

Graphical Abstract
Keywords: Atorvastatin calcium, AC-PLGA-NPs, Probe ultrasonication and evaporation method, Oral bioavailability
Abstract
A biodegradable poly(lactic-co-glycolic acid) loading atorvastatin calcium (AC) nanoparticles (AC-PLGA-NPs) were prepared by probe ultrasonication and evaporation method aiming at improving the oral bioavailability of AC. The effects of experimental parameters, including stabilizer species, stabilizer concentration and pH of aqueous phase, on particle size were also evaluated. The resultant nanoparticles were in spherical shape with an average diameter of 174.7 nm and a narrow particle size distribution. And the drug loading and encapsulation efficiency were about 8% and 71%, respectively. The particle size and polydispersion were almost unchanged in 10 days. The release curves of AC-PLGA-NPs in vitro displaying sustained release characteristics indicated that its release mechanisms were matrix erosion and diffusion. The pharmacokinetic study in vivo revealed that the Cmax and AUC0-∞ of AC-PLGA-NPs in rats were nearly 3.7-fold and 4.7-fold higher than that of pure atorvastatin calcium suspension. Our results demonstrated that the delivery of AC-PLGA-NPs could be a promising approach for the oral delivery of AC for enhanced bioavailability.
1. Introduction
Atorvastatin calcium (AC, Fig. 1), a selective, reversible and competitive inhibitor of hydroxy-methylglutaryl-coenzyme A reductase (HMG-CoA), is widely applied in the treatment of hyperlipidemia and atherosclerosis due to its superiority in reducing the levels of cholesterol, low-density lipoprotein cholesterol, and triglycerides, as well as the ability to increase high-density lipoprotein level in plasma [1], [2], [3], [4], [5]. Despite the effective anti-hyperlipidemia therapy, the oral bioavailability of AC is still restricted by its extremely poor water solubility and extensive first-pass metabolism, which hinders the further clinical application of AC [5], [6].
Fig. 1.
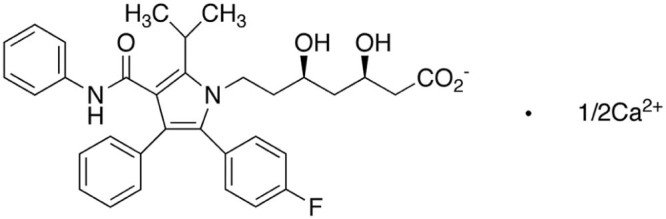
Chemical structure of atorvastatin calcium (AC).
In order to address these challenges, micronization technique and supercritical antisolvent (SAS) means were employed to enhance physicochemical properties and bioavailability through particle size reduction and generation of amorphous state [7], [8], [9]. Meanwhile, polymeric prodrug approach was also developed to improve the stability and drug release characteristic by constructing a bioadhesive nano-sized chitosan–atorvastatin conjugate and its result exhibited nearly 5-fold increase in bioavailability compared with AC suspension [10]. Recently, the focus to improve oral bioavailability moved on from AC encapsulation to novel drug delivery systems including atorvastatin hydroxypropyl-beta-cyclodextrin complex, multi-unit floating microcapsules, Eudragit nanoparticles, nanostructured lipid carriers, and self-nanoemulsifying or self-microemulsifying tablets [5], [11], [12], [13], [14], [15]. The limit of the above methods was mainly attributed to unsatisfied particle size, non-biodegradable drug carrier and troublesome preparation process. Currently, poly(lactic-co-glycolic acid) (PLGA) approved by FDA were intensively investigated because of its established safety, biodegradation and biocompatibility [16], [17]. And there is little available literature about PLGA nanoparticles encapsulating AC by peroral administration to enhance its bioavailability.
Based on the above considerations, the study mainly aimed to enhance oral bioavailability of AC by developing PLGA nanoparticles, which consisted of biodegradable PLGA and pure drug AC. AC-PLGA-NPs were prepared by the probe ultrasonication and evaporation method coupled with pH adjustment of aqueous phase at 4. The in vitro characterization and in vivo evaluation of the AC-PLGA-NPs, including in vitro stability, drug release and in vivo oral pharmacokinetics, were evaluated.
2. Materials and methods
2.1. Materials
Atorvastatin calcium was provided by Zhejiang New Donggang Pharmaceutical Co., Ltd. (China). PLGA (50/50, Mr38000) was purchased from Jinan Daigang Biological Engineering Co., Ltd. (China). F-68 was obtained from BASF Co., Ltd. (Shanghai, China). PVA, glucose, lactose, sucrose, maltose and PEG400 were supplied by Tianjin Bodi Chemical Co., Ltd. (China). Tween 80 was acquired from Hunan Erkang Pharmaceutical Co., Ltd. (China). Glucan setting G50 was purchased from Regal Biotechnology Company (Anhui, China). Hydrochloric acid, calcium chloride, citric acid, ammonium citrate, ammonia water, and potassium dihydrogen phosphate were of analytical grade and used without further purification.
2.2. Preparation of the atorvastatin calcium PLGA nanoparticles formulation
Atorvastatin calcium PLGA nanoparticles were prepared by probe ultrasonication and evaporation method. Briefly, AC dispersed in PEG400 was mixed with PLGA solution which was dissolved in acetone to form organic phase. And 1.5% w/v F-68 solution (pH 4.0) was utilized as water phase. Then the organic phase was added to the water phase by stirring, and the mixture was ultrasound using probe-type sonifier (JY92-2D, Scientz, China) for 5 min in ice bath. Next, acetone was removed by rotary vacuum evaporation. Then the initial nanoparticles solution was centrifuged (13,500 rpm, 5 min) to obtain AC-PLGA-NPs. Blank nanoparticles were prepared in the same way except AC [18].
2.3. Characterization of AC-PLGA-NPs
2.3.1. Size distribution and zeta potential
Size distribution and zeta potential of AC-PLGA-NPs were measured by dynamic light scattering (DLS) method with a Zetasizer instrument (Nano ZS, Malvern Co., UK) [19].
2.3.2. Morphology observation
The morphological images of AC-PLGA-NPs were obtained by transmission electron microscope (TEM) (H-600, Hitachi, Japan). A drop of AC-PLGA-NPs solution was deposited on a carbon-coated copper grid and excess solution was tapped with filter papers. Then the thin-film solution was dried at room temperature, stained with 0.2% phosphotungstic acid aqueous solutions for 1 min before observation under TEM [20].
2.4. Drug loading and encapsulation efficiency
Entrapment efficiency of AC-PLGA-NPs was determined by high performance liquid chromatography (HITACHI L2000) equipped with a C18 column kept at 35 °C (Phenomene Ultremex C18 250 mm × 4.6 mm, 5 µm) at an absorption wavelength of 244 nm. In brief, SephadexG-50 was swelled overnight and packed in a 5 mL syringe with filter paper in the bottom to obtain gel column. Then 200 µL AC-PLGA-NPs was added onto the gel column and centrifuged (13,000 rpm, 20 min) to collect the elution. Next, the eluent and 200 µL AC-PLGA-NPs were dissolved in a 25 mL volumetric flask with acetonitrile to determine the content of AC in nanoparticle and total AC weight in the nanoparticles formulation, respectively. The entrapment efficiency and drug loading were calculated as:
2.5. Stability of AC-PLGA-NPs
The stability study of AC-PLGA-NPs was evaluated at 37 °C for 10 days. And the changes in particle size were selected to assess the stability of nanoparticles.
2.6. In vitro releasing
An in vitro release study was estimated in pH 7.4 PBS by dialysis method. Briefly, 1.5 mL atorvastatin calcium suspension containing PEG400 as suspending agent and atorvastatin calcium nanoparticles solution were sealed in a dialysis bag (MWCO = 14 kDa, Spectrum Laboratories, USA) and incubated in 100 mL of release medium at 37 °C under orbital shaking (100 rpm). At designated intervals, 2 mL samples were removed for analysis and replaced with the same volume of fresh medium. The AC content was determined under the same HPLC conditions as described above. And the drug release profiles were evaluated by zero-order, first-order, Higuchi, Weibull and Ritger–Peppas model, respectively [21].
2.7. Preparation of calibration, plasma samples and quality control (QC) samples
The stock solution of AC (1.0 mg/mL) was prepared in methanol and then diluted to achieve a series of in vitro working solutions at concentrations of 10, 20, 60, 200, 600, 400, 800, 1600 and 2000 ng/mL with methanol, and IS at concentration of 500 ng/mL. The plasma samples spiked with different concentrations of AC were processed by liquid–liquid extraction to prepare calibration standards. Briefly, a 50 µL aliquot of the IS solution (500 ng/mL), 50 µL of HCl solution (1 mmol) and 50 µL of different concentrations of AC were added to 100 µL of blank plasma sample. The mixture was vortex mixed for 2 min. Then, a 3 mL aliquot of the methyl tert-butyl ether (MTBE) was added and vortex mixed for another 5 min. After centrifuging at 3000 rpm for 10 min, the upper organic layer was transferred to test tube and dried with a gentle stream of nitrogen at 30 °C. The residue was resolved with 100 µL dilute solution (acetonitrile: water – 70:30) and vortexed for 3 min. A 5 µL aliquot of the solution was injected into the UPLC-MS/MS system [22], [23]. Plasma samples were prepared in the same way except the addition of different concentrations of AC.
QC samples were prepared in the same way with three concentrations of AC at 800 ng/mL (high), 100 ng/mL (medium) and 10 ng/mL (low). All working solutions were stored at 4 °C until the analysis. And the method employed to determine the concentration of AC was validated before the study of bioavailability.
2.8. Pharmacokinetic study
The animal studies were approved by the Shenyang Pharmaceutical University Animal Care and Use Committee. Wistar rats weighing 220 ± 20 g were randomly separated into 2 groups and fasted overnight prior to the experiment. Two groups were administrated orally with atorvastatin calcium PLGA nanoparticles formulation and AC suspension mentioned-above separately at a single dose of 5 mg/kg. Blood samples (0.3 mL) were collected, centrifuged at 13,000 rpm for 10 min, and then plasma were frozen at −20 °C. The concentration of AC in the blood samples was determined by UPLC-MS/MS method, with gliclazide as internal standard. The chromatographic separations were acquired on an ACQUITY UPLCTM system (Waters Corp., Milford, MA) and BEH C18 column (50 mm × 2.1 mm, 1.7 µm; Waters Corp.) with a mobile phase consisting of 70% acetonitrile and 30% water (containing 0.1%, v/v formic acid). The compounds were analyzed by multiple reaction monitoring (MRM) of the transitions of m/z 559.0→440.0 for atorvastatin calcium and m/z 324.0→127.0 for gliclazide. The concentration of each sample was calculated and referred to a calibration curve with a concentration range from 5 to 1000 ng/mL, with a correlation coefficient of 0.998. The related pharmacokinetic parameters were achieved using DAS 2.0 software [18].
2.9. Statistical data analysis
The results were listed as mean or mean ± standard deviation (SD). Statistical data analysis was carried out using one-way ANOVA. Difference was set as significance at a level of P < 0.05, and a high significance was considered as P < 0.01.
3. Results and discussion
3.1. Preparation and characterization of AC-PLGA-NPs
The AC was encapsulated into AC-PLGA-NPs by probe ultrasonication and evaporation method. Surfactants or polymers were widely applied as stabilizers for their superiority in preventing spontaneous particle aggregation, decreasing the potential interactions between drug and PLGA, as well as neutralizing the acidity generated during the polymer degradation [24]. Therefore, the preliminary study, including the kind of stabilizer, the concentration of stabilizer and pH of water phase, was also optimized. As shown in Fig. 2, the stabilizer F68 could form a smaller particle size compared with PVA and Tween 80. Next, the investigation of F68 concentrations coating on PLGA nanoparticles (Fig. 3) demonstrated that a bell shape of size change with F68 concentration in the range from 0.5% to 2.0% (w/v). Table 1 illustrates the equilibrium solubility of atorvastatin calcium in a medium of different pH at 37 °C, which might indicated that pH may be an influential factor to AC-PLGA-NPs size and encapsulation efficiency. Expectedly, Fig. 4 exhibited that AC-PLGA-NPs size was immune from pH of the aqueous phase at pH 6 or below, but the performance of encapsulation efficiency in different pH revealed that the aqueous phase of pH 4 or below could significantly improve encapsulation efficiency compared with pH 6 (about 71.5% to 17.1%). The high encapsulation efficiency might result from the insolubility of AC entering into the hydrophobic internal core of PLGA nanoparticles when pH was below 4. However, when pH was 2 or below, the extensive acidity contributed to unstable lactonization of AC [25]. Based on the results, the concentration of 1.5% F68 and pH 4 water phase were selected in our next study.
Fig. 2.
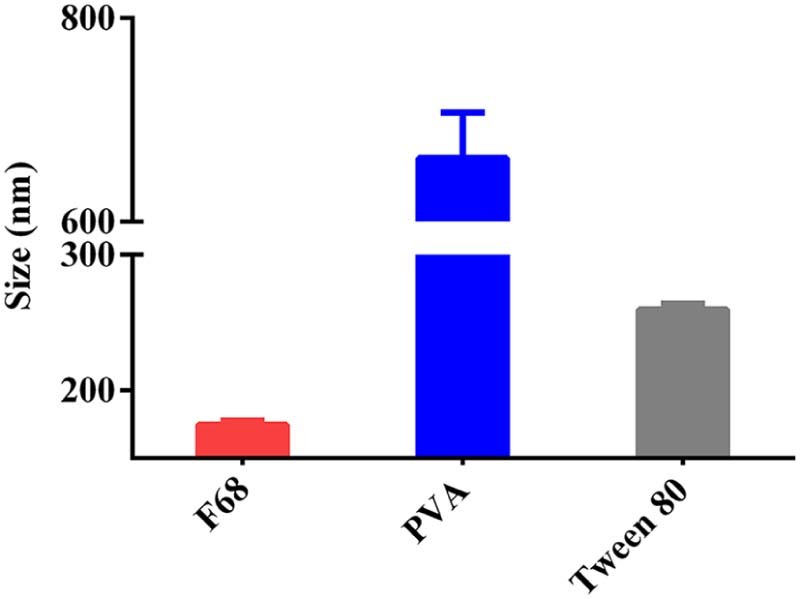
The influence of surfactants on the size of AC-PLGA-NPs.
Fig. 3.
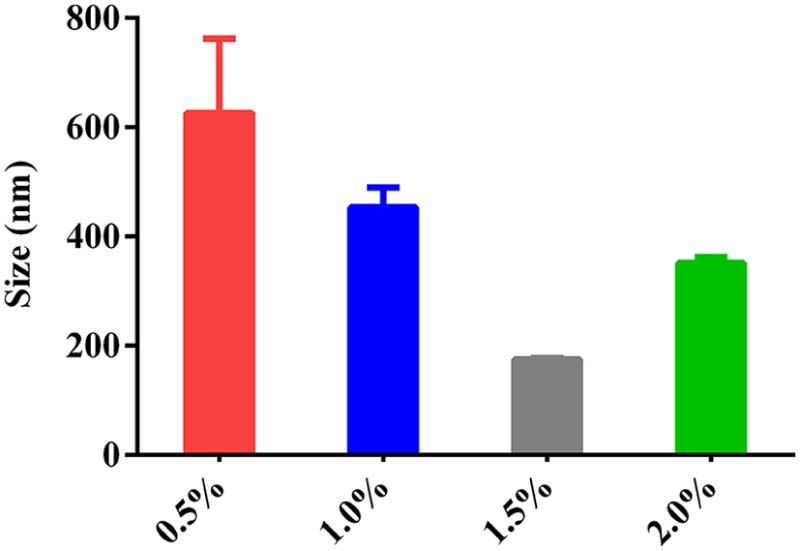
The influence of concentration of surfactant on the size of AC-PLGA-NPs.
Table 1.
The equilibrium solubility of atorvastatin calcium in a medium of different pH at 37 °C (mean + SD, n = 3).
| Medium | 0.1M HCl | pH 4.0 | pH 7.4 |
|---|---|---|---|
| Sapp (µg/mL) | 0.25 ± 0.07 | 1.40 ± 0.14 | 716.30 ± 41.29 |
Fig. 4.
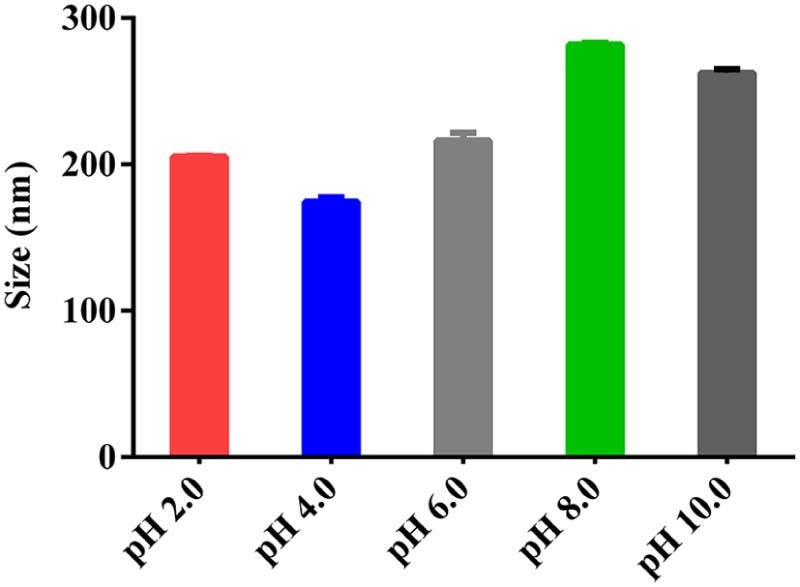
The influence of pH of aqueous phase on the size of AC-PLGA-NPs.
The characterization of size and zeta detailed that blank nanoparticles were 193.0 ± 2.7 nm with zeta potential of −4.55 ± 0.14 mv, while the size of AC-PLGA-NPs were smaller (about 174.7 ± 3.0 nm) with reduced zeta potential (-8.64 ± 0.34 mv). As shown in Fig. 5, blank nanoparticles and AC-PLGA-NPs were in good spherical shape. Then, the drug loading content and encapsulation efficiency were 8.30 ± 0.10 and 71.49 ± 0.10, respectively. Meanwhile, stability study indicated that AC-PLGA-NPs were stable at room temperature for 10 days. As implied in Fig. 6, the size of AC-PLGA-NPs had almost no change due to the protection of PLGA shell and stabilizer F68.
Fig. 5.

Intensity-size distribution of blank nanoparticles (A) and AC-PLGA-NPs (B), and transmission electron microscopy (TEM) images of blank nanoparticles (C) and AC-PLGA-NPs (D), respectively.
Fig. 6.
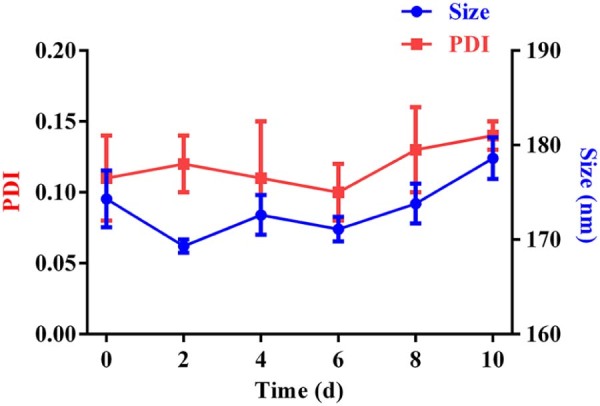
Size and encapsulation efficiency changes of AC-PLGA-NPs at 37 °C for 10 days (n = 3).
3.2. Releasing tests in vitro
The releasing profiles of AC suspension and AC-PLGA-NPs in pH 7.4 phosphate buffer were presented in Fig. 7. In contrast with the AC suspension, there was a sustained release characteristic that approximates 88.3% of AC released from the AC-PLGA-NPs after 24 h. Furthermore, the burst release of AC suspension was lower at only 22.6% in AC-PLGA-NPs at the initial 2 h. Many factors including polymer–drug interactions, drug–drug interactions and water absorption could have influenced the rate of drug diffusion and PLGA degradation kinetics [26]. Table 2 illustrated the release kinetics of AC suspension and AC-PLGA-NPs. Both the release data were best fitted to first order model. According to the Ritger–Peppas model, the release mechanism of AC suspension was Fickian diffusion, while that of AC-PLGA-NPs might involve a combination of Fickian diffusion and matrix erosion [21], [27].
Fig. 7.
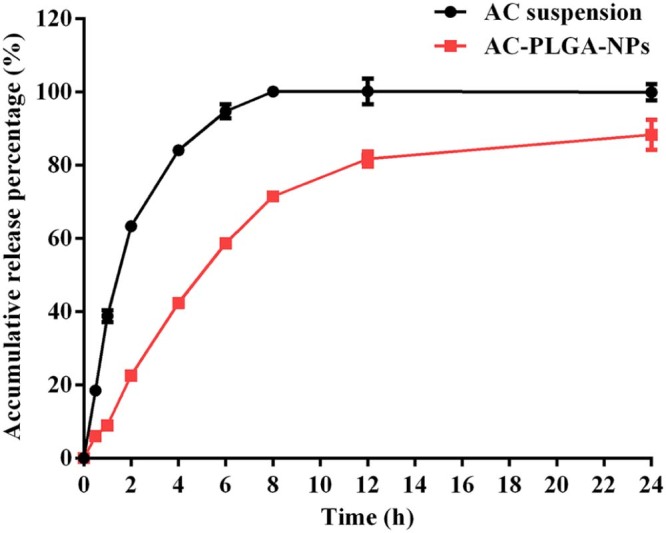
The cumulative release rate curve of AC-PLGA-NPs in pH 7.4 PBS (n = 3).
Table 2.
Release kinetics of AC suspension and AC-PLGA-NPs.
| Models | AC suspension | AC-PLGA-NPs | ||
|---|---|---|---|---|
| Zero order | ||||
| First order | ||||
| Higuchi | ||||
| Weibull | ||||
| Ritger–Peppas |
3.3. Pharmacokinetic study in vivo
Two groups of rats were orally administrated with pure AC suspension and AC-PLGA-NPs at a dose of 5 mg/kg. The methodology of AC by UPLC-MS/MS conformed to standards, with excellent extraction recovery and matrix effects, and the tested linearity ranged from 5 to 1000 ng/mL with correlation coefficient of 0.9979 and standard curve of Y = 0.1252X + 0.4234. Moreover, the acceptable precision, accuracy, selectivity and stability were also achieved with the determined method [28], [29].
As shown in Table 3 and Fig. 8, the Cmax and AUC0-∞ of NPs were significantly increased approximately 3.7-fold and 4.7-fold than that of reference suspensions. Moreover, the RSD of AUC0-∞ of AC-PLGA-NPs was around 27.1%, a decrease in comparison with commercial capsules (about 70.0%). Therefore, it might be inferred that AC-PLGA-NPs can improve oral bioavailability of AC and reduce the variability of gastrointestinal absorption, which might lie in gastrointestinal pH, shell protection of PLGA nanoparticle and endocytosis pathways [30]. AC belongs to Class II of the Biopharmaceutical Classification System (BCS); it possesses high permeability in intestinal tract and is reported to be insoluble at pH 4 and below, leading to most drug precipitation in the stomach, which might fail to provide sufficient dosage for absorption in the intestine when administrated by oral route [31], [32]. Meanwhile, the AC absorption might be also lowered by the lactonization of AC caused by the gastric juice acid catalysis [25]. Compared with AC suspension, the presence of protective shells composed of F68 and PLGA polymer might minimize the destruction of drug nanoparticles that resulted from gastrointestinal pH and enzymes. Moreover, AC-PLGA-NPs might be endocytosed through nanoparticle endocytosis pathways on account of involvement in the active endocytosis in intestinal tract.
Table 3.
Pharmacokinetic parameters of AC in rats after oral administration of AC suspension and AC-PLGA-NPs at a dose of 5 mg/kg (mean + SD, n = 5).
| Preparations | t1/2 (h) | Cmax (ng/mL) | Tmax (h) | AUC (ng/mL*h) |
|---|---|---|---|---|
| AC suspension | 0.68 ± 0.29 | 49.37 ± 26.05 | 0.30 ± 0.14 | 45.32 ± 30.80 |
| AC-PLGA-NPs | 1.08 ± 0.34 | 183.81 ± 77.28** | 0.37 ± 0.14 | 214.04 ± 58.02*** |
Fig. 8.
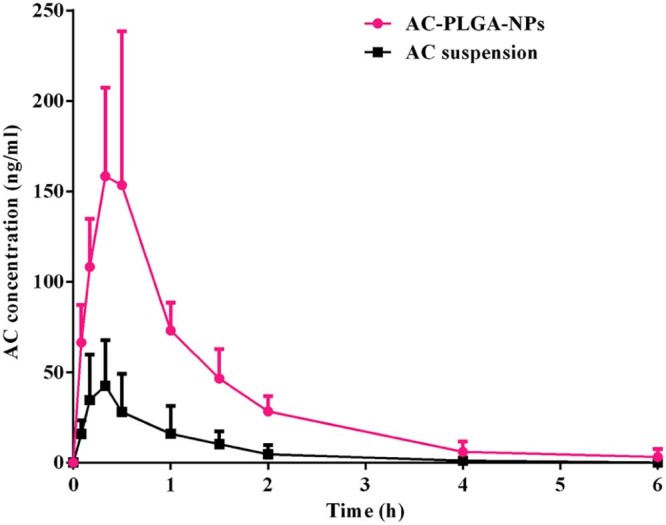
Mean plasma concentration−time curves of AC in rats after oral administration of AC suspension and AC-PLGA-NPs at a dose of 5 mg/kg AC (mean + SD, n = 5).
4. Conclusion
The aim of our investigation was to improve the oral bioavailability of atorvastatin calcium (AC) by loading AC in poly(lactic-co-glycolic acid) (PLGA) nanoparticles (NPs). The AC-PLGA-NPs were prepared by probe ultrasonication and evaporation method and characterized by evaluating the size, drug loading and encapsulation efficiency. The resultant nanoparticles displayed a mean size about 174.7 nm, and a drug loading and encapsulation efficiency about 8% and 71%, respectively. The release curves of AC-PLGA-NPs in vitro display sustained release characteristics due to its matrix erosion and diffusion release mechanism. The pharmacokinetic study in vivo revealed that the Cmax and AUC0-∞ of AC-PLGA-NPs in rats were nearly 3.7-fold and 4.7-fold higher than that of pure atorvastatin calcium suspension, separately. Therefore, it is reasonable to demonstrate that AC-PLGA-NPs facilitated to enhance the oral bioavailability of AC. The mechanism of the improved oral bioavailability of AC-PLGA-NPs is unclear, which will be investigated in our next study continuously.
Acknowledgements
This work was financially supported by the Science and Technology Research Project of Liaoning Provincial Education Department L2013390.
Footnotes
Peer review under responsibility of Shenyang Pharmaceutical University.
Contributor Information
Yinghua Sun, Email: sunyinghua77@aliyun.com.
Jin Sun, Email: sunjin66@21cn.com.
References
- 1.Nawrocki J.W., Weiss S.R., Davidson M.H. Reduction of LDL cholesterol by 25% to 60% in patients with primary hypercholesterolemia by atorvastatin, a new HMG-CoA reductase inhibitor. Arterioscler Thromb Vasc Biol. 1995;15:678–682. doi: 10.1161/01.atv.15.5.678. [DOI] [PubMed] [Google Scholar]
- 2.Bakker-Arkema R.G., Davidson M.H., Goldstein R.J. Efficacy and safety of a new HMG-CoA reductase inhibitor, atorvastatin, in patients with hypertriglyceridemia. JAMA. 1996;275:128–133. [PubMed] [Google Scholar]
- 3.Lea A.P., McTavish D. Atorvastatin. A review of its pharmacology and therapeutic potential in the management of hyperlipidaemias. Drugs. 1997;53:828–847. doi: 10.2165/00003495-199753050-00011. [DOI] [PubMed] [Google Scholar]
- 4.Gordon D.J., Probstfield J.L., Garrison R.J. High-density lipoprotein cholesterol and cardiovascular disease. Four prospective American studies. Circulation. 1989;79:8–15. doi: 10.1161/01.cir.79.1.8. [DOI] [PubMed] [Google Scholar]
- 5.Kumar N., Chaurasia S., Patel R.R. Atorvastatin calcium encapsulated Eudragit nanoparticles with enhanced oral bioavailability, safety and efficacy profile. Pharm Dev Technol. 2015:1–12. doi: 10.3109/10837450.2015.1108983. [DOI] [PubMed] [Google Scholar]
- 6.Lennernas H. Clinical pharmacokinetics of atorvastatin. Clin Pharmacokinet. 2003;42:1141–1160. doi: 10.2165/00003088-200342130-00005. [DOI] [PubMed] [Google Scholar]
- 7.Zhang H.X., Wang J.X., Zhang Z.B. Micronization of atorvastatin calcium by antisolvent precipitation process. Int J Pharm. 2009;374:106–113. doi: 10.1016/j.ijpharm.2009.02.015. [DOI] [PubMed] [Google Scholar]
- 8.Kim J.S., Kim M.S., Park H.J. Physicochemical properties and oral bioavailability of amorphous atorvastatin hemi-calcium using spray-drying and SAS process. Int J Pharm. 2008;359:211–219. doi: 10.1016/j.ijpharm.2008.04.006. [DOI] [PubMed] [Google Scholar]
- 9.Kim M.S., Jin S.J., Kim J.S. Preparation, characterization and in vivo evaluation of amorphous atorvastatin calcium nanoparticles using supercritical antisolvent (SAS) process. Eur J Pharm Biopharm. 2008;69:454–465. doi: 10.1016/j.ejpb.2008.01.007. [DOI] [PubMed] [Google Scholar]
- 10.Anwar M., Warsi M.H., Mallick N. Enhanced bioavailability of nano-sized chitosan-atorvastatin conjugate after oral administration to rats. Eur J Pharm Sci. 2011;44:241–249. doi: 10.1016/j.ejps.2011.08.001. [DOI] [PubMed] [Google Scholar]
- 11.Khan F., Islam M.S., Roni M.A. Systematic development of self-emulsifying drug delivery systems of atorvastatin with improved bioavailability potential. Sci Pharm. 2012;80:1027–1043. doi: 10.3797/scipharm.1201-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lv H.X., Zhang Z.H., Hui J. Preparation, physicochemical characteristics and bioavailability studies of an atorvastatin hydroxypropyl-beta-cyclodextrin complex. Pharmazie. 2012;67:46–53. [PubMed] [Google Scholar]
- 13.Biswal P.K., Pani N.R., Dixit P.K. Effects of carbohydrate polymers in self-microemulsified tablets on the bioavailability of atorvastatin: in vitro-in vivo study. Life Sci. 2015;135:92–100. doi: 10.1016/j.lfs.2015.05.026. [DOI] [PubMed] [Google Scholar]
- 14.Sharma K., Hallan S.S., Lal B. Development and characterization of floating spheroids of atorvastatin calcium loaded NLC for enhancement of oral bioavailability. Artif Cells Nanomed Biotechnol. 2015:1–9. doi: 10.3109/21691401.2015.1041637. [DOI] [PubMed] [Google Scholar]
- 15.Yeom D.W., Song Y.S., Kim S.R. Development and optimization of a self-microemulsifying drug delivery system for atorvastatin calcium by using D-optimal mixture design. Int J Nanomedicine. 2015;10:3865–3877. doi: 10.2147/IJN.S83520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Semete B., Booysen L., Lemmer Y. In vivo evaluation of the biodistribution and safety of PLGA nanoparticles as drug delivery systems. Nanomedicine (Lond) 2010;6:662–671. doi: 10.1016/j.nano.2010.02.002. [DOI] [PubMed] [Google Scholar]
- 17.Shive M.S., Anderson J.M. Biodegradation and biocompatibility of PLA and PLGA microspheres. Adv Drug Deliv Rev. 1997;28:5–24. doi: 10.1016/s0169-409x(97)00048-3. [DOI] [PubMed] [Google Scholar]
- 18.Li Z., Han X., Zhai Y. Critical determinant of intestinal permeability and oral bioavailability of pegylated all trans-retinoic acid prodrug-based nanomicelles: chain length of poly (ethylene glycol) corona. Colloids Surf B Biointerfaces. 2015;130:133–140. doi: 10.1016/j.colsurfb.2015.03.036. [DOI] [PubMed] [Google Scholar]
- 19.Wang J., Sun J., Chen Q. Star-shape copolymer of lysine-linked di-tocopherol polyethylene glycol 2000 succinate for doxorubicin delivery with reversal of multidrug resistance. Biomaterials. 2012;33:6877–6888. doi: 10.1016/j.biomaterials.2012.06.019. [DOI] [PubMed] [Google Scholar]
- 20.Han X., Li Z., Sun J. Stealth CD44-targeted hyaluronic acid supramolecular nanoassemblies for doxorubicin delivery: probing the effect of uncovalent pegylation degree on cellular uptake and blood long circulation. J Control Release. 2015;197:29–40. doi: 10.1016/j.jconrel.2014.10.024. [DOI] [PubMed] [Google Scholar]
- 21.Qi X., Qin X., Yang R. Intra-articular administration of chitosan thermosensitive in situ hydrogels combined with diclofenac sodium-loaded alginate microspheres. J Pharm Sci. 2016;105:122–130. doi: 10.1016/j.xphs.2015.11.019. [DOI] [PubMed] [Google Scholar]
- 22.Geng Y., Zhao L., Zhao J. Development of a supercritical fluid chromatography-tandem mass spectrometry method for the determination of lacidipine in beagle dog plasma and its application to a bioavailability study. J Chromatogr B Analyt Technol Biomed Life Sci. 2014;945–946:121–126. doi: 10.1016/j.jchromb.2013.11.029. [DOI] [PubMed] [Google Scholar]
- 23.Li D., Zhang T., Kou L. Development of a supercritical fluid chromatography-tandem mass spectrometry method for the determination of azacitidine in rat plasma and its application to a bioavailability study. Molecules. 2013;19:342–351. doi: 10.3390/molecules19010342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Santander-Ortega M.J., Jodar-Reyes A.B., Csaba N. Colloidal stability of pluronic F68-coated PLGA nanoparticles: a variety of stabilisation mechanisms. J Colloid Interface Sci. 2006;302:522–529. doi: 10.1016/j.jcis.2006.07.031. [DOI] [PubMed] [Google Scholar]
- 25.Khan F.N., Dehghan M.H. Enhanced bioavailability of atorvastatin calcium from stabilized gastric resident formulation. AAPS PharmSciTech. 2011;12:1077–1086. doi: 10.1208/s12249-011-9673-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fredenberg S., Wahlgren M., Reslow M. The mechanisms of drug release in poly(lactic-co-glycolic acid)-based drug delivery systems–a review. Int J Pharm. 2011;415:34–52. doi: 10.1016/j.ijpharm.2011.05.049. [DOI] [PubMed] [Google Scholar]
- 27.Zhou J., Zhou D. Improvement of oral bioavailability of lovastatin by using nanostructured lipid carriers. Drug Des Devel Ther. 2015;9:5269–5275. doi: 10.2147/DDDT.S90016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jiang Q., Liu W., Li X. Detection of related substances in polyene phosphatidyl choline extracted from soybean and in its commercial capsule by comprehensive supercritical fluid chromatography with mass spectrometry compared with HPLC with evaporative light scattering detection. J Sep Sci. 2016;39:350–357. doi: 10.1002/jssc.201500954. [DOI] [PubMed] [Google Scholar]
- 29.Zhang Q., Sun J., Lu T. A rapid and sensitive LC-MS/MS method for evaluation of the absolute oral bioavailability of a novel c-Met tyrosine kinase inhibitor QBH-196 in rats. Biomed Chromatogr. 2015;29:1650–1656. doi: 10.1002/bmc.3474. [DOI] [PubMed] [Google Scholar]
- 30.Luo C., Sun J., Du Y. Emerging integrated nanohybrid drug delivery systems to facilitate the intravenous-to-oral switch in cancer chemotherapy. J Control Release. 2014;176:94–103. doi: 10.1016/j.jconrel.2013.12.030. [DOI] [PubMed] [Google Scholar]
- 31.Amidon G.L., Lennernas H., Shah V.P. A theoretical basis for a biopharmaceutic drug classification: the correlation of in vitro drug product dissolution and in vivo bioavailability. Pharm Res. 1995;12:413–420. doi: 10.1023/a:1016212804288. [DOI] [PubMed] [Google Scholar]
- 32.Lennernas H. Human jejunal effective permeability and its correlation with preclinical drug absorption models. J Pharm Pharmacol. 1997;49:627–638. doi: 10.1111/j.2042-7158.1997.tb06084.x. [DOI] [PubMed] [Google Scholar]