

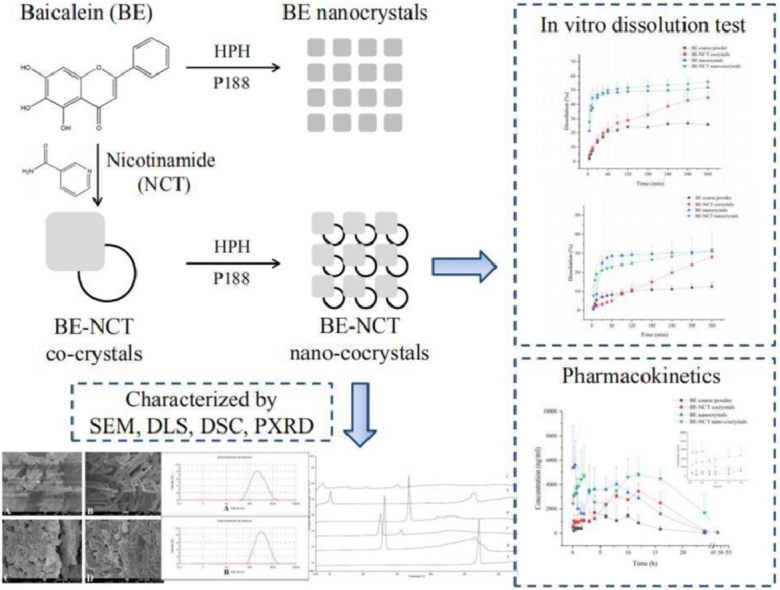
Abstract
Baicalein (BE) is one of the main active flavonoids representing the variety of pharmacological effects including anticancer, anti-inflammatory and cardiovascular protective activities, but it's very low solubility, dissolution rate and poor oral absorption limit the therapeutic applications. In this work, a nano-cocrystal strategy was successfully applied to improve the dissolution rate and bioavailability of BE. Baicalein-nicotinamide (BE-NCT) nano-cocrystals were prepared by high pressure homogenization and evaluated both in vitro and in vivo. Physical characterization results including scanning electron microscopy, dynamic light scattering, powder X-ray diffraction and differential scanning calorimetry demonstrated that BE-NCT nano-cocrystals were changed into amorphous state with mean particle size of 251.53 nm. In the dissolution test, the BE-NCT nano-cocrystals performed 2.17-fold and 2.54-fold enhancement than BE coarse powder in FaSSIF-V2 and FaSSGF. Upon oral administration, the integrated AUC0 − t of BE-NCT nano-cocrystals (6.02-fold) was significantly higher than BE coarse powder (1-fold), BE-NCT cocrystals (2.87-fold) and BE nanocrystals (3.32-fold). Compared with BE coarse powder, BE-NCT cocrystals and BE nanocrystals, BE-NCT nano-cocrystals possessed excellent performance both in vitro and in vivo evaluations. Thus, it can be seen that nano-cocrystal is an appropriate novel strategy for improving dissolution rate and bioavailability of poor soluble natural products such as BE.
Keywords: Baicalein, Nano-cocrystals, Cocrystals, Bioavailability
Graphical abstract
1. Introduction
As a significant problem, the poor aqueous solubility of nearly 40% of the marketed drugs and up to 70% of the drug candidates causes the potential risks of low oral bioavailability and exposure issues in pharmacological and toxicological studies, is important [1]. Using different methods such as suitable crystal engineering technologies or absorption-enhanced formulations to improve the solubility, dissolution, and absorption of poorly soluble compounds has a great future potential.
Several pharmaceutical methods for improving solubility or dissolution-limited drug absorption have been developed, such as solubilization, solvent mixtures, inclusion compounds, complexation and so on [2], [3], [4]. Among them, cocrystals and nanocrystals are doing well in overcoming the solubility problem.
Pharmaceutical cocrystals are multicomponent systems that composed of two or more molecules at a stoichiometric ratio and held together by H-bonding [5], [6]. Pharmaceutical cocrystals incorporate pharmaceutically acceptable conformers and the drug substance into the same crystal lattice to provide a new composition of the active pharmaceutical ingredients (APIs). According to the recent reclassification, cocrystals are considered as drug polymorph rather a new APIs which has a significant impact on drug development. Cocrystallization offers an expedient way to alter the physicochemical and biopharmaceutical properties of APIs including dissolution rate, intrinsic solubility, melting point, hygroscopicity, compressibility, bulk density and also bioavailability [7], [8], [9], [10], [11].
Different from cocrystals, nanocrystals always could improve the solubility of poorly soluble drugs. Nanocrystals, which are defined as nano-sized drug particles stabilized by surface stabilizers, have been widely studied and discussed on their saturation solubility and dissolution rate enhancement, improved oral bioavailability and bioactivities. In general, nanocrystals are prepared by top-down or bottom-up approaches. Top-down technologies consist of wet media milling and high pressure homogenization while bottom-up technologies involve anti-solvent precipitation. All those technologies are widely applied to obtain nanosized crystals. The increase in dissolution rate corresponding with the decrease in the particle size was attributed to the resultant enhancement in the surface area and decrease in the diffusion layer thickness [12]. Nanocrystal is a suitable formulation to successfully enhance the bioavailability of drugs where the dissolution rate is the rate limiting step in the absorption [13], [14], [15], [16]; however, it is not adequate to drugs whose saturation solubility is the rate limiting step because saturation solubility of drugs is not significantly improved by nanonization [17], [18].
Combining the benefits of cocrystal and nanocrystal technologies, nano-cocrystal formulations were proposed as a novel approach to achieve improving dissolution rate and oral bioavailability. However, to our knowledge, only a few nano-crystal researches had been reported, referred to the itraconazole-adipic acid [19], indomethcin-saccharin, furosemide-caffeine [20] and myricetin-nicotinamide nano-cocrystal formulations [21]. De Smet et al. reported that the itraconazole-adipic acid nano-cocrystal formulation could significantly enhance the oral bioavailability than that of amorphous formulation [19]. Recently, phenazopyridine-phthalimide nano-crystals were developed and demonstrated 2.44-fold oral bioavailability improvement than coarse suspension [22].
Baicalein (BE, shown in Fig. 1) is one of the main active flavonoids from natural product Scutellariae Radix. In spite of the variety of pharmacological effects, such as anticancer [23], anti-inflammatory [24] and cardiovascular protective activities [25], its therapeutic applications are greatly limited by its very low solubility and poor oral absorption. Although, till now, cocrystal and nanocrystal formulations of BE were achieved the solubility and bioavailability improvement to some extent [10], [15], [26], the nano-cocrystal strategy of BE was still lack of research.
Fig. 1.

The structures of BE (A) and BA (B).
Therefore, in this work, where BE was selected as the typical poorly soluble model drug, baicalein-nicotinamide (BE-NCT) nano-cocrystals were developed by the typical top-down method and evaluated both in vitro and in vivo to assess the potential of nanosized cocrystals for poorly soluble drugs as a new insight to solve the problem of insoluble drug oral absorption in advance of only cocrystals or nanocrystals.
2. Materials and methods
2.1. Materials and animals
Baicalein (BE, > 98% purity) was purchased from Nanjing Zelang Medical Technology Co. Ltd. (Nanjing, China). Poloxamer 188 was kindly gifted by BASF Co. (Ludwigshafen, Germany). Nicotinamide (NCT) was purchased from J&K Scientific Ltd. (Beijing, China). BE (98.5% purity, 111 595–201 306), baicalin (BA, 93.5% purity, 110 715–201 619) and carbamazepine (CBZ, 98.5% purity, 110 723–201 413) as internal standard (IS) were purchased from National Institute for the Control of Pharmaceutical and Biological Products (Beijing, China). Acetonitrile and methanol of HPLC grade were offered from Fisher Scientific Co. (Fair Lawn, New Jersey, USA). Formic acid was supplied from Fluka (CA, USA). All agents were analytical grade and purified Milli-Q water (Millipore, Billerica, MA, USA) was used throughout all experiments.
Sprague-Dawley rats (weighting 250 ± 20 g; Shanchuanhong Experimental Animal Tech Co. Ltd; Tianjin, China; Licence: SCXK 2014-0001, Tianjin, China) were housed at room temperature with free access to drinking water but fasted overnight before the administration of drugs. The protocol and any amendments or procedures involving the care or use of animals in this study were in accordance with the regulations for animal experimentation issued by the State Committee of Science and Technology of China and approved by TJUTCM's Institutional Animal Care and Use Committee (Document number TCMLAEC 20170020).
2.2. Preparation of cocrystals and nano-cocrystals
2.2.1. Preparation of BE-NCT cocrystals
The BE-NCT cocrystals were formed according to the previous work [10]. Briefly, 1 mol of BE and 3 mol of NCT were dissolved into 450 ml of ethyl acetate. The resulting solution was then stirred for 8 h at room temperature. After standing the suspension over night, BE-NCT cocrystals were harvested.
2.2.2. Preparation of BE nanocrystals and BE-NCT nano-cocrystals
BE nanocrystals and BE-NCT nano-cocrystals were prepared using the high pressure homogenization method (HPH) with poloxamer 188 as stabilizer [13]. The formulation containing 2% (w/w) BE (BE-NCT cocrystals) and 0.4% (w/w) poloxamer 188, was firstly homogenized at 15 000 rpm for 10 min by a lab scale high shear dispersing emulsifier (FA25, FLUKO, Germany). High-pressure homogenization at 900 bar was applied for 20 homogenization cycles (AH100D, ATS Engineering Inc. Canada) to obtain the nanosuspension of BE (BE-NCT cocrystals). All of the experiments were conducted at room temperature.
2.3. Scanning electron microscope (SEM)
The surface morphology of BE coarse powder, BE-NCT cocrystals and their nanocrystals were studied by SEM (JSM-7500F, JEOL Ltd., Japan) at an acceleration voltage of 10.0 kV. Samples were coated with gold on a holder and dried in vacuum and observed at different magnifications.
2.4. Particle size analysis
Dynamic light scattering (DLS) method was used to evaluate the mean particle size and polydispersity index (PDI) without any dilution at 25 °C. The measurements were carried out by Zetasizer (Nano ZS, Malvern Instruments, UK) and all samples were analyzed in triplicate.
2.5. Powder X-ray diffraction (PXRD)
PXRD patterns of BE coarse powder, BE-NCT cocrystals and their nanosized crystal samples were recorded using a Ni filtered Cu Kα radiation source (D/MAX-2550 V, Rigaku Co.,Japan). The radiation wavelength was 1.542 Å. The samples were continuously scanned at 40 kV and 40 mA from 5° to 80° (2-theta) using a scanning speed of 5 o/min. The data were analyzed by MDI Jade 6.0 software.
2.6. Differential scanning calorimetry (DSC)
The DSC thermograms of BE coarse powder, BE-NCT cocrystals and their nanosized formulations were determined by DSC (Jade DSC, PerkinElmer Inc. USA). Each sample (∼5 mg) was heated in an aluminium pan at a scanning rate of 10 °C/min in an atmosphere of nitrogen gas (30 ml/min) in the range of 30–350 °C.
2.7. In vitro dissolution test
The in vitro dissolution tests were executed to evaluate the dissolution rate of BE, BE-NCT cocrystals, BE nanocrystals and BE-NCT nano-cocrystals, using basket method with dissolution testing apparatus (DT-820, ERWEKA Co., Germany) with three replicates for each group. FaSSIF-V2 and FaSSGF were employed as dissolution medium. BE coarse powder, BE-NCT cocrystals, BE nanocrystals and BE-NCT nano-cocrystals (equivalent to 18 mg of BE) were separately filled into the hard capsules and then immersed into 900 ml dissolution medium. The temperature was maintained at 37 ± 0.5 °C and the stirring rate was 100 rpm. 2 ml of aliquots were withdrawn at 5, 10, 15, 30, 45, 60, 90, 120, 180, 240, 300 and 360 min, and at the same time, an equal volume of fresh dissolution medium were added. After filtration of 0.45 µm microporous membrane the samples were assayed by HPLC (LC-20A, Shimadzu, Japan) .
2.8. In vivo pharmacokinetics
Twenty-four healthy Sprague-Dawley rats weighing 250 ± 20 g were employed in this study and allowed free access to water but fasted overnight before administration. In pharmacokinetic study, randomized design was applied to divide all rats into four groups of six animals each. Each group was administered BE coarse powder suspension, BE-NCT cocrystal suspension, BE nanocrystals and BE-NCT nano-cocrystals at the same oral dose of 80 mg/kg. Blood samples were collected into heparinized tubes from the oculi chorioideae vein at 0.083, 0.25, 0.5, 0.75, 1, 1.5, 2, 3, 4, 6, 8, 10, 12, 16, 24, 48 h after oral administration. Then after centrifugation for 10 min at 4000 rpm, 100 µl separated plasma was transferred into another neat tube and frozen at -20 °C until determination.
Plasma samples were prepared according to the following steps: 200 µl IS solution (1200 ng/ml) was added to 100 µl plasma sample in a 1.5 ml plastic centrifuge tube. Vortex-mixed for 3 min and centrifuged at 12 000 rpm for 10 min. Then 5 µl aliquot was injected into the LC-MS/MS system for analysis by a well validated method as described below.
2.9. UPLC-MS/MS determination of BE and BA in plasma
BE and BA in plasma samples were analyzed by a UPLC-MS/MS system consisting of an Agilent series 1290 UPLC system and an Agilent 6460 triple quadrupole mass spectrometer (Agilent Technologies, USA). According to the article with some modifications [27], the method was established and validated for specificity, linearity, accuracy, precision, recovery, matrix effect and stability during the samples storage and processing procedure.
The chromatographic separation of analytes and IS was performed on an Acquity UPLC HSS T3 C18 column (50 mm × 2.1 mm, 1.8 µm, Waters, CA, USA) with the column temperature at 30 °C. Chromatographic separation was achieved with gradient elution using a mobile phase comprised of 0.05% formic acid in water (A) and acetonitrile (B). The UPLC gradient program was set as follows: 5%→32% B at 0.0–4.0 min; 32%→20% B at 4.0–4.1 min; 20%→50% B at 4.1–9.0 min; 50%→5% B at 9.0–9.5 min; 5%→5% B at 9.5–10.0 min. The column oven was maintained at 30 °C. Efficient and symmetrical peaks were obtained at a flow rate of 0.3 ml/min. A volume of 5µl prepared samples was injected into the chromatographic system with the temperature of autospmpler maintained at 4 °C.
The detection of the analytes was performed in the multiple reaction monitoring mode (MRM) using an electrospray positive ionization (ESI+). The characteristic precursors [M + H]+ to product ions transitions for quantification were m/z 271.06→122.9, 447.09→270.8 and m/z 237.1→ 193.8 for BE, BA and IS, respectively (detailed parameters were listed in Table 1). The ESI configuration was: gas temperature 350 °C; gas flow rate 11 l/min; nebulizer 20 psi; capillary 4000 V. The fragmentor was optimized as 160 V and 100 V meanwhile the collision energy of 14 eV and 38 eV for BE and BA, respectively. The acquired data were processed by MassHunter analysis for QQQ (Version 4.1, Agilent, USA).
Table 1.
Mass parameters for the assay of BE and BA in rat plasma.
| Compound | Mode | Ionization | Fragm-entor | MS1→MS2 | Collision energy |
|---|---|---|---|---|---|
| CBZ (IS) | MRM | ESI+ | 140 | 237.10→193.80 | 18 |
| BE | MRM | ESI+ | 160 | 271.06→122.90 | 14 |
| BA | MRM | ESI+ | 100 | 447.09→270.80 | 38 |
2.10. Statistical analysis
The pharmacokinetic parameters of BE or BA were calculated according to non-compartment model using Phoenix Winnonlin 6.4 (Certara Co., USA). To investigate and compare the total exposure after oral administration of four BE formulations, integrated pharmacokinetic parameters were calculated based on the previously reported AUC based weighting approach [28]. Specifically, the weighting coefficient (ω) was calculated using Eq. 1 or 2. Then the integrated concentrations were calculated by Eq. 3, where Ct represents the plasma concentration at each time point.
| (1) |
| (2) |
| (3) |
All experimental data were expressed as the means ± SD. Statistical significance was estimated by one-way ANOVA.
3. Results and discussion
3.1. Preparation of cocrystals and nano-cocrystals
Many technologies have been reported for cocrystal preparation, such as an array of solid state, mechanochemical and liquid assisted techniques [29], [30]. We prepared BE and NCT oversaturated solutions to precipitate BE-NCT cocrystals. Compared with melting method, the BE-NCT cocrystal obtained by precipitation had higher purity and better crystalline state. The satisfactory yield on the premise of ensuring quality of cocrystals was achieved by optimizing the concentration of BE in ethyl acetate and the ratio of BE and NCT. Thus, gram-scaled cocrystal sample was prepared in our work.
Generally, drug nanocrystal techniques are classified as ‘‘bottom up’’ methods and ‘‘top down’’ methods. The ‘‘bottom up’’ methods, such as the precipitation technique, means that nanocrystal can be constructed from the molecules; however, the ‘‘top down’’ methods, such as the pearl milling and homogenization techniques, mean that the nanocrystals can be disintegrated step by step from the coarse powder [31]. In this study, HPH was employed to prepare BE-NCT nano-cocrystals and BE nanocrystals. The stabilizer poloxamer 188 can contribute to the stability of nanosuspensions.
3.2. Surface morphology
The surface morphology of the coarse powder and the nanocrystals of BE and BE-NCT were presented in Fig. 2. BE coarse powder (Fig. 2A) appeared as brick-shaped crystalline, whereas the BE-NCT cocrystals (Fig. 2B) showed a remarkable change into rod shape. The BE nanocrystals (Fig. 2C) and BE-NCT nano-cocrystals (Fig. 2D) appeared as more small and narrow particles with short rod shape with the mean particle size of less than 300 nm.
Fig. 2.

SEM images of (A) BE coarse powder, (B) BE-NCT cocrystals, (C) BE nanocrystals and (D) BE-NCT nano-cocrystals at different magnification.
3.3. Particle size and zeta potential
The particle size and PDI were shown in Fig. 3. Mean particle size of BE nanocrystals was 207.33 ± 1.62 nm and PDI was 0.26 ± 0.01. For BE-NCT nano-cocrystals, the particle size was found to be 251.53 ± 9.00 nm with PDI of 0.29 ± 0.02. The zeta potential values of BE nanocrystals and BE-NCT nano-cocrystals were −0.39 ± −0.05 mV and −0.68 ± −0.23 mV, respectively.
Fig. 3.

Particle size of (A) BE nanocrystals and (B) BE-NCT nano-cocrystals.
3.4. Crystalline state
It should be borne in mind that the crystalline state is one of the factors affecting the dissolution behavior of drugs [32]. The evaluation of the crystalline state is necessary to understand the polymorphic changes that a drug might undergo when subjected to nanosizing.
XRD and DSC are widely used to evaluate the crystalline structure of the drug nanocrystals and were employed in the study to assess the crystal forms and crystallinity of BE coarse powder, BE-NCT cocrystals, BE nanocrystals and BE-NCT nano-cocrystals. The XRD patterns of different powders were shown in Fig. 4. The BE coarse powder (Fig. 4A) exhibited sharp, distinctive peaks at 2θ values of 10.09°, 11.37°, 13.20°, 15.31°, 16.18°, 16.73°, 18.30° 21.78°, 22.95°, 23.82°, 24.19°, 25.27°, 26.22°, 27.80°, 34.02° and 41.28°, indicating the crystalline nature of BE. The XRD pattern of NCT (Fig. 4B) was also shown some sharp peaks at 2θ values of 11.12°, 14.64°, 18.90°, 19.38°, 19.72°, 22.04°, 23.18° 24.54°, 25.18°, 25.68°, 27.14°, 36.78° and 38.46°. The characteristic peaks of BE were evident in BE-NCT mixture (Fig. 4C), revealing no crystal-state changes during the mixing process. It was obvious that new peaks at 2θ values of 5.84° and 11.74° in the XRD pattern of cocrystals (Fig. 4D) which were different from BE, NCT and their physical mixture. However, once nanonized during high pressure homogenizing, the cocrystal crystalline state showed a significant change from crystalline to amorphous state. No characteristic intense peak was found in the XRD pattern of BE-NCT nano-cocrystals (Fig. 4F) compared with that of cocrystal coarse powder. In addition, in contrast with BE coarse powder, the response of BE nanocrystals (Fig. 4E) was clearly weaker.
Fig. 4.

PXRD spectra of (A) BE coarse powder, (B) NCT, (C) the physical mixture of BE and NCT, (D) BE-NCT cocrystals, (E) BE nanocrystals and (F) BE-NCT nano-cocrystals.
The DSC curves of different powders were merged into one picture and illustrated in Fig. 5. The BE coarse powder (Fig. 5A) and NCT coarse powder (Fig. 5B) exhibited a sharp endothermic peak at 271.40 °C and 132.30 °C respectively, attributed to its crystalline nature. A new peak in the DSC curves of BE-NCT cocrystals (Fig. 5D), indicating the difference from BE, NCT and their physical mixture (Fig. 5C), was seen as the evidence of a new crystal formation [33]. The curves of BE nanocrystals (Fig. 5E) and BE-NCT nano-cocrystals (Fig. 5F) showed a broad but weak peak at further reduced temperature of 264.64 °C and 151.76 °C, indicating an amount of BE and cocrystals transferred from crystalline state into amorphous state during HPH process.
Fig. 5.

DSC curves of (A) BE coarse powder, (B) NCT, (C) the physical mixture of BE and NCT, (D) BE-NCT cocrystals, (E) BE nanocrystals and (F) BE-NCT nano-cocrystals.
It was reported that some drugs retained their crystalline state during homogenization, such as danazol nifedipine and ucb-35440-3 [34]. However, for some other drugs, such as BE and BE-NCT cocrystals, the results of DSC and X-ray showed that the amorphous state was generated during homogenization. Drugs in the amorphous state possess higher solubility and faster dissolution rate due to the higher inner energy. Therefore, when dosed through the oral route, the drug nanosuspensions in amorphous rate would show more significant effects on enhancing bioavailability than the crystalline nanosuspensions provided the high energy state could be kept in the gastrointestinal tract [35] .
3.5. In vitro dissolution test
An important feature of drug nanocrystals is the increase of the saturation solubility and dissolution velocity [36], which makes drug nanocrystals amenable to many applications. Therefore, the in vitro dissolution test was carried out to display the dissolution profiles of nanocrystals in FaSSIF-V2 and FaSSGF . The results shown in Figs. 6 and 7 exhibited a significant enhancement of BE-NCT cocrystals, nanosized BE crystals and nanosized cocrystals in both media. In FaSSIF-V2, the BE-NCT cocrystals, BE nanocrystals and BE-NCT nano-cocrystals demonstrated 1.74-fold, 2.01-fold and 2.17-fold increase in 360 min dissolution rate (44.97% ± 4.35% of cocrystals, 51.76% ± 3.59% of nanocrystals and 55.90% ± 3.81% of nano-cocrystals) compared with 25.80% ± 1.51% dissolution of BE coarse powder. In FaSSGF, the BE coarse powder showed 12.60% ± 1.93% while a 2.22-fold increase 27.95% ± 1.08% of dissolution rate was observed by BE-NCT cocrystals. When BE coarse powder and BE-NCT cocrystals were homogenized into ∼200 nm scale, the dissolution rate of their nano-forms profiled much better than coarse powder. In the first 30 min, attributed to the resultant enhancement in the surface area and decrease in the diffusion layer thickness [37]. 31.04% ± 1.31% of BE nanocrystals were dissolved at 360 min, and 31.94% ± 8.93% for BE-NCT nano-cocrystals, which exhibited 2.46-fold and 2.54-fold enhancement than BE coarse powder.
Fig. 6.

In vitro release profiles of BE coarse powder, BE-NCT cocrystals, BE nanocrystals and BE-NCT nano-cocrystals in FaSSIF-V2 (n = 3, Mean ± SD).
Fig. 7.

In vitro release profiles of BE coarse powder, BE-NCT cocrystals, BE nanocrystals and BE-NCT nano-cocrystals in FaSSGF (n = 3, Mean ± SD).
3.6. Methodology verification of plasma sample analysis
Typical chromatograms of blank plasma, blank plasma spiked with the analytes and plasma samples demonstrated that no endogenous interference was observed at the retention time of the analytes and IS in blank plasma, which proved the assay specificity. The retention time of BE, BA and IS is 8.10, 4.81 and 7.27 min respectively. Regression equations showed good linearity with correlation coefficients greater than 0.99. Intra-day and inter-day precision were less than 4.02% and 3.81%, and the accuracy was within ± 6.59% in plasma, respectively, indicating the overall reproducibility of the method. Average extraction recoveries of BE and BA at three levels ranged 67.81%–72.52% and 65.57%–70.50%, respectively. Satisfactory efficiency of extraction recovery was obtained within the acceptable limit. Matrix effect values ranged 68.08%–75.76% for BE, and 67.57%–71.79% for BA at three levels, respectively. All analytes in rat plasma were stable on storage at ambient temperature for 6 h, on storage for 30 d under -20 °C, in freeze-thaw stability and on storage in the autosampler at 4 °C for 24 h, respectively. The stability would satisfy the requirements of a routine phar macokinetic study. More details were supplied as attachment.
3.7. In vivo pharmacokinetics
Owing to the metabolic transformation from BE into BA after oral administration [38], both BE and BA was detected in rat plasma to evaluate the pharmacokinetics after oral administration of different BE formulations. The mean plasma concentration versus time plots of BE and BA after oral BE coarse powder suspension, BE-NCT cocrystal suspension, BE nanocrystals and BE-NCT nano-cocrystals at the same dose of 80 mg/kg are presented in Fig. 8 and 9. The corresponding major pharmacokinetic parameters of non-compartment model for BE and BA in rats are listed in Table 2.
Fig. 8.

Mean baicalein plasma concentration-time profiles after oral administration of BE coarse powder, BE-NCT cocrystals, BE nanocrystals and BE-NCT nano-cocrystals suspension to rats (n = 6, Mean ± SD).
Fig. 9.

Mean baicalin plasma concentration–time profiles after oral administration of BE coarse powder, BE-NCT cocrystals, BE nanocrystals and BE-NCT nano-cocrystals suspension to rats (n = 6, mean ± SD).
Table 2.
Main pharmacokinetic parameters of BE and BA in plasma after oral BE (Mean ± SD, n = 6).
| BE coarse powder |
BE-NCT cocrystals |
BE nanocrystals |
BE-NCT nano-cocrystals |
||||||
|---|---|---|---|---|---|---|---|---|---|
| Parameters | Unit | BE | BA | BE | BA | BE | BA | BE | BA |
| Cmax | ng/ml | 63.81 ± 24.22 | 2917.29 ± 785.00 | 96.42 ± 39.04 | 4165.77 ± 386.23 | 86.72 ± 57.97 | 8044.62 ± 1265.57 | 156.86 ± 56.32 | 6728.56 ± 1260.12 |
| Tmax1 | h | 0.92 ± 1.17 | 0.53 ± 0.54 | 0.78 ± 0.95 | 2.17 ± 2.93 | 0.37 ± 0.40 | 0.28 ± 0.19 | 0.90 ± 0.92 | 1.58 ± 0.98 |
| Tmax2 | h | 5.17 ± 1.83 | 6.50 ± 2.66 | 10.67 ± 2.07 | 9.67 ± 2.66 | 8.33 ± 3.20 | 4.67 ± 1.97 | 11.67 ± 3.20 | 12.67 ± 2.73 |
| AUC0−t | h·µg/ml | 0.23 ± 0.10 | 17.95 ± 5.24 | 1.54 ± 0.22 | 52.32 ± 9.27 | 0.38 ± 0.19 | 59.21 ± 6.95 | 2.70 ± 0.56 | 109.34 ± 18.73 |
| Lz | 1/h | 0.50 ± 0.41 | 0.28 ± 0.05 | 0.03 ± 0.01 | 0.11 ± 0.01 | 8.58 ± 6.31 | 0.14 ± 0.06 | 0.05 ± 0.01 | 0.14 ± 0.01 |
| AUMC0−t | h·h·µg/ml | 1.55 ± 1.32 | 147.17 ± 61.91 | 30.43 ± 3.16 | 637.42 ± 141.14 | 4.45 ± 2.36 | 563.17 ± 53.13 | 49.19 ± 10.57 | 1521.77 ± 509.50 |
| T1/2 | h | 4.61 ± 6.25 | 2.49 ± 0.40 | 26.77 ± 7.76 | 6.29 ± 0.74 | 8.58 ± 6.31 | 6.10 ± 2.64 | 15.72 ± 4.34 | 4.93 ± 0.27 |
| Vz/F | l/kg | 1704.59 ± 1988.60 | 17.86 ± 8.53 | 1318.81 ± 355.02 | 14.13 ± 3.24 | 2485.51 ± 1774.77 | 12.08 ± 5.92 | 586.06 ± 242.25 | 5.37 ± 1.24 |
| CL/F | l/h/kg | 383.82 ± 190.68 | 4.83 ± 1.68 | 34.44 ± 2.80 | 1.56 ± 0.34 | 227.92 ± 91.53 | 1.35 ± 0.15 | 25.18 ± 3.56 | 0.75 ± 0.14 |
| MRT | h | 5.72 ± 2.53 | 7.88 ± 1.53 | 19.91 ± 1.14 | 12.15 ± 1.21 | 11.67 ± 4.95 | 9.58 ± 1.00 | 18.20 ± 1.10 | 13.64 ± 2.50 |
The concentration of BA showed much larger than BE, which evidenced the first-pass metabolism in the intestine [39]. Following the oral administration of BE, BE-NCT cocrystals, BE nanocrystals or BE-NCT nano-cocrystals, the double peak shown in the mean plasma concentration versus time plots of both BE and BA was typical according to the intestinal metabolism mentioned in the references [40], [41]. The AUC of BE group was 17.95 ± 5.24 h·µg/ml. The AUCs of cocrystals and nanocrystals were 52.32 ± 9.27 h·µg/ml and 59.21 ± 6.95 h·µg/ml which represented 2.91-fold and 3.30-fold than that of BE. The highest AUC was observed by BE-NCT nano-cocrystals 109.34 ± 18.73 h·µg/ml among the four groups which was 6.09-fold than BE.
To further investigate the total exposure after oral administration of BE formulations, the integrated pharmacokinetic approach was carried out with the AUC-based weighting method and the total drug concentration method [28], [42] and integral concentration in plasma and integral pharmacokinetic parameters were thus calculated.
The integrated plasma concentration–time profiles were presented in Fig. 10. The AUC weighted factors (ω) of the BE and BA in rat plasma after oral administration of BE coarse powder, BE-NCT cocrystals, BE nanocrystals and BE-NCT nano-cocrystals were shown in Table 3. The integral pharmacokinetic parameters for oral BE were summarized in Table 4. Following the oral administration of BE coarse suspension, the peak concentration reached at 2880.83 ± 775.45 ng/ml. After administration of the same dose of cocrystals, nanocrystals, or nano-cocrystals, the plasma drug concentration peaks increased into 4048.49 ± 374.71 ng/ml, 7993.59 ± 1257.51 ng/ml and 6568.72 ± 1229.92 ng/ml, respectively. BE coarse powder group gained the first peak at 1.14 h and the second at 6.50 h while for cocrystals at 1.04 h and 9.00 h. BE nanocrystals gained the first peak at 0.28 h, much earlier than the other three groups. BE-NCT nano-cocrystals gained the first peak at 1.58 h and the second at 12.67 h, which may be related to the special property of BE-NCT nano-cocrystals. From the comparison of AUCs shown in Table 4, we could notice that AUC0 − t significantly increased from 17.73 ± 5.18 h·µg/ml for BE coarse suspension to 50.87 ± 9.01 h·µg/ml for BE-NCT cocrystals, 58.84 ± 6.91 h·µg/ml for BE nanocrystals and 106.77 ± 18.28 h·µg/ml for BE-NCT nano-cocrystals (P < 0.01), indicating that the relative bioavailability enhancement of cocrystals, nanocrystals and nano-cocrystals were 2.87, 3.32 and 6.02-fold (the ratio between AUC0-t) than BE coarse suspension, respectively.
Fig. 10.

Integrated plasma concentration–time profiles after oral administration of BE coarse powder, BE-NCT cocrystals, BE nanocrystals and BE-NCT nano-cocrystals suspension to rats (n = 6, Mean ± SD).
Table 3.
AUC0 − t and AUC weighted factors (ω) of the BE and BA in rat plasma after oral administration of BE coarse powder, BE-NCT cocrystals, BE nanocrystals and BE-NCT nano-cocrystals (Mean ± SD, n = 6).
| BE coarse powder |
BE-NCT cocrystals |
BE nanocrystals |
BE-NCT nano-cocrystals |
||||||
|---|---|---|---|---|---|---|---|---|---|
| Parameters | Unit | BE | BA | BE | BA | BE | BA | BE | BA |
| AUC0−t | h·µg/ml | 0.23 ± 0.10 | 17.95 ± 5.24 | 1.54 ± 0.22 | 52.32 ± 9.27 | 0.38 ± 0.19 | 59.21 ± 6.95 | 2.70 ± 0.56 | 109.34 ± 18.73 |
| ω | 0.01 | 0.99 | 0.03 | 0.97 | 0.01 | 0.99 | 0.02 | 0.98 | |
Table 4.
Integrated pharmacokinetic parameters of BE and BA in plasma after oral BE (Mean ± SD, n = 6).
| Parameters | Unit | BE coarse powder | BE-NCT cocrystals | BE nanocrystals | BE-NCT nano-cocrystals |
|---|---|---|---|---|---|
| Cmax | ng/ml | 2880.83 ± 775.45 | 4048.49 ± 374.71⁎⁎ | 7993.59 ± 1257.51⁎⁎ | 6568.72 ± 1229.92**,△△ |
| Tmax1 | h | 1.14 ± 0.81 | 1.04 ± 1.18 | 0.28 ± 0.19* | 1.58 ± 0.98▪ |
| Tmax2 | h | 6.50 ± 2.66 | 9.00 ± 2.45 | 9.00 ± 2.45 | 12.67 ± 2.73**,△,▪ |
| AUC0−t | h·µg/ml | 17.73 ± 5.18 | 50.87 ± 9.01⁎⁎ | 58.84 ± 6.91⁎⁎ | 106.77 ± 18.28**,△△,▪▪ |
| Lz | 1/h | 0.28 ± 0.05 | 0.11 ± 0.01⁎⁎ | 0.20 ± 0.02⁎⁎ | 0.14 ± 0.01**,△△,▪▪ |
| AUMC0−t | h·h·µg/ml | 145.33 ± 61.13 | 620.06 ± 137.17⁎⁎ | 559.61 ± 52.79⁎⁎ | 1486.28 ± 497.40**,△△,▪▪ |
| T1/2 | h | 2.49 ± 0.40 | 6.31 ± 0.74⁎⁎ | 3.54 ± 0.36⁎⁎ | 4.87 ± 0.33**,△△,▪▪ |
| Vz/F | l/kg | 18.09 ± 8.64 | 14.58 ± 3.33 | 6.98 ± 1.02* | 5.43 ± 1.33**,△△,▪ |
| CL/F | l/h/kg | 4.89 ± 1.70 | 1.61 ± 0.35⁎⁎ | 1.37 ± 0.16⁎⁎ | 0.77 ± 0.14**,△△,▪▪ |
| MRT | h | 7.88 ± 1.53 | 12.16 ± 1.21⁎⁎ | 9.58 ± 1.00* | 13.64 ± 2.49**,▪▪ |
*, P < 0.05, **, P < 0.01, compared with BE coarse powder; △, P < 0.05; △△, P < 0.01, compared with BE-NCT cocrystals.
▪, P < 0.05; ▪▪, P < 0.01, compared with BE nanocrystals.
The results of integral pharmacokinetic parameters exhibited nanocrystallizaton form as well as cocrystallization form could improve the absorption of BE, which refers to their enhanced dissolution rate. In the first two hours after administration, the integral concentration in plasma and AUC of BE nanocrystals, which were much higher than BE coarse powder, revealed the advantages of drug nanocrystals on the oral drug delivery of poorly soluble drugs. Mechanisms contributed for the improved absorption could be majorly summarized as two points: i) improved solubility and dissolution rate; and ii) bioadhesion to the intestinal wall [43]. Different from BE and BE nanocrystals, the pharmacokinetic profile of BE-NCT cocrystals exhibited the second peak at 10 h larger than the first peak at 1 h, which revealed that BE-NCT cocrystal form improved the bioavailability and modified the absorption or metabolic process to some extent, attributed to the unique properties of cocrystals beyond to our knowledge. Compared with nanocrystal and cocrystal formulation, AUC of BE-NCT nano-cocrystal from integral concentration exhibited significant increase (P < 0.01). Nano-sized BE-NCT cocrystals simultaneously possessed the advantages of both BE nanocrystals with a rapid and successful absorption in the first two hours and BE-NCT cocrystals with a rising absorption at 10 h. Therefore, BE-NCT nano-cocrystals were proved for its high potential to improve the oral absorption and bioavailability of BE in contrast with BE nanocrystals and BE-NCT cocrystals. When the particle size is reduced to nano-scale, the thickness of the diffusion layer decreases and the dissolution rate increases significantly. In addition, nanoparticles are more easily attached to the intestinal mucosa, thereby increasing drug residence time in the gastrointestinal tract and further promoting the absorption of poorly soluble drugs. This provides more sufficient time for the dissociation, dissolution and absorption of the drug cocrystal molecules. This may also be an important reason for the synergistic increase of the total exposure of BE based on the increasing oral absorption mechanism of nanocrystal and cocrystal. However, the specific mechanism of nano-cocrystal promoting oral absorption has not yet been clearly confirmed, and further intensive study is needed.
4. Conclusion
In this study, the BE-NCT nano-cocrystals were successfully prepared and characterized. Compared with BE coarse powder, BE-NCT cocrystals and BE nanocrystals, BE-NCT nano-cocrystals exhibited significantly-increasing performance both in vitro and in vivo evaluations, suggesting that the nano-cocrystals could be proposed as an advanced strategy for dissolution rate and bioavailability enhancement of poor soluble natural products such as BE.
Conflicts of interest
The authors declare that there is no conflicts of interest.
Footnotes
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.ajps.2018.04.009.
Contributor Information
Jiaxin Pi, Email: ppstar9999@sina.com.
Zhidong Liu, Email: lonerliuzd@163.com.
Appendix. Supplementary materials
References
- 1.Takagi T, Ramachandran C, Bermejo M. A provisional biopharmaceutical classification of the top 200 oral drug products in the United States, Great Britain, Spain, and Japan. Mol Pharm. 2006;3(6):631–643. doi: 10.1021/mp0600182. [DOI] [PubMed] [Google Scholar]
- 2.Ashour EA, Majumdar S, Alsheteli A. Hot melt extrusion as an approach to improve solubility, permeability and oral absorption of a psychoactive natural product, piperine. J Pharm Pharmacol. 2016;68(8):989–998. doi: 10.1111/jphp.12579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hao Y, Wang L, Li J. Enhancement of solubility, transport across Madin-Darby Canine Kidney monolayers and oral absorption of Pranlukast through preparation of a Pranlukast-phospholipid complex. J Biomed Nanotechnol. 2015;11(3):469–477. doi: 10.1166/jbn.2015.1914. [DOI] [PubMed] [Google Scholar]
- 4.Kim MS, Kim JS, Cho WK, Hwang SJ. Enhanced solubility and oral absorption of sirolimus using D-alpha-tocopheryl polyethylene glycol succinate micelles. Artif Cells Nanomed Biotechnol. 2013;41(2):85–91. doi: 10.3109/21691401.2012.742100. [DOI] [PubMed] [Google Scholar]
- 5.Qiao N, Li M, Schlindwein W. Pharmaceutical cocrystals: an overview. Int J Pharm. 2011;419(1-2):1–11. doi: 10.1016/j.ijpharm.2011.07.037. [DOI] [PubMed] [Google Scholar]
- 6.Thakuria R, Delori A, Jones W. Pharmaceutical cocrystals and poorly soluble drugs. Int J Pharm. 2013;453(1):101–125. doi: 10.1016/j.ijpharm.2012.10.043. [DOI] [PubMed] [Google Scholar]
- 7.Rahman Z, Agarabi C, Zidan AS, Khan SR, Khan MA. Physico-mechanical and stability evaluation of carbamazepine cocrystal with nicotinamide. AAPS PharmSciTech. 2011;12(2):693–704. doi: 10.1208/s12249-011-9603-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tomaszewska I, Karki S, Shur J, Price R, Fotaki N. Pharmaceutical characterisation and evaluation of cocrystals: importance of in vitro dissolution conditions and type of coformer. Int J Pharm. 2013;453(2):380–388. doi: 10.1016/j.ijpharm.2013.05.048. [DOI] [PubMed] [Google Scholar]
- 9.Hong C, Xie Y, Yao Y. A novel strategy for pharmaceutical cocrystal generation without knowledge of stoichiometric ratio: myricetin cocrystals and a ternary phase diagram. Pharm Res. 2015;32(1):47–60. doi: 10.1007/s11095-014-1443-y. [DOI] [PubMed] [Google Scholar]
- 10.Huang Y, Zhang B, Gao Y, Zhang J, Shi L. Baicalein-nicotinamide cocrystal with enhanced solubility, dissolution, and oral bioavailability. J Pharm Sci. 2014;103(8):2330–2337. doi: 10.1002/jps.24048. [DOI] [PubMed] [Google Scholar]
- 11.McNamara DP, Childs SL, Giordano J. Use of a glutaric acid cocrystal to improve oral bioavailability of a low solubility API. Pharm Res. 2006;23(8):1888–1897. doi: 10.1007/s11095-006-9032-3. [DOI] [PubMed] [Google Scholar]
- 12.Müller RH, Keck CM. Twenty years of drug nanocrystals: Where are we, and where do we go? Eur J Pharm Biopharm. 2012;80(1):1–3. doi: 10.1016/j.ejpb.2011.09.012. [DOI] [PubMed] [Google Scholar]
- 13.Pi J, Liu Z, Wang H. Ursolic acid nanocrystals for dissolution rate and bioavailability enhancement: influence of different particle size. Curr Drug Deliv. 2016;13(8):1358–1366. doi: 10.2174/1567201813666160307142757. [DOI] [PubMed] [Google Scholar]
- 14.Zuo B, Sun Y, Li H. Preparation and in vitro/in vivo evaluation of fenofibrate nanocrystals. Int J Pharm. 2013;455(1-2):267–275. doi: 10.1016/j.ijpharm.2013.07.021. [DOI] [PubMed] [Google Scholar]
- 15.Zhang J, Lv H, Jiang K, Gao Y. Enhanced bioavailability after oral and pulmonary administration of baicalein nanocrystal. Int J Pharm. 2011;420(1):180–188. doi: 10.1016/j.ijpharm.2011.08.023. [DOI] [PubMed] [Google Scholar]
- 16.Hao L, Wang X, Zhang D. Studies on the preparation, characterization and pharmacokinetics of Amoitone B nanocrystals. Int J Pharm. 2012;433(1-2):157–164. doi: 10.1016/j.ijpharm.2012.05.002. [DOI] [PubMed] [Google Scholar]
- 17.Kesisoglou F, Panmai S, Wu Y. Nanosizing-oral formulation development and biopharmaceutical evaluation. Adv Drug Deliv Rev. 2007;59(7):631–644. doi: 10.1016/j.addr.2007.05.003. [DOI] [PubMed] [Google Scholar]
- 18.Junghanns JU, Muller RH. Nanocrystal technology, drug delivery and clinical applications. Int J Nanomedicine. 2008;3(3):295–309. doi: 10.2147/ijn.s595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.De Smet L, Saerens L, De Beer T. Formulation of itraconazole nanococrystals and evaluation of their bioavailability in dogs. Eur J Pharm Biopharm. 2014;87(1):107–113. doi: 10.1016/j.ejpb.2013.12.016. [DOI] [PubMed] [Google Scholar]
- 20.Karashima M, Kimoto K, Yamamoto K, Kojima T, Ikeda Y. A novel solubilization technique for poorly soluble drugs through the integration of nanocrystal and cocrystal technologies. Eur J Pharm Biopharm. 2016;107:142–150. doi: 10.1016/j.ejpb.2016.07.006. [DOI] [PubMed] [Google Scholar]
- 21.Liu M, Hong C, Li G, Ma P, Xie Y. The generation of myricetin-nicotinamide nanococrystals by top down and bottom up technologies. Nanotechnology. 2016;27(39) doi: 10.1088/0957-4484/27/39/395601. [DOI] [PubMed] [Google Scholar]
- 22.Huang Y, Li JM, Lai ZH. Phenazopyridine-phthalimide nano-cocrystal: Release rate and oral bioavailability enhancement. Eur J Pharm Sci. 2017;109:581–586. doi: 10.1016/j.ejps.2017.09.020. [DOI] [PubMed] [Google Scholar]
- 23.Miocinovic R, McCabe NP, Keck RW. In vivo and in vitro effect of baicalein on human prostate cancer cells. Int J Oncol. 2005;26(1):241–246. [PubMed] [Google Scholar]
- 24.Fan GW, Zhang Y, Jiang X. Anti-inflammatory activity of baicalein in LPS-stimulated RAW264.7 macrophages via estrogen receptor and NF-kappaB-dependent pathways. Inflammation. 2013;36(6):1584–1591. doi: 10.1007/s10753-013-9703-2. [DOI] [PubMed] [Google Scholar]
- 25.Huang Y, Tsang SY, Yao X, Chen ZY. Biological properties of baicalein in cardiovascular system. Curr Drug Targets Cardiovasc Haematol Disord. 2005;5(2):177–184. doi: 10.2174/1568006043586206. [DOI] [PubMed] [Google Scholar]
- 26.Sowa M, Slepokura K, Matczak-Jon E. A 1:1 cocrystal of baicalein with nicotinamide. Acta Crystallogr C. 2012;68(Pt 7):262–265. doi: 10.1107/S0108270112024456. [DOI] [PubMed] [Google Scholar]
- 27.Zeng L, Wang M, Yuan Y. Simultaneous multi-component quantitation of Chinese herbal injection Yin-zhi-huang in rat plasma by using a single-tube extraction procedure for mass spectrometry-based pharmacokinetic measurement. J Chromatogr B Analyt Technol Biomed Life Sci. 2014;967:245–254. doi: 10.1016/j.jchromb.2014.07.037. [DOI] [PubMed] [Google Scholar]
- 28.Xie Y, Hao H, Kang A. Integral pharmacokinetics of multiple lignan components in normal, CCl4-induced hepatic injury and hepatoprotective agents pretreated rats and correlations with hepatic injury biomarkers. J Ethnopharmacol. 2010;131(2):290–299. doi: 10.1016/j.jep.2010.06.038. [DOI] [PubMed] [Google Scholar]
- 29.Karki S, Friscic T, Jones W, Motherwell WD. Screening for pharmaceutical cocrystal hydrates via neat and liquid-assisted grinding. Mol Pharm. 2007;4(3):347–354. doi: 10.1021/mp0700054. [DOI] [PubMed] [Google Scholar]
- 30.Dhumal RS, Kelly AL, York P, Coates PD, Paradkar A. Cocrystalization and simultaneous agglomeration using hot melt extrusion. Pharm Res. 2010;27(12):2725–2733. doi: 10.1007/s11095-010-0273-9. [DOI] [PubMed] [Google Scholar]
- 31.Brough C, Williams RO., 3rd Amorphous solid dispersions and nano-crystal technologies for poorly water-soluble drug delivery. Int J Pharm. 2013;453(1):157–166. doi: 10.1016/j.ijpharm.2013.05.061. [DOI] [PubMed] [Google Scholar]
- 32.Hasa D, Voinovich D, Perissutti B. Reduction of melting temperature and enthalpy of drug crystals: theoretical aspects. Eur J Pharm Sci. 2013;50(1):17–28. doi: 10.1016/j.ejps.2013.03.018. [DOI] [PubMed] [Google Scholar]
- 33.Kerr HE, Softley LK, Suresh K, Hodgkinson P, Evans IR. Structure and physicochemical characterization of a naproxen-picolinamide cocrystal. Acta Crystallogr C Struct Chem. 2017;73(Pt 3):168–175. doi: 10.1107/S2053229616011980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hecq J, Deleers M, Fanara D. Preparation and in vitro/in vivo evaluation of nano-sized crystals for dissolution rate enhancement of ucb-35440-3, a highly dosed poorly water-soluble weak base. Eur J Pharm Biopharm. 2006;64(3):360–368. doi: 10.1016/j.ejpb.2006.05.008. [DOI] [PubMed] [Google Scholar]
- 35.Gao L, Liu G, Ma J. Drug nanocrystals: In vivo performances. J Control Release. 2012;160(3):418–430. doi: 10.1016/j.jconrel.2012.03.013. [DOI] [PubMed] [Google Scholar]
- 36.Shegokar R, Muller RH. Nanocrystals: industrially feasible multifunctional formulation technology for poorly soluble actives. Int J Pharm. 2010;399(1-2):129–139. doi: 10.1016/j.ijpharm.2010.07.044. [DOI] [PubMed] [Google Scholar]
- 37.Tu L, Yi Y, Wu W. Effects of particle size on the pharmacokinetics of puerarin nanocrystals and microcrystals after oral administration to rat. Int J Pharm. 2013;458(1):135–140. doi: 10.1016/j.ijpharm.2013.10.001. [DOI] [PubMed] [Google Scholar]
- 38.Akao T, Sato K, Hanada M. Hepatic contribution to a marked increase in the plasma concentration of baicalin after oral administration of its aglycone, baicalein, in multidrug resistance-associated protein 2-deficient rat. Biol Pharm Bull. 2009;32(12):2079–2082. doi: 10.1248/bpb.32.2079. [DOI] [PubMed] [Google Scholar]
- 39.Zhang L, Lin G, Chang Q, Zuo Z. Role of intestinal first-pass metabolism of baicalein in its absorption process. Pharm Res. 2005;22(7):1050–1058. doi: 10.1007/s11095-005-5303-7. [DOI] [PubMed] [Google Scholar]
- 40.Kang MJ, Ko GS, Oh DG. Role of metabolism by intestinal microbiota in pharmacokinetics of oral baicalin. Arch Pharm Res. 2014;37(3):371–378. doi: 10.1007/s12272-013-0179-2. [DOI] [PubMed] [Google Scholar]
- 41.Wang J, Pang Q, Cen W, Zhu P, Xu Y. Simultaneous determination of ten active constituents of Yankening Capsule in rat plasma by ultra high performance liquid chromatography with tandem mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci. 2015;978-979:43–53. doi: 10.1016/j.jchromb.2014.10.016. [DOI] [PubMed] [Google Scholar]
- 42.Qian P, Zhang YB, Yang YF, Xu W, Yang XW. Pharmacokinetics studies of 12 alkaloids in rat plasma after oral administration of Zuojin and Fan-Zuojin formulas. Molecules. 2017;22(2):214. doi: 10.3390/molecules22020214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Alam MA, Al-Jenoobi FI, Al-mohizea AM. Commercially bioavailable proprietary technologies and their marketed products. Drug Discov Today. 2013;18(19-20):936–949. doi: 10.1016/j.drudis.2013.05.007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.