

Abstract
Globoid cell leukodystrophy (GLD, Krabbe disease, Krabbe’s disease) is caused by genetic mutations in the gene encoding, galactosylceramidase (GALC). Deficiency of this enzyme results in central and peripheral nervous system pathology, and is characterized by loss of myelin and an infiltration of globoid cells. The canine model of GLD provides a translational model which faithfully recapitulates much of the human disease pathology. Targeted lipidomic analysis was conducted in serum and cerebrospinal fluid (CSF) over the lifetime of GLD affected and normal canines, and in brain tissue at humane endpoint to better understand disease progression and identify potential biomarkers of disease. Psychosine, a substrate of GALC and primary contributor to the pathology in GLD, was observed to be significantly elevated in the serum and CSF by 2 or 4 weeks of age, respectively, and steadily increased over the lifetime of affected animals. Importantly, psychosine concentration strongly correlated with disease severity. Galactosylceramide, glucosylceramide, and lactosylceramide were also found to be elevated in the CSF of affected animals and increased with age. Psychosine and galactosylceramide were found to be significantly increased in brain tissue at humane endpoint. This study identified several biomarkers which may be useful in the development of therapeutics for GLD.
Keywords: Globoid cell leukodystrophy, canine model, brain lipids, ceramides, sphingolipids, liquid chromatography, mass spectrometry
Introduction
Globoid cell leukodystrophy (GLD, Krabbe disease) was first described in 1916, and is characterized by rapid progression of nervous system dysfunction and severe loss of myelin, as well as decrease in oligodendrocyte numbers, astrocytic gliosis, and infiltration of the hallmark “globoid cells” (Suzuki, 2003). GLD is caused by genetic mutations in the gene encoding the hydrolytic enzyme, galactosylceramidase (GALC) (Suzuki, 2003). Although the major natural substrate for GALC is galactosylceramide, accumulation of galactosylceramide is not observed in the brain tissue of patients with GLD (Suzuki, 2003). However, some groups have noted that galactosylceramide has a unique ability to induce globoid cell infiltration when injected into the brain of rats (Andrews & Menkes, 1970; Suzuki, 1970). These infiltrates are morphologically identical to those seen in patients with GLD and suggest that although galactosylceramide has not been reported to be significantly elevated in GLD brains, it is likely accumulating nonetheless and could be contributing to notable neuropathology in particular brain regions. In light of this, another substrate of GALC, galactosylsphingosine (psychosine) is believed to be responsible for the cellular death associated with GLD and has long been utilized as marker of disease pathogenesis (Vanier & Svennerholm, 1976; Igisu & Suzuki, 1984). Recently, Escolar et al. demonstrated that psychosine measured by dried blood spot (DBS) analysis can be a sensitive and efficient way to both diagnose patients and monitor treatment effects (Escolar et al., 2017). Significant elevations in psychosine proved to be an excellent predictor of severe phenotype, with levels above 3nmol/L correlating with infantile GLD phenotypes (Escolar et al., 2017). Additionally, psychosine was sensitive to treatment by hematopoietic stem cell transplantation (HSCT), with a rapid decline in psychosine levels observed following treatment (Escolar et al., 2017).
The canine model of GLD was first reported in Cairn and West Highland White terriers in the late 1960’s (Fletcher et al., 1966; Hirth & Nielsen, 1966). In the 1990’s, following the identification of the disease causing mutation, 5 Cairn terriers heterozygous for GLD were donated to the University of Pennsylvania for the purpose of establishing a breeding colony (Wenger et al., 1999a). Recent data from the University of Pennsylvania colony indicate the onset of clinical signs in dogs occurs by 4–6 weeks of age, and pelvic limb paralysis develops warranting humane euthanasia by 15.7 ± 4.8 weeks (Bradbury et al., 2016a). In an effort to improve the overall genetics and fecundity of the colony, an effort has been undertaken to outcross the small, terrier type animals within the breeding colony to larger kennel hounds. Because of the new genetic background of these animals, a thorough longitudinal natural history has been conducted to identify and assess biomarkers over the course of disease progression. In the current study, the five GLD dogs developed pelvic limb paralysis warranting humane euthanasia by 12.7 ± 2.6 weeks of age.
In the current study, changes in a number of glycosphingolipid biomarkers were explored in the canine GLD model including the GALC substrates psychosine, galactosylceramide, and lactosylceramide, as well as glucosylsphingosine, lactosylsphingosine, and glucosylceramide (Figure 1). For the ceramide substrates, six chain lengths were examined (16:0, 18:0, 20:0, 22:0, 24:0, and 24:1) to be as inclusive as possible in this initial targeted lipidomic approach. The objective of this study was to provide a comprehensive examination of changes in glycosphingolipid biomarkers over the course of GLD in the canine model. The results of this study provide new insight into dysregulation of sphingolipids in Krabbe disease and demonstrates the utility of targeted lipidomics to serve as a reliable and effective biomarker of disease progression and therapeutic efficacy in this and other sphingolipidoses.
Figure 1.
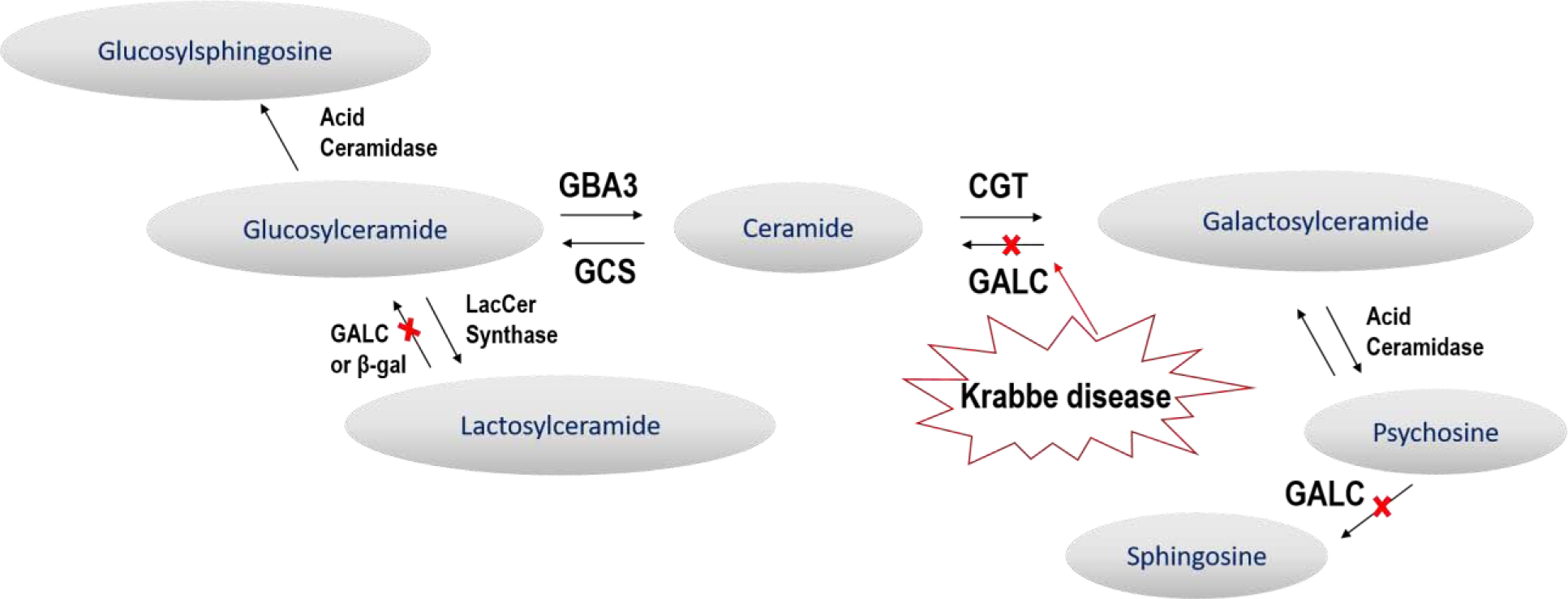
Lipid Pathway. Galactosylceramidase (GALC) can hydrolyze the amide bonds of galactosylceramide (GalCer), dihexosylceramide (DiHexCer), and galactosylsphingosine (GalSph, psychosine). In GLD, GALC is absent and psychosine has been found to be the principle substrate that accumulates. In the current study, GalCer, DiHexCer, and glucosylceramide (GluCer) were also observed to be dysregulated in cerebrospinal fluid (CSF) in the canine model of GLD.
Materials and Methods
Animals
Mixed breed dogs were raised in the Referral Center for Animal Models of Human Genetic Disease of the School of Veterinary Medicine of the University of Pennsylvania (NIH OD P40–10939) under National Institutes of Health and USDA guidelines for the care and use of animals in research. The experimental protocols were approved by the University’s Institutional Animal Care and Use Committee. An equal number of male and female dogs were use. Whole blood from dogs was tested for the GALC missense mutation, c.473A>C, p.158Y>S, using a TaqMan® real-time polymerase chain reaction-based DNA test to identify affected, normal, and heterozygote dogs as previously described (Bradbury et al. 2016a). Dogs were evaluated weekly via clinical neurological evaluations and videotaped for review. GLD dogs were euthanized at a humane endpoint defined by pelvic limb paralysis. Euthanasia was performed using an overdose of intravenous barbiturate. After sacrifice, animals were perfused through the left ventricle with 750 mL of 0.9% cold saline and tissues collected and stored at −80 degrees until analyzed.
Clinical Rating Score
A board certified veterinary neurologist (EMG) examined videos depicting affected dogs at weekly intervals. The neurologist identified neurological dysfunction and categorized them as “mild,” “moderate” or “severe.” Dysfunctions that were identified as “mild” were assigned a score of 0.5, while those were identified as “moderate” or “severe” were assigned scores of 1 or 2, respectively. Clinical signs consistent with humane endpoint, such as an animal being classified as non-ambulatory or having paralyzed pelvic limbs were assigned a 3. The clinical rating score used in the current study represents a sum of these clinical signs.
CSF collection
Dogs were anesthetized with propofol and the skin was clipped and aseptically prepared at the cerebellomedullary cistern, from which CSF was collected with a 22G spinal needle. CSF collected for lipidomics was allowed to drip into polypropylene tubes pre-coated with CHAPS (0.5 g/mL w/v) at a ratio of 20 μL of CHAPS per 500 μL of CSF for and stored at −80 degrees until analyzed. CHAPS was used to prevent absorption loss of GalSph and Glu Sph to the containers.
Psychosine and Glucosylsphingosine in Serum
Strong cation exchange solid phase extraction (SPE) with Evolute® Express CX (30 mg) (Biotage, Uppsala, Sweden) was used to extract galactosylsphingosine and its isomer, glucosylsphingosine (inactive form), from 100 μL of dog serum in the presence of deuterated internal standards (GluSph-d5 and GalSph-d5). Samples were loaded onto the Evolute® Express CX (30 mg) plates after addition of isotopic standards, and 3 volumes of 5% formic acid, washed first with 1mL 5% formic acid in water, and then 1mL formic acid in methanol. The shingosines were eluted with 2× 0.3 mL 28% NH4OH/ methanol (5/95, v/v), dried, and dissolved in 150 uL of acetonitrile/methanol/water (78/2/20, v/v/v) containing 0.0082% acetic acid and 4.1 mM ammonium acetate, and directly injected onto a Halo-HILIC for analysis byliquid chromatography and tandem mass spectrometry (LC-MS/MS), in a slight modification of the method of Sidhu et al. in a natural history of the twitcher (Krabbe) mouse (Sidhu, 2018). Extraction efficiencies of spiked labeled and unlabeled glycosphingosines was consistent across isomers and concentrations. The glycosphingosines were separated on a Halo HILIC coumn (4.6×150 mm, 2.7 μm, 90Å), maintained at 30°C, with an isocratic mobile phase, made from mixing 95/2.5/2.5 acetonitrile/methanol/water containing 0.01% acetic acid and 5mM NH4OH acetate with water at an 82:18 ratio. Each sample was run for 25 min at a flow rate of 0.5 mL/min using a Shimadzu Prominence HPLC System coupled to a Sciex 5500 Triple Quad Calibration curves were prepared in 5% bovine serum albumin in water. Quality control samples, which were prepared by spiking pooled normal Beagle dog serum (BioreclamationIVT) with glucosylsphingosine and galactosylsphingosine at 0.600/0.600, 6.00/6.00, and 35.0/35.0 ng/mL, were used to monitor the assay performance.
Lipidomics in CSF and Brain Tissue
Brain tissue was obtained when the GLD affected animals were euthanized. Brains were sectioned into 2mm coronal slices using a brain mold specifically sized for canine brains. Tissues were collected using a 3mm punch biopsy tool from the internal capsule and cerebellar white matter. Each punch biopsy was weighed to ensure tissue samples were of a consistent size. Average biopsy weight was 138.3 mg (± 15.2 mg).
The following lipid classes were analyzed: dihexosylceramides (DiHexCer) that is a mixture of lactosylceramide (LacCer) and digalactosylceramide (DiGalCer), glucosylceramides (GluCer), galactosylceramides (GalCer), glucosylsphingosine (GluSph), galactosylsphingosine (GalSph), and dihexosylsphingosine (DihexSph) that is a mixture of lactosylsphingosine (LacSph) and digalactosylsphingosine (DiGalSph). The brain tissues were homogenized in 2% CHAPS solution (4 mL/g brain) with Omini Bead Ruptor 24 (Omini International, Kennesaw GA). The CSF internal standards including d3-LacCer(16:0) (5 ng/mL) for LacCer, d5-GluCer(18:0) (5 ng/mL) for GluCer and GalCer, d5-GluSph (4 ng/mL) for GluSph, and d5-GalSph (4 ng/mL) for GalSph and LacSph were prepared in in acetonitrile solution. The brain internal standards including d3-LacCer(16:0) (250 ng/mL) for LacCer, d5-GluCer(18:0) (250 ng/mL) for GluCer and GalCer, d5-GluSph (200 ng/mL) for GluSph, and d5-GalSph (200 ng/mL) for GalSph were also prepared in acetonitrile solution. Protein precipitation was performed to extract lipids by mixing 50 μL of dog CSF or brain homogenate with 1 mL of internal standard solution in acetonitrile. The samples were vortexed for approximately 3 minutes and then centrifuged at 10,000 rpm for 10 minutes. The supernatants were transferred to 1.2 mL glass inserts (VWR, West Chester, PA) in 96 well plates. A quality control (QC) sample was prepared by pooling 20% of extracts from study samples and used to monitor the instrument performance. The crude extracts were directly injected to LC–MS/MS system for analysis of GluSph, GalSph, DihexSph in CSF and GluSph, GalSph, DihexSph, LacCer, GluCer, and GalCer in brain homogenates, and the injection volumes were 100 μL and 5 μL, respectively. For analysis of LacCer, GluCer, and GalCer in CSF, the crude extracts (1 mL) were dried and reconstituted in 100 μL of acetonitrile-methanol (9:1), and 5 μL was injected.
LC–MS/MS analysis was conducted on a Shimadzu (Columbia, MD) Prominence HPLC system coupled with an Applied Biosystems/MDS Sciex (Ontario, Canada) 4000QTRAP mass spectrometer using multiple reaction monitoring (MRM). The HPLC system consists of Prominence HPLC system with a CBM-20A system controller, 2 LC-20AD pumps, a SIL-20ACHT autosampler, and a DGU-20A5R degasser. The compartment of the autosampler was set at 4 °C.
For analysis of GluSph, GalSph, DihexSph, the chromatography was performed at ambient temperature using Ascentis® Express HILIC (4.6 × 50 mm, 2.7 μm, Supelco, Bellefonte, PA) protected with a HILIC Securityguard™ column (4 × 3.0 mm, Phenomenex, Torrance, CA) (9). The compartment of the autosampler was set at 4 oC. Mobile phase A (0.1% formic acid and 1 mM ammonium formate in water) and mobile phase B (0.1% formic acid and 1 mM ammonium formate in acetonitrile-water (95:5)) were operated with a gradient elution as follows: 0 – 0.2 min 100 – 95% B, 0.2 – 3.5 min 95% B, 3.5 – 3.9 min 95 – 90% B, 3.9 – 5.8 min 90% B, 5.8 – 5.9 min 90 – 10% B, 5.9 – 6.9 min 10% B, 6.9 – 7.0 min 10 – 100% B, and 7.0 – 8.8 min 100% B at a flow rate of 1.5 ml/min. The HPLC flow was diverted to waste except for 2.0 – 6.5 min to mass spectrometer. The ESI source temperature was 600 °C; the ESI needle was 5000 V; the declustering potentials for GalSph, GluSph, d5-GalSph, d5-GluSph, and DihexSph were 76, 76, 76, 76, and 80 V, respectively; both the entrance potential and the collision cell exit potential were 10 V for all compounds. The collision and curtain gas were set at medium and 20, respectively. Both desolvation gas and nebulizing gas were set at 45. For MRM, the collision energies for mass transitions of m/z 462.3 to 282.3 for GalSph and GluSph, m/z 624.5 to 282.3 for LacSph, and m/z 467.3 to 287.3 for d5-GalSph and d5-GluSph were 31, 26, 38, and 31 V, respectively. The dwell time was set at 50 ms for each mass transition.
For analysis of GluCer, GalCer, DihexCer, the chromatography was performed at ambient temperature using Supelcosil™ LC-Si (2.1 × 250 mm, 5 μm, Supelco, Bellefonte, PA) protected with a HILIC Securityguard™ column (4 × 3.0 mm, Phenomenex, Torrance, CA) (Shaner, 2009). Mobile phase A (5 mM ammonium acetate in water) and mobile phase B (5 mM ammonium acetate in acetonitrile-methanol-acetic acid (97:2:1)) were operated with a gradient elution as follows: 0 – 4.0 min 100 % B, 4.0 – 4.1 min 100 – 95% B, 4.1 – 6.5 min 95% B, 6.5 – 6.6 min 95 – 10% B, 6.6 – 7.6 min 10% B, 7.6 – 7.7 min 10 – 100% B, 6.9 – 7.0 min 10 – 100% B, and 7.0 – 10 min 100% B at a flow rate of 1.5 ml/min. The HPLC flow was diverted to waste except for 2.0 – 6.5 min to mass spectrometer. The ESI source temperature was 550 °C; the ESI needle was 5000 V; the declustering potentials for GluCer, GalCer, and DihexCer were 75, 75, and 80 V, respectively; both the entrance potential and the collision cell exit potential was 10 V for all the compounds. The collision and curtain gas were set at medium and 20, respectively. The desolvation gas and nebulizing gas were set at 35 and 55, respectively. For MRM, the collision energies for mass transitions of m/z 700.5 to 264.3 for GalCer(16:0) and GluCer(16:0), m/z 728.5 to 264.3 for GalCer(18:0) and GluCer(18:0), m/z 756.5 to 264.3 for GalCer(20:0) and GluCer(20:0), m/z 784.6 to 264.3 for GalCer(22:0) and GluCer(22:0), m/z 812.7 to 264.3 for GalCer(24:0) and GluCer(24:0), m/z 810.7 to 264.3 for GalCer(24:1) and GluCer(24:1), m/z 862.7 to 264.3 for DihexCer(16:0), m/z 890.7 to 264.3 for DihexCer(18:0), m/z 918.7 to 264.3 for DihexCer(20:0), m/z 946.7 to 264.3 for DihexCer(22:0), m/z 974.8 to 264.3 for DihexCer(24:0), m/z 972.7 to 264.3 for DihexCer(24:1), m/z 733.5 to 269.3 for d5-GluCer(18:0), and m/z 865.7 to 264.3 for d3- LacCer(16:0) were 50, 50, 50, 50, 50, 50, 61, 67, 67, 67, 67, 50 and 61 V, respectively. The dwell time was set at 50 ms for each mass transition. Data processing was conducted with Analyst 1.5.1 (Applied Biosystems). The signal of noise ratio of analyte less than 3 was defined as below limit of detection (LOD). The relative quantification data were obtained as peak area ratios of analytes to their internal standards, which were converted to concentrations.
Statistics
Two-tailed, unpaired t-tests and figures were conducted using Graph Pad Prism 6 (Graphpad Prism, La Jolla, CA, http://www.graphpad.com, RRID: SCR_002798). * p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.001. Linear regression was conducted using SigmaPlot (Systat Software, San Jose, CA).
Results
Clinical Rating
Historically, clinical signs appear in GLD dogs at 4–6 weeks of age and include tremor and pelvic limb weakness and progress to pelvic limb ataxia and thoracic limb dysmetria followed by tetraparesis and lastly pelvic limb paralysis, which defines humane endpoint. In the current study, five GLD dogs reached pelvic limb paralysis warranting humane euthanasia by 12.7 ± 2.6 weeks of age. Although the standard deviation of survival is relatively small (<3 weeks), there remains variability in the onset and severity of disease progression that has, until now, been impossible to predict at an early age due to a lack of reliable biomarkers of disease. In this study, the variability in disease onset and progression can be seen by comparing GLD affected dog 7A which reached humane endpoint at 9.7 weeks (Supplemental Video S1) and GLD affected dog 9A, which reached humane endpoint at 16.1 weeks of age (Supplemental Video S2). A blinded veterinary neurologist performed clinical rating evaluations of GLD dogs, and clinical rating scores for individual dogs demonstrated a precipitous decline over the course of disease progresssion (Figure 3A).
Figure 3: Progression of Clinical Signs in Affected Dogs and Temporal Correlation with Increases in CSF Psychosine.
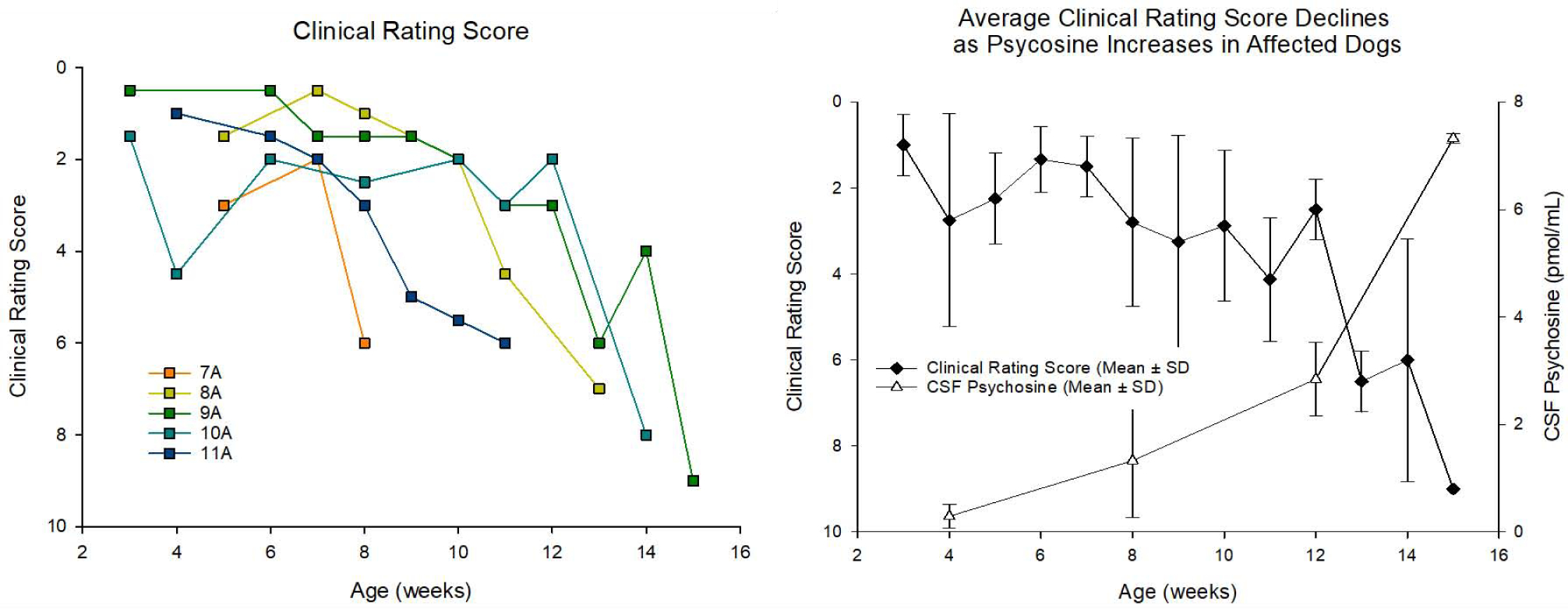
A blinded, board certified veterinary neurologist examined videos depicting GLD affected dogs at weekly intervals. The neurologist identified symptoms, categorized them as “mild,” “moderate” or “severe,” and (a) assigned an appropriate numerical score based on symptom severity. (b) Average CSF psychosine levels showed a strong inverse correlation with average clinical rating score for each dog examined (R2 = 0.86).
Serum Psychosine and Glucosylsphingosine
Serum samples from affected GLD dogs and normal age-matched control dogs were measured for psychosine concentrations (Figure 2A). Serum was collected every 2 weeks (beginning at 2 weeks of age) and at endpoint. Psychosine was significantly elevated in affected dogs by 2–4 weeks of age (p=<0.001) and continued to increase over the lifetime of affected animals. No noteworthy differences were observed in serum glucosylsphingosine (a breakdown product of GluCer) between affected and normal control dog (data not shown).
Figure 2: Longitudinal analysis of psychosine concentrations in the serum and CSF.
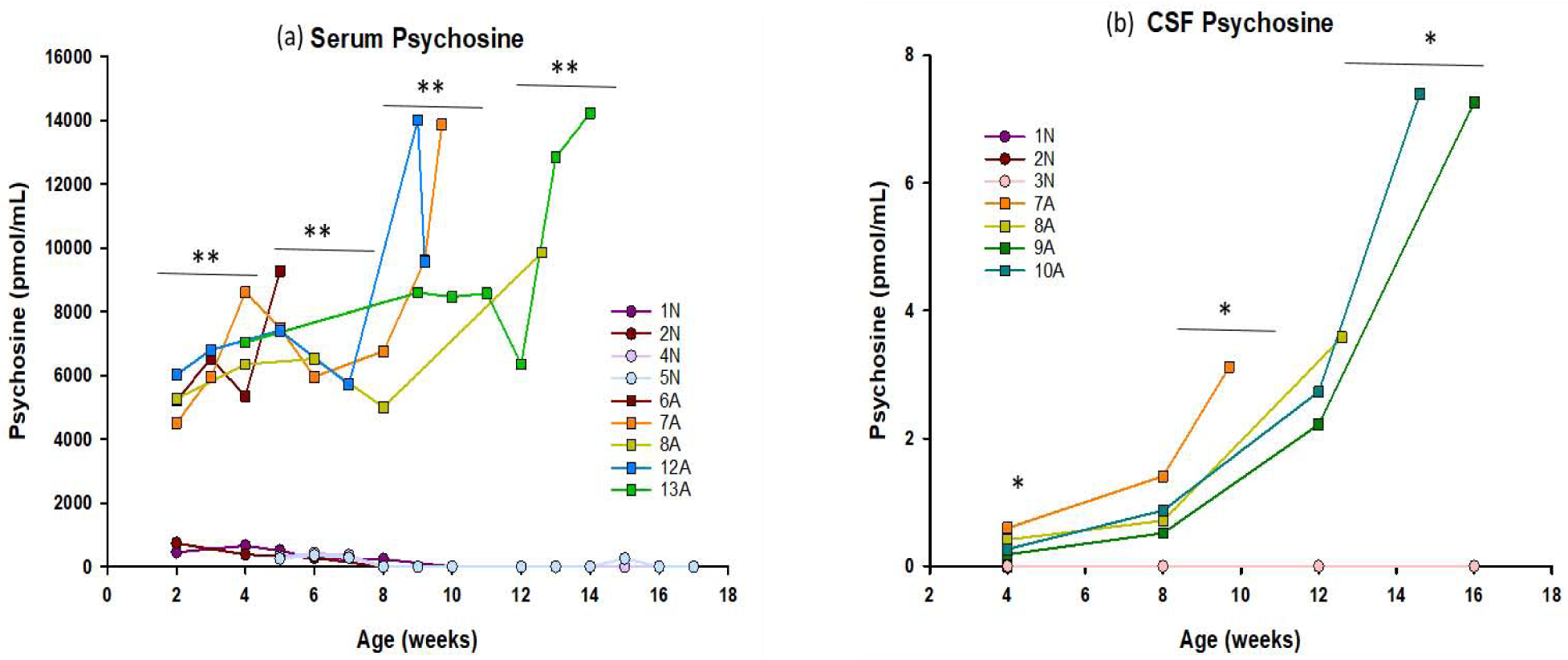
Psychosine concentrations were measured in (a) serum and (b) CSF of 3–4 normal controls (● symbols) and 4–5 GLD affected dogs (■ symbols). Serum was collected every other week, beginning at 2 weeks of age, until humane endpoint. CSF was collected monthly, beginning at 4 weeks of age, until humane endpoint. Psychosine concentrations were extremely low, or undetectable in the serum and CSF of normal animals at every age, but were significantly elevated in the serum and CSF of GLD affected animals at age group analyzed (indicated by bars) and generally increased with age/disease progression. * = p < 0.05; ** = p < 0.01.
CSF Lipidomics
CSF samples from affected GLD dogs and normal age-matched control dogs were measured for DihexCer, GluCer, GalCer, GluSph, GalSph, and DihexSph. CSF was collected every 4 weeks (beginning at 4 weeks of age) and at endpoint.
Galactosylsphingosine (psychosine)
GalSph, more commonly referred to as psychosine, is the primary cytotoxic storage product in GLD. Psychosine was significantly elevated in CSF at 4 weeks of age when compared to normal dogs (p = 0.045) and continued to increase with disease progression at 8– 10 weeks (p = 0.048) and 12 −16 weeks of age (p = 0.014) (Figure 2B). Importantly, CSF psychosine concentration correlated with clinical severity of neurological disease (Figure 3). GLD affected dog 7A (orange square, Supplemental Video S1) had the earliest onset and most severe disease progression, reaching humane endpoint at 9.7 weeks of age, and had the highest CSF psychosine at all time points analyzed. In contrast, GLD affected dog 9A (green square, Supplemental Video S2) had the slowest disease progression, reaching humane endpoint at 16.1 weeks of age and had the lowest CSF psychosine levels at each time point analyzed. All GLD dogs were evaluated by a veterinary neurologist and assigned a clinical rating score (Figure 3A). CSF psychosine levels inversely correlated with the CRS (R2 = 0.86) (Figure 3B). These results indicate that CSF psychosine can serve as an early and reliable indicator of GLD-specific disease progression.
Glucosylsphingosine and dihexosylsphingosine
GluSph and DihexSph were below the level of detection in GLD and normal dog CSF (data not shown).
Galactosylceramide
GalCer was significantly elevated in CSF of GLD dogs by 4 weeks of age for 20:0 (p=0.043) or 8 weeks of age for 16:0 (p = 0.036), 22:0 (p = 0.015), 24:1 (p=0.026), and 24:0 (p=0.019) species. GalCer species 18:0 reached significance at 12 weeks of age (P<0.001) (Figure 4). Similar to psychosine, GalCer levels correlated with disease progression, with our shortest lived affected animal (dog 7A) having the highest concentration at all time points analyzed and longest lived affected animal (dog 9A) having the lowest concentration at all time points analyzed (Figure 3a, Figure 4, Supplemental Videos S1 and S2).
Figure 4: Serial analysis of multiple species of galactosylceramide in the CSF.
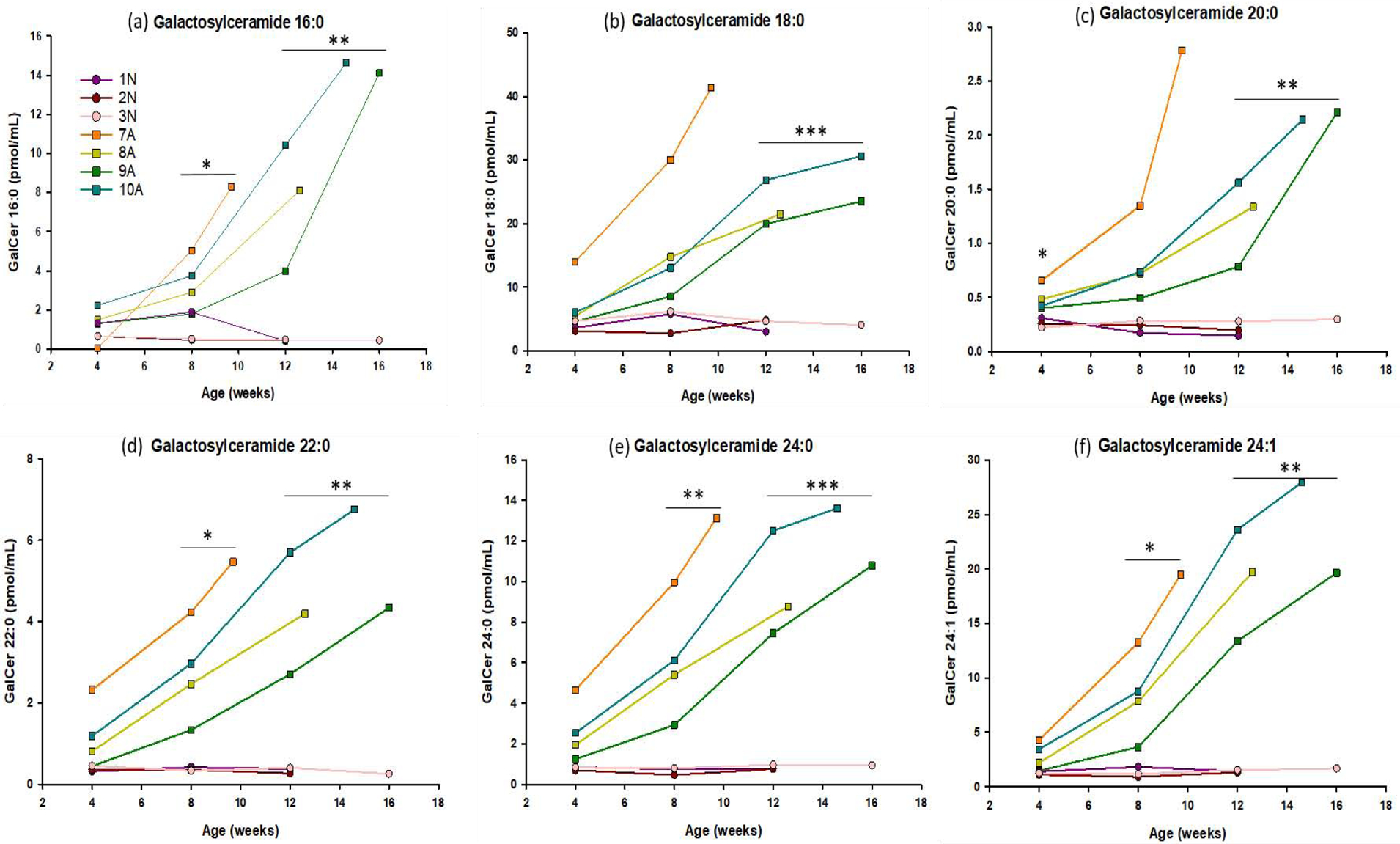
Concentrations of galactosylceramide (16:0, 18:0, 20:0, 22:0, 24:1, 24:0) were measured in CSF of 4 GLD affected dogs (■ symbols), and 3 normal littermate controls (● symbols). CSF was collected monthly, beginning at 4 weeks of age, until humane endpoint. GalCer was significantly elevated in CSF of GLD dogs by 4 weeks of age for 20:0 (p=0.043), 8 weeks of age for 16:0 (p = 0.036), 22:0 (p = 0.015), 24:1 (p=0.026), and 24:0 (p=0.019) species. GalCer species 18:0 reached significance at 12 weeks of age (p=0.0003). * = p < 0.05; ** = p < 0.01; *** = p < 0.001.
Glucosylceramide
This substrate is the same molecular weight as GalCer and can be difficult to separate via liquid chromatography/mass spectrometry. In the current study, the two substrates were baseline separated, and no carryover from GalCer was observed in the GluCer peak and vice versa (Supplemental Figure 1). GluCer 20:0 was significantly elevated in the CSF of GLD dogs at the first and last time points analyzed (4 weeks p= 0.043, 12 weeks p = 0.044) while all other species except 22:0 reached significance at 12 weeks of age. GluCer 22:0 did not reach significance at any time point, but trended upward similar to the other species (Figure 5).
Figure 5: Serial analysis of multiple species of glucosylceramide in the CSF.
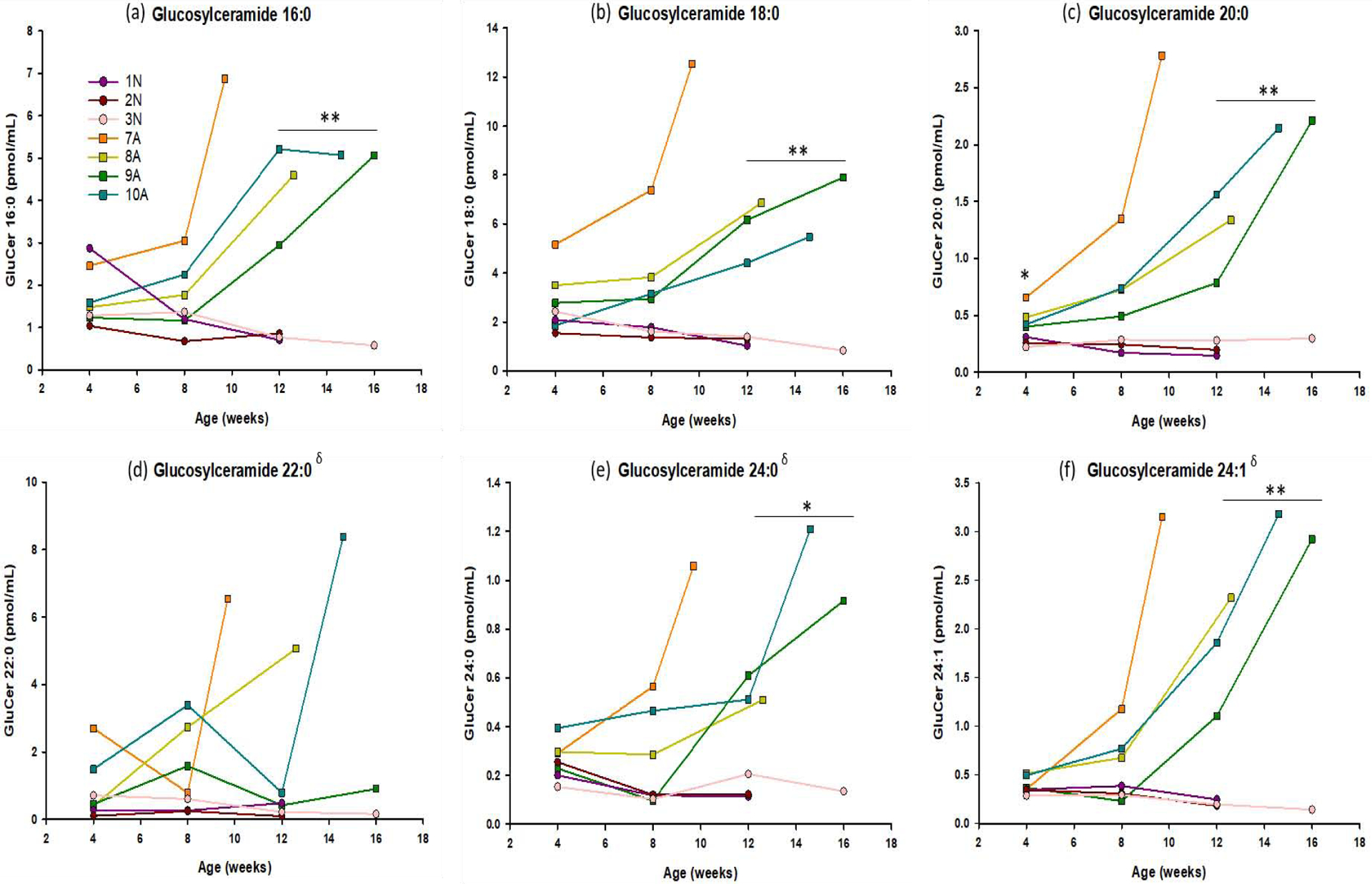
Concentrations of glucosylceramide (16:0, 18:0, 20:0, 22:0, 24:1, 24:0) were measured in CSF of 4 GLD affected dogs (■ symbols), and 3 normal littermate controls (● symbols). CSF was collected monthly, beginning at 4 weeks of age, until humane endpoint. GluCer 20:0 was significantly elevated in the CSF of GLD dogs at the first and last time points analyzed (4 weeks p= 0.043, 12 weeks p = 0.044) while all other species except 22:0 reached significance at 12 weeks of age. GluCer 22:0 did not reach significance at any time point, but trended upward similar to the other species. * = p < 0.05; ** = p < 0.01; *** = p < 0.001; δ%CV > 15.
Dihexosylceramide
DihexCer 16:0 and 22:0 were significantly elevated at all time points analyzed (4 weeks p= 0.036 and 0.038, 8 weeks p = 0.030 and 0.012, 12 weeks p = 0.005 and 0.005), while DihexCer 18:0 (p=0.009), 20:0 (0.030), and 24:1 (p=0.032) reached significantly elevated levels at 8 weeks of age. Species DihexCer 24:0 reached significance at 12 weeks of age (p=0.004) (Figure 6).
Figure 6: Serial analysis of multiple species of Dihexosylceramide in the CSF.
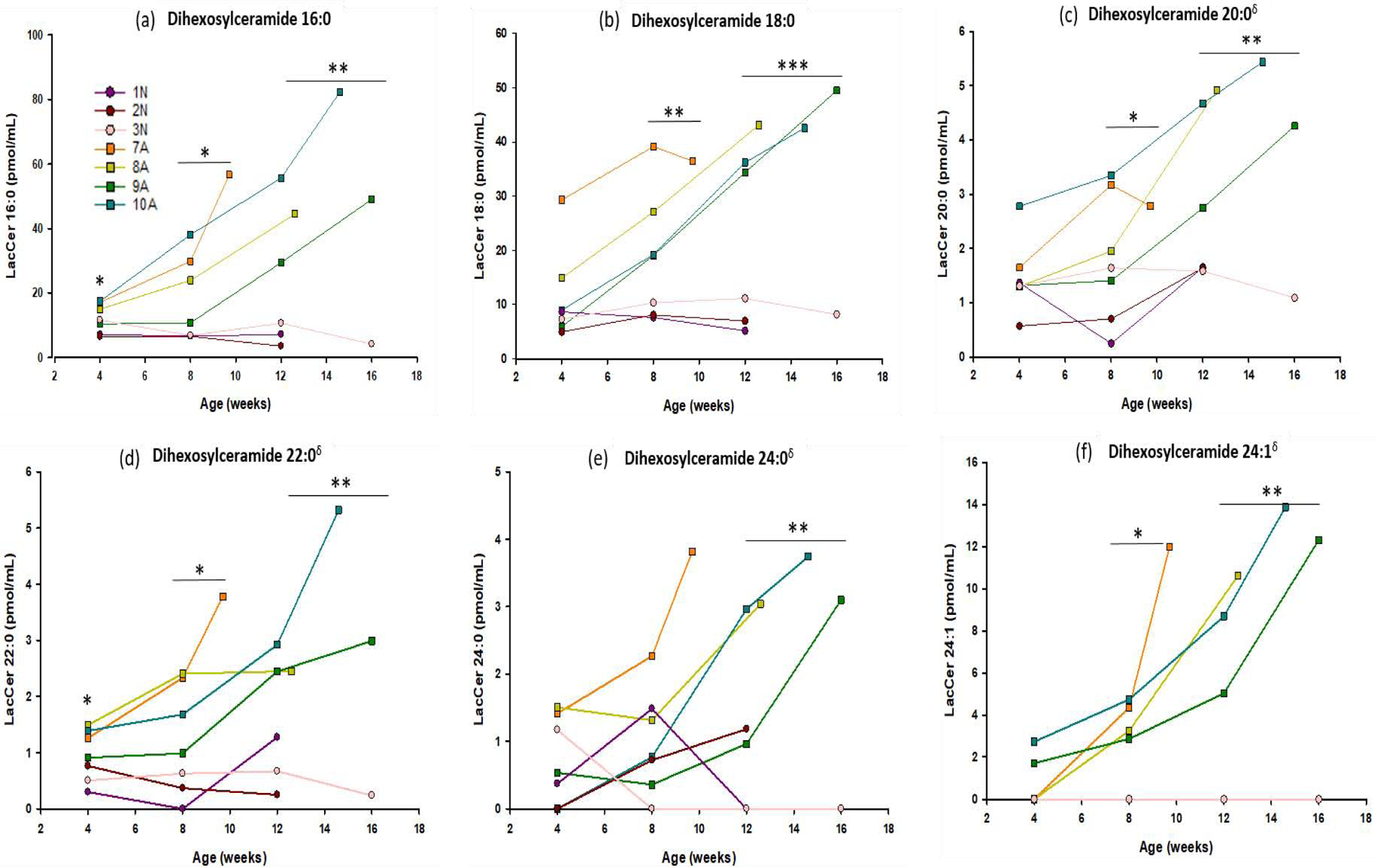
Concentrations of Dihexosylceramide (16:0, 18:0, 20:0, 22:0, 24:1, 24:0) were measured in CSF of 4 Krabbe affected dogs (■ symbols), and 3 normal littermate controls (● symbols). CSF was collected monthly, beginning at 4 weeks of age, until humane endpoint. DihexCer 16:0 and 22:0 were significant at all time points analyzed (4 weeks p= 0.036 and 0.038, 8 weeks p = 0.030 and 0.012, 12 weeks p = 0.005 and 0.005), while DihexCer 18:0 (p=0.009), 20:0 (0.030), and 24:1 (p=0.032) reached significance at 8 weeks of age. Species DihexCer 24:0 reached significance at 12 weeks of age (p=0.004). * = p < 0.05; ** = p < 0.01; *** = p < 0.001; δ%CV > 15.
Brain Tissue Lipidomics
Brain tissue was obtained when the GLD affected animals were euthanized. Tissues, in the form of punch biopsies, were collected from the internal capsule and cerebellar white matter and measured for DihexCer, GluCer, GalCer, GluSph, GalSph, and DihexSph.
Galactosylsphingosine (psychosine)
Psychosine, GalSph, was significantly elevated in the internal capsule (p=0.05) and cerebellum (p=0.002) of affected dogs at humane endpoint when compared to age-matched normal control tissue (Figure 7).
Figure 7: Lipid analysis in two brain regions at humane endpoint.
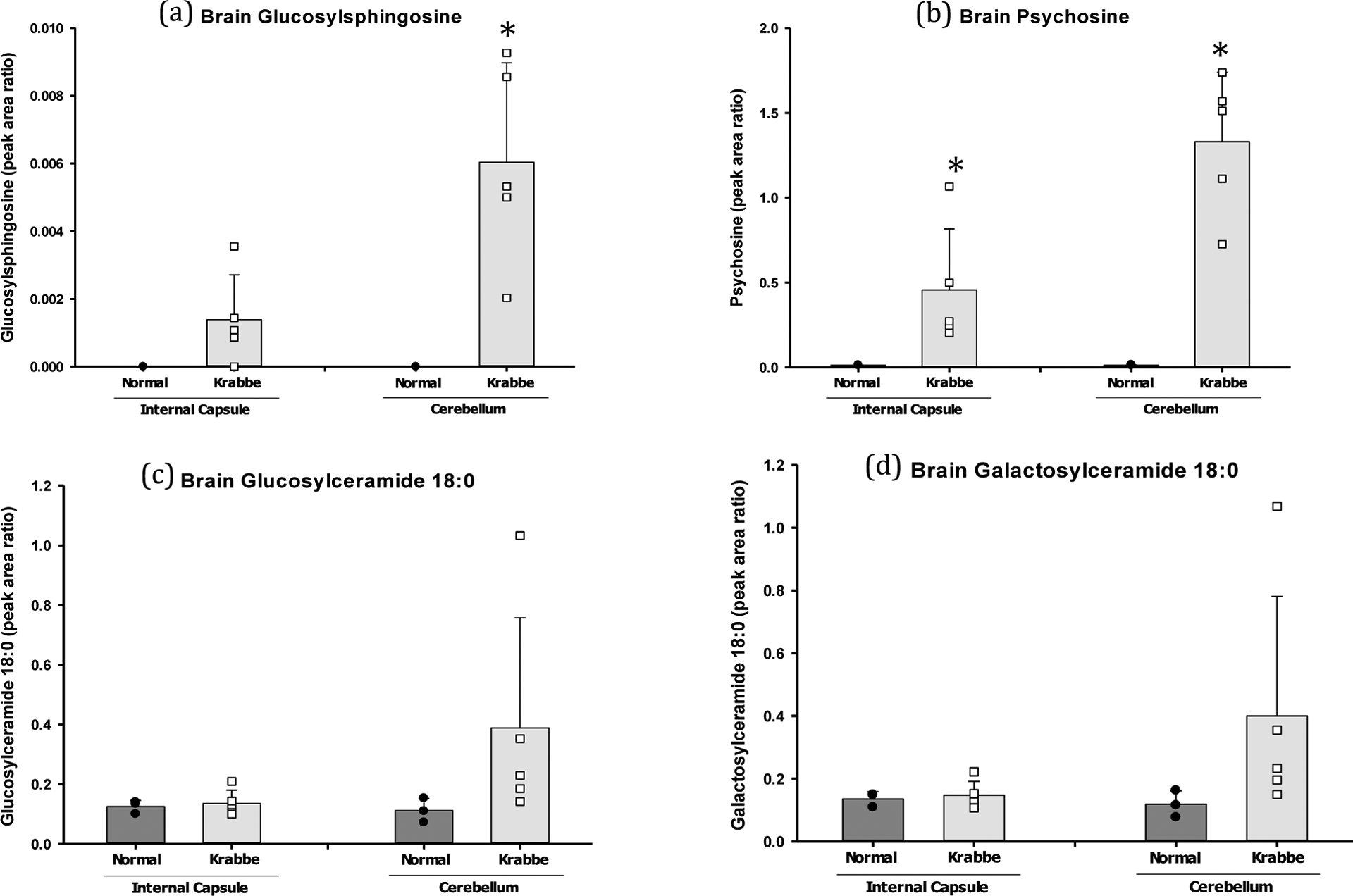
Tissue punches were collected from the internal capsule and cerebellum and measured for DiHexCer, GluCer, GalCer (16:0, 18:0, 20:0, 22:0, 24:1, 24:0), as well as GluSph, GalSph, and LacSph. a) GluSph was significantly elevated in the cerebellum (p=0.01) of GLD dogs. b) Psychosine (GalSph) was significantly elevated in the internal capsule (p=0.05) and cerebellum (p=0.002) of affected dogs. c) GluCer appeared elevated in the internal capsule and cerebellum of affected dogs, but significance was not achieved for any species [Supplemental Figure S1]. d) GalCer appeared elevated in the internal capsule and cerebellum of affected dogs at humane endpoint, but significance was achieved only for the 16:0 species in the internal capsule (p=0.0008) [Supplemental Figure S2]. * = p < 0.05; ** = p < 0.01; *** = p < 0.001.
Glucosylsphingosine
GluSph was not significantly elevated in the internal capsule, but was significantly elevated in the cerebellum (p=0.01) of affected dogs at humane endpoint when compared to age-matched normal control tissue (Figure 7).
Dihexosylsphingosine
DihexSph was below the limit of detection in the internal capsule and cerebellum samples from affected and normal dogs at humane endpoint.
Galactosylceramide
GalCer 18:0 was the most abundance species measured in canine internal capsule and cerebellum. GalCer 18:0 appeared elevated in the internal capsule and cerebellum of affected dogs at humane endpoint (Figure 7), but significance was achieved only for the 16:0 species in the internal capsule (p=0.0008) when compared to normal age-matched control tissue (Supplemental Figure S2).
Glucosylceramide
GluCer 18:0 was the most abundant species measured in canine internal capsule and cerebeluum. GluCer 18:0 appeared elevated in the internal capsule and cerebellum of affected dogs at humane endpoint (Figure 7), but significance was not achieved for any species (Supplemental Figure S3).
Dihexosylceramide
DihexCer was only detected sporadically in the internal capsule and cerebellum of affected and normal dogs at humane endpoint.
Discussion
A naturally occurring canine model of GLD closely mimics the clinical disease progression, neuropathological alterations, and biochemical abnormalities observed in human disease and provides an excellent translational model to test new therapeutics (Bradbury et al., 2016a; Bradbury et al., 2016b; Cozzi 1998; Victoria et al., 1996; Wenger et al., 1999a). Previously, biochemical analysis in a single dog showed elevation of psychosine in brain tissue at humane endpoint (Wenger et al., 1999b). More recently, reduction in GALC enzyme activity and elevation of psychosine have been reported in the cerebrum, cerebellum, spinal cord, sciatic nerve, and liver of untreated GLD dogs at disease endpoint (Bradbury, 2018). In order to further investigate the full scope and timing of sphingolipid dysregulation, targeted lipidomics was performed on serially collected samples from GLD dogs.
Although psychosine is widely accepted as the main toxic substrate in GLD, and has previously been reported to be elevated in the canine model of GLD, a thorough characterization of psychosine accumulation throughout disease progression had not been conducted in the canine model. Herein, in dogs affected with GLD, psychosine was found to be significantly elevated in both serum and CSF at the earliest time points examined, 2 weeks and 4 weeks respectively. Elevated CSF psychosine, in particular, correlated strongly with worsening clinical signs. Recently, Escolar et al. demonstrated that psychosine measured by dried blood spot (DBS) analysis can be a sensitive and efficient way to both diagnose patients and monitor treatment effects (Escolar et al., 2017). Similarly, we demonstrate here that both blood and CSF psychosine concentration serve as a strong and early predictor of disease progression.
Other lipids, which are also known substrates of GALC, including GalCer and DihexCer, were also found to be elevated in CSF early in disease progression, and increased over the lifetime of GLD affected dogs. Although GalCer should theoretically be elevated in GLD, it has been difficult to detect a significant elevation in patient tissue samples, perhaps due to a high level of background, as it is a normal constituent of myelin. No reports of GalCer elevation in patient CSF could be located in the literature. Therefore, for the first time to best of our knowledge, this study demonstrates 3 substrates of GALC: GalSph, GalCer, and DihexCer that are all significantly increased in the CSF of GLD dogs and increase with disease progression, suggesting a combination of reliable biomarkers that could be easily evaluated in a single sample. While all GalCer species were significantly elevated in CSF, 18:0 was the most abundant and would be the most relevant for future use in diagnosis or treatment monitoring. Our current method cannot separate lactosylceramide (LacCer) from digalactosylceramide (DiGalCer), and the data was reported as DihexCer that is a mixture of LacCer and DiGalCer. DiGalCer was reported to elevate in in Krabbe disease, and accounts for 10% of the DihexCer (Svenerholm et al., 1980). Therefore, the majority of elevation of DihexCer observed in this study can be attributed to LacCer.
GluCer can be formed by glucosylation of ceramide by GCS, or degalactosylation of LacCer (Figure 1), but is not a substrate of GALC and thus was not anticipated to be increased in GLD affected animals. If anything, loss of the LacCer → GluCer pathway through GALC would be expected to diminish the levels of GluCer. Interestingly, GluCer was found to be significantly elevated in the CSF of GLD affected dogs. Since GluCer is the same molecular weight as GalCer, it can be difficult to separate via mass spectrometry; however, in the current study, the two substrates were baseline separated, and artefactual elevation of GluCer would not expected to be due to carryover from GalCer (Supplemental Figure 1). As with GalCer, GluCer 18:0 was the most abundant and reliable (species 22:0, 24:0, and 24:1 had CV >15%), and would be the most relevant for future use in diagnosis or treatment monitoring. Broader dysregulation of sphingolipid lysosomal metabolic pathways by a number of factors could induce a secondary increase of GluCer; however, the exact mechanism has not been elucidated. Herein we demonstrate for the first time dysregulation of GluCer in GLD.
In the brain tissue of human patients with GLD, psychosine is the only lipid species that has been reported to be significantly dysregulated. Therefore, in this study, we evaluated GLD dog brain tissue for all lipid species analyzed in the CSF: DihexCer, GluCer, GalCer, GluSph, GalSph, and DihexSph. In recent imaging studies all (15/15) early and late infantilte patients had confluent deep cerebral, including the internal capsule, and deep cerebellar white matter signal changes (Mathusamy et al., 2019). Additionally, sampling of these large white matter tracts allowed for restricted isolation of white matter with minimal inclusion of surrpounding gray matter. Therefore, internal capsule and cerebellum were chosen as the brain regions to evaluate herein. Psychosine was significantly elevated in both the internal capsule and cerebellum. GluSph appeared elevated in both regions, but only achieved significance in the cerebellum. GluCer also appeared elevated in the brains of affected dogs at humane endpoint (Figure 7), but significance was not achieved in either region for any species [Supplemental Figure S3]. GalCer appeared elevated in the brain of affected dogs (Figure 7), but significance was achieved only for the 16:0 species in the internal capsule (p=0.0008) [Supplemental Figure S2]. Similar to the production of psychosine from GalCer, GluSph can be formed by the action of acid ceramidase on GluCer. Elevation of GluSph in brain tissue may be a natural consequence of the increased GluCer we observed. These findings suggest that elevations detected in the CSF can serve as early indicators of alterations occurring globally in the CNS. Furthermore, these findings reveal novel alterations in brain lipid species and regional brain differences in canine GLD tissue. These regional differences in disease pathology are also being evaluated in the same cohort of animals by DTI (Li et al., 2018) and histology. Further, analysis of canine GLD brain tissue by matrix assisted desorption ionization (MALDI) imaging mass spectrometry as a molecular imaging tool to map spatial distribution of these dysregulated lipids is ongoing. It is likely that the methods used to evaluate lipids in the canine CSF and brain tissue are more sensitive than those previously used to evaluate GLD patient samples. It would be of interest to evaluate human samples utilizing these same techniques described herein and determine if these same lipid species elevated in canine species are indeed also elevated in human GLD tissues.
In conclusion, as new therapeutic strategies now show promise in murine and canine models of GLD, development of biomarkers to serve as outcome measures has become increasingly important (Li & Sands, 2014;, Bradbury et al., 2018). Due to the increased size, the canine model of GLD is especially valuable in the development of biochemical, imaging, and electrodiagnostic biomarkers using techniques identical to those used in the human patient population (Bradbury, 2016a, Li & Sands, 2014,Li et al., 2018). Here we performed longitudinal, targeted lipidomic analysis of CSF from GLD affected and normal age-matched control dogs. We identified significant elevation of 4 lipid classes: GalSph, GalCer, GluCer and DihexCer in the CSF of GLD dogs. Importantly, changes were identifiable early in disease onset and correlated with disease progression. Lipid species elevated in the CSF were also found to be elevated in brain tissue at endpoint, suggesting that CSF samples serve as a good indicator of CNS disease progression. Most importantly, this study revealed numerous lipid species that could serve as biomarkers for future clinical trial design in pediatric patients.
Supplementary Material
Highlights.
CSF psychosine increased over time in GLD affected dogs and correlates with disease severity
CSF Galactosylceramide, glucosylceramide, and lactosylceramide were elevated in GLD affected dogs
Psychosine and galactosylceramide were elevated in GLD affected brain tissue at humane endpoint
Acknowledgements/Grant Support
This work was funded by BioMarin Pharmaceutical, Inc. C.H.V. is also funded through NIH OD P40-10939 and NIH R01 NS096087. A.M.B. is funded through a NRSA fellowship (F32NS093898) and The Legacy of Angels Foundation. The lipidomics in CSF and brain tissue was performed in the Washington University Metabolomics Facility (NIH P30 DK020579 and P30 DK056341). We thank the students at the University of Pennsylvania School of Veterinary Medicine for animal care.
Abbreviations
- GLD
globoid cell leukodystrophy
- GALC
galactosylceramidase
- CSF
cerebrospinalfluid
- DBS
dried blood spot
- HSCT
hematopoietic stem cell transplant
- SPE
solid phase extraction
- QC
quality control
- MRM
multiple reaction monitoring
- LacCer
lactosylceramide
- DihexCer
dihexosylceramide
- DiGalCer
digalactosylceramide
- GluCer
glucosylceramide
- GalCer
galactosylceramide
- GluSph
glucosylsphingosine
- GalSph
galactosylsphingosine
- LacSph
lactosylsphingosine
- DiGalSph
digalactosylsphingosine
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declarations of interest: None
References
- Andrews JM, Menkes JH 1970. Ultrastructure of experimentally produced globoid cells in the rat. Experimental Neurology; 29:483–493. [DOI] [PubMed] [Google Scholar]
- Bradbury AM, Bagel JH, Jiang X et al. 2016a. Clinical, electrophysiological, and biochemical markers of peripheral and central nervous system disease in canine globoid cell leukodystrophy (Krabbe’s disease). Journal of neuroscience research; 94:1007–1017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradbury A, Peterson D, Vite C et al. 2016b. Diffusion tensor imaging analysis of the brain in the canine model of Krabbe disease. Neuroradiol J; 29:417–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradbury AM, Rafi MA, Bagel JH, Brisson BK, et al. 2018. AAVrh10 gene therapy ameliorates central and peripheral nervous system disease in canine globoid cellleukodystrophy(Krabbe disease). Human Gene Therapy; doi: 10.1089/hum.2017.151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cozzi F, Vite CH, Wenger DA et al. 1998. MRI and electrophysiological abnormalities in a case of canine globoid cell leucodystrophy. The Journal of small animal practice;39:401–405. [DOI] [PubMed] [Google Scholar]
- Escolar ML, Kiely BT, Shawgo E, et al. 2017. Psychosine, a marker of Krabbe phenotype and treatment effect. Molecular Genetics and Metabolism;121(3):271–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fletcher TF, Kurtz HJ, Low DG. 1966. Globoid cell leukodystrophy (Krabbe type) in the dog. American Journal of Veterinary Medicine;149:165–172. [PubMed] [Google Scholar]
- Hirth RS and Nielsen SW. 1966. A familial canine globoid cell leukodystrophy (“Krabbe type”). Journal of Small Animal Practice;8:569–575. [DOI] [PubMed] [Google Scholar]
- Igisu H, Suzuki K. 1984. Progressive accumulation of toxic metabolite in a genetic leukodystrophy. Science; 18: 753–755. [DOI] [PubMed] [Google Scholar]
- Li Y and Sands M. 2014. Experimental Therapies in the murine model of globoid cell leukodsytrophy. Pediatric Neurology;51(5):600–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svennerholm L, Vanier MT, and MPnsson JE. 1980. Krabbe disease: a galactosylsphingosine (psychosine) 1ipidosis. J. Lipid Res.. 21: 53–64. [PubMed] [Google Scholar]
- Li J, Middleton DM, Chen S, et al. 2018. Quantitative DTI metrics in a canine model of Krabbe disease: comparisons versus age-matched controls across multiple ages. The Neuroradiology Journal. 31: 168–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muthusamy K, Sudhakar SV, Thomas M, Yoganathan S, Christudass CS, Chandran M, Panwala H, Gibikote S 2019. Revisiting magnetic resonance imaging pattern of Krabbe disease-lessons from an Indian cohort. J Clin Imaging Sci. 9: 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaner RL, Allegood JC, Park H, et al. 2009. Quantitative analysis of sphingolipids for lipidomics using triple quadrupole and quadrupole linear ion trap mass spectrometers. J Lipid Res, 50(8):1692–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidhu R, Mikulka CR, Fujiwara H, et al. 2018. A HILIC-MS/MS method for simultaneous quantification of the lysosomal disease markers galactosylsphingosine and glucosylsphingosine in mouse serum. Biomedical Chromatography; doi:10.1002bmn.4235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki K 2003. Globoid cell leukodystrophy (Krabbe’s disease): Update. Journal of Child Neurology;18(9): 595–603. [DOI] [PubMed] [Google Scholar]
- Suzuki K 1970. Ultrastructural study of experimental globoid cells. Laboratory Investigation;23:612–619. [PubMed] [Google Scholar]
- Vanier M, Svennerholm L 1976. Chemical pathology of Krabbe disease: the occurrence of psychosine and other neutrual sphingolipids. Adv Exp Med Biol; 68: 115–126. [DOI] [PubMed] [Google Scholar]
- Victoria T, Rafi MA, Wenger DA. 1996. Cloning of the canine GALC cDNA and identification of the mutation causing globoid cell leukodystrophy in West Highland White and Cairn terriers. Genomics;33:457–462. [DOI] [PubMed] [Google Scholar]
- Wenger DA, Victoria T, Rafi MA et al. 1999a. Globoid cell leukodystrophy in cairn and West Highland white terriers. J Hered;90:138–142. [DOI] [PubMed] [Google Scholar]
- Wenger DA, Victoria T, Rafi MA, et al. 1999b. Globoid cell leukodystrophy in Cairn and West Highland White Terriers. The American Genetic Association;90:138–142. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.