

Abstract
Peramivir was a novel and highly potent neuraminidase (NA) inhibitor for the treatment of influenza A and B. However, it exhibited a very low oral bioavailability (only 3%) due to the high polarity (log P of −1.4) and the low membrane permeability across the intestine. To utilize the PEPT1-mediated prodrug strategy to improve the oral absorption and develop the oral alternative, seven amino acid ester prodrugs and seven amino acid amide prodrugs have been synthesized. The permeability of these prodrugs across Caco-2 cells were screened. Peramivr-(CH2)2-l-Val and Peramivir-l-Ile were of the highest permeability in ester prodrugs and amide prodrugs, respectively, and then they were selected for further studies. Glycylsarcosine (gly-sar) uptake by Caco-2 could be inbihited by Peramivir-(CH2)2-l-Val and Peramivir-l-Ile in a concentration-dependent manner, and the IC50 was 1.34 ± 0.31 mM and 1.78 ± 0.48 mM, respectively. The direct uptake of Peramivir-(CH2)2-l-Val and Peramivir-l-Ile in MDCK-PEPT1 cells were significantly higher than in MDCK mock cells, and could be markedly inhibited by gly-sar. The uptake of Peramivir-(CH2)2-l-Val and Peramivir-l-Ile (0.01 to 50 mM) in MDCK-hPEPT1 cells conformed to Michaelis–Menten Equation. The oral bioavailability of peramivir was 65.3% and 37.3% after the oral administration of Peramivir-(CH2)2-l-Val and Peramivir-l-Ile to rats, respectively. The oral absorption and bioactivation of Peramivir-(CH2)2-l-Val was rapid and extensive, and no Peramivir-(CH2)2-l-Val was found in plasma. Because the amide bond was relatively stable, Peramivir-l-Ile could not be totally converted to the parent drug in vivo. Peramivir-(CH2)2-l-Val with good oral profiles and rapid bioactivation might be a promising prodrug for the further clinic development. The present study also corroborated the idea that the PEPT1-mediated prodrug approach has enormous promise for improving the oral absorption of poorly absorbed drug.
Keywords: Peramivir, Prodrug, Peptide transporter 1, Pharmacokinetics, Oral bioavailability
Graphical abstract
Peramivir-(CH2)2-l-Val can increase the oral bioavailability of peramivir from 4.1% to 65.3%.
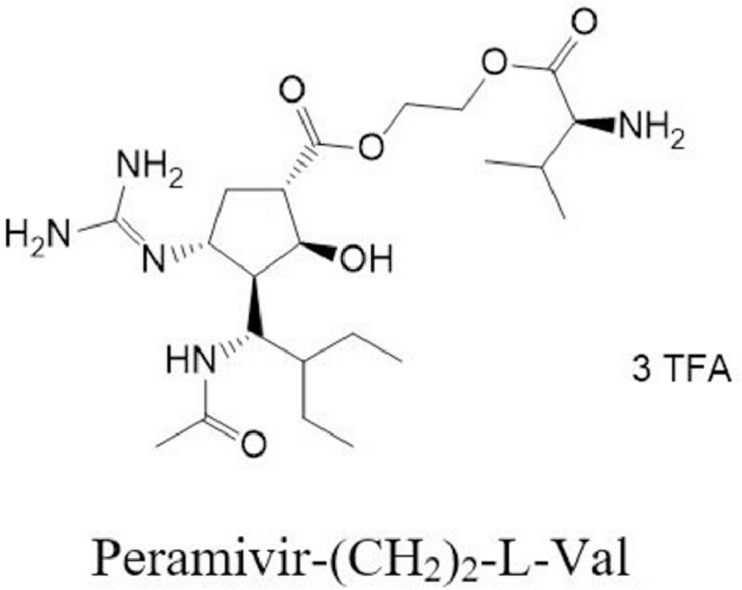
1. Introduction
Peramivir was a novel and highly potent neuraminidase inhibitor developed by BioCryst Pharmaceuticals Inc. for the treatment of influenza A and B [1]. Like oseltamivir and zanamivir, it can bind to the active site of the influenza virus neuraminidase to prevent the spread of viruses. Peramivir showed a distinct in vitro resistance profile when compared with oseltamivir and zanamivir, and some viral variants with in vitro oseltamivir and zanamivir resistance could be susceptible to peramivir [2], [3]. FDA issued an Emergency Use Authorization (EUA) on 23 October 2009, authorizing peramivir for treatment of certain hospitalized adults and children with pH1N1 in the United States till 23 June 2010, which was the first time that an investigational drug was authorized for clinical use under an EUA [4], [5]. Due to the high polarity (log P of −1.4) and the low membrane permeability across the intestine, peramivir had a very low oral bioavailability (only 3%). Oral peramivir was associated with reduced viral titers, but no significant decrease was observed to relief of symptoms, which could be attributed to a low oral bioavailability in humans [2]. Therefore, peramivir can only be given as an intravenous injection to the patients with influenza in clinic, which limited the clinical utility of peramivir to treat influenza infections. Hence, the development of an oral alternative to intravenous administration of peramivir is indispensable for reducing costs dictated by hospital treatment and improving patient compliance.
Many attempts had focused on the structural modification of peramivir with the aim to improve the oral bioavailability. Wang et al. have synthesized a series of peramivir phosphonate derivatives as orally available anti-influenza drugs [6]. But none of them were in routine clinical use at present.
With the development of molecular biology, many membrane transporters have been identified, for example, peptide transporters 1 (PEPT1) [7], amino acid transporter B0,+ (ATB0,+, SLC6A14) [8], [9], the glucose transporter GLUT1 [10]. Prodrugs targeted to membrane transporters have been extensively developed in order to improve oral bioavailability and overcome undesired biopharmaceutical properties of drugs. Thanks to the high expression on the apical membrane of the small intestine, and the broad substrate specificity, PEPT1 has been the most promising and attractive target in the prodrug design among all nutrient transporters. It has been demonstrated a posteriori that a 3 to 5-fold higher oral bioavailability of valacyclovir and valganciclovir compared with acyclovir and ganciclovir, respectively, was attributed to the transport by PEPT1 [11], [12], [13], [14]. This PEPT1-targeted prodrug strategy has also been extended to LY2140023, an N-linked methionil prodrug of LY404039. LY2140023 improved the oral bioavailability of the parent drug from 6.0% to 68.0% in human [15], [16]. In our previous study, we have synthesized s series of amino acid prodrug of cytarabine, and the candidate drug valcytarabine, the l-valyl prodrug of cytarabine, displayed a PEPT1-mediated transport across the small intestine and increased the oral bioavailability of cytarabine from 21.8% to 60.0%. Valcytarabine has been allowed to the clinic trial by CFDA in 2016 (Clinical NO 2016L10399 and 2016L10400) [17].
In order to improve the oral availability and develop the oral alternative to peramivir, the structural modification was performed on the carboxyl group, and seven amino acid ester prodrugs and seven amino acid amide prodrugs were synthesized. The transport of these prodrugs was compared with peramivir in Caco-2 cell monolayers to screen the lead compound with the highest permeability. The uptake of peramivr-(CH2)2-l-Val and peramivir-l-Ile in PEPT1-overexpressing MDCK (MDCK-PEPT1) cells was performed to delineate the role of PEPT1 in the transport of peramivr-(CH2)2-l-Val and peramivir-l-Ile across the intestinal epithelium. We also reported the chemical and enzymatic stability of the prodrugs in phosphate buffers, rat plasma, tissue homogenates and gastrointestinal fluids. Finally, the pharmacokinetics (PK) of peramivir-(CH2)2-l-Val and peramivir-l-Ile was evaluated in Sprague-Dawley rats after oral administration. All above results indicated the PEPT1-mediated prodrug strategy has successfully enhanced the oral absorption of peramivir.
2. Materials and methods
2.1. Materials
Peramivir was purchased from Hubei Xinkang Chemicals Co. Ltd (Wuhan, China). (1S, 2S, 3R, 4R)-Methyl 3-((R)-1-acetamido-2-ethylbutyl)-4-(tert‑butoxycarbonyl) amino)-2-hydroxycyclopentanecarboxylate (compound 1, CAS: 229,614-05-5) was purchased from Acesys Pharmatech Co. Ltd (Nanjing, China). 1, 3-Bis(tert‑butoxycarbonyl)-2-methylthiopseudourea (BocTU) was purchased from Shanghai Balmxy Pharmaceutic Co. Ltd (Shanghai, China). HgCl2, LiOH and 4-dimethylaminopyridine (DMAP) were purchased from Shandong Xiya Chemicals Co. Ltd (Jinan China). 3-(3-dimethylaminopropyl)-1-ethylcarbodiimide hydrochloride (EDC) was purchased from Zhejiang Kangpu Chemicals Co. Ltd (Quzhou China). 1-Hydroxybenzotriazole (HOBT) was purchased from Chengdu Best Reagents Co. Ltd (Chengdu China). All amino acids were purchsed from GL Chemical Ltd (Shanghai, China). PEPT1-overespressing MDCK cell (MDCK-PEPT1) was established by Cellbio Biology Co. Ltd (Shanghai China). All other chemicals used were of the highest purity available.
2.2. The synthesis procedure for prodrugs
2.2.1. Synthesis of N-Boc-protected peramivir (4) (Fig. 1)
Fig. 1.

Synthesis of N-Boc-protected peramivir. (i) HCl-1, 4-dioxane, ether, room temperature, overnight; (ii) triethylamine, Boc-TU, HgCl2, DMF, room temperature, 2 h; (iii) 5% NaOH, EtOH and THF, room temperature, 3 h.
To the suspension of compound 1 (4.0 g, 10 mmol) in 20 ml ether at 0 °C, 4 M HCl-1, 4-dioxane (10.0 mL, 40 mmol) and ether (40 mL) was added. The reaction mixture was stirred overnight at room temperature, and then refluxed at 50 °C for 2 h. The mixture was cooled to room temperature and filtered, and the filtrate was concentrated in vacuo to give the white solid compound 2 (3.0 g, 100%).
To the solution of compound 2 (3.0 g, 10 mmol) in 30.0 mL N, N-Dimethylformamide (DMF), triethylamine (TEA, 4.2 ml, 30 mmol), Boc-TU (3.2 g, 11 mmol) and HgCl2 (3.2 g, 11 mmol) were added under ice-cooled water. The reaction mixture was stirred at room temperature for 2 h to give a white compound 3 (4.3 g, 79%).
To the solution of compound 3 (4.3 g, 7.92 mmol) in ethanol (EtOH, 30 ml) and tetrahydrofuran (THF, 30 ml) under 0 °C, 5% NaOH solution (24 ml, 30 mmol) was added. The reaction mixture was stirred at room temperature for 3 h, and EtOH and THF were evaporated under vacuum. Water (30 ml) and glacial acetic acid (2 ml) was added to the residue and stirred to give compound 4 (3.6 g, 85.9%) as a white solid.
2.2.2. Synthesis of amino acid ester prodrug of peramivir (8a–g) (Fig. 2)
Fig. 2.

Synthesis of amino acid ester prodrugs of peramivir (8a–8g). (i) Ethylene glycol, DMAP, EDC and CH2Cl2, room temperature, 20 h; (ii) DMAP, EDC, overnight, room temperature; (iii) TFA, CH2Cl2, room temperature, 16 h.
Boc-protected amino acids (5, 29.0 mmol), ethylene glycol (9.0 g, 140 mmol) and DMAP (0.5 g, 5.8 mmol) and CH2Cl2 (150 ml) were stirred under argon at 0 °C for 10 min. Then EDC (7.2 g, 37.3 mmol) was added to the solution above and stirred for 20 h at room temperature. The organic solvent was evaporated in vacuo and the residue was purified by column chromatography (CH2Cl2/ CH3OH) to give compound 6.
Compound 4 (1.06 g, 2 mmol), compound 6 (5 mmol) and DMAP (75 mg, 0.6 mmol) were dissolved in CH2Cl2 (30 ml) and stirred under argon at room temperature for 20 min. EDC (0.58 g, 3.0 mmol) was added to the solution above and stirred overnight at room temperature. The organic solvent was evaporated in vacuo and the residue was purified by column chromatography (ethyl acetate (EtOAc)/ petroleum ether (PE)) to give a white foam-like compound 7.
Trifluoroacetic acid (TFA, 2.0 ml) was added to the solution of compound 7 (1.04 mmol) in CH2Cl2 (20 mL) and stirred at room temperature for 16 h. The organic solvent was evaporated under reduced pressure to give a white solid compound 8a–g.
2.2.3. Synthesis of amino acid amide prodrug of peramivir (11a–g) (Fig. 3)
Fig. 3.

Synthesis of amino acid amide prodrugs of peramivir (11a–g). (i) TEA, EDC, HOBt, amino acid methyl ester, DMF, room temperature, 24 h; (ii) LiOH, EtOH, THF and water, overnight, room temperature; (iii) TFA, CH2Cl2, room temperature, 16 h.
To the solution of compound 4 (1.06 g, 2.0 mmol) in N, N-dimethylformamide (DMF, 15 ml), triethylamine (TEA, 0.56 ml, 4.0 mmol), EDC (0.57 g, 3.0 mmol), HOBt (0.41 g, 3.0 mmol) were added sequentially, and stirred under argon at room temperature for 10 min. Then amino acid methyl ester (2.4 mmol) was added to the above mixture and stirred at room temperature for 24 h. TEA and DMF was evaporated under reduced pressure. The residue was diluted with EtOAc (100 ml) and successively washed with water and brine. The organic layer was condensed and the residue was purified by column chromatography (EtOAc/PE) to give the white compound 9.
The solution of LiOH (0.126 g, 3 mmol) in water (3 ml) was added to the solution of compound 9 (1.4 mmol) in EtOH (10 ml) and THF (10 ml) and stirred overnight at room temperature. The reaction was monitoring by thin-layer chromatography (TLC). EtOH and THF were evaporated under vacuum. Water (3 ml) and glacial acetic acid (3 ml) was added to the residue and stirred under ice-cooled water to give a white compound 10.
To the solution of 10 (1.27 mmol) in CH2Cl2 (20 mL), TFA (2 mL) was added and stirred at room temperature for 16 h. The reaction mixture was condensed under reduced pressure to give the white product 11a–g.
Peramivir-(CH2)2-l-Val (8a): yield 28.2%, 1H NMR (600 MHz, DMSO‑d6, δ ppm) 8.43 (s, 3H), 7.96–7.81 (m, 2H), 6.88 (d, J = 10.5 Hz, 2H), 4.52–4.46 (m, 1H), 4.40 (td, J = 10.6, 2.5 Hz, 1H), 4.34–4.24 (m, 3H), 4.21–4.14 (m, 1H), 3.85 (dd, J = 15.3, 7.9 Hz, 1H), 2.75–2.71 (m, 1H), 2.67–2.57 (m, 1H), 2.20–2.06 (m, 2H), 2.04 (s, 1H), 1.82–1.74 (m, 2H), 1.61 (dd, J = 13.3, 6.6 Hz, 0.5H), 1.49–1.24 (m, 5H), 1.11 (dd, J = 8.6, 4.7 Hz, 0.5H), 0.99–0.82 (m, 12H); 13C NMR (600 MHz, DMSO‑d6, δ ppm) 174.05, 169.38, 159.19, 156.41, 62.60, 57.66, 54.73, 50.88, 49.62, 48.69, 47.26, 43.36, 43.04, 29.83, 23.11, 21.32, 18.60, 17.82, 13.03, 12.32, 11.80.
Peramivir-(CH2)2-d-Ala (8b): yield 23.3%, 1H NMR(600 MHz, DMSO‑d6, δ ppm) 7.26 (dd, J = 10.0, 3.5 Hz, 4H), 7.19–7.18 (m, 4H), 5.61 (d, J = 4.8 Hz, 1H), 4.56 (d, J = 5.6 Hz, 1H), 4.32–4.21 (m, 4H), 4.10 (d, J = 5.3 Hz, 1H), 3.44–3.40 (m, 1H), 3.05 (d, J = 7.9 Hz, 1H), 2.88 (d, J = 7.5 Hz, 1H), 2.71 (dd, J = 8.6, 5.1 Hz, 1H), 2.60–2.56 (m, 2H), 2.07–2.01 (m, 2H), 1.90 (s, 3H), 1.66 (ddd, J = 13.7, 8.1, 3.4 Hz, 2H), 1.24 (s, 3H), 0.93–0.83 (m, 6H); 13C NMR(600 MHz, DMSO‑d6, δ ppm) 174.05, 169.38, 159.19, 156.41, 62.60, 57.66, 54.73,50.88, 49.62, 48.69, 47.26, 43.36, 29.83, 23.11, 21.32, 18.60, 17.82, 13.03, 12.32, 11.80.
Peramivir-(CH2)2-l-Ala (8c): yield 25.4%, 1H NMR (600 MHz, DMSO‑d6, δ ppm)7.26 (dd, J = 10.0, 3.5 Hz, 4H), 7.19–7.18 (m, 4H), 5.61 (d, J = 4.8 Hz, 1H), 4.56 (d, J = 5.6 Hz, 1H), 4.32–4.21 (m, 4H), 4.10 (d, J = 5.3 Hz, 1H), 3.44–3.40 (m, 1H), 3.05 (d, J = 7.9 Hz, 1H), 2.88 (d, J = 7.5 Hz, 1H), 2.71 (dd, J = 8.6, 5.1 Hz, 1H), 2.60–2.56 (m, 2H), 2.07–2.01 (m, 2H), 1.90 (s, 3H), 1.66 (ddd, J = 13.7, 8.1, 3.4 Hz, 2H), 1.24 (s, 3H), 0.93–0.83 (m, 6H); 13C NMR (600 MHz, DMSO‑d6, δ ppm) 174.05, 169.38, 159.19, 156.41, 62.60. 57.66, 54.73, 50.88, 49.62, 48.69, 47.26, 43.36, 29.83, 23.11, 21.32, 18.60, 17.82, 13.03, 12.32, 11.80.
Peramivir-(CH2)2-d-Leu (8d): yield 25.4%, 1H NMR(600 MHz, DMSO‑d6, δ ppm) 7.29–7.23 (m, 1H), 7.20–7.13 (m, 1H), 7.04 (s, 1H), 4.42–4.39 (m, 1H), 4.33–4.27 (m, 1H), 4.20–4.14 (m, 1H), 4.02–3.94 (m, 1H), 3.62 (q, J = 5.2 Hz, 1H), 3.45 (q, J = 7.0 Hz, 2H), 1.73–1.59 (m, 4H), 1.52–1.43 (m, 1H), 1.40–1.25 (m, 2H), 1.22–1.13 (m, 1H), 1.07–1.04 (m, 5H), 1.03–0.98 (m, 1H), 0.89 (ddd, J = 15.2, 8.3, 5.3 Hz, 9H); 13C NMR (600 MHz, DMSO‑d6, δ ppm) 174.05, 169.38, 159.19, 156.41, 62.60, 57.66, 54.73, 50.88, 49.62, 48.69, 47.26, 43.36, 30.35, 29.83, 23.11, 22.53, 21.32, 18.60, 17.82, 13.03, 12.32, 11.80
Peramivir-(CH2)2-l-Leu (8e): yield 20.6%, 1H NMR (600 MHz, DMSO‑d6, δ ppm) 7.29–7.23 (m, 1H), 7.20–7.13 (m, 1H), 7.04 (s, 1H), 4.42–4.39 (m, 1H), 4.33–4.27 (m, 1H), 4.20–4.14 (m, 1H), 4.02–3.94 (m, 1H), 3.62 (q, J = 5.2 Hz, 1H), 3.45 (q, J = 7.0 Hz, 2H), 1.73–1.59 (m, 4H), 1.52–1.43 (m, 1H), 1.40–1.25 (m, 2H), 1.22–1.13 (m, 1H), 1.07–1.04 (m, 5H), 1.03–0.98 (m, 1H), 0.89 (ddd, J = 15.2, 8.3, 5.3 Hz, 9H); 13C NMR (600 MHz, DMSO‑d6, δ ppm) 174.05, 169.38, 159.19, 156.41, 62.60.57.66, 54.73, 50.88, 49.62, 48.69, 47.26, 43.36, 30.35, 29.83, 23.11, 22.53, 21.32, 18.60, 17.82, 13.03, 12.32, 11.80.
Peramivir-(CH2)2-l-Phe (8f): yield 17.8%, 1H NMR (600 MHz, DMSO‑d6, δ ppm) 7.32–7.13 (m, 5H), δ 7.26 (dd, J = 10.0, 3.5 Hz, 4H), 7.19–7.18 (m, 4H), 5.61 (d, J = 4.8 Hz, 1H), 4.56 (d, J = 5.6 Hz, 1H), 4.32–4.21 (m, 4H), 4.10 (d, J = 5.3 Hz, 1H), 3.44–3.40 (m, 1H), 3.29 (d, J = 7.9 Hz, 2H),3.05 (d, J = 7.9 Hz, 1H), 2.88 (d, J = 7.5 Hz, 1H), 2.71 (dd, J = 8.6, 5.1 Hz, 1H), 2.60–2.56 (m, 2H), 2.07–2.01 (m, 2H), 1.90 (s, 3H), 1.66 (ddd, J = 13.7, 8.1, 3.4 Hz, 2H), 0.93–0.83 (m, 6H); 13C NMR (600 MHz, DMSO‑d6, δ ppm) 174.05, 169.38, 159.19, 156.41, 141.69, 137.34, 136.31, 129.82, 128.75, 126.28, 62.60, 57.66, 54.73, 50.88, 49.62, 48.69, 47.26, 43.36, 30.35, 29.83, 23.11, 22.53, 21.32, 18.60, 17.82, 13.03, 12.32, 11.80.
Peramivir-(CH2)2-l-Pro (8g): yield 23.7%, 1H NMR (600 MHz, DMSO‑d6, δ ppm) 7.26 (dd, J = 10.0, 3.5 Hz, 4H), 7.19–7.18 (m, 4H), 5.61 (d, J = 4.8 Hz, 1H), 4.56 (d, J = 5.6 Hz, 1H), 4.32–4.21 (m, 4H), 4.10 (d, J = 5.3 Hz, 1H), 3.44–3.40 (m, 1H), 3.05 (d, J = 7.9 Hz, 1H), 2.88 (d, J = 7.5 Hz, 1H), 2.80 (m, 2H), 2.71 (dd, J = 8.6, 5.1 Hz, 1H), 2.60–2.56 (m, 2H), 2.07–2.01 (m, 2H), 1.96 (m, 2H), 1.90 (s, 3H), 1.66 (ddd, J = 13.7, 8.1, 3.4 Hz, 2H), 1.64 (m, 2H), 0.93–0.83 (m, 6H); 13C NMR (600 MHz, DMSO‑d6, δ ppm) 174.05, 169.38, 159.19, 156.41, 62.60, 57.66, 54.73,50.88, 49.62, 48.69, 47.26, 46.87, 43.36, 30.30, 29.83, 25.31, 23.11, 21.32, 18.60, 17.82, 13.03, 12.32, 11.80.
Peramivr-l-Val (11a): yield 45.2%; 1H NMR (600 MHz, DMSO‑d6, δ ppm) 12.51 (s, 1H), 8.13 (dd, J = 36.3, 8.5 Hz, 1H), 7.82 (s, 1H), 7.48 (dd, J = 79.6, 8.1 Hz, 1H), 7.26 (s, 1H), 6.79–6.68 (m, 1H), 5.15 (d, J = 4.9 Hz, 0.5H), 4.37 (dd, J = 10.5, 8.4 Hz, 0.5H), 4.16–4.11 (m, 1H), 3.84 (dd, J = 8.4, 6.1 Hz, 0.5H), 3.39–3.33 (m, 0.5H), 2.93–2.78 (m, 1H), 2.75–2.70 (m, 0.5H), 2.58–2.53 (m, 0.5H), 2.16 (td, J = 10.4, 4.5 Hz, 0.5H), 2.14–1.99 (m, 3H), 1.76 (s, 1H), 1.56–1.02 (m, 6H), 0.99 (dt, J = 14.8, 7.2 Hz, 1H), 0.95–0.77 (m, 12H); 13C NMR (600 MHz, DMSO‑d6, δ ppm) 173.50, 169.97, 159.06, 156.17, 74.48, 59.48, 48.85, 48.08, 47.63, 43.39, 30.25, 23.16, 21.33, 19.60, 12.40.
Peramivir-l-Phe (11b): yield 39.4%; 1H NMR (600 MHz, DMSO‑d6, δ ppm) 12.51 (s, 1H), 7.32–7.13 (m, 5H), δ 7.26 (dd, J = 10.0, 3.5 Hz, 4H), 7.19–7.18 (m, 4H), 5.61 (d, J = 4.8 Hz, 1H), 4.56 (d, J = 5.6 Hz, 1H), 4.10 (d, J = 5.3 Hz, 1H), 3.44–3.40 (m, 1H), 3.29 (d, J = 7.9 Hz, 2H),3.05 (d, J = 7.9 Hz, 1H), 2.88 (d, J = 7.5 Hz, 1H), 2.71 (dd, J = 8.6, 5.1 Hz, 1H), 2.60–2.56 (m, 2H), 2.07–2.01 (m, 2H), 1.90 (s, 3H), 1.66 (ddd, J = 13.7, 8.1, 3.4 Hz, 2H), 0.93–0.83 (m, 6H); 13C NMR (600 MHz, DMSO‑d6, δ ppm) 174.05, 169.38, 159.19, 156.41, 141.69, 137.34, 136.31, 129.82, 128.75, 126.28, 57.66, 54.73, 49.62, 48.69, 47.26, 43.36, 30.35, 29.83, 23.11, 22.53, 21.32, 18.60, 17.82, 13.03, 12.32, 11.80.
Peramivir-d-Phe (11c): yield 36.1%, 1H NMR (600 MHz, DMSO‑d6, δ ppm) 12.51 (s, 1H), 7.32–7.13 (m, 5H), δ 7.26 (dd, J = 10.0, 3.5 Hz, 4H), 7.19–7.18 (m, 4H), 5.61 (d, J = 4.8 Hz, 1H), 4.56 (d, J = 5.6 Hz, 1H), 4.10 (d, J = 5.3 Hz, 1H), 3.44–3.40 (m, 1H), 3.29 (d, J = 7.9 Hz, 2H),3.05 (d, J = 7.9 Hz, 1H), 2.88 (d, J = 7.5 Hz, 1H), 2.71 (dd, J = 8.6, 5.1 Hz, 1H), 2.60–2.56 (m, 2H), 2.07–2.01 (m, 2H), 1.90 (s, 3H), 1.66 (ddd, J = 13.7, 8.1, 3.4 Hz, 2H), 0.93–0.83 (m, 6H).13C NMR (600 MHz, DMSO‑d6, δ ppm) 174.05, 169.38, 159.19, 156.41, 141.69, 137.34, 136.31, 129.82, 128.75, 126.28, 57.66, 54.73, 49.62, 48.69, 47.26, 43.36, 30.35, 29.83, 23.11, 22.53, 21.32, 18.60, 17.82, 13.03, 12.32, 11.80.
Peramivir-l-Ile (11d): yield 29.7%, 1H NMR (600 MHz, DMSO‑d6, δ ppm) 12.51 (s, 1H), 8.26 (dd, J = 7.8, 6.5 Hz, 1H), 7.82 (s, 1H), 7.57 (d, J = 7.3 Hz, 1H), 7.33 (dd, J = 8.9, 7.0 Hz, 1H), 6.82–6.70 (m, 1H), 5.12–5.03 (m, 1H), 4.37 (t, J = 10.6 Hz, 1H), 4.27–4.24 (m, 1H), 3.86 (dd, J = 8.6, 5.6 Hz, 1H), 2.74–2.67 (m, 1H), 2.05 (d, J = 5.5 Hz, 3H), 1.76 (s, 2H), 1.67–1.56 (m, 1H), 1.55–1.44 (m, 2H), 1.35 (ddd, J = 14.4, 9.0, 3.5 Hz, 2H), 1.31–1.24 (m, 1H), 1.18 (dt, J = 11.5, 7.1 Hz, 1H), 1.13–1.05 (m, 1H), 0.98 (dd, J = 14.0, 8.1 Hz, 1H), 0.91–0.82 (m, 12H). 13C NMR (600 MHz, DMSO‑d6, δ ppm) 174.74, 172.02, 169.74, 159.20, 65.91, 55.18, 48.75, 47.91, 46.86, 42.69, 24.77, 23.35, 21.75, 20.86, 12.38.
Peramivir-l-Pro (11e): yield 23.1%, 1H NMR(600 MHz, DMSO‑d6, δ ppm) 174.05, 7.26 (dd, J = 10.0, 3.5 Hz, 4H), 7.19–7.18 (m, 4H), 5.61 (d, J = 4.8 Hz, 1H), 4.56 (d, J = 5.6 Hz, 1H), 4.10 (d, J = 5.3 Hz, 1H), 3.44–3.40 (m, 1H), 3.05 (d, J = 7.9 Hz, 1H), 2.88 (d, J = 7.5 Hz, 1H), 2.80 (m, 2H), 2.71 (dd, J = 8.6, 5.1 Hz, 1H), 2.60–2.56 (m, 2H), 2.07–2.01 (m, 2H), 1.96 (m, 2H), 1.90 (s, 3H), 1.66 (ddd, J = 13.7, 8.1, 3.4 Hz, 2H), 1.64 (m, 2H), 0.93–0.83 (m, 6H); 13C NMR (600 MHz, DMSO‑d6, δ ppm) 174.05, 169.38, 159.19, 156.41, 57.66, 54.73, 49.62, 48.69, 47.26, 46.87, 43.36, 30.30, 29.83, 25.31, 23.11, 21.32, 18.60, 17.82, 13.03, 12.32, 11.80.
Peramivir-d-Leu (11f): yield 30.6%, 1H NMR (600 MHz, DMSO‑d6, δ ppm) 12.51 (s, 1H), 8.26 (dd, J = 7.8, 6.5 Hz, 1H), 7.82 (s, 1H), 7.57 (d, J = 7.3 Hz, 1H), 7.33 (dd, J = 8.9, 7.0 Hz, 1H), 6.82–6.70 (m, 1H), 5.12–5.03 (m, 1H), 4.37 (t, J = 10.6 Hz, 1H), 4.27–4.24 (m, 1H), 3.86 (dd, J = 8.6, 5.6 Hz, 1H), 2.74–2.67 (m, 1H), 2.05 (d, J = 5.5 Hz, 3H), 1.76 (s, 2H), 1.67–1.56 (m, 1H), 1.55–1.44 (m, 2H), 1.35 (ddd, J = 14.4, 9.0, 3.5 Hz, 2H), 1.31–1.24 (m, 1H), 1.18 (dt, J = 11.5, 7.1 Hz, 1H), 1.13–1.05 (m, 1H), 0.98 (dd, J = 14.0, 8.1 Hz, 1H), 0.91–0.82 (m, 12H); 13C NMR (600 MHz, DMSO‑d6, δ ppm) 174.74, 172.02, 169.74, 159.20, 65.91, 55.18, 48.75, 47.91, 46.86, 42.69, 24.77, 23.35, 21.75, 20.86, 12.38.
Peramivir-l-Leu (11 g): yield 43.1%, 1H NMR(600 MHz, DMSO‑d6, δ ppm) 12.51 (s, 1H), δ 8.32–8.01 (m, 1H), 7.82 (s, 1H), 7.46 (d, J = 8.2 Hz, 1H), 7.24 (s, 1H), 6.76–6.66 (m, 1H), 4.48–4.35 (m, 1H), 4.23–4.19 (m, 1H), 4.12–4.06 (m, 1H), 3.84 (dt, J = 14.5, 8.7 Hz, 1H), 2.82 (m, 1H), 2.74–2.65 (m, 1H), 2.07–2.03 (m, 1H), 1.86–1.71 (m, 2H), 1.65–1.45 (m, 5H), 1.40–1.35 (m, 1H), 1.33–1.28 (m, 1H), 1.26 (dd, J = 9.9, 8.3 Hz, 1H), 1.21–1.14 (m, 1H), 1.13–1.03 (m, 2H), 1.03–0.94 (m, 1H), 0.94–0.80 (m, 12H); 13C NMR(600 MHz, DMSO‑d6, δ ppm) 174.59, 172.64, 169.73, 159.92, 74.51, 56.48, 50.69, 48.09, 47.32, 43.00, 27.66, 24.84, 21.30, 19.45, 12.40.
2.3. Caco-2 culture
Caco-2 cells were obtained from the American Tissue Culture Collection (Rockville, MD) and were grown routinely on 75 cm2 culture flasks in Dulbecco's modified Eagle's medium (DMEM, 4500 mg/L glucose) as previously described [17].
2.4. Caco-2 permeability
For transport experiments, Caco-2 cells were seeded onto polycarbonate membrane (0.6 cm2 growth area, 0.4 µm pore size, Millipore, MA) at a density about 1.0 × 105 cells/cm2 and allowed to grow for 21–25 d [17]. Permeability studies were conducted with the monolayers that developed the transepithelial electrical resistance (TEER) values above 250 Ω cm2. Samples (100 µl) at 15, 30, 45, 60, 90, 120 min were taken from the basolateral (BL) solution, and the volume was replaced with prewarmed Hanks’ balanced salt solution (HBSS). Because the amino acid prodrugs were partially metabolized to peramivir and amino acids during the transport across the Caco-2 monolayers, the prodrugs transported was calculated as the total sum of the unchanged prodrugs and peramivir.
2.5. Gly-Sar uptake inhibition
For uptake experiments, Caco-2 cells were seeded onto 24-well plastic cluster trays at about 1.0 × 105 cells/ cm2 for 15 days after seeding. After washing twice with HBSS buffer, Caco-2 were incubated with 20 µM Gly-Sar along with various concentrations of peramivir-(CH2)2-l-Val or peramivir-l-Ile (0.05 − 10 mM) at 37 °C for 10 min [18], [19]. After 10 min, the medium was removed, and the cells were rapidly rinsed twice with 1 ml of ice-cold uptake buffer (pH 6.0). The cells were collected and homogenized in 0.25 ml water. The homogenates were centrifuged at 1500 g for 6 min, and gly-sar in the supernatant was analyzed by HPLC/MS/MS [18]. The protein concentration of each sample was determined by Coomassie Brilliant Blue assay using a bovine serum albumin as standard. IC50 was determined using nonlinear data fitting.
2.6. Uptake by MDCK-hPEPT1 cells
MDCK-PEPT1 and MDCK mock cells were grown in DMEM as described previously [19]. The uptake of peramivir-(CH2)2-l-Val or peramivir-l-Ile by MDCK-PEPT1 cells and MDCK cells in the presence or in the absence of inhibitor was evaluated to study the role of PEPT1. The samples were analyzed by HPLC.
The concentration-dependent uptake of peramivir-(CH2)2-l-Val or peramivir-l-Ile was also studied at pH 6 over a concentration range of 0.01–50 mM. The mock cells and the MDCK-PEPT1 cells were incubated with each individual substrate for 10 min. The mock cell line kinetics values were subtracted from those observed in MDCK-PEPT1 cells [19].
2.7. Hydrolysis stability study
2.7.1. Chemical study
The chemical study of peramivir-(CH2)2-l-Val or peramivir-l-Ile was determined in pH 1.2 hydrochloric acid solution and phosphate buffers (pH 4.5, 6.8, 7.4) at 37 °C for 4 h. At every time point, 100 µL of the samples was taken and analyzed by HPLC.
2.7.2. Hydrolysis in rat gastric juices and intestinal fluids
The gastric juices and intestinal fluids for stability studies were collected from male Sprague-Dawley rats. The experiments were carried out by adding 200 µl of a stock solution of compound peramivir-(CH2)2-l-Val or peramivir-l-Ile to 1.8 ml of gastric juices or intestinal fluids preheated to 37 °C, and the concentration of peramivir-(CH2)2-l-Val or peramivir-l-Ile in the biological media was about 80 µg/ml and 100 µg/ml, respectively. Hydrolysis of the prodrugs was studied at 37 °C for a period of 4 h. Samples (100 µl) were taken at various time points and quenched with 300 µl of ice-cold methanol, then centrifuged at 2500 g and 4 °C for 10 min. The supernatants were analyzed by HPLC.
2.7.3. Stability in intestinal and liver homogenates
The jejunum segment and liver were removed from the euthanized Sprague-Dawley rat and washed with ice-bath buffer C (25 mM KCl, 5 mM MgCl2 and 10 mM HEPES, pH 7.4) several times to remove blood, then homogenized with a tissue homogenizer and centrifuged at 2000 g and 4 °C for 10 min. The resulting supernatant was collected, and the total protein amount was determined as above. The hydrolysis experiment was carried out by addition of drug solutions to the homogenates at 37 °C. Peramivir-(CH2)2-l-Val, peramivir-l-Ile and protein concentrations in the mixture were 100 µg/ml, 100 µg/ml, and 200 µg/ml, respectively. The sample was pretreated as above, and the supernatant was analyzed using HPLC.
2.7.4. Stability in rat plasma
Plasma was obtained from rat after centrifugation at 2500 g for 10 min. One volume of drug stock solution was mixed with nine volumes of plasma preheated to 37 °C. Aliquot samples were collected at various time points (0, 30, 60, 90, 120 min). Extraction and analysis methods were similar to those for the gastrointestinal fluids experiment.
2.8. Pharmacokinetics study
Male Sprague-Dawley rats ranging from 180 to 220 g were used for the pharmacokinetic study. The animal experiment was approved by the University Committee on Use and Care of Animals, Jiangxi University of Traditional Chinese Medicines. Animals were housed under standard conditions of temperature (25 ± 2 °C), humidity (65 ± 10%) and light. The rats were allowed free movement and had access to food and water for 7 d before the experiments. Rats were fasted overnight with free access to water before the day of the experiment. The rats (n = 5 to 6 per treatment) were administered by gavage of peramivir, peramivir-(CH2)2-l-Val or peramivir-l-Ile (160 mg/kg calculated as peramivir) in aqueous solution, respectively. Serial blood samples (0.2 ml) were obtained from orbital plexus at 5, 15, 30, 45 min and 1, 1.5, 2, 3, 4, 6, 8, 10, 24, 48 h after oral administration. The solution of peramivir was also intravenously administered to 6 rats at 8 mg/kg. During sampling, rats were anesthetized with ether. The blood samples were centrifugated at 1200 g and 4 °C for 10 min, and the plasma was collected and frozen at −40 °C until analytes were determined by HPLC/MS/MS.
2.9. Analytical method
(a). HPLC Analysis. Peramivir and amino acid prodrugs were analyzed on SHIMADZU LC-20AT HPLC system consisting of a SPD-M20A DAD, a SIL-20A autosampler and a LabSolution workstation. The analytes were separated on ODS C18 (4.6 mm × 250 mm, 5 µm) with a wavelength of 210 nm and the mobile phase was the mixture of acetonitrile: water (containing 0.1% glacial acetic acid). The column temperature was set at 25 °C.
(b). HPLC-MS/MS analysis. The analytes were determined by SHIMADZU LCMS-8050 liquid chromatograph mass spectrometer system. HPLC-MS/MS method was fully validated in selectivity, linearity, precision and accuracy, extraction recovery, matrix effect and stability according to Guidance for Industry, Bioanalytical, Method Validation US Food and Drug Administration. Quantification was achieved by constructing a calibration curve with the least-square linear regression.
For determination of peramivir, peramivir and internal standard berberine were extracted from rat plasma with the protein precipitation. A reversed-phase C18 column (Phenomenex, 2.1 mm × 50 mm, 1.7 µm) was used to retain and separate the analytes from the endogenous components. The column was eluted by the gradient of acetonitrile and water containing 0.1% formic acid. ESI was set in positive ionization mode. Quantification was performed using multiple reaction monitoring (MRM) and the optimized MRM transitions were 329.1→270.1 for peramivir and 336.2→292.0 for berberine.
The determination of peramivir-l-Ile was similar to peramivir, but peramivir-l-Ile was pretreated from the plasma with the liquid-liquid extraction. The optimized MRM transition of 442.2→383.3 was used for peramivir-l-Ile.
2.10. Data analysis
- (a). The permeability coefficient (Papp) was calculated using the following equation:
where dQ/dt is the steady-state appearance rate of the test compound on the receiver side, C0 is the initial concentration of the test compound on the donor side, and A is the monolayer growth surface area. (b). Plasma pharmacokinetic parameters were calculated by noncompartmental analysis. The plasma concentration at different times was expressed as mean ± standard deviation (S.D.), and the mean concentration-time profiles were plotted. The Cmax and Tmax were observed directly from the concentration–time curves. The area under the plasma concentration–time profiles (AUC) was calculated using the linear-trapezoidal rule.
(c). Statistical Analysis. The statistical differences were tested using a one-tailed Student t test at the P < 0.05 level.
3. Results and discussion
3.1. Synthesis of amino acid prodrugs of peramivir
To avoid the potential side reactions, Boc-peramivir was synthesized and then used as the starting materials for prodrug synthesis (Figs. 1–3). The structures of all prodrugs were confirmed by NMR and MS.
3.2. Caco-2 permeability
The apical to basolateral permeability for peramivir and 14 prodrugs was in Fig. 4A. All prodrugs were of higher permeability than the parent drug, peramivir. Peramivir-(CH2)2-l-Val exhibited the highest permeability in all ester prodrugs and its permeability was approximately 10.9-fold higher than peramivir. The permeability of Peramivir-l-Ile was the highest in all amide prodrugs, which was about 9.1-fold higher than peramivir. Therefore, in the subsequent experiments, peramivir-(CH2)2-l-Val and peramivir-l-Ile were selected as the representative compounds for further study.
Fig. 4.

(A) The apical-to-basolateral permeability (Papp) for the amino acid prodrugs (0.5 mM) in Caco-2 cells (mean ± SD, n = 3). *, P < 0.05, compared with peramivir. (B) Percent prodrug remaining intact in receiver side at 120 min across Caco-2 cell monolayer (mean ± SD, n = 3).
The extent of prodrug hydrolysis across Caco-2 monolayer was listed in Fig. 4B. The amide prodrugs were all more stable than the ester prodrugs. The amino acid promoiety and the stereochemistry had a really important effect on the hydrolysis rate of the ester prodrugs.
3.3. Inhibition of gly-sar uptake by Caco-2 cells
An inhibition effect of peramivir-(CH2)2-l-Val and peramivir-l-Ile for PEPT1 was determined by studying their ability to inhibit the uptake of gly-sar by Caco-2 cells. The initial rate time point (10 min) for the uptake of gly-sar was selected because the accumulation of gly-sar was linear up to 20 min after incubation in Caco-2 cells (data not shown). As shown in Fig. 5, gly-sar uptake was inhibited by Peramivir-(CH2)2-l-Val and Peramivir-l-Ile in a concentration-dependent manner, and the IC50 was 1.34 ± 0.31 mM and 1.78 ± 0.48 mM, respectively.
Fig. 5.

Inhibition of Peramivir-(CH2)2-l-Val and Peramivir-l-Ile on gly-sar uptake by Caco-2 cells. Caco-2 cells were incubated with 20 µM gly-sar at 37 °C for 10 min in the presence of various concentrations of peramivir-(CH2)2-l-Val or peramivir-l-Ile (0.05 − 10 mM). After the incubation, the concentrations of gly-sar were determined by HPLC-MS/MS. Data are presented as mean ± SD, n = 3.
3.4. Uptake of peramivir-(CH2)2-l-val and peramivir-l-ile by MDCK-hPEPT1 cells
To further confirm that Peramivir-(CH2)2-l-Val and Peramivir-l-Ile were the substrates of PEPT1, the uptake of the two compounds by MDCK-hPEPT1 cell and MDCK mock cell was investigated. Peramivir-(CH2)2-l-Val and Peramivir-l-Ile showed 5.5-fold and 4.6-fold increase in uptake by MDCK-hPEPT1 cells when compared with the mock cells, respectively. This kind of uptake can be inhibited by excess of gly-sar, a well-known substrate of PEPT1. In contrast, no PEPT1-mediated transport of peramivir was observed (Fig. 6A).
Fig. 6.

(A) Uptake of Peramivir-(CH2)2-l-Val and Peramivir-l-Ile by MDCK-hPEPT1 cells. The MDCK-hPEPT1 cells were incubated with Peramivir-(CH2)2-l-Val or Peramivir-l-Ile (0.5 mM) for 10 min, pH 6.0, in the presence or absence of gly-sar (10 mM). After incubation, the concentration of Peramivir-(CH2)2-l-Val and Peramivir-l-Ile was determined by HPLC. *, P < 0.05, compared with the presence of gly-sar. (B) Concentration-dependent uptake of Peramivir-(CH2)2-l-Val and Peramivir-l-Ile in MDCK-hPEPT1 cells. Each value was calculated after subtracting the endogenous transporters contributions observed in the mock MDCK cells. Data are presented as mean ± SD, n = 3.
Concentration-dependent profiles of Peramivir-(CH2)2-l-Val and Peramivir-l-Ile determined in MDCK-hPEPT1 cells after correcting for mock cells were shown in Fig. 6B. Vmax values for Peramivir-(CH2)2-l-Val and Peramivir-l-Ile were 14.89 ± 0.65 and 14.07 ± 0.54 nmol/mg protein/10 min, respectively. Km values for Peramivir-(CH2)2-l-Val and Peramivir-l-Ile were 4.23 ± 0.11 and 5.92 ± 0.14 mM, respectively.
3.5. Stability studies
The stability of Peramivir-(CH2)2-l-Val and Peramivir-l-Ile was studied in pH 1.2 hydrochloric acid solution, different pH phosphate buffers, intestinal and liver homogenates, gastric and intestinal fluids, rat plasma. The estimated half-lives (t1/2) were calculated from linear regression of pseudo-first-order plots of the concentration vs. times. From Table 1, Peramivir-l-Ile was more stable than Peramivir-(CH2)2-l-Val under all conditions, which may be that the amide bond was more stable than the ester bond. The chemical hydrolysis of two prodrugs was in a pH-dependent manner, and they were more stable in the acidic environment than at the neutral and alkaline pH. In the biological fluids, Peramivir-(CH2)2-l-Val and Peramivir-l-Ile hydrolyzed at a higher speed (shorter t1/2) than in the phosphate buffer with the same pH, which indicated that a catalytic effect of enzyme may arise from these biological fluids.
Table 1.
The Stability of Peramivir-(CH2)2-l-Val and Peramivir-l-Ile in Different Phosphate buffer, in Rat Tissue Homogenate, Plasma, Gastric and Intestinal fluids (Mean ± SD, n = 3).
| Medium | pH |
t1/2 |
|
|---|---|---|---|
| Peramivir-(CH2)2-l-Val (min) | Peramivir-l-Ile (h) | ||
| Phosphate buffer | 1.2 | 306 ± 23 | 43.2 ± 1.3 |
| 4.5 | 246 ± 34 | 37.5 ± 1.2 | |
| 6.8 | 214 ± 31 | 33.1 ± 0.5 | |
| 7.4 | 178 ± 22 | 25.7 ± 0.8 | |
| Intestinal homogenate | 7.4 | 78 ± 29 | 28.4 ± 0.2 |
| Liver homogenate | 7.4 | 65 ± 13 | 8.3 ± 0.1 |
| Gastric fluids | 1.3 | 203 ± 15 | 27.6 ± 0.6 |
| Intestinal fluids | 6.6 | 161 ± 12 | 9.4 ± 0.1 |
| Rat plasma | 75 ± 13 | 5.3 ± 0.8 | |
3.6. Pharmacokinetics study
The pharmacokinetics performance of Peramivir-(CH2)2-l-Val and Peramivir-l-Ile was studied in rats to determine whether the amino acid prodrug strategy could improve the oral bioavailability of peramivir in vivo. The aqueous solution of peramivir, Peramivir-(CH2)2-l-Val and Peramivir-l-Ile (all 160 mg/kg calculated as peramivir) were orally administered to rats, respectively. Peramivir was also intravenously injected to rats at a dose of 8 mg/kg. PK parameters were presented in Table 2 and plasma-concentration curves were showed in Fig. 7.
Table 2.
Mean pharmacokinetic parameters of peramivir after oral administration of Peramivir-(CH2)2-l-VAL, Peramivir-l-Ile and Peramivr (160 mg/kg calculated as peramivir) and intravenous injection (8 mg/kg) to the Male Sprague-Dawley rats, respectively (n = 5 to 6).
| Dosing | Cmax (ng/ml) | Tmax (h) | t1/2 (h) | AUC0-∞ (ng•h/ml) | F(%) |
|---|---|---|---|---|---|
| Peramivir-(CH2)2-l-VAL | 16,101.9 ± 3467.1 | 1.0 | 4.3 | 47,107.7 | 65.3 |
| Peramivir-l-Ile | 9227.1 ± 1675.2 | 1.0 | 3.5 | 26,914.5 | 37.3 |
| Peramivir (p.o.) | 771.6 ± 278.3 | 1.0 | 3.2 | 2955.9 | 4.1 |
| Peramivir (i.v.) | 3.9 | 3604.8 |
Fig. 7.

Mean (± SD) plasma concentration-time of peramivir and Peramivir-l-Ile (n = 5 to 6). ( ,
, ,
, ): the concentration of peramivir after oral administration of Peramivir-(CH2)2-l-Val, Peramivir and Peramivir-l-Ile, respectively, (160 mg/kg calculated as peramivir); (
): the concentration of peramivir after oral administration of Peramivir-(CH2)2-l-Val, Peramivir and Peramivir-l-Ile, respectively, (160 mg/kg calculated as peramivir); ( ): the concentration of Peramivir-l-Ile after oral administration of Peramivir-l-Ile (160 mg/kg calculated as peramivir).
): the concentration of Peramivir-l-Ile after oral administration of Peramivir-l-Ile (160 mg/kg calculated as peramivir).
After Peramivir-(CH2)2-l-Val was orally administered to the rats, Peramivir-(CH2)2-l-Val could not be found in plasma because of the rapid hydrolysis. As shown in Table 2, the AUC for peramivir after dosing of Peramivir-(CH2)2-l-Val and peramivir was 47,107.7 and 2955.9 ng h/ml respectively. The absolute oral bioavailability of peramivir following oral administration of peramivir-(CH2)2-l-Val and peramivir was 65.3% and 4.1%, respectively.
After Peramivir-l-Ile was administered to the rats, only portion of Peramivir-l-Ile was hydrolyzed to peramivir. The AUC for peramivir-l-Ile and peramivir was 20,030.3 and 26,914.5 ng h/ml, respectively. Therefore, the oral bioavailability of peramivir after dosing of peramivir-l-Ile was 37.3%, which was 9.24-fold increase when compared with oral administration of peramivir.
3.7. Discussion
Peramivir has been approved for the treatment of influenza A and B by many countries. But the intravenous administration limited the patients’ compliance because of the high polarity and low oral bioavailability resulting from the carboxyl and the guanidine group. The intestinal peptide transporter, PEPT1, was abundant in the epithelium of the gastrointestinal tract, and it was a promising target for oral drug delivery strategy for the broad substrate specificity [20], [21], [22], [23]. Therefore, a lot of amino acid prodrugs of peramivir targeted to PEPT1 have been synthesized with the aim to improve the oral bioavailability and develop the oral alternative to peramivir in the present study.
Amino acid amide prodrugs targeted to PEPT1 have been applied to many successful cases, for example LY2140023 (methionine prodrug of LY404039) [15], [16], lisdexamfetamine dimesylate (the lysine amide of d-amphetamine) [24], [25], LY544344 [26], [27] and midodrine [28]. The amide linkage could provide sufficient stability in the gastrointestinal tract when compared with the ester bond, and it may exhibit potentially better oral absorption. Therefore, seven amino acid amide prodrugs were also synthesized in the present study, except for seven amino acid ester prodrugs. The transport experiment across Caco-2 cells was performed to evaluate their transport across the intestinal membranes. Peramivir-(CH2)2-l-Val and Peramivir-l-Ile were of the highest permeability in all ester prodrugs and in all amide prodrugs, respectively. The l-valine prodrug was the most permeable drug in most cases, which could be attributed to that the l-valine may have the optimal combination of chain length and branch at the β-C position of amino acid for the intestinal transport [13]. But as the amide prodrugs were concerned in the present study, Peramivir-l-Ile had the highest permeability, and it was slightly higher than Peramivir-l-val. Therefore, peramivir-(CH2)2-l-Val and peramivir-l-Ile were selected as the candidate compounds for the next studies.
From gly-sar uptake inhibition by Caco-2 cells, it can be concluded that Peramivir-(CH2)2-l-Val and Peramivir-l-Ile competed with gly-sar to interact with PEPT1. In the MDCK-hPEPT1 cells, the direct uptake of Peramivir-(CH2)2-l-Val and Peramivir-l-Ile were 5.5-fold and 4.6-fold higher than in mock MDCK cells, respectively, and this uptake can be inhibited by excess of gly-sar. The uptake of peramivir was not (statistically) significant different between MDCK-hPEPT1 and MDCK mock cells [19]. These results showed hPEPT1 contributed to the uptake of these two prodrugs by the small intestine. The uptake of Peramivir-(CH2)2-l-Val and Peramivir-l-Ile in MDCK-hPEPT1 cells conformed to Michaelis–Menten Equation, which further confirmed the transport and uptake of two prodrugs was a PEPT1-mediated process. Km values for peramivir-(CH2)2-l-Val were smaller than peramivir-l-Ile (4.23 mM vs 5.92 mM), which showed peramivir-(CH2)2-l-Val had a higher affinity to PEPT1 than peramivir-l-Ile, and these results was consistent with the gly-sar uptake inhibition by Caco-2 cells and the direct uptake by MDCK-hPEPT1 cells.
In the pharmacokinetics studies, no Peramivir-(CH2)2-l-Val could be found in the plasma, which might result from rapid degradation during the first-pass process [29]. These were consistent with the stability experiment result. The t1/2 in the intestinal homogenate, liver homogenate and rat plasma was less than 80 min, which suggested the first-pass metabolism might be striking. After oral administration of Peramivir-(CH2)2-l-Val, the oral bioavailability of peramivir was 65.3%, which was 15.9-fold higher than the oral doing of peramivir. The oral availability of peramivir after oral administration of Peramivir-l-Ile was 37.3%, which was 9.24-fold increase when compared with oral administration of peramivir. But the AUC for Peramivir-l-Ile was 20,030.3 ng h/ml, which was comparable to the AUC of peramivir (26,914.5 ng h/ml). Though the affinity of Peramivir-l-Ile to PEPT1 was similar (slightly smaller) to Peramivir-(CH2)2-l-Val, Peramivir-l-Ile could not improve the oral bioavailability of peramivir to the same extent as Peramivir-(CH2)2-l-Val because of the limited in vivo bioactivation, which can be forecasted from the stability results of Peramivir-l-Ile. Therefore, to improve the oral bioavailability of peramivir with amide prodrug, more amino acids prodrugs, such as some unnatural amino acids, must be attempted in the future to combine the improve of permeability, the stability in the gastrointestinal tract with the rapid bioactivation.
4. Conclusions
In summary, the prodrug strategy targeted to PEPT1 described in the present study has been very successfully in improving the oral bioavailability of peramivir and developing its oral alternative. Peramivir-(CH2)2-l-Val with good oral profiles and rapid conversion to the parent drug might be a promising prodrug for the further clinic development. The present studies also corroborated the idea that the PEPT1-mediated prodrug approach has enormous promise for improving the oral absorption of poorly absorbed drug [21], [22].
Acknowledgments
Conflict of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of this article.
Acknowledgments
This work was supported by National Natural Science Foundation of China (81360485 and 81560577), and National Natural Science Foundation of Jiangxi (20132BAB215023).
Contributor Information
Lvjiang Hu, Email: hulvjiangmail@163.com.
Yi Jin, Email: jinyipharm@163.com.
References
- 1.Wester A, Shetty AK. Peramivir injection in the treatment of acute influenza: a review of the literature. Infent Drug Resist. 2016;9:201–214. doi: 10.2147/IDR.S86460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sweet C, Jakeman KJ, Bush K. Oral administration of cyclopentane neuraminidase inhibitors protects ferrets against influenza virus infection. Antimicrob Agents Chemother. 2002;45:749–757. doi: 10.1128/AAC.46.4.996-1004.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bantia S, Parker CD, Ananth SL. Comparison of the anti-influenza virus activity of RWJ-270201 with those of oseltamivir and zanamivir. Antimicrob Agents Chemother. 2001;45:1162–1167. doi: 10.1128/AAC.45.4.1162-1167.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hollister AS, Sheridan WP. The emergency use authorization of peramivir IV: a view from the manufacturer. Clin Pharmacol Ther. 2011;89:172–174. doi: 10.1038/clpt.2010.278. [DOI] [PubMed] [Google Scholar]
- 5.Arya V, Carter WW, Robertson SM. The role of clinical pharmacology in supporting the emergency use authorization of an unapproved anti-influenza drug, peramivir. Clin Pharmacol Ther. 2010;88:587–589. doi: 10.1038/clpt.2010.187. [DOI] [PubMed] [Google Scholar]
- 6.Wang PC, Fang JM, Tsai KC. Peramivir phosphonate derivatives as influenza neuraminidase inhibitors. J Med Chem. 2016;59:5297–5310. doi: 10.1021/acs.jmedchem.6b00029. [DOI] [PubMed] [Google Scholar]
- 7.Liang R, Fei YJ, Prasad PD. Human intestinal H+/ peptide cotransporter: cloning, function, expression, and chromosomal location. J Biol Chem. 1995;270:6456–6463. doi: 10.1074/jbc.270.12.6456. [DOI] [PubMed] [Google Scholar]
- 8.Bhutia YD, Babu E, Prasad PD, Ganapathy V. The amino acid transporter SLC6A14 in cancer and its potential use in chemotherapy. Asian J Pharm Sci. 2014;9:293–303. [Google Scholar]
- 9.Karunakaran S, Ramachandran S, Coothankandaswamy V. SLC6A14 (ATB0,+) protein, a highly concentrative and broad specific amino acid transporter, is a novel and effective drug target for treatment of estrogen receptor-positive breast cancer. J Biol Chem. 2011;286:31830–31838. doi: 10.1074/jbc.M111.229518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Deng D, Xu C, Sun P. Crystal structure of the human glucose transporter GLUT1. Nature. 2014;510:121–125. doi: 10.1038/nature13306. [DOI] [PubMed] [Google Scholar]
- 11.Beauchamp LM, Orr GF, De Miranda P, Burnette T, Krenitsky TA. Amino acid ester prodrugs of acyclovir. Antiviral Chem Chemother. 1992;3:157–164. [Google Scholar]
- 12.Beutner KR. Valacyclovir: a review of its antiviral activity, pharmacokinetic properties, and clinical efficacy. Antiviral Res. 1995;28:281–290. doi: 10.1016/0166-3542(95)00066-6. [DOI] [PubMed] [Google Scholar]
- 13.Balimane PV, Tamai I, Guo AL. Direct evidence for peptide transporter (PepT1)-mediated uptake of a nonpeptide prodrug, valacyclovir. Biochem Biophys Res Commun. 1998;250:246–251. doi: 10.1006/bbrc.1998.9298. [DOI] [PubMed] [Google Scholar]
- 14.Sugarawa M, Huang W, Fei YJ, Leibach FH, Ganapathy V, Ganapathy ME. Transport of valganciclovir, a ganciclovir prodrug, via peptide transporters PEPT1 and PEPT2. J Pharm Sci. 2000;89:781–789. doi: 10.1002/(SICI)1520-6017(200006)89:6<781::AID-JPS10>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 15.Pak YA, Long AJ, Annes WF. In vitro and clinical evaluations of the drug-drug interaction potential of a metabotropic glutamate 2/3 receptor agonist prodrug with intestinal peptide transporter 1. Drug Metab Dispos. 2017;45:137–144. doi: 10.1124/dmd.116.071118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Annes WF, Long A, Witcher JW. Relative contributions of presystemic and systemic peptidases to oral exposure of a novel metabotropic glutamate 2/3 receptor agonist (LY404039) after oral administration of prodrug pomaglumetad methionil (LY2140023) J Pharm Sci. 2015;104:207–214. doi: 10.1002/jps.24226. [DOI] [PubMed] [Google Scholar]
- 17.Sun YB, Sun J, Shi SL. Synthesis, transport and pharmacokinetics of 5′-amino acid ester prodrugs of 1-β-d-arabinofuranosylcytosine. Mol Pharmmaceutics. 2009;6:315–325. doi: 10.1021/mp800200a. [DOI] [PubMed] [Google Scholar]
- 18.Sun YB, Sun J, Liu JF. Rapid and sensitive hydrophilic interaction chromatography/tandem mass spectrometry method for the determination of glycyl-sarcosine in cell homogenates. J Chromatogr B. 2009;877:649–652. doi: 10.1016/j.jchromb.2009.01.019. [DOI] [PubMed] [Google Scholar]
- 19.Bhardwaj RK, Herrera-Ruiz D, Sinko PJ, Gudmundsson OS, Gregory Knipp G. Delineation of human peptide transporter 1 (hPEPT1)-mediated and transport of substrates with varying transporter affinities utilizing stably transfected hPEPT1/Madin-Darby Canine Kindey clones and Caco-2 cells. J Pharmacol Exp Ther. 2005;314:1093–1100. doi: 10.1124/jpet.105.087148. [DOI] [PubMed] [Google Scholar]
- 20.Alper J. Drug delivery: breaching the membrane. Science. 2002;296:838–839. doi: 10.1126/science.296.5569.838. [DOI] [PubMed] [Google Scholar]
- 21.Zhang Y, Sun J, Sun Y, Wang YJ, He ZG. Prodrug design targeting intestinal PepT1 for improved oral absorption: design and performance. Curr Drug Metab. 2013;14:675–687. doi: 10.2174/1389200211314060004. [DOI] [PubMed] [Google Scholar]
- 22.Vig BS, Huttunen KM, Laine K, Rautio J. Amino acids as promoieties in prodrug design and development. Adv Drug Delivery Rev. 2013;65:1370–1385. doi: 10.1016/j.addr.2012.10.001. [DOI] [PubMed] [Google Scholar]
- 23.Song XT, Sun YH, Zhao CY, He ZG. The effect of cephalexin in influencing the pharmacokinetics of a novel drug 5′-valyl-cytarabinehydrochloride. Asian J Pharm Sci. 2017;12:143–148. doi: 10.1016/j.ajps.2016.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Michael P. Absorption of lisdexamfetamine dimesylate and its enzymatic conversion to d-ampjetamine. Neuropsychiatric Dis Treat. 2010;6:317–327. doi: 10.2147/ndt.s9749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Elia J, Easley C, Kirkpatrick P. lisdexamfetamine dimesylate. Nat Rev Drug Discov. 2007;6:343–344. doi: 10.1038/nrd2315. [DOI] [PubMed] [Google Scholar]
- 26.Bueno AB, Collado I, De Dios A. Dipeptides as effective prodrugs of unnatural amino acid (+)-2-aminobicyclo[3.1.0]hexane-2, 6-dicarboxylic acid (LY354740), a select group П metabotropic glutamate receptor agonist. J Med Chem. 2005;48:5305–5320. doi: 10.1021/jm050235r. [DOI] [PubMed] [Google Scholar]
- 27.Perkins EJ, Abraham T. Pharmacokinetics, metabolism, and excretion of the intestinal peptide transporter 1 (SLC15A1)-targeted prodrug (1S, 2S, 5R, 6S)-2-[(2’S)-(2-amino)propionyl] aminobicyclo[3.1.0.] hexen-2, 6-dicarboxylic acid (LY544344) in rats and dogs: assessment of first-pass bioactivation and dose linearity. Drug Metab Dispos. 2007;35:1903–1909. doi: 10.1124/dmd.107.016154. [DOI] [PubMed] [Google Scholar]
- 28.Masahiro T, Tomohiro T, Megumi I. Transport Characteristics of a novel peptide transporter 1 substrates, antihypotensive drug midodrine, and its amino acid derivatives. J Pharmacol Exp Ther. 2006;318:455–460. doi: 10.1124/jpet.106.102830. [DOI] [PubMed] [Google Scholar]
- 29.Tao WH, Zhao DY, Sun MC. Intestinal absorption and activation of decitabine amino acid ester prodrugs mediated by peptide transporter PEPT1 and enterocyte enzymes. Int J Pharm. 2018;541:64–71. doi: 10.1016/j.ijpharm.2018.02.033. [DOI] [PubMed] [Google Scholar]