- 7.Aguiar A.J., Zelmer A.J., Kinkel A.W. Deaggregation behavior of a relatively insoluble substituted benzoic acid and its sodium salt. J Pharm Sci. 1979;56:1243–1252. doi: 10.1002/jps.2600561006. [DOI] [PubMed] [Google Scholar]
- 8.Craig D.Q.M. The mechanisms of drug release from solid dispersions in water-soluble polymers. Int J Pharm. 2002;231:131–144. doi: 10.1016/s0378-5173(01)00891-2. [DOI] [PubMed] [Google Scholar]
- 9.Ebert W.R. Soft gelatin capsules: unique dosage form. Pharm Tech. 1977;1:44–50. [Google Scholar]
- 10.Hiremath S.N., Raghavendra R.K., Sunil F. Dissolution enhancement of gliclazide by preparation of inclusion complexes with β-cyclodextrin. Asian J Pharm. 2008;2:73–76. [Google Scholar]
- 11.Li D.X., Oh Y.K., Lim S.J. Novel gelatin microcapsule with bioavailability enhancement of ibuprofen using spray-drying technique. Int J Pharm. 2008;355:277–284. doi: 10.1016/j.ijpharm.2007.12.020. [DOI] [PubMed] [Google Scholar]
- 12.Kocbek P., Baumgartner S., Kristl J. Preparation and evaluation of nanosuspensions for enhancing the dissolution of poorly soluble drugs. Int J Pharm. 2006;312:179–186. doi: 10.1016/j.ijpharm.2006.01.008. [DOI] [PubMed] [Google Scholar]
- 13.Mahmoud E.B., Nazrul H., Gihan F. Solubility and dissolution enhancement of tadalafil using self-nanoemulsifying drug delivery system. J Oleo Sci. 2014;63:567–576. doi: 10.5650/jos.ess13236. [DOI] [PubMed] [Google Scholar]
- 14.Müller R.H., Runge S., Ravelli V. Oral bioavailability of cyclosporine: solid lipid nanoparticles (SLN®) versus drug nanocrystals. Int J Pharm. 2006;317:82–89. doi: 10.1016/j.ijpharm.2006.02.045. [DOI] [PubMed] [Google Scholar]
- 15.Javadzadeh Y., Shariati H., Movahhed-Danesh E. Effect of some commercial grades of microcrystalline cellulose on flowability, compressibility, and dissolution profile of piroxicam liquisolid compacts. Drug Dev Ind Pharm. 2009;35:243–251. doi: 10.1080/03639040802277672. [DOI] [PubMed] [Google Scholar]
- 16.Elkordy A.A., Tan X.N., Essa E.A. Spironolactone release from liquisolid formulations prepared with Capryol™ 90, Solutol® HS-15 and Kollicoat® SR 30 D as non-volatile liquid vehicles. Eur J Pharm Biopharm. 2013;83:203–223. doi: 10.1016/j.ejpb.2012.08.004. [DOI] [PubMed] [Google Scholar]
- 17.Suliman A.S., Anderson R.J., Elkordy A.A. Norfloxacin as a model hydrophobic drug with unique release from liquisolid formulations prepared with PEG 200 and Synperonic PE/L-61 non-volatile liquid vehicles. Powder Technol. 2014;257:156–167. [Google Scholar]
- 18.Spireas S., Bolton S.M. 1999. Liquisolid systems and methods of preparing same. US5968550. [Google Scholar]
- 19.Spireas S. 2002. Liquisolid system and method of preparing same. U.S Patent; 6423339B1. [Google Scholar]
- 20.Spireas S.S., Jarowski C.I., Rohera B.D. Powdered solution technology: principles and mechanism. Pharm Res. 1992;9:1351–1358. doi: 10.1023/a:1015877905988. [DOI] [PubMed] [Google Scholar]
- 21.Spireas S., Sadu S. Enhancement of prednisolone dissolution properties using liquisolid compacts. Int J Pharm. 1998;166:177–188. [Google Scholar]
- 22.Fahmy R.H., Kassem M.A. Enhancement of famotidine dissolution rate through liquisolid tablets formulation: in vitro and in vivo evaluation. Eur J Pharm Biopharm. 2008;69:993–1003. doi: 10.1016/j.ejpb.2008.02.017. [DOI] [PubMed] [Google Scholar]
- 23.Komala D.R., Janga K.Y., Jukanti R. Competence of raloxifene hydrochloride loaded liquisolid compacts for improved dissolution and intestinal permeation. J Drug Deliv Sci Technol. 2015;30:232–241. [Google Scholar]
- 24.Sanka K., Poienti S., Mohd A.B. Improved oral delivery of clonazepam through liquisolid powder compact formulations: in-vitro and ex-vivo characterization. Powder Technol. 2014;256:336–344. [Google Scholar]
- 25.Javadzadeh Y., Musaalrezaei L., Nokhodchi A. Liquisolid technique as a new approach to sustain propranolol hydrochloride release from tablet matrices. Int J Pharm. 2008;362:102–108. doi: 10.1016/j.ijpharm.2008.06.022. [DOI] [PubMed] [Google Scholar]
- 26.Nokhodchi A., Aliakbar R., Desai S. Liquisolid compacts: the effect of cosolvent and HPMC on theophylline release. Colloids Surf B Biointerfaces. 2010;79:262–269. doi: 10.1016/j.colsurfb.2010.04.008. [DOI] [PubMed] [Google Scholar]
- 27.Pavani E., Noman S., Syed I.A. Liquisolid technique based sustained release tablet of trimetazidine dihydrochloride. Drug Invention Today. 2013;5:302–310. [Google Scholar]
- 28.El-Hammadi M., Awad N. Investigating the use of liquisolid compacts technique to minimize the influence of pH variations on loratadine release. AAPS PharmSciTech. 2012;13:53–58. doi: 10.1208/s12249-011-9719-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Badawy M.A., Kamel A.O., Sammour O.A. Use of biorelevant media for assessment of a poorly soluble weakly basic drug in the form of liquisolid compacts: in vitro and in vivo study. Drug Deliv. 2016;23:818–827. doi: 10.3109/10717544.2014.917442. [DOI] [PubMed] [Google Scholar]
- 30.Khames A. Liquisolid technique: a promising alternative to conventional coating for improve ment of drug photostability in solid dosage forms. Drug Deliv. 2013;10:1335–1343. doi: 10.1517/17425247.2013.798297. [DOI] [PubMed] [Google Scholar]
- 31.Javadzadeh Y., Siahi M.R., Asnaashari S. An investigation of physicochemical properties of piroxicam liquisolid compacts. Pharm Dev Technol. 2007;12:337–343. doi: 10.1080/10837450701247574. [DOI] [PubMed] [Google Scholar]
- 32.Javadzadeh Y., Siahi M.R., Asnaashari S. Liquisolid technique as a tool for enhancement of poorly water-soluble drugs and evaluation of their physicochemical properties. Acta Pharm. 2007;57:99–109. doi: 10.2478/v10007-007-0008-6. [DOI] [PubMed] [Google Scholar]
- 33.Chella N., Narra N., Rama R.T. Preparation and characterization of liquisolid compacts for improved dissolution of telmisartan. J Drug Deliv. 2014;2014:1–10. doi: 10.1155/2014/692793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tayel S.A., Soliman I.I., Louis D. Improvement of dissolution properties of carbamazepine through application of the liquisolid tablet technique. Eur J Pharm Biopharm. 2008;69:342–347. doi: 10.1016/j.ejpb.2007.09.003. [DOI] [PubMed] [Google Scholar]
- 35.Gubbi S.R., Jarag R. Formulation and characterization of atorvastatin calcium liquisolid compacts. Asian J Pharm Sci. 2010;5:50–60. [Google Scholar]
- 36.Karmarkar A.B., Gonjari I.D., Hosmani A.H. Evaluation of in vitro dissolution profile comparison methods of sustained release tramadol hydrochloride liquisolid compact formulations with marketed sustained release tablets. Drug Discov Ther. 2010;4:26–32. [PubMed] [Google Scholar]
- 37.Singh S.K., Srinivasan K.K., Gowthamarajan K. Influence of formulation parameters on dissolution rate enhancement of glyburide using liquisolid technique. Drug Dev Ind Pharm. 2012;38:961–970. doi: 10.3109/03639045.2011.634810. [DOI] [PubMed] [Google Scholar]
- 38.Javadzadeh Y., Jafari-Navimipour B., Nokhodchi A. Liquisolid technique for dissolution rate enhancement of a high dose water-insoluble drug (carbamazepine) Int J Pharm. 2007;341:26–34. doi: 10.1016/j.ijpharm.2007.03.034. [DOI] [PubMed] [Google Scholar]
- 39.Charman S.A., Charman W.N. Oral modified release delivery systems. In: Rathbone M.J., Hadgraftb J., Roberts M.S., editors. Modified release drug delivery technology. 2003. pp. 1–9. New York. [Google Scholar]
- 40.Saeedi M., Akbari J., Morteza-Semnani K. Enhancement of dissolution rate of indomethacin using liquisolid compacts. Iran J Pharm Res. 2011;10:25–34. [PMC free article] [PubMed] [Google Scholar]
- 41.Karmarkar A.B., Gonjari I.D., Hosmani A.H. Dissolution rate enhancement of fenofibrate using liquisolid tablet technique. Lat Am J Pharm. 2009;28:219–225. [Google Scholar]
- 42.Hentzschel C.M., Sakmann A., Leopold C.S. Suitability of various excipients as carrier and coating materials for liquisolid compacts. Drug Dev Ind Pharm. 2011;37:1200–1207. doi: 10.3109/03639045.2011.564184. [DOI] [PubMed] [Google Scholar]
- 43.Nokhodchi A., Hentzschel C.M., Leopold C.S. Drug release from liquisolid systems: speed it up, slow it down. Expert Opin Drug Deliv. 2011;8:191–205. doi: 10.1517/17425247.2011.548801. [DOI] [PubMed] [Google Scholar]
- 44.Fuji Chemical Industry Co., Ltd. Fujicalin® – The unique DCPA. 2011. http://www.fujicalin.com/product/general_properties.php Available from. Accessed 28 March 2016.
- 45.Fuji Chemical Industry Co., Ltd. Neusilin ® – The specialty excipient. 2009. http://www.neusilin.com/product/general_properties.php Available from. Accessed 28 March 2016.
- 46.Vranikova B., Gajdziok J., Vetchy D. Determination of flowable liquid retention potential of aluminometasilicate carrier for liquisolid systems preparation. Pharm Dev Technol. 2015;20:839–844. doi: 10.3109/10837450.2014.926921. [DOI] [PubMed] [Google Scholar]
- 47.Hentzschel C.M., Alnaief M., Smirnova I. Enhancement of griseofulvin release from liquisolid compacts. Eur J Pharm Biopharm. 2012;80:130–135. doi: 10.1016/j.ejpb.2011.08.001. [DOI] [PubMed] [Google Scholar]
- 48.Speybroeck V.M., Mellaerts R., Mols R. Enhanced absorption of the poorly soluble drug fenofibrate by tuning its release rate from ordered mesoporous silica. Eur J Pharm Sci. 2010;41:623–630. doi: 10.1016/j.ejps.2010.09.002. [DOI] [PubMed] [Google Scholar]
- 49.Chen B., Quan G., Wang Z. Hollow mesoporous silicas as a drug solution delivery system for insoluble drugs. Powder Technol. 2013;240:48–53. [Google Scholar]
- 50.Gavali S.M., Pacharane S.S., Sankpal S.V. Liquisolid compact: a new technique for enhancement of drug dissolution. Int J Res Pharm Chem. 2011;1:705–713. [Google Scholar]
- 51.Yadav V.B., Yadav A.V. Improvement of solubility and dissolution of indomethacin by liquisolid and compaction granulation technique. J Pharm Sci Res. 2009;1:44–51. [Google Scholar]
- 52.Adibkia K., Shokri J., Barzegar-Jalali M. Effect of solvent type on retardation properties of diltiazem HCl form liquisolid tablets. Colloids Surf B Biointerfaces. 2014;113:10–14. doi: 10.1016/j.colsurfb.2013.08.017. [DOI] [PubMed] [Google Scholar]
- 53.Chellaa N., Shastria N., Tadikondab R.R. Use of the liquisolid compact technique for improvement of the dissolution rate of valsartan. Acta Pharm Sin B. 2012;2:502–508. [Google Scholar]
- 54.Vittal G.V., Deveswaran R., Bharath S. Formulation and characterization of ketoprofen liquisolid compacts by Box-Behnken design. Int J Pharm Investig. 2012;2:150–156. doi: 10.4103/2230-973X.104398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bonthagarala B., Sai P.D.L., Venkata S.K. Enhancement of dissolution rate of clofibrate (BCS Class – II drug) by using liquisolid compact technology. Int J Biomed Adv & Res. 2015;6:288–298. [Google Scholar]
- 56.Pezzini B.R., Beringhs A.O., Ferraz H.G. Liquisolid technology applied to pellets: evaluation of the feasibility and dissolution performance using felodipine as a model drug. Chem Eng Res Des. 2016 [Google Scholar]
- 57.Khan A., Iqbal Z., Shah Y. Enhancement of dissolution rate of class II drugs (hydrochlorothiazide); a comparative study of the two novel approaches; solid dispersion and liquid-solid techniques. Saudi Pharm J. 2015;23:650–657. doi: 10.1016/j.jsps.2015.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Khaled K.A., Asiri Y.A., El-Sayed Y.M. In vivo evaluation of hydrochlorothiazide liquisolid tablets in beagle dogs. Int J Pharm. 2001;222:1–6. doi: 10.1016/s0378-5173(01)00633-0. [DOI] [PubMed] [Google Scholar]
- 59.Nokhodchi A., Javadzadeh Y., Siahi-Shadbad M.R. The effect of type and concentration of vehicles on the dissolution rate of a poorly soluble drug (indomethacin) from liquisolid compacts. J Pharm Sci. 2005;8:18–25. [PubMed] [Google Scholar]
- 60.Yadav V.B., Yadav A.V. Liquisolid granulation technique for tablet manufacturing: an overview. J Pharm Res. 2009;2:670–674. [Google Scholar]
- 61.Lu M., Xing H.N., Yang T.Z. Dissolution enhancement of tadalafil by liquisolid technique. Pharm Dev Technol. 2016 doi: 10.1080/10837450.2016.1189563. Accepted. [DOI] [PubMed] [Google Scholar]
- 62.Elkordy A.A., Essa E.A., Dhuppad S. Liquisolid technique to enhance and to sustain griseofulvin dissolution: effect of choice of non-volatile liquid vehicles. Int J Pharm. 2012;434:122–132. doi: 10.1016/j.ijpharm.2012.05.072. [DOI] [PubMed] [Google Scholar]
- 63.Gonjari I.D., Karmarkar A.B., Hosmani A.H. Evaluation of in vitro dissolution profile comparison methods of sustained release tramadol hydrochloride liquisolid compact formulations with marketed sustained release tablets. Dig J Nanomater Bios. 2009;4:651–661. [PubMed] [Google Scholar]
- 64.Khanfar M., Salem M.S., Kaddour F. Preparation of sustained-release dosage form of Venlafaxine HCl using liquisolid technique. Pharm Dev Technol. 2014;19:103–115. doi: 10.3109/10837450.2012.757785. [DOI] [PubMed] [Google Scholar]
- 65.Ramenskaia G.V., Shokhin I.E., Savchenko A.I. The dissolution test in biorelevant media as a prognostic tool for modeling of drug behavior in vivo. Biomed Khim. 2011;57:482–489. doi: 10.18097/pbmc20115705482. [DOI] [PubMed] [Google Scholar]
- 66.Tonnensen H.H. Formulation and stability testing of photolabile drugs. Int J Pharm. 2001;225:1–14. doi: 10.1016/s0378-5173(01)00746-3. [DOI] [PubMed] [Google Scholar]