

Abstract
Covalent modification of LC3 and GABARAP proteins to phosphatidylethanolamine (PE) in the double-membrane phagophore is a key event in the early phase of macroautophagy, but can also occur on single membrane structures. In both cases this involves transfer of LC3/GABARAP from ATG3 to PE at the target membrane. Here we have purified the full-length human ATG12–5–16L1 complex and show its essential role in LC3B/GABARAP lipidation in vitro. We have identified two functionally distinct membrane-binding regions in ATG16L1. An N-terminal membrane-binding amphipathic helix is required for LC3B lipidation under all conditions tested. In contrast, the C-terminal membrane-binding region is dispensable for canonical autophagy, but essential for VPS34-independent LC3B-lipidation at perturbed endosomes. We further show that the ATG16L1 C-terminus can compensate for WIPI2 depletion to sustain lipidation during starvation. Remarkably, the C-terminal membrane-binding region comprises the β-isoform-specific sequence of ATG16L1, showing that ATG16L1 isoforms mechanistically distinguish LC3B-lipidation under different cellular conditions.
Keywords: Autophagy, ATG16L1, ATG12-5–16L1, ATG7, ATG3, LC3, WIPI2b
Introduction
Macroautophagy (hereafter autophagy) involves sequestration of intracellular constituents into double membrane autophagosomes that fuse with lysosomes to allow cargo degradation. The LC3 and GABARAP subfamilies comprise ubiquitin-like proteins that become covalently attached to phosphatidylethanolamine (PE) in the autophagosomal membrane1 where they function in autophagosome biogenesis, recruitment of cargo and fusion with the lysosome. Recently, it has become evident that LC3 also can be conjugated to single-layer endomembrane structures in response to cellular stress as infection or vesicle damage2.
Prior to lipidation, LC3/GABARAPs are cleaved by ATG4 to expose a C-terminal glycine, which becomes adenylated and conjugated to a catalytic cysteine on the E1-like ATG73–5 and then transferred onto a cysteine residue of the E2-like ATG3. The final transfer to PE requires the E3-like ATG12-5–16L1 complex6. It is proposed that ATG12-5–16L1 targets ATG3 to the site of lipidation through an interaction between ATG3 and ATG127 and membrane targeting by ATG16L18, but the exact function of the ATG12-5–16L1 complex is poorly described. During amino acid or glucose starvation, membrane recruitment of ATG16L1 involves its binding to WIPI2b11, an effector protein of phosphatidyl 3-phosphate (PI(3)P) or PI(5)P12, 13. However, ATG16L1 targeting to membranes may be independent of WIPI2b during endomembrane LC3 lipidation14–16.
Here we demonstrate that ATG16L1 is able to bind directly to membranes through separate N-terminal and C-terminal membrane-binding regions. Using a purified human full-length ATG12-5–16L1 complex, we show that both membrane interaction regions are required for efficient lipidation of LC3 in vitro. Interestingly, while the N-terminal membrane-binding region is essential for LC3B lipidation under all conditions tested in vivo, the β-isoform-specific C-terminal region is required for VPS34-independent LC3-lipidation on perturbed endosomes, but dispensable for canonical autophagy. We propose that distinct membrane-binding by ATG16L1 isoforms mechanistically distinguish LC3-lipidation on different intracellular membrane compartments.
Results
ATG16L1 has two membrane binding regions
As the ATG12-5–16L1 complex supports LC3 lipidation at both WIPI2b-positive11 and -negative compartments14,15, we speculated that it can associate with membrane through a direct interaction with lipids. To unravel potential membrane binding site(s) in ATG16L1, N- and C-terminal deletions were tested in a liposome co‐sedimentation assay. Whereas the first 28 aa, required for ATG5 interaction (Fig. 1a), were dispensable for membrane binding, aa 28–44 were required for binding to liposomes (Fig. 1b). These amino acids constitute a highly conserved amphipathic α-helix (denoted helix2) (Fig. 1c). Mutation of one or more residues on the hydrophobic face of helix2 (F32, I35 and I36) to alanine reduced membrane binding (Supplementary Fig. 1a,b), while the triple mutant F32A/I35A/I36A (referred to as FII) lost all membrane interaction (Fig. 1d). Helix2 was found to associate with ATG5 in two crystal structures of ATG12-5–ATG16L117,18. The hydrophobic face of helix2 interacted as a homodimer in both structures, indicating that helix2 can accommodate other interactions than the one with ATG5. We propose that in close proximity to membrane, a slight change in the torsion angle of the hinge between helix1 and helix2 (Q30, A31) leads to exposure and membrane insertion of the non-polar side chains of helix2 (Supplementary Fig. 1c). As expected, the FII mutant retained full interaction with ATG5 (Supplementary Fig. 1d,g).
Fig. 1: Identification of two membrane binding regions in ATG16L1.

a, Schematic overview of ATG16L1 isoforms, domains and protein interaction sites. b, Co-sedimentation assay of recombinant ATG16L1 aa 28–229 and aa 44–229 with liposomes containing 20% (w/w) Brain PC, 40% (w/w) Brain PS and 40% (w/w) Brain PE (See also Supplementary Figure 1a, b). S: supernatant, P: pellet. Coomassie stained gels are representative of n=3 independent experiments. c, Sequence alignment of ATG16L1 from different vertebrate species corresponding to aa 29–46 (Helix2) of human ATG16L1. Φ indicates conserved hydrophobic residues. A helical wheel representation of Helix2. Color coding: yellow, hydrophobic; purple, serine and threonine; blue, basic; red, acidic; pink, asparagine and glutamine; grey, alanine and glycine. d, Binding of recombinant ATG16L1 aa 28–229 WT or F32A/I35A/I36A (FII mut) to liposomes as described in (b) (See also Supplementary Fig. 1a, b). Coomassie stained gels are representative of n=3 independent experiments. e, Illustration of C-terminal ATG16L1 deletion mutants used in the liposome sedimentation assay as described in (b). Liposome binding of the various recombinant proteins was quantified as the percentage of the total protein found in the pellet fraction (See also Supplementary Figure 1e,f). Data is presented as mean±SEM from n=4 independent experiments. Statistical analyses were performed by Two-way Anova followed by Bonferonis multiple comparison test. f, Illustration of the ATG16L1 isoforms and the C-terminal membrane binding mutants used for the co-sedimentation assay. Stars above the sequences denote identical amino acids in the species indicated in (c). Liposome binding was quantified as percent of total protein in the pellet. Data is presented as mean±SEM from n=3 independent experiments. Unprocessed gels are shown in Supplementary Figure 4. Numerical source data can be found in Source data Suppl. Table 1.
Truncation analysis also revealed a more C-terminal membrane-binding activity, comprising two short motifs in the region spanning amino acids 266–319 (Fig. 1e and Supplementary Fig. 1e,f). One (aa 266–284) encompasses the distinguishing feature between the α and β isoforms of ATG16L1 (present only in the β isoform) and when deleted (319α) reduced liposome binding. Deletion of aa 306–319 in the β isoform (306β) also reduced liposome binding. Indeed, combining only the deletion of aa 266–284 (hereafter α) with a triple alanine mutation in aa 306–319 (V308A,R309A,V310A, hereafter VRV) was sufficient to completely abolish membrane binding (Fig. 1f). As expected, this αVRV double mutant retained full interaction with ATG5 in cells (Supplementary Fig. 1g). Taken together, our data show that ATG16L1 can bind directly to membranes through specific motifs in both the N-and C-terminal parts of the protein, suggesting a mechanism whereby close membrane proximity could be achieved for the ATG12–5 conjugate.
Purification of full-length human ATG12-5–16L1 complex for in vitro assays
To clarify whether membrane binding of ATG16L1 is important for lipidation of LC3/GABARAP proteins, the full-length human ATG12-5–16L1 complex was purified and used for in vitro lipidation assays. Successful production of active full-length human ATG12-5–16L1 complex was achieved in HEK293-F suspension cells (Supplementary Fig. 2a) and required co-expression of exogenous ATG10, which was otherwise limiting (Fig. 2a). Importantly, by using SUMOstar tag fusion proteins in the final purification procedure, all residues upstream of the desired product were removed by the SUMOstar protease, resulting in proteins that resembled endogenous proteins (Supplementary Table 1). The final purified product eluted as a single complex of ~600 kDa by size exclusion chromatography (Fig. 2b), in line with what was previously described to be a dimer of the complex19. We also purified a complex containing ATG16L1 lacking the entire C-terminal region (ATG16L1 aa 1-249) with and without the FII mutation and found that only the complex containing WT ATG16L1 (aa 1-249) retained association with liposomes when incubated with sonicated liposomes and subjected to liposome floatation analysis, confirming the relevance of helix2 for membrane binding even in the presence of ATG12-5 (Fig. 2c,d).
Fig. 2: The ATG12-5–16L1 complex is required for in vitro LC3B lipidation.
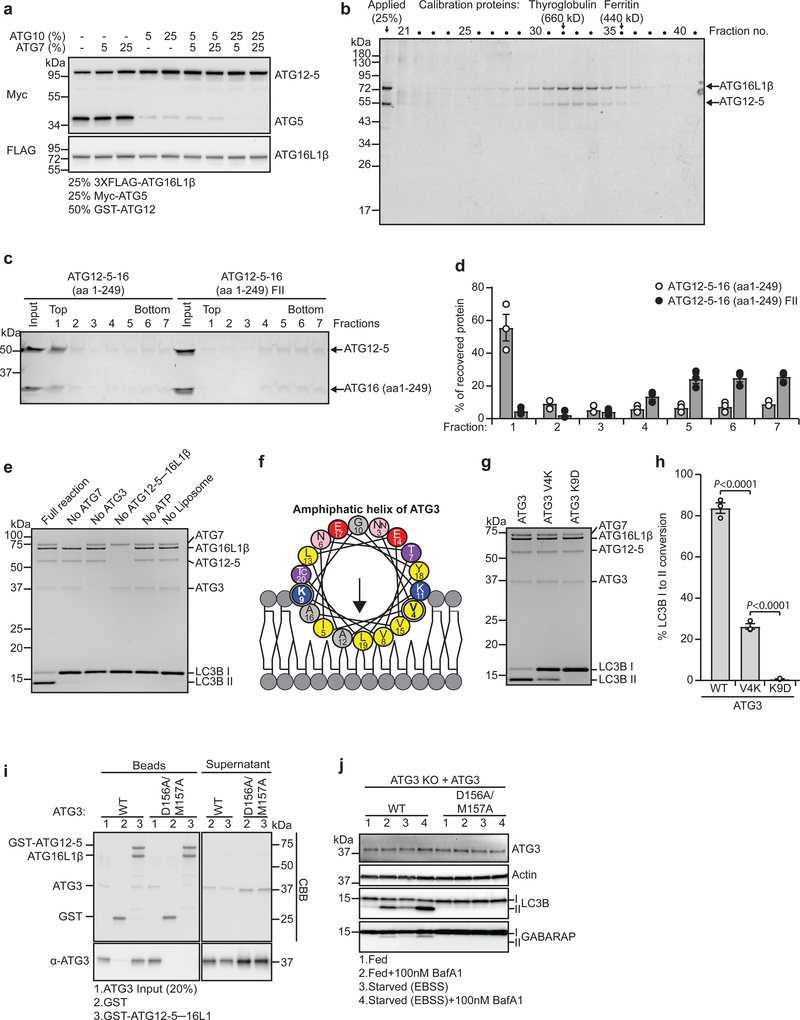
a, 3 ml HEK-F cell cultures were transfected with 1 μg plasmid/1×106 cells (25% myc-ATG5: 50% GST-ATG12: 25% 3XFLAG-ATG16L1), with or without untagged ATG7 and ATG10 (% extra plasmid). Cell lysate immunoblot is representative of n=3 independent experiments. b, Size-exclusion chromatography fractions of purified ATG12-5–16L1 (Supplementary Figure 2a) are visualized by Coomassie blue stain (molecular mass standards are indicated) (n=1 experiment). c, Liposome floatation assay using sonicated liposomes (69.5 mol% POPC, 30 mol% DOPS, 0.5 mol% DOPE-Rhod) incubated with either ATG12–5–16 (aa 1–249) WT or F32A/I35A/I36A mutant protein complex. Input sample and fractions (top fraction contains liposomes) were subjected to SDS–PAGE and Coomassie blue stain. d, Quantification of protein recovery in fractions from (c) based on n=3 independent experiments presented as mean±SEM. e, In vitro lipidation reaction containing 0.5 μM ATG7, 1 μM ATG3, 0.25 μM ATG12-5–16L1β, 10 μM LC3B, 3 mM lipid (sonicated liposomes: 10 mol% bl-PI, 50 mol% DOPE and 40 mol% POPC), 1 mM dithiothreitol and 1 mM ATP were run at 30°C for 90 min. Reactions were subjected to SDS–PAGE and Coomassie blue stain (n=1 experiment). f, Helical wheel representation of the ATG3 amphipathic helix. Color coding as in Fig.1c. g, In vitro LC3B lipidation reactions as described in (e-full reaction) were run with the indicated ATG3 mutants. h, Quantification of LC3B-II formation in (g) plotted as percentage of total LC3B based on n=3 independent experiments presented as mean±SEM. Statistical analyses were performed by One-way Anova followed by Bonferonis multiple comparison test. i, ATG3 WT or D156A/M157A mutant were incubated with either GST or GST-ATG12-5–16L1β immobilized on glutathione sepharose beads. ATG3 input, pulled-down proteins on beads and supernatant were subjected to SDS-PAGE and Coomassie blue staining or immunoblotting against ATG3. (n=1 experiment) j, LC3B/GABARAP lipidation in ATG3 KO HEK293 cells, rescued with ATG3 WT or D156A/M157A mutant, treated for 2 h as indicated. Cell lysates were immunoblotted against the indicated proteins. (n=1 experiment). Unprocessed immunoblots and gels are shown in Supplementary Figure 4. Numerical source data can be found in Source data Suppl. Table 1.
The ATG12-5–16L1β complex is required for vitro LC3B lipidation
To assess the functionality of the purified ATG12-5–16L1β complex, lipidation of LC3B was reconstituted in vitro and analyzed by quantification of LC3B-I to LC3B-II conversion. As expected, the reaction displayed an absolute requirement for ATG7, ATG3, ATP and liposomes (Fig. 2e). Importantly, the ATG12-5–16L1β complex was also required for efficient LC3B lipidation (Fig. 2e). The N-terminal membrane binding amphipathic helix of ATG310 was essential for its function in LC3B lipidation, even in the presence of ATG12–5─16L1β (Fig. 2f–h). Moreover, an interaction between ATG3 and ATG12 (Fig. 2i), previously found to be important for LC3B lipidation7, was required for lipidation of both LC3B and GABARAP in vivo (Fig. 2j). Thus, ATG3 activity depends upon both its own membrane-binding helix and direct interaction with the ATG12-5–16L1 complex.
ATG16L1 membrane binding is required for efficient in vitro lipidation
To test the importance of the ATG16L1 membrane binding domains in lipidation, ATG125–16L1 complexes containing WT or mutated (β FII, αVRV or α FII VRV) ATG16L1 were purified from HEK293-F cells (Supplementary Fig. 2a). The ability of WT and mutant ATG16L1 proteins to support lipidation was evaluated in the context of both LC3B and GABARAP proteins, using liposomes of varying PE content (50mol%PE or 20mol%PE) and curvature (sonicated ~25–65nm or extruded ~400nm) (Fig. 3). In all cases, lipidation efficiency was highest with strong curvature (sonication) and high PE. On such membranes, nearly all LC3B was converted to LC3B-II when ATG12-5–16L1β was present and this was strongly reduced by the FII mutant (Fig. 3a,c). In contrast, the αVRV mutant was only mildly inhibitory. Intriguingly, on flatter (extruded) membranes where LC3B lipidation was less efficient, mutations at either site were inhibitory (Fig. 3a–c). Purified ATG12–5 was used as a control and did not support lipidation under any condition tested.
Fig. 3: ATG16L1 membrane binding is required for efficient in vitro LC3 and GABARAP lipidation.

a, In vitro lipidation reactions containing 0.5 μM ATG7, 1 μM ATG3, 0.5 μM ATG12–5 or 0.25 μM ATG12-5–16L1β/β FII/α VRV/α FII VRV, 10 μM LC3B, 3 mM lipid (liposomes composed of 10 mol% bl-PI, 50 mol% DOPE and 40 mol% POPC were sonicated or extruded to 400 nm), 1 mM dithiothreitol and 1 mM ATP were incubated at 30 °C for 90 min. Reactions were subjected to SDS–PAGE and Coomassie blue stain. b, Lipidation reactions were performed as in (a) but using sonicated or extruded liposomes composed of 10 mol% b1PI, 20 mol% DOPE and 70 mol% POPC. c, The extent of LC3B-II formation in (a) and (b) was determined and plotted as percentage of total LC3B based on n=3 independent experiments and presented as mean±SEM. Statistical analyses was performed by Two-way Anova followed by Bonferonis multiple comparison test. d, In vitro lipidation reactions containing 0.5 μM ATG7, 1 μM ATG3, 0.2 μM ATG12–5 or 0.1 μM ATG12-5–16L1β/β FII/α VRV/α FII VRV, 10 μM GABARAP, 3 mM lipid (liposomes composed of 10 mol% bl-PI, 50 mol% DOPE and 40 mol% POPC were sonicated or extruded to 400 nm), 1 mM dithiothreitol and 1 mM ATP were incubated at 30 °C for 90 min. Reactions were subjected to SDS–PAGE and Coomassie blue stain. e, Lipidation reactions were performed as in (d) but using sonicated or extruded liposomes composed of 10 mol% bl-PI, 20 mol% DOPE and 70 mol% POPC. Note the presence of accumulated GABARAP-AMP. f, The extent of GABARAP-II formation in (d) and (e) was determined and plotted as percentage of total GABARAP based on n=2 independent experiments. Note that the proteins containing the αVRV mutation co-purify with an additional protein* of 70kDa. Unprocessed gels are shown in Supplementary Figure 4. Numerical source data can be found in Source data Suppl. Table 1.
Interestingly, in vitro lipidation of GABARAP displayed a reduced dependence of ATG12-5–16L1, showing some lipidation on sonicated liposomes even in the absence of ATG12-5–16L1 (Fig. 3d–f). In contrast, GABARAP lipidation on liposomes with low curvature and high PE required the ATG12-5–16L1 complex and was reduced with the FII or αVRV mutant (Fig. 3d,f). No GABARAP lipidation was observed using low curvature liposomes with low PE content (Fig. 3e,f). Similar results were obtained when using GABARAPL1 and GABARAPL2 (Supplementary Fig. 2b). Together, our results show that LC3B lipidation generally require the N-terminal membrane-binding motif of ATG16L1, while the C-terminal motif is dispensable on highly curved membranes, but becomes important when membrane curvature is low.
The ATG16L1 N-terminal membrane-binding domain is required for non-selective and selective types of autophagy
The importance of ATG16L1 membrane binding for its function in autophagy was analyzed in HEK293 cells depleted of both α and β isoforms of ATG16L1 (ATG16L1 knock-out (KO)) and transduced with lentivirus for stable expression of ATG16L1β WT, FII mutant, aa 1–249 or the α VRV mutant (Fig. 4a and Supplementary Fig. 2d). The cells were incubated in rich medium (fed) or EBSS (starve) in the absence or presence of the lysosomal inhibitor Bafilomycin A1 (Baf.A1), followed by immunoblotting to measure autophagic flux. As expected, LC3B and GABARAP lipidation, as well as degradation of long-lived proteins, was nearly abolished in ATG16L1 KO cells and could be efficiently rescued in cells expressing ATG16L1β WT, aa 1–249 or ATG16L1α VRV, but not in cells expressing the ATG16L1β FII mutant (Fig. 4a–c and Supplementary Fig. 2c–e).
Fig. 4: Membrane binding of ATG16L1 in autophagy and during lipidation on perturbed endosomes reveals isoform specific functions.

a, LC3B/GABARAP lipidation in WT and ATG16L1 KO HEK293 cells rescued or not with ATG16L1β, ATG16L1β F32A/I35A/I36A (β FII) or ATG16L1 (aa 1–249), treated for 2 h as indicated. Cell lysates were immunoblotted against the indicated proteins. b, Levels of LC3B-II/GAPDH quantified from immunoblots in (a) and normalized to fed WT cells. c, Degradation of long-lived proteins in HEK293 WT and in ATG16L1 KO cells rescued or not with ATG16L1β, ATG16L1β F32A/I35A/I36A (β FII) or ATG16L1 (aa 1–249) was quantified based on the Baf.A1 sensitive release of 14C-valine after 4 h in indicated conditions. d, Confocal images of Zymosan-Alexa594-containing phagosomes counter-stained for LC3B in ATG16L1 KO RAW264.7 cells rescued or not with ATG16L1β, ATG16L1β F32A/I35A/I36A (β FII) or ATG16L1α V308A/R309A/V310A (α VRV). Scale bars: 10μm. The images are representative of n=2 independent experiments. e, LC3B lipidation in ATG16L1 KO HEK293 cells rescued or not with ATG16L1β, ATG16L1α V308A/R309A/V310A (α VRV) or ATG16L1 (aa 1–249). Cells were treated or not with monensin for 1 h or EBSS for 2 h in the presence or absence of Baf.A1. Co-treatment with VPS34 inhibitor VPS34IN1 and ULK1/2 inhibitor MRT68921 was performed where indicated. Cell lysates were immunoblotted against the indicated proteins. f, The levels of LC3B-II/GAPDH were quantified from immunoblots in (e) and normalized to cells rescued with ATG16L1β in the starved condition. g, LC3B lipidation in ATG16L1 KO HEK293 cells rescued or not with ATG16L1β, ATG16L1α, ATG16L1β V308A/R309A/V310A (β VRV), ATG16L1α V308A/R309A/V310A (α VRV) or ATG16L1 (aa 1–249), treated as indicated for 2h. Cell lysates were immunoblotted against the indicated proteins. h, The levels of LC3BII/GAPDH were quantified from immunoblots in (g) and normalized to cells rescued with ATG16L1β WT. All quantification in this figure are presented as mean±SEM from n=3 independent experiments. Statistical analyses were performed by Two-way Anova followed by Bonferonis multiple comparison test. Note the presence of a degradation product* upon expression of exogenous ATG16L1. Unprocessed immunoblots are shown in Supplementary Figure 4. Numerical source data can be found in Source data Suppl. Table 1.
ATG16L1 helix 2 membrane binding was also required for selective autophagy of mitochondria, as demonstrated by induction of mitophagy with the iron-chelator deferiprone (DFP) in U2OS cells with inducible expression of a mitochondrial matrix localized EGFP-mCHERRY fusion protein, which can be visualized as red-only puncta in lysosomes (where EGFP is quenched) (Supplementary Fig. 2f,g). As for starvation-induced autophagy, the ATG16L1α VRV mutant and ATG16L1 (aa 1–249) could rescue mitophagy, while the FII mutant could not (Supplementary Fig. 2g).
Thus, membrane binding of ATG16L1 via helix2 is required for the function of the ATG12-5–16L1 complex in starvation-induced lipidation of LC3B/GABARAP and for autophagic degradation of long-lived proteins and damaged mitochondria. The C-terminal membrane binding domain is however dispensable for these types of autophagy.
Distinct roles of ATG16L1 isoforms in LAP and LC3 lipidation on perturbed endosomes
Conjugation of LC3 to single-membrane structures, including perturbed endosomes and phagosomes in a process referred to as LC3-associated phagocytosis (LAP)22,23, has been found to require the C-terminal part of ATG16L1 and we therefore set out to investigate whether ATG16L1 membrane binding is required for these processes15,24.
The mouse macrophage cell line RAW264.7 was depleted of ATG16L1 and rescued with ATG16L1β WT or various ATG16L1 mutants (Supplementary Fig. 3a). Recruitment of endogenous LC3B to Alexa594 positive compartments after phagocytosis of Alexa594-labeled Zymosan beads was blocked in ATG16L1 KO cells and in KO cells expressing the ATG16L1β FII mutant, but readily observed in KO cells expressing ATG16L1β WT and the ATG16L1α VRV mutant (Fig. 4d). In line with a recent report15, the N-terminal ATG16L1 protein (aa 1–249) did not rescue LC3B lipidation onto Zymosan positive compartments (Supplementary Fig. 3b).
To investigate a possible role of ATG16L1 membrane binding in conjugation of LC3 to perturbed endosomes, ATG16L1 KO HEK293 cells expressing different ATG16L1 mutants were co-treated with the sodium/proton ionophore monensin and inhibitors targeting VPS34 (VPS34IN1) and ULK1/2 (MRT68921), as this is a WIPI2b and ULK1/2 independent process15,24. Monensin-induced LC3B lipidation was blocked in ATG16L1 KO cells and could be rescued with WT ATG16L1β (Fig. 4e–h). As expected15, monensin-induced LC3B lipidation was significantly reduced in cells lacking the C-terminal part of ATG16L1 (aa 1–249). Most interestingly, the ATG16L1αVRV mutant was not able to rescue monensin-induced LC3B lipidation, although starvation-induced LC3 lipidation was fully rescued (Fig. 4e,f). Similar results were obtained when cells were treated with chloroquine or ammonium chloride in addition to VPS34 and ULK1/2 inhibitors (Supplementary Fig. 3c). Remarkably, monensin-induced LC3B lipidation was strongly inhibited in cells expressing ATG16L1α, at comparable levels to ATG16L1αVRV, while the ATG16L1 VRV mutant alone had no effect (Fig. 4g,h). Thus, our data identify a unique functional difference of the ATG16L1 α and β isoforms; while both isoforms can support LC3B lipidation during autophagy and LAP, only the β isoform is able to facilitate VPS34-independent LC3 lipidation on perturbed endosomes.
ATG16L1 membrane binding is not required for recruitment to WIPI2b puncta
Given that WIPI2b facilitates recruitment of ATG12-5–16L1 for LC3 lipidation during starvation-induced autophagy11 we speculated that ATG16L1 membrane binding may be important for its recruitment and/or stabilization at WIPI2b positive structures. Colocalization analysis of WT and mutant EGFP-ATG16L1 with WIPI2b in HEK293 ATG16L1 KO and rescue cells showed that both EGFP-ATG16L1β WT and the two membrane-binding deficient mutants (β FII and α) localized to WIPI2 positive structures upon starvation (Fig. 5a). Interestingly, there were no overall differences in the area or number of ATG16L1 or WIPI2 puncta formed or in their colocalization (Fig. 5b–f). In contrast, both the area of LC3B positive structures and the extent of LC3B lipidation were reduced in cells expressing the EGFP-ATG16L1β FII mutant compared to cells expressing EGFP-ATG16L1β WT or EGFP-ATG16L1α (Fig. 5a,g and Supplementary Fig. 3d). Thus, the N-terminal membrane-binding site of ATG16L1 is required to support lipidation of LC3B, but is dispensable for recruitment to WIPI2 positive puncta.
Fig. 5: ATG16L1 membrane binding is not required for recruitment to WIPI2b puncta.
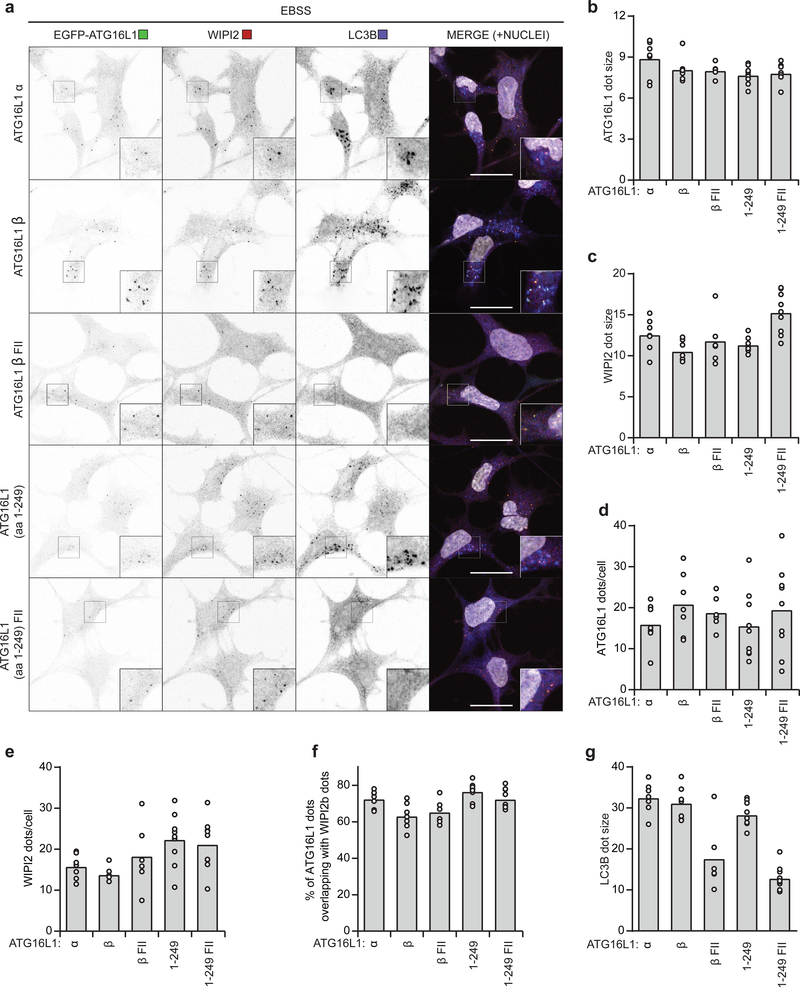
a, Maximum projections of ATG16L1 KO HEK293 cells rescued with EGFP-ATG16L1α, EGFP-ATG16L1β, EGFP-ATG16L1β F32A/I35A/I36A (β FII), EGFP-ATG16L1 (aa 1–249) or EGFP-ATG16L1 (aa 1–249) F32A/I35A/I36A and starved for 1h in EBSS before fixation and immunostaining for WIPI2 and LC3B. Nuclei were counterstained with Hoechst 33342. Scale bars: 20μm. ATG16L1 (b) and WIPI2 (c) dot size (represented as average dot area in pixels) as well as number of ATG16L1 (d) and WIPI2 (e) dots per cell and %ATG16L1 dots overlapping with WIPI2 dots (f) and LC3B (g) dot size in (a) were quantified by automated analysis using CellProfiler (individual data points correspond to single images analyzed from n=2 independent experiments, corresponding to a total of 56 (α), 68 (β), 39 (β-FII), 81 (aa 1249) and 67 (aa 1–249 FII) cells). Numerical source data can be found in Source data Suppl. Table 1.
The ATG16L1 C-terminal end can compensate for WIPI2b function
As the C-terminal membrane-binding region of ATG16L1 was dispensable for starvation-induced and selective autophagy, but important for LC3B lipidation on flat membranes in vitro (Fig. 3) and for WIPI2b/VPS34-independent LC3B lipidation onto perturbed endosomes (Fig. 4e–h and Supplementary figure 3c), we speculated that WIPI2b might substitute the function of this domain under specific conditions. To address this, we performed in vitro LC3B lipidation using liposomes with or without PI(3)P, in the absence or presence of purified WIPI2b WT protein or the ATG16L1 binding deficient WIPI2 mutant (R108E/R125E)11 (Fig. 6a–c). Addition of PI(3)P stimulated LC3B lipidation using the WT ATG12-5–16L1 complex, but did not rescue the lipidation defects seen with the C-terminal membrane-binding mutant of ATG16L1 (Fig. 6b,c). Most interestingly, WIPI2b further stimulated LC3B lipidation in the presence of ATG16L1 WT (β) and the α VRV mutant, but had no effect in the absence of PI(3)P. The WIPI2 R108E/R125E mutant was unable to promote LC3B lipidation under any condition (Fig. 6b,c). Together, these data indicate that WIPI2b binding overlaps in function with the C-terminal membrane-binding domain, providing an explanation to why ATG16L1 αVRV and ATG16L1 (aa 1–249) can rescue starvation-induced LC3B lipidation on WIPI2 positive membranes.
Fig. 6: The C-terminal end of ATG16L1 can compensate for WIPI2b function.

a, Size-exclusion chromatography fractions of purified WIPI2b visualized by Coomassie blue stain (molecular mass standard is indicated). (n=2 independent experiments). b, In vitro lipidation reactions containing 0.5 μM ATG7, 1 μM ATG3, 0.1 μM ATG12-5–16L1β or ATG12-5–16L1α VRV, 10 μM LC3B, 0.3 mM lipid (liposomes composed of 10 mol% bl-PI, 50 mol% DOPE, with/without 10 mol% PI(3)P and 40/30 mol% POPC extruded to 400nm), 1 mM dithiothreitol and 1 mM ATP were incubated at 30°C for 90 min in the absence or presence of 1 μM WIPI2b or WIPI2b R108E/R125E (EE). Reactions were subjected to SDS–PAGE and Coomassie blue stain. c, The extent of LC3B-II formation in (b) was determined and plotted as percentage of total LC3B from n=3 independent experiments and presented as mean±SEM. Statistical analyses were performed by Two-way Anova followed by Bonferonis multiple comparison test. d, LC3B/GABARAP lipidation in WT, WIPI2 KO, ATG16L1 KO or WIPI2/ATG16L1 double KO HEK293 cells rescued or not with EGFP-tagged ATG16L1 (aa 1–249), ATG16L1 (aa 1–249) F32A/I35A/I36A, ATG16L1α or ATG16L1β, treated as indicated for 2h. Cell lysates were immunoblotted against the indicated proteins. e, The levels of LC3B-II/Actin were quantified from immunoblots in (d) and normalized to WT cells in the starvation with Baf.A1 condition. Data is based on n=3 independent experiments and presented as mean±SEM. Statistical analyses were performed by Two-way Anova followed by Bonferonis multiple comparison test. f, LC3B/GABARAP lipidation in ATG16L1 KO or WIPI2/ATG16L1 double KO HEK293 cells rescued with EGFP-ATG16L1α or EGFP-ATG16L1β, treated as indicated for 2h. Cell lysates were immunoblotted against the indicated proteins (n=1 experiment). g, LC3B/GABARAP lipidation in WT and WIPI2/ATG16L1 double KO HEK293 cells, rescued or not with EGFP-ATG16L1 (aa 1–249), mCHERRY-WIPI2b WT, mCHERRY-WIPI2b FTTG (TT) or mCHERRY-WIPI2b R108E/R125E (EE), treated as indicated for 2h. Cell lysates were immunoblotted against indicated proteins. (n=1 experiment). Unprocessed immunoblots and gels are shown in Supplementary Figure 4. Numerical source data can be found in Source data Suppl. Table 1.
To further corroborate this finding, ATG16L1/WIPI2b double KO (DKO) cells were rescued with various ATG16L1 proteins. Interestingly, while ATG16L1 (aa1–249) rescued starvation-induced lipidation in ATG16L1 KO cells, it did not sustain LC3B/GABARAP lipidation in the DKO cells (Fig. 6d,e). In contrast, both full length ATG16L1 α and β were able to sustain LC3B /GABARAP lipidation in the absence of WIPI2b, indicating that the C-terminal part of ATG16L1 can overcome the lack of WIPI2b during starvation-induced autophagy. This might also explain why single WIPI2b KO has little or no effect on LC3B/GABARAP lipidation during starvation-induced autophagy (Fig. 6d,e). Most interestingly, the ability of full length ATG16L1 to rescue lipidation in DKO cells was VPS34-dependent (Fig. 6f), indicating that ATG16L1 can bind PI(3)P or that its C-terminus binds to another PI(3)P effector protein. Importantly, the LC3/GABARAP lipidation defect in DKO cells expressing ATG16L1 aa1–249 could be rescued with WIPI2b WT, but not with the PI(3)P (TT) or ATG16L1 (EE) binding deficient mutants (Fig. 6g).
The ATG16L1 β-specific membrane-binding domain is required for its activity at perturbed endosomes
To further assess the function of the C-terminal membrane-binding domain of ATG16L1, ATG16L1 KO HEK293 cells expressing mTAG2BFP-LC3B were rescued with either EGFP-ATG16L1α or -β (Fig. 7a–c). Live cell imaging of cells treated with a combination of monensin and VPS34IN1 showed an accumulation of large vesicular structures that were positive for EGFP-ATG16L1β, but not for EGFP-ATG16L1α (Fig.7a–c). The enlarged EGFP-ATG16L1β positive vesicles were also positive for BFP-LC3B, whereas little or no BFP-LC3B staining was detected in the EGFP-ATG16L1α cells (Fig. 7a–c). Likewise, while EGFP-ATG16L1β could restore LC3B lipidation under these conditions, this was not the case for EGFP-ATG16L1α, the β FII mutant or the previously described β F467A mutant15 (Fig. 7d,e). As expected WIPI2 KO did not prevent LC3 lipidation on perturbed endosomes (Fig. 7f). In line with a differential membrane binding capacity of the ATG16L1 α and β isoforms, the β isoform was enriched in the membrane fraction after a crude cytosol/membrane fractionation (Fig. 7g,h)
Fig. 7: The β-specific sequence in ATG16L1 is required for recruitment and LC3B lipidation on perturbed endosomes.

a, Confocal images of ATG16L1 KO HEK293 cells expressing mTAG2BFP-LC3B in addition to being rescued with EGFP-ATG16L1α or EGFP-ATG16L1β treated for 1h as indicated before live cell imaging was performed. Scale bars: 20μm. The % of enlarged vesicles in (a) positive for ATG16L1 (b) and LC3B (c) was quantified (individual data points represent single images analyzed from n=2 independent experiments, corresponding to a total of 111 and 75 cells for EGFP-ATG16L1α and EGFP-ATG16L1β, respectively). d, LC3B lipidation in ATG16L1 KO HEK293 cells rescued or not with EGFP-ATG16L1α, ATG16L1β, ATG16L1β F32A/I35A/I36A (β FII) or ATG16L1β F467A, treated for 1h as indicated. Cell lysates were immunoblotted against the indicated proteins. e, The levels of LC3B-II/Actin were quantified from immunoblots in (d) and normalized to the rescue with EGFPATG16L1α cells. The data is presented as mean±SEM from n=3 independent experiments. Statistical analyses were performed by Two-way Anova followed by Bonferonis multiple comparison test. f, WT and WIPI2 KO HEK293 cells were treated for 1 h as indicated. Cell lysates were immunoblotted against the indicated proteins. The immunoblots are representative of n=3 independent experiments. g, HeLa cells were subjected to fractionation of cytosol and membrane followed by Western blotting. h, The ratio between isoforms in cytosol/membrane fractions was quantified from immunoblot in (g) from n=2 independent experiments. i, Schematic representation of ATG16L1 domains and interactions facilitating LC3B-lipidation and membrane targeting during canonical autophagy and upon perturbation of endosomes. Unprocessed immunoblots are shown in Supplementary Figure 4. Numerical source data can be found in Source data Suppl. Table 1.
Discussion
In this study we have identified two membrane-binding regions in ATG16L1 with distinct functions in lipidation of LC3B and GABARAP proteins. Our data show that an N-terminal membrane-binding amphipathic helix (helix2) in ATG16L1 is important for LC3B or GABARAP lipidation under all conditions tested in vitro and in vivo, including non-selective and selective types of autophagy, as well as LAP and endosome perturbation. In contrast, the C-terminal β isoform-specific membrane-binding region is dispensable for canonical autophagy, but is required for ATG16L1 membrane recruitment and LC3B lipidation on perturbed endosomes in cells treated with monensin and the VPS34 inhibitor VPS34IN1 (Fig. 7i). Thus, we propose that distinct membrane binding by the two isoforms of ATG16L1 mechanistically distinguish lipidation of LC3 and GABARAP proteins at distinct cellular locations and/or under different cellular conditions.
The transfer of LC3 and GABARAP proteins from ATG7 via ATG3 to PE in the membrane necessitates close contact between the components and accessibility to the lipid head groups. Using a purified full length human ATG12-5–16L1 complex for in vitro lipidation assays, we show that ATG12–16L1 is essential for ATG3 function. In line with previous findings10, the N-terminal amphipathic helix of ATG3 is also required for efficient in vitro LC3B lipidation. Although ATG16L1 helix2 enables membrane binding, it is not required for targeting and localization of the ATG12-5–16L1 complex to WIPI2b positive structures. We therefore propose that the main function of ATG16L1 helix2 is to provide close proximity contact of the complex and ATG3 with the membrane, promoting membrane insertion of ATG3’s amphipathic helix and subsequent lipidation of LC3B/GABARAP proteins. It is also possible that helix embedment in the outer layer of a bilayer membrane produces local membrane distortions26, thus promoting binding of the ATG3 amphipathic helix. It was recently reported that incubation of yeast Atg12–5–16 with giant liposomes caused Atg16-dependent deformations of the membrane25, suggesting that yeast Atg16 may contain a similar amphipathic helix. It has also been reported that Atg5 may facilitate membrane binding of the Atg12–5–16 complex in yeast27. Interestingly, while Atg12–5 was unable to bind membrane, membrane binding was regained in the presence of Atg16, suggesting that Atg16 also contributes to membrane binding in yeast.
The C-terminal β-isoform-specific membrane-binding region of ATG16L1 was not required for starvation-induced LC3B/GABARAP-lipidation. This is in line with earlier studies showing that the C-terminus of ATG16L1 is dispensable for starvation-induced autophagy19 and consistent with the fact that yeast Atg16 lacks the C-terminal domains of human ATG16L128. Intriguingly, we found that the ability of the N-terminal ATG16L1 (aa 1–249) protein to mediate LC3B/GABARAP lipidation was dependent of the presence of WIPI2 and that the C-terminal part of ATG16L1 could support autophagic flux in the absence of WIPI2. Moreover, the inability of the C-terminal membrane binding mutant to mediate in vitro LC3B lipidation onto low-curvature liposomes was rescued by adding PI(3)P and WIPI2b to the reaction, indicating that ATG16L1 C-terminal membrane binding is redundant with WIPI2b in targeting the ATG12-5–16L1 complex to membranes. In support of such a model, we show that the ATG16L1 β isoform-specific membrane-binding sequence is required for LC3-lipidation onto endosomal membranes after their perturbation by lysosomotrophic agents or ionophores, in an ULK1/2 and VPS34-independent process, known to also require the WD40 domain of ATG16L115. The mechanisms involved in targeting of the ATG12-5–16L1 complex to damaged endosomes is not clear, but likely involves both C-terminal membrane binding and protein interactions (Fig. 7i). While both WIPI2b and FIP200 are reported to recruit the ATG12-5–16L1 complex to sites of autophagosome formation during starvation-induced autophagy11,29,30 other proteins may facilitate ATG12-5–16L1 complex targeting to perturbed endosomes, as the endosomal membrane proteins TMEM59 and TMEM166/EVA1, found to interact with the WD-repeat domain in ATG16L131,32. Moreover, ubiquitin binds to the WD-repeat domain of ATG16L1 to enhance the autophagic response to leaky endosomes during bacterial infection33. The C-terminus of ATG16L1 has likely evolved to permit additional functions in metazoans, such as defense against invading pathogens and LC3-decoration of non-autophagic low-curvature membranes, whereas LC3 lipidation on the phagophore is more promiscuous in this sense because the curvature and lipid composition is different. A third isoform of ATG16L1 expressed in mouse brain has been described (the γ-isoform)34 and possesses an even longer sequence (35 aa) at the site of the β-specific sequence, but its role in human is uncertain and it was not included in our study.
Interestingly, the C-terminal membrane binding region surrounds the Crohńs disease related mutation T300A and the Caspase 3 cleavage site (amino acids 296–299)35. Whether membrane binding somehow regulates accessibility of the caspase cleavage site remains to be investigated, but the region encompassing the C-terminal membrane binding domain is clearly a hub for regulation as it is the most heavily phosphorylated region in ATG16L1 (PhosphoSitePlus, https://www.phosphosite.org/).
In conclusion, we here identify two membrane-binding regions in ATG16L1 and unravel distinct functions of the α and β isoforms of ATG16L1 in LC3B lipidation. Our findings pave the way for a better understanding of the physiological functions of the two ATG16L1 isoforms and provide mechanistic insight into their role in canonical autophagy and other applications utilizing LC3/GABARAP lipidation.
Methods
Plasmid constructs
Plasmid constructs are listed in Supplementary Table 2 and construction details are available upon request. All plasmids were verified by DNA sequencing (GATC Biotech or MWG-Biotech AG).
Antibodies and reagents
Immunoblotting antibodies:
ATG16L1 mouse mAb (MBL, M150–3, 1:1000), ATG5 rabbit pAb (CST, 2630, 1:1000), ATG3 rabbit pAb (CST, 3415, 1:1000), Beta-ACTIN (8H10D10) mouse mAb (CST, 3700, 1:5000), GAPDH rabbit mAb (CST, 5174, 1:5000), GABARAP (E1J4E) rabbit mAb (CST, 13733, 1:1000), LC3B (D11) XP rabbit mAb (CST, 3868, 1:1000), WIPI2 mouse mAb [2A2] (Abcam, ab105459, 1:2000), FLAG rabbit pAb (Sigma, F7425, 1:1000), Myc rabbit pAb (CST, 2272, 1:1000). DyLight-680 and −800 conjugated secondary antibodies were obtained from Invitrogen (SA5–10170 and SA5–10044, 1:5000), Starbright Blue 700 secondary antibodies were obtained from Bio-Rad (12004158 and 12004161), HRP-conjugated antibodies were obtained from Jackson ImmunoResearch (115-035-003 and 111-035-144).
Immunofluorescence antibodies:
WIPI2 mouse mAb [2A2] (Abcam, ab105459, 1:2000), LC3B rabbit pAB (MBL, PM036, 1:500). Cy3, Cy5 and DyLight649-conjugated secondary antibodies were obtained from Jackson ImmunoResearch (Cy3: 715-165-151 and 711-165-152, Cy5: 711-175-152, DyLight649: 711-495-152, 1:500 for all).
Reagents:
All lipids were purchased pre-dissolved in chloroform, except for PI(3)P, from Avanti Polar Lipids. Lipids used in Fig. 1 and Supplementary Fig. 1 are: L-α-phosphatidylethanolamine (840022C) (BrainPE), L-α-phosphatidylserine (840032C) (Brain PS) and L-α-phosphatidylcholine (840053C) (Brain PC). Lipids used in Fig. 2, 3 and 6 are: 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (850725C) (DOPE), 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-(lissamine rhodamine B sulfonyl) (810150C) (Rhod-PE), 1palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (850457C) (POPC), 1,2-dioleoyl-sn-glycero3-phospho-(1’-myo-inositol-3’-phosphate) (850150P) (PI(3)P) and L-α-phosphatidylinositol (Liver, Bovine) (840042C) (PI). Adenosine-5’-triphosphate (A2383, Sigma) (ATP), Bafilomycin A1 (BML-CM110, AH diagnostics) (BafA1), Deferiprone (379409, Sigma) (DFP), Fibronectin (F2006–1MG, Sigma), Hoechst 33342 (H1399, Thermo Fisher Scientific), Hygromycin b (400052–20), Monensin sodium salt (M5273, Sigma), MRT68921 (S7949, Selleckchem), Puromycin dihydrochloride (P7255, Sigma), VPS34-IN1 (S7980, Selleckchem), Zeocin (45–0430, Invitrogen), Zymosan A particles (Alexa 594 Z23374 and Alexa 488 Z23373, Thermo Fischer Scientific).
Production of stable lentiviral transduced cell lines
WT or mutants of human ATG16L1 were subcloned into lentiviral vector pLENTI (PGK). Lentiviral packaging (psPAX2) and envelope (pCMV-VSV-G) plasmids (from Addgene) were used for lentivirus production in HEK293FT cells (Invitrogen). Briefly, 90% confluent HEK293FT cells in 10 cm plates were co-transfected with 5μg of the aforementioned plasmids (at 1:1:1 ratio) using XtremeGene9 (Roche). Medium was replaced the following day, and collected on days 2–4 after transfection. The virus-containing medium was centrifuged at 600 x g for 5 min to remove cellular debris and filtered through a 0.45 μm pore syringe filter. Viruses were 10x enriched using the Lenti-X concentrator reagent (Clontech) following the manufacturer’s instructions. For transduction, 10 cm plate with 30% confluent corresponding KO cells incubated with 10 ml medium plus 100 μl viral solution and 8 μg/m1 Polybrene. The following day, medium was replaced with new medium without Polybrene and selected with 2 μg/ml puromycin, 200 μg/ml zeocin or 100 μg/ml Hygromycin B for 1–4 days to generate stable polyclonal cell lines.
Purification of proteins for in vitro assays
Full-length ATG5–12–ATG16L1 complex was expressed and purified from HEK suspension cells (HEK-F, Invitrogen) in 200 ml scale. Cells were grown to 2–3 × 106 cells/ml on a shaker (160 rpm) at 37°C with 8% CO2 in 4 mM glutamine supplemented BalanCD medium (Irvine Scientific). A total of 1 μg per 1 × 106 cells of the following plasmids was mixed with a threefold excess (w/w) of polyethyleneimine “MAX” (40 kDa, Polysciences, Inc.) in 8 ml OptiPro (Invitrogen): pCMV-3xFLAG-SUMOstar-hATG16L1, pCMV-hATG5, pCMV-GST-hATG12, in amount ratio 1:2:2. To this was added 10% (w/w) of plasmid pCMV-hATG10, and the mixture was incubated for 20 min at room temperature before added to the cells. Cells were grown for three days with addition of 5% BalanCD Feed (Irvine Scientific) after 1 and 2 days. Cells were centrifuged at 350 × g for 5 min, washed with 30 ml PBS, and the cell pellet was lysed with 22 ml PBS containing 1% Nonidet P40 (Pierce), 1 mM EDTA, and cOmplete ULTRA protease inhibitors (Roche). After 15 min incubation on ice, lysed cells were centrifuged at 350 × g for 5 min and the supernatant was collected, snap frozen in liquid nitrogen, and stored at −80°C.
Lysate was thawed and centrifuged at 20.000 × g for 10 min and the supernatant was added to 3 ml of anti-FLAG (M2)-agarose (Sigma), and incubated 5h in the cold by end-to-end rotation. The gel matrix was transferred to a column and washed step-wise by at least 5 column volumes of NT350 (350 mM NaCl, 20 mM Tris-HCl, pH 7.4). The gel was resuspended in 1 ml NT350 to which was added 2μl SUMOstar protease (20 U, Life Sensors) and the closed column was incubated in the refrigerator overnight. Cleaved protein was eluted by step-wise 1-ml additions of NT350 and fractions with highest amount of protein were pooled and added to 0.5 ml of glutathione-Sepharose (GE Healthcare), equilibrated with NT350. After 5 h incubation by end-to-end rotation in the cold, the gel matrix was washed three times with 1 ml NT350 by centrifugation and resuspended in 1 ml NT350 with 20 μg GST-HRV 3C protease (produced in-house at 4 mg/ml by expression from a pGEX plasmid in E. coli). The gel was incubated at 4°C by end-to-end rotation overnight, pelleted and the cleaved complex was collected in the supernatant, snap-frozen in liquid nitrogen, and stored at −80°C.
A 50 cm column with Sepharose 4B was equilibrated with NT350 and calibrated with thyroglobulin (669 kDa) and ferritin (440 kDa) (GE Healthcare). Gel filtration was performed at a 6 ml/h flow rate, with 1 ml fractions collected.
Carboxyl-terminal fragments of ATG16L1 (used in Fig. 1e,f and Supplementary Fig. 1e) were produced from HEK-F cells similarly as described above (100-ml scale), except for plasmids lacking the SUMOstar encoding sequence. Elution from the M2-agarose was performed by step-wise addition of 1 ml portions of 0.1 mg/ml 3xFLAG peptide (Sigma) in NT350, and the pooled eluted fraction was used directly for liposome-binding experiments or stored at −80°C.
Full-length ATG7 and ATG3 were expressed and purified from HEK-F suspension cells in 100-ml scale by a similar procedure to that described for ATG12-5–16L1, with the second purification step omitted. ATG12–5 was purified from 100 ml HEK-F cells where GST-ATG12 was expressed together with 3XFLAG-SUMOstar-ATG5 to enable similar two-step purification as used to purify ATG12-5–16L1 complex.
Full-length human WIPI2b WT and mutant R108E/R125E were expressed with 3xFLAG-SUMOstar N-terminal tags in 200 ml scale HEK-F suspension cells, similarly as described above. After binding to anti-FLAG (M2)-agarose, beads were washed with NT350 and incubated overnight in the cold with 5 mM ATP in NT350 (to remove excess chaperone). Next day, the column was washed extensively with NT350 and incubated overnight with SUMOstar protease in NT350. WIPI2b was eluted with NT350, concentrated by VivaSpin 4 (10 kDa cut-off, Sartorius), and applied to a column (40 × 1 cm) of Sephacryl S-300HR equilibrated with NT350. Flow rate was 7 ml/h and 1 ml fractions were collected. Fractions with the highest protein amount were pooled, concentrated with VivaSpin, snap-frozen in liquid nitrogen, and stored at −80°C.
Amino-terminal fragments of ATG16L1 (used in Fig. 1b,d and Supplementary 1b) were produced in E. coli using plasmid pGEX-6P. Transformed E. coli (DE3) pLysS cells were grown in 250 ml LB medium at 37°C and expression was induced with 0.05 mM IPTG at OD 0.5. Cells were harvested after 3 h and lysed by freezing/thawing in NH150 (150 mM NaCl, 25 mM Hepes-OH, pH 7.4) containing protease inhibitors. After 50.000 × g centrifugation for 30 min, the supernatant was incubated 5 h with 0.5 ml glutathione-Sepharose and the gel matrix was transferred to a column and washed with 2 ml of 0.1% Nonidet P40 in NH150 and 10 ml of NT350. The gel was resuspended in 0.25 ml NT350 containing 30 μg GST-HRV 3C protease and the closed column was incubated overnight at 4°C. The column was eluted by step-wise additions of 0.25-ml portions of NT350 and fractions with highest amount of protein were pooled, snap-frozen in liquid nitrogen, and stored at −80°C.
To purify LC3B, GABARAPL1 and GABARAPL2, pGEX-2T plasmid containing the corresponding cDNA was transformed into BL21-Gold (DE3) E. coli. Cells were grown at 37°C to an OD of 0.6–0.8 before induction with 0.5mM IPTG. Cells were then grown for 3 additional hours before they were collected by centrifugation. Cells were resuspended in thrombin buffer (20mM Tris pH 7.6, 100mM NaCl, 5mM MgCl2, 2mM CaCl2, 1mM DTT) supplemented with a Roche Complete Protease inhibitor, lysed using a cell disruptor and cleared by centrifugation. The supernatant was incubated at 4°C with Glutathione Beads (Sigma) for 4 h. Beads were collected and washed twice with thrombin buffer before thrombin was added and allowed to cut at 4°C overnight. The next morning, protein fractions were collected and stored at −80°C in 20% glycerol.
GABARAP was expressed in BL21-pLysS E. coli cells from a pGEX-6P plasmid by induction at OD 0.5 with 0.1 mM IPTG at 37°C. Cells were harvested after 3 h, washed and distrupted by freeze/thaw in 50 mM Tris-HCl pH 7.4, 150 mM NaCl, 1 mM DTT (TND buffer) supplemented with 1 mM EDTA and Complete Protease inhibitor, centrifuged at 50.000 × g for 30 min, and the supernatant was applied to glutathione-Sepharose. After 2 h incubation, the beads were washed with TND buffer and incubated overnight with GST-HRV 3C protease. GABARAP was eluted with TND buffer and applied to a Sephacryl S-300 column (50 × 1 cm) equilibrated with TND buffer. Eluted fractions with the highest protein amount were snap-frozen in liquid nitrogen, and stored at −80°C
Liposome preparation
Lipids were mixed in the indicated compositions and dried to a thin film under N2-gas. The lipid film was further dried under vacuum for 1 hour. Lipids were reconstituted in NT350 buffer (350mM NaCl, 20mM Tris-HCl pH 7.4) and subjected to 7 cycles of flash-freezing in liquid nitrogen and thawing in a 37°C bath. Liposomes were then extruded 21 times through a polycarbonate membrane with a 400 nm pore size (Mini-Extruder, Avanti Polar Lipids). To obtain smaller and more highly curved liposomes extruded liposomes were treated with tip sonicator immediately prior to lipidation reaction. For liposome experiments in Fig. 1 and Supplementary Fig. 1, after hydration of dried lipids in NT350 buffer, liposomes were formed by 2–5 min bath-sonication and used within 2 days.
Protonation of PI(3)P
PI(3)P (100 μg powder) was dissolved in 100 μl chloroform, dried by a nitrogen stream, desiccated for 1h, and dissolved in 100 μl chloroform/50 μl methanol with 0.5 μl 1 M HCl. After 15 min incubation at room temperature, the sample was dried as above followed by two cycles of drying from 75 μl chloroform/25 μl methanol and 400 μl chloroform, respectively. Finally, the lipid was dissolved in 100 μl chloroform and stored under nitrogen at −20°C.
In vitro lipidation assay
Constructs used to prepare proteins for in vitro lipidation experiments are listed in Supplementary Table 3 along with the N-terminal residue resulting from their purification procedure. LC3B, GABARAP, GABARAPL1, or GABARAPL2 (10 μM), ATG3 (1 μM), ATG7 (0.5 μM), ATG12–5–16 (0.25 μM or 0.1 μM) and sonicated or extruded liposomes (3 mM) were mixed in NT350 buffer containing 1 mM DTT in a total volume of 8μl. Lipidation was initiated by adding 1 mM ATP and reactions were incubated at 30°C for 90 minutes. After completion, the lipidation reaction was subjected to SDS-PAGE and Coomassie Blue staining and analyzed with Image Lab (Biorad).
Liposome floatation assay
Liposome floatation assays were performed by incubating 0.1 μM protein with 2 mM total lipid and 1 mM DTT in NT350 buffer in a total volume of 150μl for 20 min at 25°C. To separate liposomes and bound protein from free protein, the mixture was subjected to a Nycodenz liposome floatation assay as in reference10. Gradients were centrifuged at 48,000 r.p.m. (280,000g) in a SW55Ti rotor (Beckman) for 4 h at 4°C and seven fractions were recovered, starting with the liposomes and bound protein extracted from the top 80 μl of the gradient. Fractions were subjected to SDS-PAGE and Coomassie Blue staining and analyzed with Image Lab (BioRad).
CRISPR/Cas9 knockout of human and mouse ATG16L1
ATG16L1 knockout was generated in HEK293, U2OS and Raw264.7 cells, using two independent sgRNA guides designed against the 5’ end of both human and mouse genes using the CRISPR Design Tool through MIT (crispr.mit.edu) (Supplementary Table 4). The sgRNA guides were synthesized (Sigma) and cloned into the pSpCas9 (BB)-2A-Puro (pX459) V2.0 plasmid (Addgene). Cells were transfected using XtremeGene9 (Roche) according to the manufacturer’s protocol. After 24 h, cells were treated with selection medium containing 2.0 μg/ml puromycin and fresh selection medium was provided every 2–3 days. After 7 days puromycin resistant cells were seeded as single cell/well in a 96-well plate through serial dilution. Individual clones were isolated and expanded before knockouts were confirmed via western blotting.
Cell fractionation
HeLa cells were harvested by trypsinization and resuspended in 2–5 volumes of KSHM buffer (100 mM potassium acetate, 85 mM sucrose, 20 mM HEPES-KOH, pH 7.4, and 1 mM magnesium acetate) containing 1 mM PMSF and protease inhibitor cocktail (EMD Millipore) before a quick freeze/thaw cycle to open up the plasma membrane36. Cracked cells were centrifuged at 1,500 g for 5 min, and the supernatant was further centrifuged at 70,000 g for 30 min where cytosol (supernatant) was collected. The pellets from the high- and low-speed centrifugations were combined and solubilized in KSHM containing 1% NP-40 and protease inhibitors, and centrifuged at 70,000 g for 30 min to obtain membranes (supernatant). Equal proportions of cytosol and membrane were analyzed by immunoblotting.
Long-lived protein degradation
Proteins were labelled with 0.125 μCi/ ml 14C-L-valine (Perkin Elmer) for 24 h in GIBCO-RPMI 1640 medium (Invitrogen) containing 10% FBS. Cells were washed and then chased for 16 h in nonradioactive Dulbecco’s modified Eagle’s medium (Invitrogen) containing 10% FBS and 10 mM valine (Sigma), to allow degradation of short-lived proteins. Cells were then washed twice with EBSS (Invitrogen), and treated with EBSS or complete medium containing 10 mM valine with or without 100 nm Bafilomycin A1 for 4 h (also in the presence of 10 mM valine). Then, medium was collected and mixed with 50% trichloroacetic acid followed by 2 h end-to-end rotation. Meanwhile the cells were treated with 0.2 M KOH and left to dissolve for 2h. Ultima Gold LSC cocktail (Perkin Elmer) was added to both medium and cell samples and protein degradation was determined by measuring the ratio of radioactivity in the medium relative to the total radioactivity detected by a liquid scintillation analyser (Tri-Carb 3100TR, Perkin Elmer), counting 3 min per sample.
Starvation-induced autophagy
ATG16L1 HEK293 knockout cells were rescued by lentiviral transduction with N-terminal EGFP-tagged ATG16L1β, ATG16L1β FII, ATG16L1α, ATG16L1 (aa 1–249) and ATG16L1 (aa 1–249) FII. Pooled populations were subjected to cell sorter in order to homogenize the EGFP expression levels (cells with ~7000 mean EGFP expression were gated and expanded). Cells were seeded on 10 μg/ml fibronectin coated glass coverslips. The following day cells were washed twice with EBSS prior to 1h EBSS incubation. After starvation treatment, cells were fixed on ice for 15 min with 4% paraformaldehyde (PFA; Polysciences, 18814–10), washed with PBS and immunostained with the indicated antibodies (diluted in 0.05% saponin PBS) for 1 h at room temperature. Cell nuclei were counterstained using 1 μg/ml Hoechst 33342 diluted in PBS and coverslips were mounted in ProLong Diamond Antifade Mountant (Invitrogen, p36965). Cells were imaged using a Zeiss 710 confocal microscope with a 63× objective lens, 5-image stacks covering a 1.4 micron thick section were acquired. A stack max intensity Z projection was generated and used for quantitation of punctate structures and colocalization analysis using CellProfiler open-source software37. Data mining and analysis was performed using KNIME38.
LC3B lipidation on perturbed endosomes
ATG16L1 HEK293 knockout cells were rescued with N-terminal EGFP-tagged ATG16L1β and ATG16L1α by lentiviral transduction. Cells with high expression of EGFP-ATG16L1 (~50000 mean EGFP expression) were sorted, expanded, transduced with lentiviral particles for expression of mTagBFP2-LC3B and sorted based on high levels of mTagBFP2-LC3B expression (~80000 mean mTagBFP2 expression). Cell expressing high levels of both EGFP-ATG16L1 and mTagBFP2-LC3 were then seeded on 10μg/ml fibronectin coated 8-well chambers (155409, Thermo Fisher Scientific). The following day cells were washed with FlouroBrite DMEM (Thermo Fisher Scientific) supplemented with 25 mM Hepes pH 7.4 and incubated for 1 h with 5 μM VPS34IN1 and 100 μM Monensin. Live image acquisition (with cells kept in a 37ºC environmental chamber) was performed using a Zeiss 710 confocal microscope with a 63× objective lens.
Mitophagy
A doxycycline inducible U2OS FlpIn cell line expressing EGFP-CHERRY with an N-terminal mitochondrial targeting motif (aa 1–53 of NIPSNAP1, mediates transport to the mitochondrial matrix) was generated. To monitor mitophagy, cells were seeded in glass bottom chamber slides and the next day 500 ng/ml doxycycline was added for 24 h to induce expression of mitochondrial targeted EGFP-CHERRY. Doxycycline was removed and cells were treated for 24 h with 1 mM deferiprone (DFP). washed with PBS and fixed with 4% PFA at 37°C before analysis by confocal microscopy.
LAP
RAW264.7 cells were plated on glass coverslips. The following day cells were fed 8 Zymosan particles/cell and after 1h of incubation cells were washed and fixed before staining for endogenous LC3 and analysis by confocal microscopy.
Western blotting
To demonstrate specific protein knockdown and to monitor LC3 and GABARAP levels, cells were lysed in either a SDS-lysis buffer (2%SDS, 50mM Tris pH 8.0, 1mM EDTA) or a Triton-lysis buffer (50mM Tris-HCl pH 7.4, 150mM NaCl, 1mM EDTA and 1% Triton-X 100) supplemented with complete protease inhibitor (Roche) and centrifuged. Supernatant protein concentration was measured by Biorad Protein Assay and equal amounts of cell lysates were subjected to SDS–polyacrylamide gel electrophoresis (PAGE) and western blotting using specified primary antibodies and far-red fluorophore or HRP-conjugated secondary antibodies. Detection and analysis was performed by LI-COR Odyssey imaging or by BioRad’s ChemiDoc system.
Code availability
Code used to perform image analysis is available from the corresponding authors upon request.
Statistics and Reproducibility
The findings of all key experiments were reliably reproduced at least three times. The names of statistical tests, measure of centre and definition of error bars, as well as the precise number of independent experiments are indicated in the figure legends. Fig 2b, 2e, 2i–j, Fig 6f–g, Fig S1d, S1g, Fig S2b–d, S2f, Fig S3a and S3c–d were performed once. Fig 3d–f, Fig 4d, Fig 5a–g, Fig 6a, Fig 7a–c and Fig 7g–h were performed twice.
Data availability
Source data for Fig. 1e and f, 2d and h, 3c and f, 4b,c,f and h, 5b–g, 6c and e, and 7b,c,e and h and Supplementary Fig. 1b and f, and 2e have been provided as Supplementary Table 1. All other data supporting the findings of this study are available from the corresponding authors upon reasonable request.
Supplementary Material
Acknowledgements
We thank Toshiharu Fujita for help with the generation of HEK293 ATG16L1 KO cells and Gunnveig Toft Bjørndal and Nagham Theres Asp for help with cell culture. This work was partly supported by the Research Council of Norway (project number 221831) and through its Centres of Excellence funding scheme (project number 262652), as well as the Norwegian Cancer Society (project number 171318). Support for T.M. was provided by NIH grant (GM1000930), and support for K.K. was provided by Cellular and Molecular Biology NIH Training grant T32-GM007223. The research leading to these results has also received funding from the European Union Seventh Framework Programme (FP7-PEOPLE-2013-COFUND) under grant agreement n° 609020 - Scientia Fellows.
Footnotes
Declaration of Interests
The authors have no conflict of interest.
References
- 1.Kabeya Y. et al. LC3, GABARAP and GATE16 localize to autophagosomal membrane depending on form-II formation. J Cell Sci 117, 2805–2812 (2004). [DOI] [PubMed] [Google Scholar]
- 2.Florey O. & Overholtzer M. Autophagy proteins in macroendocytic engulfment. Trends Cell Biol 22, 374–380 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li M. et al. Kinetics comparisons of mammalian Atg4 homologues indicate selective preferences toward diverse Atg8 substrates. The Journal of biological chemistry 286, 7327–7338 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tanida I, Tanida-Miyake E, Ueno T & Kominami E. The human homolog of Saccharomyces cerevisiae Apg7p is a Protein-activating enzyme for multiple substrates including human Apg12p, GATE-16, GABARAP, and MAP-LC3. The Journal of biological chemistry 276, 1701–1706 (2001). [DOI] [PubMed] [Google Scholar]
- 5.Tanida I. et al. HsAtg4B/HsApg4B/autophagin-1 cleaves the carboxyl termini of three human Atg8 homologues and delipidates microtubule-associated protein light chain 3- and GABAA receptor-associated protein-phospholipid conjugates. The Journal of biological chemistry 279, 36268–36276 (2004). [DOI] [PubMed] [Google Scholar]
- 6.Tanida I, Tanida-Miyake E, Komatsu M, Ueno T. & Kominami E. Human Apg3p/Aut1p homologue is an authentic E2 enzyme for multiple substrates, GATE-16, GABARAP, and MAP-LC3, and facilitates the conjugation of hApg12p to hApg5p. The Journal of biological chemistry 277, 13739–13744 (2002). [DOI] [PubMed] [Google Scholar]
- 7.Metlagel Z, Otomo C, Takaesu G. & Otomo T. Structural basis of ATG3 recognition by the autophagic ubiquitin-like protein ATG12. Proceedings of the National Academy of Sciences of the United States of America 110, 18844–18849 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fujita N. et al. The Atg16L complex specifies the site of LC3 lipidation for membrane biogenesis in autophagy. Molecular biology of the cell 19, 2092–2100 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sakoh-Nakatogawa M. et al. Atg12-Atg5 conjugate enhances E2 activity of Atg3 by rearranging its catalytic site. Nat Struct Mol Biol 20, 433–439 (2013). [DOI] [PubMed] [Google Scholar]
- 10.Nath S. et al. Lipidation of the LC3/GABARAP family of autophagy proteins relies on a membrane-curvature-sensing domain in Atg3. Nat Cell Biol 16, 415–424 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dooley HC et al. WIPI2 links LC3 conjugation with PI3P, autophagosome formation, and pathogen clearance by recruiting Atg12–5-16L1. Mol Cell 55, 238–252 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Itakura E, Kishi C, Inoue K & Mizushima N. Beclin 1 forms two distinct phosphatidylinositol 3-kinase complexes with mammalian Atg14 and UVRAG. Molecular biology of the cell 19, 5360–5372 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vicinanza M. et al. PI(5)P regulates autophagosome biogenesis. Mol Cell 57, 219–234 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gao Y. et al. Golgi-associated LC3 lipidation requires V-ATPase in noncanonical autophagy. Cell Death Dis 7, e2330 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fletcher K. et al. The WD40 domain of ATG16L1 is required for its non-canonical role in lipidation of LC3 at single membranes. EMBO J (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Martinez-Martin N. et al. A switch from canonical to noncanonical autophagy shapes B cell responses. Science 355, 641–647 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kim JH et al. Insights into autophagosome maturation revealed by the structures of ATG5 with its interacting partners. Autophagy 11, 75–87 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Otomo C, Metlagel Z, Takaesu G & Otomo T. Structure of the human ATG12~ATG5 conjugate required for LC3 lipidation in autophagy. Nat Struct Mol Biol 20, 59–66 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fujita N. et al. Differential Involvement of Atg16L1 in Crohn Disease and Canonical Autophagy ANALYSIS OF THE ORGANIZATION OF THE Atg16L1 COMPLEX IN FIBROBLASTS. Journal of Biological Chemistry 284, 32602–32609 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sou YS, Tanida I, Komatsu M, Ueno T. & Kominami E. Phosphatidylserine in addition to phosphatidylethanolamine is an in vitro target of the mammalian Atg8 modifiers, LC3, GABARAP, and GATE-16. The Journal of biological chemistry 281, 3017–3024 (2006). [DOI] [PubMed] [Google Scholar]
- 21.Dupont N, Leroy C, Hamai A. & Codogno P. Long-Lived Protein Degradation During Autophagy. Methods in enzymology 588, 31–40 (2017). [DOI] [PubMed] [Google Scholar]
- 22.Sanjuan MA et al. Toll-like receptor signalling in macrophages links the autophagy pathway to phagocytosis. Nature 450, 1253–1257 (2007). [DOI] [PubMed] [Google Scholar]
- 23.Sanjuan MA, Milasta S. & Green DR Toll-like receptor signaling in the lysosomal pathways. Immunol Rev 227, 203–220 (2009). [DOI] [PubMed] [Google Scholar]
- 24.Florey O, Gammoh N, Kim SE, Jiang X. & Overholtzer M. V-ATPase and osmotic imbalances activate endolysosomal LC3 lipidation. Autophagy 11, 88–99 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kaufmann A, Beier V, Franquelim HG & Wollert T. Molecular mechanism of autophagic membrane-scaffold assembly and disassembly. Cell 156, 469–481 (2014). [DOI] [PubMed] [Google Scholar]
- 26.Jarsch IK, Daste F. & Gallop JL Membrane curvature in cell biology: An integration of molecular mechanisms. J Cell Biol 214, 375–387 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Romanov J. et al. Mechanism and functions of membrane binding by the Atg5Atg12/Atg16 complex during autophagosome formation. EMBO J 31, 4304–4317 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mizushima N, Noda T. & Ohsumi Y. Apg16p is required for the function of the Apg12p-Apg5p conjugate in the yeast autophagy pathway. EMBO J 18, 3888–3896 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gammoh N, Florey O, Overholtzer M. & Jiang X. Interaction between FIP200 and ATG16L1 distinguishes ULK1 complex-dependent and -independent autophagy. Nat Struct Mol Biol 20, 144–149 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nishimura T. et al. FIP200 regulates targeting of Atg16L1 to the isolation membrane. EMBO reports 14, 284–291 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Boada-Romero E. et al. TMEM59 defines a novel ATG16L1-binding motif that promotes local activation of LC3. EMBO J 32, 566–582 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hu J. et al. TMEM166/EVA1A interacts with ATG16L1 and induces autophagosome formation and cell death. Cell Death Dis 7, e2323 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fujita N. et al. Recruitment of the autophagic machinery to endosomes during infection is mediated by ubiquitin. J Cell Biol 203, 115–128 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mizushima N. et al. Mouse Apg16L, a novel WD-repeat protein, targets to the autophagic isolation membrane with the Apg12-Apg5 conjugate. J Cell Sci 116, 1679–1688 (2003). [DOI] [PubMed] [Google Scholar]
- 35.Murthy A. et al. A Crohn’s disease variant in Atg16l1 enhances its degradation by caspase 3. Nature 506, 456–462 (2014). [DOI] [PubMed] [Google Scholar]
- 36.Lundmark R. & Carlsson SR Sorting nexin 9 participates in clathrin-mediated endocytosis through interactions with the core components. The Journal of biological chemistry 278, 46772–46781 (2003). [DOI] [PubMed] [Google Scholar]
- 37.Carpenter AE et al. CellProfiler: image analysis software for identifying and quantifying cell phenotypes. Genome biology 7, R100 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Berthold MR et al. KNIME: The Konstanz Information Miner. Stud Class Data Anal, 319–326 (2008). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Source data for Fig. 1e and f, 2d and h, 3c and f, 4b,c,f and h, 5b–g, 6c and e, and 7b,c,e and h and Supplementary Fig. 1b and f, and 2e have been provided as Supplementary Table 1. All other data supporting the findings of this study are available from the corresponding authors upon reasonable request.