

Graphical Abstract
Terms and tools of Quality by Design (QbD) in drug development.
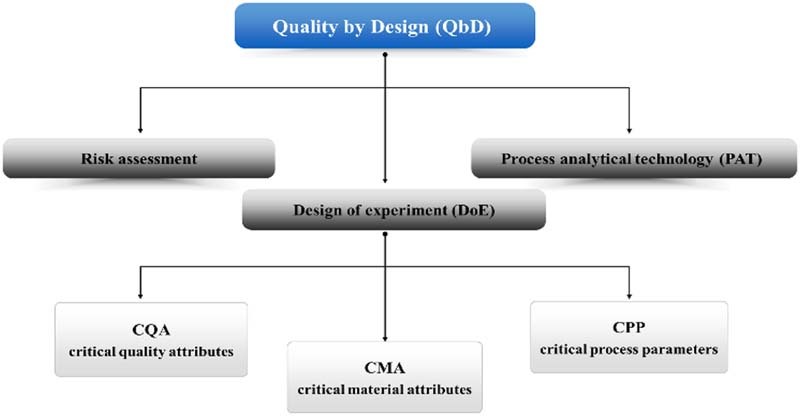
Keywords: Quality by design (QbD), Process analytical technology (PAT), Critical quality attributes (CQA), Design of experiment (DoE), Risk assessment, Near infrared (NIR) spectroscopy
Abstract
Quality by Test was the only way to guarantee quality of drug products before FDA launched current Good Manufacturing Practice. To clearly understand the manufacture processes, FDA generalized Quality by Design (QbD) in the field of pharmacy, which is based on the thorough understanding of how materials and process parameters affect the quality profile of final products. The application of QbD in drug formulation and process design is based on a good understanding of the sources of variability and the manufacture process. In this paper, the basic knowledge of QbD, the elements of QbD, steps and tools for QbD implementation in pharmaceutics field, including risk assessment, design of experiment, and process analytical technology (PAT), are introduced briefly. Moreover, the concrete applications of QbD in various pharmaceutical related unit operations are summarized and presented.
1. Introduction
While medicine is well known as special goods, the development of pharmaceutical industry is based on innovation and manufacturing. However, there are lots of complaints from pharmaceutical industry about the strict rules. In current quality by test (QbT) system (Fig. 1a), product quality is ensured by following a sequence of steps, including raw material testing, fixed drug product manufacturing process, and end product testing. It is only when all the specifications of the FDA or other standards are complied with that the materials can be used for manufacturing or come into market. If not, they need to be reprocessed. Root causes for failure are usually not well understood due to the poor process understanding and uncertainty about how characteristics of substances impacts target product profile. As a result, the manufacturers have to restart the procedure until the root causes of failure are understood and addressed or FDA approves supplements to revise (e.g., widen) the acceptance criteria to pass the previously failed batches [1]. This causes poor cost-efficiency and product variation, which may lead to poor drug safety.
Fig. 1.
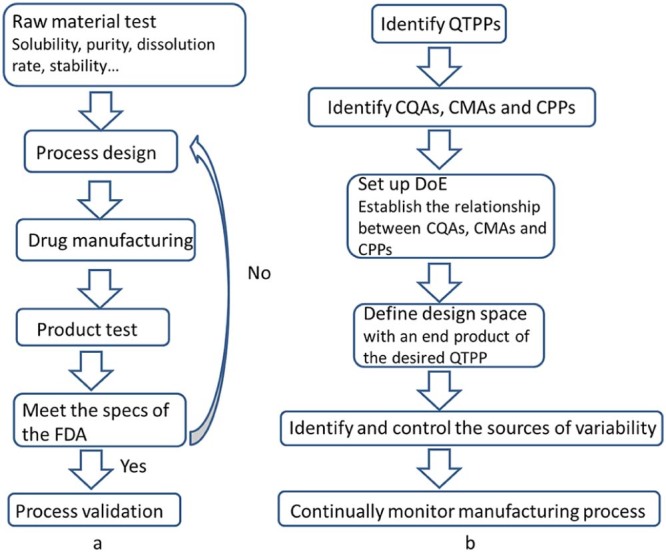
Comparison between QbT (a) and QbD (b). (QbT: quality by test; QbD: quality by design; QTPP: quality target product profile; CQA: critical quality attributes; CMA: critical material attributes; CPP: critical process parameters; DoE: design of experiments).
Fortunately, with the development of the concept “Quality by Design (QbD)”, there will be a significant transformation in pharmaceutical quality regulation, from an empirical process to a more scientific and risk-based approach. QbD (Fig. 1b) is a systematic risk-based, proactive approach to pharmaceutical development that begins with predefined objectives and emphasizes product and process understanding and process control based on sound science and quality risk management. Comparison between QbT and QbD procedures is shown in Fig. 1.
However, although there are some reviews about the theory of QbD [1], [2], the papers about application of different analytical tools, such as Raman spectroscopy and near-infrared spectroscopy, in research and development of pharmaceutical dosage forms are also available [3], [4], [5], [6]. References about the application of different analytical methods as monitoring tools in the framework of QbD are not available to the best of our knowledge. Nevertheless, good implementation of QbD in formulation and process design in pharmaceutical field is highly dependent on a good understanding of the sources of variability and the manufacture process, and Process Analytical Technology (PAT) is an indispensible tool in the QbD system. Therefore, the objective of this paper is to provide a whole picture about the application of QbD in pharmaceutical field by using PAT as a tool. Except for the basic knowledge of QbD, the elements of QbD, steps and tools for implementation of QbD in the field of pharmaceutics, and the applications of QbD in various dosage forms, which are summarized and presented as guidance. Moreover, PAT tools applied in different manufacturing processes in the QbD system have been summarized to provide an insight in the continuous manufacturing process.
2. Understanding pharmaceutical QbD
To overcome the limitation of GMP, FDA launched cGMP in 2002 [7], [8]. cGMP places emphasis on the “software” during the manufacturing, namely management level, and specifies staff's responsibility strictly and clearly. In contrast, GMP attaches a great importance on the qualification and training details of the staff instead of their duties, and these relatively lower requirements are still broadly used in many developing countries. After the cGMP was carried out, there is still another problem, that is, in comparison with other industries, such as automobile, aircraft and electronic industries, the specification of pharmaceutical industry is much more rigid and fixed. However, it is almost impossible to keep all the parameters of the whole conditions constant and the environment may vary in small degrees inevitably. Then, the problem is in the approval documents for a new product to be handed over to FDA, the company can only write fixed number in the report, as ‘details’ and ‘the authenticity of the process’ are quite critical in cGMP, it may happen that batches of products fail to meet the rigid specifications. To solve this problem, the International Conference on Harmonization (ICH) and FDA began to learn from the other industries, and QbD was introduced into the chemical manufacturing control (CMC) review pilot program in 2004 with the objective of improving pharmaceutical drug quality and safety to achieve a desired state for pharmaceutical manufacturing on the basis of scientific and engineering knowledge. The function of QbD, Design Space and real-time release had been evaluated through the CMC project. Years later, a series of guidelines was published by ICH: ICH Q8 Pharmaceutical Development [9], ICH Q9 Quality Risk Management [10], ICH Q10 Pharmaceutical Quality System [11], and the ICH Q11 Development and Manufacture of Drug Substances [12].
Quality by Design (QbD) is defined in the ICH Q8 guideline as ‘a systematic approach to development that begins with predefined objectives and emphasizes product and process understanding and process control, based on sound science and quality risk management’ [9], which is in accordance with FDA's current drug quality system ideology of ‘quality cannot be tested into products; it should be built-in or should be by design.’ [13]
2.1. Elements of QbD
There are several statements about the elements of QbD, the most widely accepted is that, QbD consists of the following parameters [2], [9]:
Quality Target Product Profile (QTPP): including dosage form, delivery systems, dosage strength(s), etc. It is a prospective summary of quality characteristics of a drug product to be achieved, taking into account dosage strength(s) and container closure system of the drug product, together with the attributes affecting pharmacokinetic characteristics (e.g., dissolution, aerodynamic performance) and drug product quality criteria (e.g., sterility, purity, stability and drug release) appropriate for the intended marketed product.
Critical Quality Attributes (CQAs): including physical, chemical, biological, or microbiological properties or characteristics of an output material including finished drug product. Potential drug product CQAs derived from the QTPP and/or prior knowledge are used to guide the product and process development and they should be within an appropriate limit, range, or distribution to ensure the desired product quality.
Critical Material Attributes (CMAs): including physical, chemical, biological, or microbiological properties or characteristics of an input material. CMAs should be within an appropriate limit, range, or distribution to ensure the desired quality of that drug substance, excipient, or in-process material.
Critical Process Parameters (CPPs): parameters monitored before or in process that influence the appearance, impurity, and yield of final product significantly.
During the QbD process, product design and understanding include the identification of CMAs, which are different from CQAs. CQAs are for output materials while CMAs are for input materials including drug substance, excipients, in-process materials. The CQA of an intermediate may become a CMA of the same intermediate for a downstream manufacturing step. While process design and understanding include the identification of CPPs and a thorough understanding of scale-up principles, linking CMAs and CPPs to CQAs is of special importance. From the viewpoint of QbD, CMAs and CPPs can vary within the established Design Space without significant influence on CQAs, and as a result, the quality of the final product will meet the QTPP.
2.2. Steps for Pharmaceutical QbD implementation
As a general rule, the practical implementation of QbD in the development of new pharmaceutical products can go through the following steps [1], [14], [15]:
-
1.
Define the desired performances of the product and identify the QTPPs;
-
2.
Identification of the CQAs;
-
3.
Identification of possible CMAs and CPPs;
-
4.
Setup and execution of DoE to link CMAs and CPPs to CQAs and get enough information of how these parameters impact QTPP. Thereafter, a process Design Space should be defined, leading to an end product with desired QTPP;
-
5.
Identify and control the sources of variability from the raw materials and the manufacturing process;
-
6.
Continually monitor and improve the manufacturing process to assure consistent product quality.
So far, most of the pharmaceutical unit operation processes can be optimized by applying the concept of QbD [7]. Each unit operation has its own input material attributes, process parameters and quality attributes, such as during spray drying, hot melt extrusion, roller compaction and homogenization process, as summarized in Table 1.
Table 1.
Representative applications of Quality by Design in pharmaceutical unit operations and dosage forms.
| Pharmaceutical unit operations | Dosage form | Model drug | Design of experiment (DoE) | Critical material attributes (CMA) | Critical process parameters (CPP) | Critical quality attributes (CQA) |
|---|---|---|---|---|---|---|
| Fluid bed granulation | Tablets | Not mentioned | Fractional factorial design (screening) Central composite design (optimization) |
Viscosity, temperature and concentration of the binder aqueous dispersion | Inlet air temperature, binder spray rate and air flow rate | Particle size distribution (PSD), bulk and tapped densities, flowability and angle of repose [16] |
| Roller compaction | Tablets | Not mentioned | Fractional factorial statistical design | API composition, API excipient ratio | API flow rate, lubricant flow rate, pre-compression pressure | Tablet weight, tablet dissolution, hardness, ribbon density [17] |
| Film coating | Coated tablets | Placebo tablets | Central composite – face centered – response surface design | Solid percent of the coating dispersion | Inlet air temperature, air flow rate, solid level, coating pan speed, spray rate | Appearance (coating defects, gloss, and color uniformity), disintegration time (dissolution of the film coating) [18] |
| Spray drying | Solid nano-crystalline dry powders | Indomethacin | Full factorial design | NA | Inlet temperature, flow rate, aspiration rate | Particle size, moisture content, percent yield, crystallinity [19] |
| Hot-melt extrusion (HME) | Solid lipid nanoparticles (SLN) | Fenofibrate (FBT) | Plackett–Burman (PB) screening design | Lipid concentration, surfactant concentration | Screw speed, barrel temperature, zone of liquid addition | Particle size, polydispersibility index, zeta potential, entrapment efficiency [20] |
| Homogenization | Nanoparticles | Paclitaxel | Box–Behnken design | Surfactant concentration in aqueous phase (%) | Homogenization rate | Average particle size, zeta potential, encapsulation efficiency [21] |
| Solid lipid nanoparticle (SLN) | Rivastigmine | Factorial design | Drug: lipid ratio, surfactant concentration | Homogenization time | Size, PDI, entrapment efficiency [22] | |
| O/W emulsification–solvent evaporation | Nanoparticles | Cyclosporine A (CyA) | Plackett–Burman (PB) design | Type of solvent organic to aqueous phase ratio, drug concentration, polymer concentration, surfactant concentration, O/W ratio | Stirring rate | Encapsulation efficiency, particle size, zeta potential, burst release and dissolution efficiency [23] |
| Physical mixture, solvent evaporation | Controlled-release tablets | Felodipine | Box–Behnken design | Amount of polymer HPMC Amount of polymeric surfactants, amount of Pluronic F127 |
Preparation technique | Maximum solubility after 30 min, equilibrium solubility after 24 h, dissolution efficiency [24] |
| Homogenate membrane method | Orodispersible films | Theophylline | Central composite design | Percentage of HPMC, percentage of glycerol | Drying temperature | Tensile strength, elongation at break, Young's modulus, disintegration time [25] |
NA, not available.
3. Tools of QbD
The concept of QbD has two components – the science underlying the design and the science of manufacturing. Upon understanding the elements of QbD and the steps for QbD implementation, it is important to be familiar with the commonly used tools in QbD, including risk assessment, design of experiment (DoE), and process analytical technology (PAT) [9].
3.1. Risk assessment
Risk assessment is a systematic process of organizing information to support a risk decision to be made within a risk management process. It consists of the identification of hazards and the analysis and evaluation of risks associated with exposure to those hazards. It is the first step of quality risk management process; the other two steps are risk control and risk review. Risk control includes decision making to reduce and/or accept risks. The purpose of risk control is to reduce the risk to an acceptable level. At the final stage, the output/results of the risk management process should be reviewed to take into account new knowledge and experience. Throughout the risk management process, risk communication, the sharing of information about risk and risk management between the parties (including regulators and industry, industry and the patient, within a company, industry or regulatory authority, etc.), should be ongoing at any stage of the risk management process. The included information might relate to the existence, nature, form, probability, severity, acceptability, control, treatment, detectability or other aspects of risks to quality [10].
There are three components of risk assessment, that is, risk identification, risk analysis and risk evaluation. (1) Risk Identification: The systematic use of information to identify potential sources of harm (hazards) that are referring to the risk question or problem description, which can include historical data, theoretical analysis, informed opinions, and the concerns of stakeholders; (2) Risk Analysis: The estimation of the risk associated with the identified hazards; (3) Risk Evaluation: The comparison of the estimated risk to given risk criteria using a quantitative or qualitative scale to determine the significance of the risk.
The above components aim at giving answers to the following three questions in the pre-formulation study, (1) What might go wrong? (2) What is the likelihood (probability) it will go wrong? (3) What are the consequences (severity)? The evaluation of the risk to quality should be based on scientific knowledge and ultimately link to the protection of the patient, the level of effort and formality.
ICH Q9 provides a non-exhaustive list of 9 common risk management tools as follows [10]: (1) Basic risk management facilitation methods (Ishikawa fishbone diagram, flowcharts, check sheets, etc.); (2) Fault tree analysis; (3) Risk ranking and filtering; (4) Preliminary hazard analysis; (5) Hazard analysis and critical control points; (6) Failure mode and effects analysis (FMEA); (7) Failure mode, effects, and criticality analysis (FMECA); (8) Hazard operability analysis; (9) Supporting statistical tools.
According to the implementation of QbD, risk assessment has the priority over DoE. Among the tools, Ishikawa fishbone diagram [26] and FMEA are widely used approaches for risk assessment, either separately [27] or in combination [28]. Taking the preparation of extruded particles as an example, the Ishikawa diagram is shown in Fig. 2. The risk factors in the fishbone diagram are classified into broad categories, while the FMEA could identify the failure modes that have the greatest chance of causing product failure, which means each of the factors in the Ishikawa fishbone diagrams will be ranked later in the FMEA analysis. The FMEA method can be used to perform the quantitative risk assessment, identifying the CQAs that have the greatest chance of causing product failure. The outcome of an FMEA are risk priority numbers (RPN) for each combination of failure mode severity, occurrence probability, and likelihood of detection. The RPN is defined as [29]:
where Occurrence probability (O), Severity (S), and Detectability (D) are all expressed with scale 1–5. For Occurrence probability (O), the number 5 represents likely to occur; number 3 for 50:50 chance of occurring, and number 1 for unlikely to occur. The Severity (S) is a measure of how severe of an effect a given failure mode would cause, number 5 means severe effect, 3 for moderate effect, and 1 for no effect. The Detectability is denoted by parameter D, the more detectable a failure mode is, the less risk it presents to product quality. For D, similar to parameter O and S, number 1 means easily detectable, number 3 for moderately detectable and number 5 represents hard to detect.
Fig. 2.

Ishikawa diagram for preparation of extruded particles [12]
(reproduced with permission from Elsevier).
3.2. Design of experiment (DoE)
To carry out the design of experiment, the risk assessment should be taken into function first. A structured, organized method for determining the relationship between factors affecting a process and the output of that process is known as “Design of Experiments” (DoE). DoE is an excellent tool that allows pharmaceutical scientists to systematically manipulate factors according to a pre-specified design. A good design is based on sound cognition of product and effective management of whole process during manufacturing. DoE studies work together with mechanism-based studies to achieve better product and process understanding.
DoE is a reasonable method to determine the relationship between the inputs and outputs of a process. It can help identify optimal conditions, CMAs, CPPs, and, ultimately, the Design Space. It is wise to establish a Design Space through DoE for multivariate experiments. ICH Q8 defines the Design Space as “the multidimensional combination and interaction of input variables (e.g., material attributes) and process parameters that have been demonstrated to provide assurance of quality” [9]. It has been reported that there is no need to hand over supplements to revise (e.g., widen) the acceptance criteria to FDA if the changes are within the Design Space.
So far, a number of studies have been launched in the drug delivery systems after QbD initiative was claimed, as summarized in Table 1. It has been demonstrated that DoE is effective in the design of different dosage forms and unit operations, it can be used more broadly in the near future to guarantee high research efficiency with improved product quality.
3.3. PAT as an important tool of QbD
PAT is defined as “Tools and systems that utilize real-time measurements, or rapid measurements during processing, of evolving quality and performance attributes of in-process materials to provide information to ensure optimal processing to produce final product that consistently conforms to established quality and performance standards” [10]. ICH Q8 [9] identifies the use of PAT to ensure that the process remains within an established Design Space.
The concept originates from the desire of the regulators to shift control of product quality toward a science-based approach that explicitly attempts to reduce the risk to patients by controlling the manufacturing based on understanding of the process.
From a PAT standpoint, a process is considered well understood when [26], [30]:
-
(1)
All critical sources of variability are identified and explained;
-
(2)
Variability is managed by the process; and
-
(3)
Product quality attributes can be accurately and reliably predicted.
3.3.1. PAT steps
With the combination of guideline [13] and literatures of Read et al. [31], [32], there is a three-step-process in the design and optimization of drug formulations and manufacturing processes, namely design, analyze and control.
In the design step, experimentation is performed to understand which quality attributes are related to a given unit operation and which process parameters and raw material attributes have the most impact on the final product quality. This knowledge is then used to identify the QTPP, CPP and CQA, which are needed for consideration in the design of an effective PAT based control scheme for the process.
In the analysis step, to identify the chosen quality attribute and process parameters and the raw material attributes, a process measurement system allows for real time (or near real time) monitoring of all CQAs and CPPs, using direct or indirect analytical methods with appropriate analytical tools.
Finally, control strategies provide adjustments to ensure control of all critical attributes, and set up the understanding of relationships among CQAs, CPPs and QTPPs so as to decide what action to take in case the process performance deviates from the optimal path or product quality from the desired attributes [33].
3.3.2. PAT tools
For the sake of understanding scientific, risk-managed pharmaceutical development, manufacture, and quality assurance, many tools are available in the PAT framework. They can be categorized into four classes according to the PAT guidance [13]:
-
(1)
Multivariate tools for design, data acquisition and analysis;
-
(2)
Process analyzers;
-
(3)
Process control tools;
-
(4)
Continuous improvement and knowledge management tools.
As defined by FDA's PAT guidance document, whether to remove the sample or not, process analysis can be divided into three categories, namely at-line, on-line and in-line [13]: (1) At-line: Measurement where the sample is removed, isolated from, and analyzed in close proximity to the process stream; (2) On-line: Measurement where the sample is diverted from the manufacturing process, and may be returned to the process stream; (3) In-line: Measurement where the sample is not removed from the process stream and can be invasive or noninvasive.
It is obvious that PAT is definitely effective in helping QbD implement. It can do a job of real-time monitoring of process without interruption to get technological parameters and material parameters on-line. PAT enhances understanding of technology (including the relationship between CQA and CPP), which leads to accomplishment of quality improvement and register simplification.
3.3.3. Application of PAT
In most cases, spectroscopic techniques, including Raman spectroscopy, UV–VIS spectroscopy, and nuclear magnetic resonance (NMR), are commonly used. Besides, other PAT analytical methods, such as Near Infrared spectroscopy (NIR), focused beam reflectance measurements (FBRM), nanometric temperature measurement (MTM), tunable diode laser absorption spectroscopy (TDLAS), are widely applied in the pharmaceutical manufacturing field and play important roles in the real-time monitoring of the processes, as summarized in Table 2.
Table 2.
Representatives of some monitoring tools used in pharmaceutical processes (2011–2015).
| Processes | Monitoring tool | Attributes measured | Major outcome |
|---|---|---|---|
| Co-precipitation process | Lasentec particle vision microscopy system PVM819 | Nucleation and crystal growth | Obtain direct information about the morphology and size of the co-precipitates [34] |
| Mammalian cell culture process | Raman spectroscopy | Glycoprotein product yield | Selecting which small scale batches are progressed to large-scale manufacture, improving process efficiency significantly [35] |
| Chinese hamster ovary (CHO) cell fed-batch process | Fluorescence excitation–emission matrix (EEM) spectroscopy | Key fluorophores (e.g. tyrosine, tryptophan, and the glycoprotein product) | Quantitative predictive analysis of recombinant glycoprotein production [36] |
| Fluid bed granulation | Microwave resonance technology (MRT) | Determine moisture, temperature and density of the granules | Predict information about the final granule size [16], [37] |
| Pan coating process | New real-time monitoring tool (PyroButtons) | Record and data in real-time | Move with the tablets providing information on the thermodynamic conditions (microenvironment) [38] |
| Continuous direct compaction tablet manufacturing process | Near infrared (NIR) spectroscopy | Powder blend bulk density | The NIR spectra are sent to a real time prediction engine that utilizes the NIR calibration models for blend density and drug concentration and a real time prediction tool (OLUPX) to generate the signals for the control variables in real time [39] |
Among those PAT tools, NIR has drawn great attention in the pharmaceutical industry, it is a rapid, non-invasive analytical technique and there is no need for extensive sample preparation. NIR has been described in both the United States and the European Pharmacopeia. It is the most commonly used device in the manufacturing process, and it has been used for the identification and characterization of raw materials and intermediates, analysis of dosage forms manufacturing, and prediction of one or more variables in process streams or final product streams (composition) on the basis of on-line, in-line or at-line spectroscopic measurements [40]. Its concrete applications in different unit operations are exemplified in Table 3.
Table 3.
Representative applications of near infrared spectroscopy (NIR) in representative unit operations.
| Unit operations | Parameters | Description |
|---|---|---|
| Crystallization | Polymorphism and particle size of the indomethacin powder | In-line [41] |
| Determine API and residual solvent contents | On-line [42] | |
| Co-precipitation | Turbidity monitoring, and in situ crystal size monitoring | On-line [43] |
| Freeze-drying | Moisture content analysis | In-line [44] |
| Hot-melt extrusion | Screw speed and drug loading | In-line [45] |
| Powder mixing | Monitor blending uniformity | In-line [28] |
| Compression | Content uniformity | On-line [46] |
| Continuous granulation process | Show the variation in solid state (transform anhydrous theophylline to theophylline monohydrate) | In-line [47] |
| Fluidized bed granulation | Determine the moisture content, size distribution, and bulk density | In-line [48] |
| Fluid-bed coating | Film thickness on pharmaceutical pellets | In-line [49] |
Due to the complexity of pharmaceutical product-process design, an efficient and systematic understanding coupled with an inference system is essential. The real-time monitoring tools have increasingly attracted the interests of pharmaceutical manufacturers. So far, the continuous manufacturing and real-time monitoring are mostly used in the tablet manufacturing processes. With the successful application in the tablets, the PAT tools in other dosage forms manufacturing will soon be in use.
4. Conclusion
The fast growth of interest in QbD and its tools indicates that the approaches are not fashionable phenomena but responses to the demands of modern manufacturing process. QbD is a cost and time efficient approach in design and manufacturing, with DoE, risk assessment, and PAT as its tools to achieve a better understanding on the materials and processes, which make the QbD available and feasible to the pharmaceutical field. With its broad implementation in the pharmaceutical manufacture, drug products with high and reproducible quality can be anticipated. Moreover, QbD has become a broadly applicable manufacturing model and is going far beyond pharmaceutical (or related) areas.
Acknowledgements
This project is financially supported by Talents Project of Liaoning Province, China (LR2013047).
Footnotes
Peer review under responsibility of Shenyang Pharmaceutical University.
References
- 1.Yu L.X. Pharmaceutical quality by design: product and process development, understanding, and control. Pharm Res. 2008;25(4):781–791. doi: 10.1007/s11095-007-9511-1. [DOI] [PubMed] [Google Scholar]
- 2.Yu L.X., Amidon G., Khan M.A. Understanding pharmaceutical quality by design. AAPS J. 2014;16(4):771–783. doi: 10.1208/s12248-014-9598-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.De Beer T., Burggraeve A., Fonteyne M. Near infrared and Raman spectroscopy for the in-process monitoring of pharmaceutical production processes. Int J Pharm. 2011;417(1–2):32–47. doi: 10.1016/j.ijpharm.2010.12.012. [DOI] [PubMed] [Google Scholar]
- 4.Jamrogiewicz M. Application of the near-infrared spectroscopy in the pharmaceutical technology. J Pharm Biomed Anal. 2012;66:1–10. doi: 10.1016/j.jpba.2012.03.009. [DOI] [PubMed] [Google Scholar]
- 5.Paudel A., Raijada D., Rantanen J. Raman spectroscopy in pharmaceutical product design. Adv Drug Deliv Rev. 2015;89:3–20. doi: 10.1016/j.addr.2015.04.003. [DOI] [PubMed] [Google Scholar]
- 6.Gordon K.C., McGoverin C.M. Raman mapping of pharmaceuticals. Int J Pharm. 2011;417(1–2):151–162. doi: 10.1016/j.ijpharm.2010.12.030. [DOI] [PubMed] [Google Scholar]
- 7.Food and Drug Administration . 2002. Pharmaceutical current Good Manufacturing Practices (cGMPs) for the 21st century – A risk-based approach. [Google Scholar]
- 8.Food and Drug Administration . 2003. Pharmaceutical cGMPs for the 21st century – A risk based approach: second progress report and implementation plan. [Google Scholar]
- 9.International Conference on Harmonization (ICH) 2009. Guidance for industry: Q8 (R2) pharmaceutical development, ICH harmonised tripartite guideline, step 4. [Google Scholar]
- 10.International Conference on Harmonization (ICH) 2005. Guidance for industry: Q9 quality risk management, ICH harmonised tripartite guideline, step 4. [Google Scholar]
- 11.International Conference on Harmonization (ICH) 2008. Guidance for industry: Q10 quality systems approach to pharmaceutical CGMP regulations, ICH harmonised tripartite guideline, step 4. [Google Scholar]
- 12.International Conference on Harmonization (ICH) 2012. Guidance for industry: Q11 development and manufacture of drug substances (chemical entities and biotechnological/biological entities), ICH harmonised tripartite guideline, step 4. [Google Scholar]
- 13.US Department of Health and Human Services, Food and Drug Administration (FDA), Center for Drug Evaluation and Research (CDER) 2004. PAT guidance for industry – A framework for innovative pharmaceutical development, manufacturing and quality assurance. [Google Scholar]
- 14.Tomba E., Facco P., Bezzo F. Latent variable modeling to assist the implementation of quality-by-design paradigms in pharmaceutical development and manufacturing: a review. Int J Pharm. 2013;457(1):283–297. doi: 10.1016/j.ijpharm.2013.08.074. [DOI] [PubMed] [Google Scholar]
- 15.Rathore A.S. Roadmap for implementation of quality by design (QbD) for biotechnology products. Trends Biotechnol. 2009;27(9):546–553. doi: 10.1016/j.tibtech.2009.06.006. [DOI] [PubMed] [Google Scholar]
- 16.Lourenço V., Lochmann D., Reich G. A quality by design study applied to an industrial pharmaceutical fluid bed granulation. Eur J Pharm Biopharm. 2012;81(2):438–447. doi: 10.1016/j.ejpb.2012.03.003. [DOI] [PubMed] [Google Scholar]
- 17.Singh R., Ierapetritou M., Ramachandran R. An engineering study on the enhanced control and operation of continuous manufacturing of pharmaceutical tablets via roller compaction. Int J Pharm. 2012;438(1–2):307–326. doi: 10.1016/j.ijpharm.2012.09.009. [DOI] [PubMed] [Google Scholar]
- 18.Teckoe J., Mascaro T., Farrell T.P. Process optimization of a novel immediate release film coating system using QbD principles. AAPS PharmSciTech. 2013;14(2):531–540. doi: 10.1208/s12249-013-9935-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kumar S., Gokhale R., Burgess D.J. Quality by design approach to spray drying processing of crystalline nanosuspensions. Int J Pharm. 2014;464(1–2):234–242. doi: 10.1016/j.ijpharm.2013.12.039. [DOI] [PubMed] [Google Scholar]
- 20.Patil H., Feng X., Ye X. Continuous production of fenofibrate solid lipid nanoparticles by hot-melt extrusion technology: a systematic study based on a quality by design approach. AAPS J. 2015;17(1):194–205. doi: 10.1208/s12248-014-9674-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yerlikaya F., Ozgen A., Vural I. Development and evaluation of paclitaxel nanoparticles using a Quality-by-design approach. J Pharm Sci. 2013;102(10):3748–3761. doi: 10.1002/jps.23686. [DOI] [PubMed] [Google Scholar]
- 22.Shah B., Khunt D., Bhatt H. Application of quality by design approach for intranasal delivery of rivastigmine loaded solid lipid nanoparticles: effect on formulation and characterization parameters. Eur J Pharm Sci. 2015;78:54–66. doi: 10.1016/j.ejps.2015.07.002. [DOI] [PubMed] [Google Scholar]
- 23.Rahman Z., Zidan A.S., Habib M.J. Understanding the quality of protein loaded PLGA nanoparticles variability by Plackett–Burman design. Int J Pharm. 2010;389(1–2):186–194. doi: 10.1016/j.ijpharm.2009.12.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Basalious E.B., El-Sebaie W., El-Gazayerly O. Application of pharmaceutical QbD for enhancement of the solubility and dissolution of a Class II BCS drug using polymeric surfactants and crystallization inhibitors: development of controlled-release tablets. AAPS PharmSciTech. 2011;12(3):799–810. doi: 10.1208/s12249-011-9646-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Visser J.C., Dohmen W.M.C., Hinrichs W.L.J. Quality by design approach for optimizing the formulation and physical properties of extemporaneously prepared orodispersible films. Int J Pharm. 2015;485(1–2):70–76. doi: 10.1016/j.ijpharm.2015.03.005. [DOI] [PubMed] [Google Scholar]
- 26.Patwardhan K., Asgarzadeh F., Dassinger T. A quality by design approach to understand formulation and process variability in pharmaceutical melt extrusion processes. J Pharm Pharmacol. 2015;67(5):673–684. doi: 10.1111/jphp.12370. [DOI] [PubMed] [Google Scholar]
- 27.Bousses C., Ferey L., Vedrines E. Using an innovative combination of quality-by-design and green analytical chemistry approaches for the development of a stability indicating UHPLC method in pharmaceutical products. J Pharm Biomed Anal. 2015;115:114–122. doi: 10.1016/j.jpba.2015.07.003. [DOI] [PubMed] [Google Scholar]
- 28.Corredor C.C., Lozano R., Bu X. Analytical method quality by design for an on-line near-infrared method to monitor blend potency and uniformity. J Pharm Innov. 2015;10(1):47–55. [Google Scholar]
- 29.Fahmy R., Kona R., Dandu R. Quality by design I: application of failure mode effect analysis (FMEA) and Plackett–Burman design of experiments in the identification of “main factors” in the formulation and process design space for roller-compacted ciprofloxacin hydrochloride immediate-release tablets. AAPS PharmSciTech. 2012;13(4):1243–1254. doi: 10.1208/s12249-012-9844-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rao G., Moreira A., Brorson K. Disposable bioprocessing: the future has arrived. Biotechnol Bioeng. 2009;102(2):348–356. doi: 10.1002/bit.22192. [DOI] [PubMed] [Google Scholar]
- 31.Read E.K., Park J.T., Shah R.B. Process analytical technology (PAT) for biopharmaceutical products: part I. Concepts and applications. Biotechnol Bioeng. 2010;105(2):276–284. doi: 10.1002/bit.22528. [DOI] [PubMed] [Google Scholar]
- 32.Read E.K., Shah R.B., Riley B.S. Process analytical technology (PAT) for biopharmaceutical products: part II. Concepts and applications. Biotechnol Bioeng. 2010;105(2):285–295. doi: 10.1002/bit.22529. [DOI] [PubMed] [Google Scholar]
- 33.Orlandini S., Pinzauti S., Furlanetto S. Application of quality by design to the development of analytical separation methods. Anal Bioanal Chem. 2013;405(2–3):443–450. doi: 10.1007/s00216-012-6302-2. [DOI] [PubMed] [Google Scholar]
- 34.Wu H., White M., Khan M.A. Quality-by-Design (QbD): an integrated process analytical technology (PAT) approach for a dynamic pharmaceutical co-precipitation process characterization and process design space development. Int J Pharm. 2011;405(1–2):63–78. doi: 10.1016/j.ijpharm.2010.11.045. [DOI] [PubMed] [Google Scholar]
- 35.Li B., Ray B.H., Leister K.J. Performance monitoring of a mammalian cell based bioprocess using Raman spectroscopy. Anal Chim Acta. 2013;796:84–91. doi: 10.1016/j.aca.2013.07.058. [DOI] [PubMed] [Google Scholar]
- 36.Li B., Shanahan M., Calvet A. Comprehensive, quantitative bioprocess productivity monitoring using fluorescence EEM spectroscopy and chemometrics. Analyst. 2014;139(7):1661–1671. doi: 10.1039/c4an00007b. [DOI] [PubMed] [Google Scholar]
- 37.Lourenco V., Herdling T., Reich G. Combining microwave resonance technology to multivariate data analysis as a novel PAT tool to improve process understanding in fluid bed granulation. Eur J Pharm Biopharm. 2011;78(3):513–521. doi: 10.1016/j.ejpb.2011.02.008. [DOI] [PubMed] [Google Scholar]
- 38.Pandey P., Bindra D.S. A commentary on scale-up of pan coating process using microenvironmental control. J Pharm Sci. 2014;103(11):3412–3415. doi: 10.1002/jps.24191. [DOI] [PubMed] [Google Scholar]
- 39.Singh R., Roman-Ospino A.D., Romanach R.J. Real time monitoring of powder blend bulk density for coupled feed-forward/feed-back control of a continuous direct compaction tablet manufacturing process. Int J Pharm. 2015;495(1):612–625. doi: 10.1016/j.ijpharm.2015.09.029. [DOI] [PubMed] [Google Scholar]
- 40.Reich G. Near-infrared spectroscopy and imaging: basic principles and pharmaceutical applications. Adv Drug Deliv Rev. 2005;57:1109–1143. doi: 10.1016/j.addr.2005.01.020. [DOI] [PubMed] [Google Scholar]
- 41.Lee H.-E., Lee M.-J., Kim W.-S. In-line monitoring and interpretation of an indomethacin anti-solvent crystallization process by near-infrared spectroscopy (NIRS) Int J Pharm. 2011;420(2):274–281. doi: 10.1016/j.ijpharm.2011.08.044. [DOI] [PubMed] [Google Scholar]
- 42.Schaefer C., Clicq D., Lecomte C. A process analytical technology (PAT) approach to control a new API manufacturing process: development, validation and implementation. Talanta. 2014;120:114–125. doi: 10.1016/j.talanta.2013.11.072. [DOI] [PubMed] [Google Scholar]
- 43.Wu H., Khan M.A. Quality-by-design (QbD): an integrated process analytical technology (PAT) approach for real-time monitoring and mapping the state of a pharmaceutical coprecipitation process. J Pharm Sci. 2010;99(3):1516–1534. doi: 10.1002/jps.21923. [DOI] [PubMed] [Google Scholar]
- 44.Kauppinen A., Toiviainen M., Lehtonen M. Validation of a multipoint near-infrared spectroscopy method for in-line moisture content analysis during freeze-drying. J Pharm Biomed Anal. 2014;95:229–237. doi: 10.1016/j.jpba.2014.03.008. [DOI] [PubMed] [Google Scholar]
- 45.Islam M.T., Maniruzzaman M., Halsey S.A. Development of sustained-release formulations processed by hot-melt extrusion by using a quality-by-design approach. Drug Deliv Transl Res. 2014;4(4):377–387. doi: 10.1007/s13346-014-0197-8. [DOI] [PubMed] [Google Scholar]
- 46.Sulub Y., LoBrutto R., Vivilecchia R. Content uniformity determination of pharmaceutical tablets using five near-infrared reflectance spectrometers: a process analytical technology (PAT) approach using robust multivariate calibration transfer algorithms. Anal Chim Acta. 2008;611(2):143–150. doi: 10.1016/j.aca.2008.02.016. [DOI] [PubMed] [Google Scholar]
- 47.Fonteyne M., Vercruysse J., Diaz D.C. Real-time assessment of critical quality attributes of a continuous granulation process. Pharm Dev Technol. 2013;18(1):85–97. doi: 10.3109/10837450.2011.627869. [DOI] [PubMed] [Google Scholar]
- 48.Burggraeve A., Monteyne T., Vervaet C. Process analytical tools for monitoring, understanding, and control of pharmaceutical fluidized bed granulation: a review. Eur J Pharm Biopharm. 2013;83(1):2–15. doi: 10.1016/j.ejpb.2012.09.008. [DOI] [PubMed] [Google Scholar]
- 49.Lee M.J., Seo D.Y., Lee H.E. In line NIR quantification of film thickness on pharmaceutical pellets during a fluid bed coating process. Int J Pharm. 2011;403(1–2):66–72. doi: 10.1016/j.ijpharm.2010.10.022. [DOI] [PubMed] [Google Scholar]