

Graphical Abstract
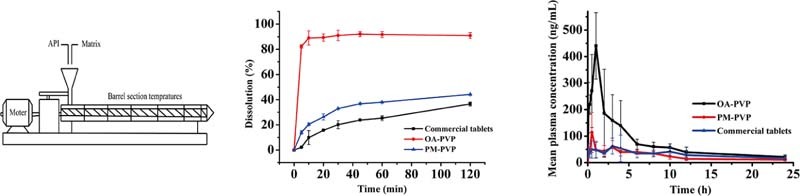
Keywords: Hot melt extrusion, Solid dispersion, Oleanolic acid, Dissolution rate, Oral bioavailability
Abstract
The aim of this study was to improve the in vitro dissolution rate and oral bioavailability of oleanolic acid (OA), a water insoluble drug belonging to BCS class IV. Hot melt extrusion (HME) was applied to develop OA amorphous solid dispersions. The characterizations of the optimal formulation were performed by differential scanning calorimetry, X-ray powder diffraction, Fourier transform infrared spectroscopy and in vitro dissolution test. The in vivo pharmacokinetic study was conducted in rats. As a result, OA solid dispersion based on PVP VA 64 (OA-PVP) was successfully prepared. In the dissolution medium containing 0.3% SDS, OA-PVP dramatically increased the releasing rate of OA compared with the physical mixture (PM-PVP) and commercial tablet. Furthermore, OA-PVP exhibited higher AUC (P < 0.05) and Cmax (P < 0.05) than PM-PVP and commercial tablet. The superior dissolution property and bioavailability of OA-PVP mainly attributed to the amorphous state of OA in PVP VA64 and the well dispersion caused by thermal melting and shearing. Overall, hot melt extrusion was an efficient strategy to enhance the dissolution rate and oral bioavailability of OA.
1. Introduction
Oleanolic acid (OA) is a bioactive pentacyclic triterpenoid compound widely existing in Asian herbs [1]. Its clinical pharmacology activities mainly include hepatoprotective, anti-inflammatory, antibacterial and antiulcer effect [2]. As a natural origin drug, OA owes a good application prospect due to its low toxicity. However, it exhibits low bioavailability after oral administration on account of the poor aqueous solubility (<1 µg/ml) [3] and poor permeability (Papp = 1.1–1.3 × 10−6 cm/sec in the apical-to-basolateral direction at 10 and 20 µM) [4]. Jeong et al. reported that the absolute oral bioavailability of OA was 0.7% for oral doses of 25 and 50 mg/kg. The very low oral bioavailability of OA could be due to a poor absorption and extensive metabolic clearance [4].
Many attempts were made to improve the solubility of OA, such as solid lipid nanoparticles [5], nanosuspensions [6], [7], [8], β-cyclodextrin inclusion compounds [9], self-nanoemulsified formulations [10], phospholipid complexes [11] and solid dispersions [12], [13]. However, the complicated unavailable scale-up process and the presence of organic solvents limit their application in the production process.
In recent years, hot melt extrusion (HME) has gained great interest in pharmaceutical field due to many advantages, such as scalable continuous processing, solvent-free and environmentally friendly [14], [15]. In the HME process, drug and polymers are mixed thoroughly under the controlled temperature and pumped with a rotating screw through a die plate, yielding solid dispersions. To obtain a desirable product, the extrusion temperature is typically set 30–60 °C higher than the Tg (glass transition temperature) or Tm (melting temperature) of polymers, which will soften the mixture and provide a good flowability [16], [17], [18]. In addition, polymers play an important role in stabilizing the metastable solid, inhibiting crystallization [19] and maintaining supersaturation during dissolution [20], [21]. Several hydrophilic polymers are developed for the HME process, for example, polyvinyl caprolactampolyvinyl acetate-polyethylene glycol graft copolymer (Soluplus®), polyvinylpyrrolidone-vinyl acetate copolymer (PVP VA64), polyethylene glycol (PEG), Eudragit® EPO, and hypromellose acetate succinate (HPMCAS). However, the miscibility of drug molecular and polymers needs to be investigated.
In the present study, we developed a HME method to improving the dissolution and oral bioavailability of OA by preparation of solid dispersions. Hydrophilic polymers, drug-to-carrier ratio and extrusion temperature were optimized. Thermal gravimetric analysis was used to detect weight loss due to degradation of OA. Differential scanning calorimetry (DSC), X-ray powder diffraction (XRPD), Fourier transform infrared (FT-IR) spectroscopy and in vitro dissolution were carried out to characterize the optimal formulation. Furthermore, the pharmacokinetic study was evaluated in rats.
2. Materials and methods
2.1. Materials
OA was purchased from Wuhan Dahua Pharmaceutical Co., Ltd. (Wuhan, Hubei). Soluplus® and PVP VA64 were purchased from BASF Co., Ltd. (Shanghai, China). PEG 6000 was obtained from Beijing Fengli Jingqiu Pharmaceutical Co., Ltd. (Beijing, China). Sodium dodecyl sulfate (SDS) was purchased from Tianjin Bodi Chemical Holding Co., Ltd. (Tianjin, China). H3PO4 was obtained from Tianjin Kemiou Chemical Reagent Co., Ltd. (Tianjin, China). Glycyrrhetinic acid was obtained from China's food and drug administration. Methanol, Methyl tert-Butyl Ether of HPLC grade was purchased from Thermo Fisher Scientific (Shanghai, China). Ammonium acetate (HPLC grade) was purchased from Dikma Technology Inc. (Beijing, China).
2.2. Preparation of hot-melt extrudates
OA was mixed with the hydrophilic carriers at a ratio of 1:10 (drug: carrier, w/w) to obtain the physical mixtures (PMs) by the method of increment by equal quantity. Then the PMs were extruded by a co-rotating twin-screw extruder (AK-26; Coperion Keya Co., Najing, China) at temperature of 160 °C. The screw rate was set at 20 rpm. The extrudates were collected from the die and cooled at ambient temperature. A universal high-speed smashing machine (FW400A, Zhongxingweiye instrument Co., Ltd, Beijing, China) was used to crushing the banded product into powder. After passing through an 80-mesh screen, the obtained solid dispersions powder was stored in dry dark conditions for further in vitro dissolution test and in vivo pharmacokinetic study.
2.3. Characterization of solid dispersions
2.3.1. Thermal analysis
The thermal stability of OA was determined on a thermal gravimetric analyzer (TGA-50, Shimadzu, Japan). Samples were heated from 30 to 400 °C at a rate of 10 °C/min with an empty aluminum pan as the reference. Nitrogen was used as the purge gas at a flow rate of 40 ml/min. Plots of weight versus temperature were recorded.
Differential scanning calorimetry (DSC) thermograms of the samples were conducted using a DSC1 STAR® system (Mettler Tolelo, Switzerland) equipped with a cooling system. Samples (3–5 mg) were weighted accurately and loaded in the aluminum pans. The DSC cell was purged by using nitrogen gas at flow rate of 40 ml/min. A heating rate of 20 °C/min was used to scan from 40 to 330 °C. All results were analyzed using the STARe software.
2.3.2. XRPD
XRPD analysis was performed on a D\Max-2400 X-Ray powder diffractometer (Rigaku, Japan) at ambient temperature. Monochromatic Cu-Kα radiation (λ = 1.5406 Å) was used in the 2θ angle range from 3° to 50° with a step width of 0.05°.The voltage and current of the equipment were 30 mA and 40 kV.
2.3.3. FT-IR
FT-IR spectra of samples were measured using an EQUINOX55 Fourier infrared spectrometer (Bruker, Germany) instrument. The samples were mixed with KBr using a mortar and pestle, and then compressed to prepare a disk. The spectra were recorded over a wavenumber range of 4000 cm−1 to 400 cm−1, with a resolution of 4 cm−1.
2.3.4. In vitro dissolution testing
The in vitro drug release from samples was tested according method 2 as described in the Chinese Pharmacopeia 2010 using a dissolution tester (ZRS-8G, Tianda Tianfa Science and Technology Ltd., Tianjin, China). Samples equivalent to 20 mg of OA were loaded into 00 capsule shells and sunk into 900 ml of deionized water containing 0.3% (w/v) SDS at a temperature of 37 ± 0.2 °C. The paddle rotation speed was set to 100 rpm. 5 ml of samples were withdrawn at predetermined time intervals 5, 10, 20, 30, 45, 60 and 120 min and filtered through a 0.45 µm microfiltration membrane. The same volume of fresh medium was replaced at each time point. Then, 20 µl of each subsequent filtrate was injected into HPLC for analysis. All experiments were performed in triplicate.
A HITACHI HPLC system equipment equipped a UV Detector 5430 and a pump 5110 (HITACHI Ltd, Japan) was used to determine the concentrations of OA. The separation of OA was conducted on a Cosmosil C18 (150 µm × 4.6 µm, 5 µm) column. The mobile phase was methanol and purified water containing 0.1% phosphoric acid (90:10, v/v) with a flow rate of 1 ml/min. The column temperature was set at 25 °C and the detector was operated at 210 nm.
2.4. In vivo pharmacokinetic study
The pharmacokinetic study was performed in male Sprague Dawley (SD) rats (250 ± 5 g). The experiment was conducted according to the “Guidelines for the Care and Use of Laboratory Animals” and approved by the Animal Ethics Committee of Shenyang Pharmaceutical University (Shenyang, China). SD rats (n = 15) were randomly divided into three groups, and fasted overnight before dosing and fed 6 h after dosing. The optimized OA solid dispersion, physical mixture of OA and carrier (1:10, w/w) and commercial tablet were suspended in deionizer water and orally administered to the animals at a dosage of 50 mg/kg. Blood samples (0.2 ml) were collected from the orbit before dosing and at 0.08, 0.25, 0.5, 1, 2, 3, 4, 6, 8, 10, 12 and 24 h after dosing. After centrifuged at 13,000 rpm for 10 min, the plasma samples were obtained and stored at −20 °C until analysis.
2.5. Sample preparation
Frozen plasma samples were thawed at room temperature, and 50 µl of each sample was transformed into a 10 ml polypropylene tube. A liquid–liquid extraction procedure was applied to extract the analyte. 50 µl internal standard (IS) solution containing glycyrrhetinic acid 1 µg/ml and 50 µl methanol-water (85:15, v/v) was added to the samples and vortexed for 1 min. Then, 3 ml of methyl tert-butyl ether was added and vortexed for another 3 min. After centrifuged at 3500 rpm for 10 min, the supernatant was transformed and dried at 37 °C under a soft nitrogen gas flow. The residue was reconstituted with 100 µl methanol-water (85:15, v/v) and 5 µl was injected into the Acquity UPLC™ system (Waters Co., Ltd., Milford, Ma, USA).
2.6. UPLC-MS/MS analysis
OA was separated using a Phenomenex XB-C18 (50 × 2.1 mm, 2.6 µm) column. The mobile phase consisted of menthol and water with 10 Mm ammonium acetate (85:15, v/v). The flow rate was 0.2 ml/min at the first two minutes, and changed to 0.25 ml/min within 0.01 min. After maintaining there from 2.01 to 3.99 min, the flow rate returned to 0.2 ml/min in 0.01 min. The column temperature was maintained at 30 °C.
Detection of OA was performed in negative ion mode with an ESI source. Selective ion reaction (SIR) was applied to monitor the transitions of m/z 455.5 and m/z 469.5 for OA and IS, respectively, with a scan time of 0.2 sec. The capillary voltage was set at 2.54 kV. The cone voltage was 55 and 35 V for OA and IS, respectively. The ion source temperature and desolvation temperature was set at 120 and 500 °C. The method showed good linearity over the concentration range of 10–1000 ng/ml for OA. The pharmacokinetic parameters were calculated using DAS 2.1.1 software and the values were expressed as mean ± S.D.
2.7. Statistical analysis
Statistical analysis was carried out using Student's t-test by IBM SPSS statistics 21.0 software. Differences were considered statistically significant if the P value was less than 0.05 (P < 0.05).
3. Results and discussion
3.1. Preparation of OA solid dispersions
Hot melt extrusion was applied to prepare OA amorphous solid dispersions. However, amorphous drug substances are physically unstable on account of their high energy state and tend to recrystallize. Therefore, in the present study, three polymer carriers Soluplus®, PVP VA64 and PEG 6000 were used to prepare OA solid dispersions (OA-SOL, OA-PVP and OA-PEG) at a drug-to-carrier ratio of 1:10 (w/w). The extrusion temperature was set at 160 °C for OA-SOL and OA-PVP, and 80 °C for OA-PEG. From Fig. 1A, the cumulative released quantities were 40% and 80% for OA-PEG and OA-SOL after 2 h, while over 90% of OA was dissolved for OA-PVP within 10 min. The results indicated that PVP VA64 obviously enhanced the dissolution of OA, which was chosen as the efficient carrier.
Fig. 1.

Dissolution profiles of: A. OA solid dispersion with Soluplus®, PVP VA64 and PEG 6000 as carriers; B. OA solid dispersion with PVP VA64 as carrier at different drug-to-carrier ratio of 1:3, 1:5, 1:7, 1:10 and 1:15; C. OA-PVP extruded at extrusion temperature of 140 °C, 150 °C, 160 °C and 170 °C; D. OA-PVP, PM-PVP and commercial tablet.
Subsequently, the effect of drug-to-carrier ratio ranging from 1:3 to 1:15 on dissolution of OA solid dispersion was investigated. As shown in Fig. 1B, the extrudates with lower percentage of PVP VA64 demonstrated lower dissolution. This may be due to the low solubility of the OA in the dissolution medium. However, at a drug-to-carrier ratio of 1:10, over 90% of the drug released from the hydrophilic matrix within the first 10 min. No further increase was obtained with extra amount of PVP VA64 (1:15), thus the optimal ratio of drug to PVP VA64 was 1:10.
Hot melt extrusion technology is a thermal processing technique, which increases the risk of thermal degradation of drugs. In addition, the extrusion temperature greatly influences the miscibility of drug and carrier and the rheological properties of a molten formulation. Thermo gravimetric analysis (Fig. 2) showed that OA was thermally stable and no weight lost before 250 °C. Meanwhile, in the preliminary experiment, the extruder barrels stopped below 140 °C due to high torque, and the extrudates became more molten and dark yellow above 170 °C. Hence, the temperature range was investigated from 140 to 170 °C. As can be seen in Fig. 1C, an increasing dissolution was obtained with the increasing temperature. This is because, higher extrusion temperature lowered the melt viscosity, which made the material flow faster inside the extruder and dispersed homogeneously in the carrier. The extrudates from 160 °C and 170 °C showed similar dissolution behavior, nevertheless, the degradation of OA at 170 °C was higher than at 160 °C (1.43% vs 0.22%). Therefore, the extrusion temperature was controlled at 160 °C.
Fig. 2.
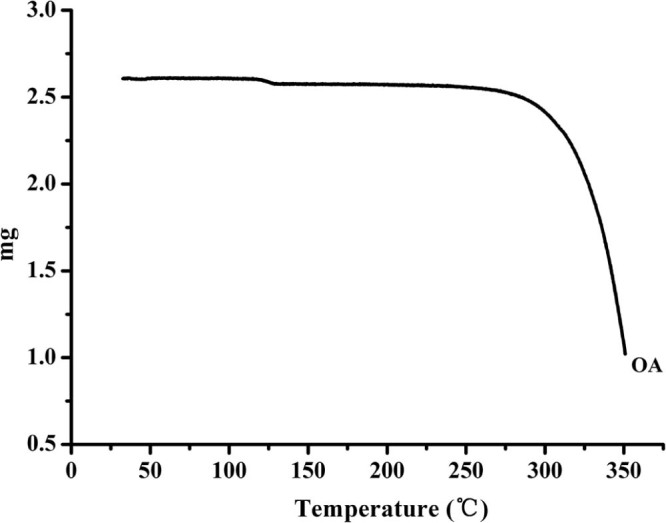
TGA thermogram of OA.
3.2. Thermal analysis
The DSC curves of OA, PVP VA64, physical mixture and OA-PVP were presented in Fig. 3. The pure OA exhibited a first endothermic peak at 130 °C, which was probably due to the loss of methanol from the OA methanol crystallization solvate [3], [22]. An exothermic peak at 190 °C indicated a transformation of OA from metastable state to crystalline state, which was also observed by Sun et al. [5]. The sharp endotherm peak at 310 °C was corresponding to the melting point of OA. The physical mixture of OA and PVP VA64 also showed the weakened endothermic peak of OA. However, the disappearance of endothermic peak in OA-PVP indicated that OA existed as amorphous state.
Fig. 3.

DSC thermograms of A. OA, B. PM-PVP, C. PVP VA64, and D. OA-PVP.
3.3. XRPD
XRPD was used to confirm the crystal form of OA in OA-PVP. The XRPD patterns of OA, PVP VA64, physical mixture and OA-PVP were shown in Fig. 4. The pure OA exhibited strong sharp peaks at 8.43, 12.99, 13.68, 15.36, 19.56 and 20.97°, indicating the crystalline state of the drug. However, in the pattern of OA-PVP, no distinct diffraction peaks were observed, indicating that the drug was in an amorphous state. The results of XRPD were in agreement with that of DSC.
Fig. 4.
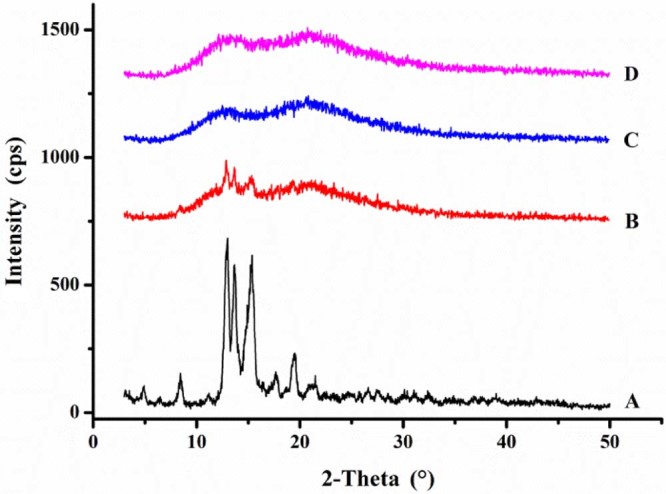
XRPD patterns of A. OA, B. PM-PVP, C. PVP VA64, and D. OA-PVP.
3.4. FT-IR
The interaction of OA and PVP VA64 was detected by FT-IR spectroscopy and the results were demonstrated in Fig. 5. The characteristic absorption peaks at 1697.6 and 3432.4 cm−1 were the stretch vibration of —C O and —OH in OA. PVP VA64 exhibited peaks of —OH stretch vibration (3463.5 cm−1), —C O stretch vibration (1738.8 cm−1) in ester group and —C O stretch vibration (1678.7 cm−1) in amide group. The FT-IR spectra of physical mixture showed a summation of drug and PVP VA64. However, after the hot melt extrusion process, the peak of —C O of OA shifted to 1678.8 cm−1, and the peak of —OH stretch vibration of PVP VA64 shifted to 3457.8 cm−1. This was indicative of hydrogen bonds interactions between OA and PVP VA64.
Fig. 5.
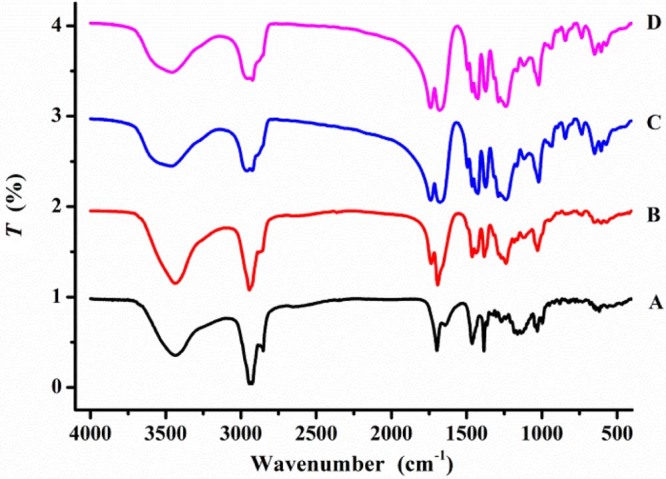
FT-IR spectrogram of A. OA, B. PM-PVP, C. PVP VA64, and D. OA-PVP.
3.5. In vitro dissolution testing
The in vitro dissolution profiles of commercial tablet, physical mixture and OA-PVP solid dispersion were shown in Fig. 1D. A dissolution medium containing 0.3% (w/v) SDS was used to provide sink condition. From the results, the solid dispersion OA-PVP dramatically improved the dissolution rate of OA, and about 90% of the drug was released within 10 min. The improvement of OA-PVP extrudate was attributed to the transformation of crystalline drug into amorphous state that no energy was required to break up the crystal lattice of OA during the dissolution process [23]. Furthermore, the solubility and wettability of OA can be improved by PVP VA64 due to its excellent solubilizing property [24]. The dissolution of physical mixture (the accumulative dissolution was about 45% after 2 h) showed slightly enhancement compared with commercial tablet (the accumulative dissolution was about 37% after 2 h). This may be for the reason that the solubilization of PVP VA64 created a hydrodynamic layer surrounding the drug particles and inhibited the recrystallization of the drug in aqueous environment [24].
3.6. In vivo pharmacokinetic study
Oral absorption of OA from OA-PVP, PM-PVP and commercial tablet was evaluated in rats at a dosage of 50 mg/kg. The mean plasma concentration-time profiles were shown in Fig. 6 and the main pharmacokinetic parameters were summarized in Table 1. As shown, the AUC0–24 (1840 ± 381.8 ng⋅h/ml) of OA-PVP was 3.0-fold and 2.4-fold higher than that of PM-PVP and commercial tablet (P < 0.05), while the Cmax (498.7 ± 120.8 ng/ml) of OA-PVP was found to be 3.7-fold and 5.6-fold higher, respectively (P < 0.05). The results indicated that OA-PVP exhibited superior oral bioavailability, which may be attributed to the enhanced dissolution after preparation of solid dispersion with PVP VA64. The Cmax of PM-PVP was slightly higher than the commercial tablet. This may be due to solubilization of PVP VA64 surrounding the OA crystalline particles improved its dissolution in the gastrointestinal tract. However, no significant difference was observed between these two formulations. Thus, solid dispersion prepared by hot melt extrusion method was a potential strategy for improving the oral bioavailability of OA.
Fig. 6.

Plasma concentration-time curves of OA after oral administration of OA-PVP, PM-PVP, and commercial tablet to SD rats (data are mean ± S.D., n = 5).
Table 1.
Pharmacokinetic parameters of OA after oral administration of OA-PVP, PM-PVP, and commercial tablet to SD rats (data are mean ± S.D., n = 5).
| Pharmacokinetic parameters | OA-PVP | PM-PVP | Commercial tablet |
|---|---|---|---|
| AUC0–24 (ng⋅h/mL) | 1840 ± 381.8* | 615.1 ± 115.5 | 761.8 ± 272.2 |
| AUC0–∞ (ng⋅h/mL) | 2039.5 ± 483.4* | 696.8 ± 151.6 | 875.08 ± 292.1 |
| Cmax (ng/mL) | 498.7 ± 120.8* | 134.4 ± 47.4 | 89.1 ± 33.1 |
| Tmax (h) | 1.1 ± 0.5 | 1.1 ± 1.0 | 1.8 ± 1.6 |
| F (%) | 241.53 | 80.74 | 100 |
Note: P < 0.05 (*) versus the commercial tablet as the control.
4. Conclusions
OA amorphous solid dispersion based on PVP VA64 was developed using hot melt extrusion method. The drug-to-carrier ratios and extrusion temperatures were optimized according to the dissolution of the extrudates. The crystal form of the optimal formulation, OA-PVP, was characterized by DSC and XRPD, which indicated that OA existed in amorphous state and dispersed homogenously in PVP VA64. FT-IR spectra revealed the formation of hydrogen bond between OA and the polymer molecules. The in vitro and in vivo evaluation showed a remarkable improvement of OA-PVP compared with PM-PVP and commercial tablet. These results indicated that the amorphization of OA by HME process was a potential method to enhancing its oral absorption.
Acknowledgments
This work was financially supported by National Natural Science Foundation of China (No. 81502993), Doctoral Research Funding of Liaoning Province (No. 20141066), General Project in Department of Education of Liaoning Province (No. L2014379), and Career Development Program for Young Teachers in Shenyang Pharmaceutical University.
Footnotes
Peer review under responsibility of Shenyang Pharmaceutical University.
Contributor Information
Qiang Fu, Email: graham_pharm@aliyun.com.
Zhonggui He, Email: hezhonggui@vip.163.com.
References
- 1.Pollier J., Goossens A. Oleanolic acid. Phytochemistry. 2012;77:10–15. doi: 10.1016/j.phytochem.2011.12.022. [DOI] [PubMed] [Google Scholar]
- 2.Liu J. Pharmacology of oleanolic acid and ursolic acid. J Ethnopharmacol. 1995;49:57–68. doi: 10.1016/0378-8741(95)90032-2. [DOI] [PubMed] [Google Scholar]
- 3.Tong H.H.Y., Wu H.B., Zheng Y., et al. Physical characterization of oleanolic acid nonsolvate and solvates prepared by solvent recrystallization. Int J Pharm. 2008;355:195–202. doi: 10.1016/j.ijpharm.2007.12.005. [DOI] [PubMed] [Google Scholar]
- 4.Dong W.J., Kim Y.H., Hui H.K., et al. Dose-linear pharmacokinetics of oleanolic acid after intravenous and oral administration in rats. Biopharm Drug Dispos. 2007;28:51–57. doi: 10.1002/bdd.530. [DOI] [PubMed] [Google Scholar]
- 5.Sun H., Zhang X.H., Wang S., et al. Preparation and characterization of oleanolic acid-loaded solid lipid nanoparticles for oral administration. J Chin Pharm Sci. 2011;20:259–265. [Google Scholar]
- 6.Li W., Ng K.Y., Heng P.W. Development and evaluation of optimized sucrose ester stabilized oleanolic acid nanosuspensions prepared by wet ball milling with design of experiments. Biol Pharm Bull. 2014;37:926–937. doi: 10.1248/bpb.b13-00864. [DOI] [PubMed] [Google Scholar]
- 7.Li W., Das S., Ng K.Y., et al. Formulation, biological and pharmacokinetic studies of sucrose ester-stabilized nanosuspensions of oleanolic Acid. Pharm Res. 2011;28:2020–2033. doi: 10.1007/s11095-011-0428-3. [DOI] [PubMed] [Google Scholar]
- 8.Chen Y.J., Liu J., Yang X.L., et al. Oleanolic acid nanosuspensions: preparation, in-vitro characterization and enhanced hepatoprotective effect. J Pharm Pharmacol. 2005;57:259–264. doi: 10.1211/0022357055407. [DOI] [PubMed] [Google Scholar]
- 9.Chen Q.S., Wu J., Li W., et al. Extraction of oleanolic acid from leaves of Chaenomeles speciosa and processing techniques of its HP-beta-cyclodextrin inclusion compound. Zhong Yao Cai. 2010;33:804–807. [PubMed] [Google Scholar]
- 10.Xi J., Chang Q., Chan C.K., et al. Formulation development and bioavailability evaluation of a self-nanoemulsified drug delivery system of oleanolic acid. AAPS PharmSciTech. 2009;10:172–182. doi: 10.1208/s12249-009-9190-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jiang Q.K., Yang X.X., Du P., et al. Dual strategies to improve oral bioavailability of oleanolic acid: enhancing water-solubility, permeability and inhibiting cytochrome P450 isozymes. Eur J Pharm Biopharm. 2016;99:65–72. doi: 10.1016/j.ejpb.2015.11.013. [DOI] [PubMed] [Google Scholar]
- 12.Liu L.X., Wang X.C. Improved dissolution of oleanolic acid with ternary solid dispersions. AAPS PharmSciTech. 2007;8:267–271. doi: 10.1208/pt0801016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tong H.H.Y., Du Z., Wang G.N., et al. Spray freeze drying with polyvinylpyrrolidone and sodium caprate for improved dissolution and oral bioavailability of oleanolic acid, a BCS Class IV compound. Int J Pharm. 2011;404:148–158. doi: 10.1016/j.ijpharm.2010.11.027. [DOI] [PubMed] [Google Scholar]
- 14.Agrawal A.M., Dudhedia M.S., Zimny E. Hot melt extrusion: development of an amorphous solid dispersion for an insoluble drug from mini-scale to clinical scale. AAPS PharmSciTech. 2016;17:133–147. doi: 10.1208/s12249-015-0425-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Madan S., Madan S. Hot melt extrusion and its pharmaceutical applications. Asian J Pharm Sci. 2012;7:123–133. [Google Scholar]
- 16.Chan S.Y., Qi S., Craig D.Q.M. An investigation into the influence of drug–polymer interactions on the miscibility, processability and structure of polyvinylpyrrolidone-based hot melt extrusion formulations. Int J Pharm. 2015;496:95–106. doi: 10.1016/j.ijpharm.2015.09.063. [DOI] [PubMed] [Google Scholar]
- 17.Chokshi R.J., Sandhu H.K., Iyer R.M., et al. Characterization of physico-mechanical properties of indomethacin and polymers to assess their suitability for hot-melt extrusion process as a means to manufacture solid dispersion/solution. Braz Dent J. 2005;94:2463–2474. doi: 10.1002/jps.20385. [DOI] [PubMed] [Google Scholar]
- 18.Li Y.C., Pang H.S., Guo Z.H., et al. Interactions between drugs and polymers influencing hot melt extrusion. J Pharm Pharmacol. 2014;66:148–166. doi: 10.1111/jphp.12183. [DOI] [PubMed] [Google Scholar]
- 19.Gao Y., Olsen K.W. Drug–polymer interactions at water-crystal interfaces and implications for crystallization inhibition: molecular dynamics simulations of amphiphilic block copolymer interactions with tolazamide crystals. J Pharm Sci. 2015;104:2132–2141. doi: 10.1002/jps.24442. [DOI] [PubMed] [Google Scholar]
- 20.Alonzo D.E., Gao Y., Zhou D., et al. Dissolution and precipitation behavior of amorphous solid dispersions. J Pharm Sci. 2011;100:3316–3331. doi: 10.1002/jps.22579. [DOI] [PubMed] [Google Scholar]
- 21.Alonzo D.E., Zhang G.G., Zhou D., et al. Understanding the behavior of amorphous pharmaceutical systems during dissolution. Pharm Res. 2010;27:608–618. doi: 10.1007/s11095-009-0021-1. [DOI] [PubMed] [Google Scholar]
- 22.Li Y.Y., Liu H.Z., Guo B., et al. Enhancement of dissolution rate and oral bioavailability in beagle dogs of oleanolic acid by adsorbing onto porous silica using supercritical carbon dioxide. J Drug Deliv Sci Technol. 2014;24:380–385. [Google Scholar]
- 23.Shinde V.R., Shelake M.R., Shetty S.S., et al. Enhanced solubility and dissolution rate of lamotrigine by inclusion complexation and solid dispersion technique. J Pharm Pharmacol. 2008;60:1121–1129. doi: 10.1211/jpp.60.9.0002. [DOI] [PubMed] [Google Scholar]
- 24.Fei Y., An K., Shan J.J., et al. Preparation of osthole-polymer solid dispersions by hot-melt extrusion for dissolution and bioavailability enhancement. Int J Pharm. 2014;465:436–443. doi: 10.1016/j.ijpharm.2014.02.040. [DOI] [PubMed] [Google Scholar]