

Abstract
Background
Limited evidence supports a hypothesis suggesting that the symptoms of schizophrenia may be the result of altered neuronal membrane structure and metabolism. The structure and metabolism is dependent on blood plasma levels of certain essential fatty acids and their metabolites.
Objectives
To assess the effects of polyunsaturated fatty acids for people with schizophrenia.
Search methods
We have updated the initial searches of 1998, 2002 and 2005 with a search of the Cochrane Schizophrenia Group's Register, November 2008, which is based on regular searches of CINAHL, EMBASE, MEDLINE and PsycINFO.
Where necessary, we contacted authors and relevant pharmaceutical companies for additional information.
Selection criteria
We included all randomised controlled trials of polyunsaturated fatty acid treatment for schizophrenia.
Data collection and analysis
Working independently, we selected studies for quality assessment and extracted relevant data. We analysed on an intention‐to‐treat basis. Where possible and appropriate we calculated the Relative Risk (RR) and their 95% confidence intervals (CI) and estimated the number needed to treat (NNT). For continuous data we calculated weighted mean differences (WMD) and their 95% confidence intervals. We also inspected the data for heterogeneity.
Main results
Eight studies are now included in this review. When any dose omega‐3 (E‐EPA or EPA) is compared with placebo, small short trials suggest that the need for neuroleptics appears to be reduced for people allocated omega‐3 supplementation (n=30, 1 RCT, RR 0.73 CI 0.54 to 1.00) and mental state may improve (n=30, 1 RCT, RR not gaining 25% change in PANSS scores 0.54 CI 0.30 to 0.96, NNT 3 CI 2 to 29). There are no differences in the number of people leaving the study early (n=595, 6 RCTs, RR 0.86 CI 0.50 to 1.48). There are few data on the comparison of any dose omega‐6 (GLA) with placebo. For movement disorder outcomes, the one small study we found does not show any difference for average short‐term endpoint AIMS score (n=16, 1 RCT, WMD 1.30 CI ‐1.96 to 4.56). When any dose omega‐3 (E‐EPA or EPA) is compared with any dose omega‐3 (DHA) there is no significant difference for mental state outcome of not gaining 25% change in PANSS scores (n=31, 1 RCT, RR 0.66 CI 0.39 to 1.11). When different doses of omega‐3 (E‐EPA) are compared with placebo there are no differences in measures of global and mental state between the studies. For the outcome of 'experiencing at least one adverse effect' no differences between groups are found for any dose (1 g/day E‐EPA vs placebo n=63, 1 RCT, RR 0.97 CI 0.60 to 1.56; 2 g/day E‐EPA vs placebo n=63, 1 RCT, RR 0.67 CI 0.37 to 1.20; 4 g/day E‐EPA vs placebo n=58, 1 RCT, RR 1.15 CI 0.72 to 1.82).
Authors' conclusions
Three updates of this review have resulted in more included studies and more people randomised but still relatively little useful additional data. The results remain inconclusive. The new trials all compare the omega‐3 polyunsaturated fatty acids, in particular eicosapentaenoic acid and its ester, ethyl‐eicosapentaenoic acid. The use of omega‐3 polyunsaturated fatty acids for schizophrenia still remains experimental and this review highlights the need for large, well designed, conducted and reported studies.
Plain language summary
Polyunsaturated fatty acid supplementation for schizophrenia
Schizophrenia is a serious mental health problem that affects about one percent of any population. For some people it can become an illness that they have to live with for their entire life. Early research has suggested that supplementing the diet with omega 3 or omega 6 fatty acids may have a positive effect on the symptoms of schizophrenia. This review looks at randomised control trials where omega 3 or omega 6 were used in combination with antipsychotic medication, or as a treatment in their own right for schizophrenia. Eight studies were found which included a total of 517 people who had a diagnosis of schizophrenia or schizoaffective disorder (combined symptoms of schizophrenia and a mood disorder). They ranged from six to 16 weeks in length and were in both hospital and community settings.
The majority of the trials compared two different types of omega 3 fatty acids, EPA (usually as E‐EPA) and DHA with placebo, in people with schizophrenia who are stable on antipsychotic medication. Some of these trials show some improvement in general functioning and in mental state but not to a statistically significant degree. In the longest trial there was no difference between the two groups at the end of the study. One trial compared E‐EPA with DHA and found a suggestion that E‐EPA works better than DHA, but again it was not statistically significant. Where EPA was compared to placebo as a first line treatment for schizophrenia (30 people), those taking EPA had a better overall outcome and improvement in mental state. However, this was a short trial with few people. Finally, one trial compared a type of omega 6 with placebo in men who had the movement disorder tardive dykinesia (16 people). There was no improvement in the symptoms of movement adverse effects in either group at the end of six weeks.
These trials were both small and short. In addition most of the data they reported were not able to be used, and half of the trials were funded by the group supplying the trial medication. Therefore it is still not clear whether taking manufactured omega 3 or 6 improves overall functioning or mental state in people with schizophrenia.
(Plain language summary prepared for this review by Janey Antoniou of RETHINK, UK www.rethink.org)
Background
Description of the condition
Currently 45 million people worldwide suffer from schizophrenia (WHO 1998). The lifetime prevalence for this disabling mental illness is about one per cent, irrespective of race, gender, social class or country of origin (Jablensky 1992).
Abnormal neurotransmission has been found in those who suffer from schizophrenia (Heresco‐Levy 1998). Brain activity is the result of a highly complex transmission of an infinite amount of nervous impulses. Chemicals (neurotransmitters) conduct these impulses between the nerve cells (neurones), and any alteration in this neurotransmission ultimately has far‐reaching consequences for brain function.
The mainstay of treatment for schizophrenia has been pharmacological; targeting neurotransmission within the brain. Most of the drugs used block various neurotransmitter sites within the brain. Although these compounds were first formulated in the 1950s, they are still in wide use today, and newer medications are refinements of the 'old generation' of drugs. Many people with schizophrenia have symptoms that are responsive to one or other of these drugs. Response, however, may often be partial, still leaving the sufferer disabled and unwell (Wiersma 1998).
Description of the intervention
There is now some evidence that dietary influences, particularly levels of essential fatty acids (EFAs), can also affect the occurrence and course of schizophrenia (Christensen 1998, Mellor 1995, Mellor 1996, Peet 2001a, Puri 1997). Such evidence, albeit limited, gives added support to an innovative hypothesis suggesting that the symptoms of schizophrenia may be the result of altered neuronal membrane structure and metabolism (Horrobin 1994).
There are two types of EFAs, omega‐6 and omega‐3. Those of the omega‐6 type are found in soft margarine and substances such as vegetable oil. The second type, omega‐3, is abundant in oily fish such as mackerel and sardines. Other sources of EFAs are manufactured preparations such as fish oil and evening primrose oil capsules.
How the intervention might work
Neurotransmission is dependent on the structure and metabolism of neurotransmitter sites located in the neuronal membrane. As brain activity is dependent on an immeasurable number of neurotransmissions occurring at neurotransmitter sites, even a slight change in neuronal cell membrane metabolism could profoundly alter brain functioning and promote symptoms such as those seen in schizophrenia (Horrobin 1994).
In turn, neuronal cell membrane structure and metabolism itself is dependent on blood plasma levels of certain essential fatty acids (EFAs) and their hydroxy‐metabolites (Di Marzo 1992, Nunez 1993). EFAs are polyunsaturated fatty acids that are not synthesised by the body and therefore they are only available through diet. Several studies have shown that people with schizophrenia often have low levels of the particular EFAs necessary for normal nerve cell membrane metabolism ( Glen 1994, Horrobin 1991). Dietary supplementation of these EFAs is possible, and early uncontrolled, open studies suggest such supplementation may have a direct, positive, effect on the symptoms of schizophrenia (Mellor 1995, Mellor 1996). In addition, if membrane functioning has significant effects upon nerve impulse transmission, EFA supplementation may enhance the efficacy of currently available antipsychotic drugs already known to have a beneficial affect on altered neurotransmission.
Why it is important to do this review
Recently, more trials investigating the effects of EFA supplementation for schizophrenia have been completed. As more data becomes available, hopefully more useful evidence for this relatively simple treatment can be provided.
Objectives
To assess the effects of EFA supplementation of antipsychotic treatment for schizophrenia‐like illnesses.
To investigate the evidence for a difference between the effects of omega‐3 and omega‐6 EFA supplementation of neuroleptic treatment for schizophrenia‐like illnesses.
To investigate the effects of pure preparations of EFA such as the omega‐3 eicosapentaenoic acid compared with a mixture of EFA's such as the 'fish oil' or 'evening primrose oil' preparations sold over the counter in pharmacies or drug stores.
Methods
Criteria for considering studies for this review
Types of studies
All relevant randomised controlled trials. We excluded quasi‐randomised trials, such as those where allocation is undertaken on surname. If a trial was described as double‐blind, but it was implied it had been randomised, we included these trials in a sensitivity analysis.
Randomised cross‐over studies will be eligible but only data up to the point of first cross‐over because of the instability of the problem behaviours and the likely carry‐over effects of all treatments.
Types of participants
People with schizophrenia or similar chronic mental illnesses, diagnosed by any criteria, irrespective of gender, age or race.
Types of interventions
1. Any type of polyunsaturated fatty acid supplementation of a standard neuroleptic care: any dose. There are two types of relevance.
1.1 Omega‐3 EFAs: these include eicosapentaenoic acid (EPA), its ester, ethyl‐eicosapentaenoic acid (E‐EPA) and docosahexanoic acid (DHA) 1.2 Omega‐6 EFAs: for example, gamma‐linolenic acid (GLA).
2. Standard neuroleptic care: the normal level of psychiatric care and medication provided in the area where the trial was conducted.
Types of outcome measures
Primary outcomes
1. Global state 1.1 Relapse
2. General functioning 2.1 No clinically important change in general functioning
3. Satisfaction with treatment 3.1 Leaving the study early ‐ general reason 3.2 Recipient of care not satisfied with treatment
4. Adverse effects 4.1 No clinically important general adverse effects
Secondary outcomes
1. Death ‐ suicide and natural causes
2. Global state 2.1 Time to relapse 2.2 No clinically important change in global state 2.3 Not any change in global state 2.4 Average endpoint global state score 2.5 Average change in global state scores
3. Service outcomes 3.1 Hospitalisation 3.2 Time to hospitalisation
4. Mental state 4.1 No clinically important change in general mental state 4.2 Not any change in general mental state 4.3 Average endpoint general mental state score 4.4 Average change in general mental state scores 4.5 No clinically important change in specific symptoms 4.6 Not any change in specific symptoms 4.7 Average endpoint specific symptom score 4.8 Average change in specific symptom scores
5. Satisfaction with treatment 5.1 Leaving the study early: specific reason 5.2 Recipient of care not satisfied with treatment 5.3 Recipient of care average satisfaction score 5.4 Recipient of care average change in satisfaction scores 5.6 Carer not satisfied with treatment 5.7 Carer average satisfaction score 5.8 Carer average change in satisfaction scores
6. General functioning 6.1 Average endpoint general functioning score 6.2 Average change in general functioning scores 6.3 No clinically important change in specific aspects of functioning, such as social or life skills 6.4 Not any change in specific aspects of functioning, such as social or life skills 6.5 Average endpoint specific aspects of functioning, such as social or life skills 6.6 Average change in specific aspects of functioning, such as social or life skills
7. Behaviour 7.1 No clinically important change in general behaviour 7.2 Not any change in general behaviour 7.3 Average endpoint general behaviour score 7.4 Average change in general behaviour scores 7.5 No clinically important change in specific aspects of behaviour 7.6 Not any change in specific aspects of behaviour 7.7 Average endpoint specific aspects of behaviour 7.8 Average change in specific aspects of behaviour
8. Adverse effects 8.1 Not any general adverse effects 8.2 Average endpoint general adverse effect score 8.3 Average change in general adverse effect scores 8.4 No clinically important change in specific adverse effects 8.5 Not any change in specific adverse effects 8.6 Average endpoint specific adverse effects 8.7 Average change in specific adverse effects
9. Engagement with services 9.1 No clinically important engagement 9.2 Not any engagement 9.3 Average endpoint engagement score 9.4 Average change in engagement scores
10. Quality of life 10.1 No clinically important change in quality of life 10.2 Not any change in quality of life 10.3 Average endpoint quality of life score 10.4 Average change in quality of life scores 10.5 No clinically important change in specific aspects of quality of life 10.6 Not any change in specific aspects of quality of life 10.7 Average endpoint specific aspects of quality of life 10.8 Average change in specific aspects of quality of life
11. Economic outcomes 11.1 Direct costs 11.2 Indirect costs
We selected outcome measures which provided global estimations of functioning. We did not report highly specific outcomes, such as 'sense of safety'. Such specific outcomes are rarely reported in more than one study and it is difficult to assess their relevance to the effectiveness of the treatment. We also reported other outcomes not readily falling into these categories but they are not of pre‐stated interest.
We divided outcomes into short term (less than three months) medium term (three to six months) and long term (over six months).
Search methods for identification of studies
Electronic searches
1. Update of 2008 1.1 The Cochrane Schizophrenia Group Trials Register was searched (November 2008): for details of the search phrase see Appendix 3.
2. Previous versions of this review 2.1 For details of the original search: see Appendix 1.
2.2 For details of the searches in previous updates: see Appendix 2.
Searching other resources
Where necessary, we contacted authors and relevant pharmaceutical companies for any missing information or data.
Data collection and analysis
Selection of studies
Authors CJ and RMC independently inspected citations identified from the search. We identified potentially relevant reports and ordered full papers for reassessment. Where difficulties or disputes arose we asked author LAJ for help and if it was impossible to decide, those full papers were ordered for assessment. This process was repeated for the full papers. If it was impossible to resolve disagreements these studies were added to those awaiting assessment and the authors of the papers contacted for clarification.
Data extraction and management
1. Extraction Authors CJ and RMC independently extracted data from included studies. Again, any disagreement was discussed, decisions documented and, if necessary, authors of studies were contacted for clarification. With remaining problems LAJ helped clarify issues and those final decisions were documented.
2. Management Data were extracted onto standard, simple forms.
3. Scale‐derived data A wide range of instruments are available to measure outcomes in mental health studies. These instruments vary in quality and many are not validated, or are even ad hoc. We included continuous data from rating scales only if the measuring instrument had been described in a peer‐reviewed journal (Marshall 2000) and the instrument is either a self‐report or completed by an independent rater or relative (not the therapist).
Assessment of risk of bias in included studies
Again working independently, CJ and RMC assessed risk of bias using the tool described in the Cochrane Collaboration Handbook (Higgins 2008). This tool encourages consideration of how the sequence was generated, how allocation was concealed, the integrity of blinding at outcome, the completeness of outcome data, selective reporting and other biases. We would not have included studies where sequence generation was at high risk of bias or where allocation was clearly not concealed.
If disputes arose as to which category a trial has to be allocated, again, resolution was made by discussion, after working with the third reviewer (LAJ).
Measures of treatment effect
1. Binary data For binary outcomes we calculated a standard estimation of the fixed‐effect risk ratio (RR) and its 95% confidence interval (CI). For statistically significant results we calculated the number needed to treat/harm statistic (NNT/H), and its 95% confidence interval (CI) using Visual Rx (http://www.nntonline.net/) taking account of the event rate in the control group.
2. Continuous data 2.1 Summary statistic For continuous outcomes we estimated a fixed‐effect weighted mean difference (WMD) between groups. We did not calculate effect size measures.
2.2 Endpoint versus change data We preferred to use scale endpoint data, which typically cannot have negative values and is easier to interpret from a clinical point of view. Change data are often not ordinal and are very problematic to interpret. If endpoint data were unavailable, we used change data.
2.3 Skewed data Continuous data on clinical and social outcomes are often not normally distributed. To avoid the pitfall of applying parametric tests to non‐parametric data, we aimed to apply the following standards to all data before inclusion: (a) standard deviations and means are reported in the paper or obtainable from the authors; (b) when a scale starts from the finite number zero, the standard deviation, when multiplied by two, is less than the mean (as otherwise the mean is unlikely to be an appropriate measure of the centre of the distribution, (Altman 1996); (c) if a scale starts from a positive value (such as PANSS which can have values from 30 to 210) the calculation described above was modified to take the scale starting point into account. In these cases skew is present if 2SD>(S‐S min), where S is the mean score and S min is the minimum score. Endpoint scores on scales often have a finite start and end point and these rules can be applied. When continuous data are presented on a scale which includes a possibility of negative values (such as change data), it is difficult to tell whether data are skewed or not. Skewed data from studies of less than 200 participants were entered in additional tables rather than into an analysis. Skewed data pose less of a problem when looking at means if the sample size is large and were entered into syntheses.
Unit of analysis issues
1. Cluster trials Studies increasingly employ 'cluster randomisation' (such as randomisation by clinician or practice) but analysis and pooling of clustered data poses problems. Firstly, authors often fail to account for intraclass correlation in clustered studies, leading to a 'unit of analysis' error (Divine 1992) whereby p values are spuriously low, confidence intervals unduly narrow and statistical significance overestimated. This causes type I errors (Bland 1997, Gulliford 1999).
Where clustering is not accounted for in primary studies, we presented data in a table, with a (*) symbol to indicate the presence of a probable unit of analysis error. In subsequent versions of this review we will seek to contact first authors of studies to obtain the intraclass correlation coefficient of their clustered data and to adjust for this by using accepted methods (Gulliford 1999). Where clustering had been incorporated into the analysis of primary studies, we present these data as if from a non‐cluster randomised study, but adjusted for the clustering effect.
We have sought statistical advice and have been advised that the binary data as presented in a report should be divided by a 'design effect'. This is calculated using the mean number of participants per cluster (m) and the intraclass correlation coefficient (ICC) [Design effect = 1+(m‐1)*ICC] (Donner 2002). If the ICC was not reported it was assumed to be 0.1 (Ukoumunne 1999).
If cluster studies has been appropriately analysed taking into account the intraclass correlation coefficient and relevant data documented in the report, synthesis with other studies would have been possible using the generic inverse variance technique.
2. Cross‐over trials A major concern of cross‐over trials is the carry‐over effect. It occurs if an effect (e.g. pharmacological, physiological or psychological) of the treatment in the first phase is carried over to the second phase. As a consequence on entry to the second phase the participants can differ systematically from their initial state despite a wash‐out phase. For the same reason cross‐over trials are not appropriate if the condition of interest is unstable (Elbourne 2002). As both effects are very likely in schizophrenia, we will only use data of the first phase of cross‐over studies.
3. Studies with multiple treatment groups Where a study involved more than two treatment groups, if relevant, the additional treatment arms were presented in comparisons. Where the additional treatment arms were not relevant, these data were not reproduced.
Dealing with missing data
1. Overall loss of credibility At some degree of loss of follow up data must lose credibility (Xia 2007). We are forced to make a judgment where this is for the short‐term trials likely to be included in this review. Should more than 30% of data be unaccounted for by the first follow up we did not reproduce these data or use them within analyses.
2. Binary In the case where attrition for a binary outcome is between 0 and 30% and outcomes of these people are described, we included these data as reported. Where these data were not clearly described, we assumed the worst primary outcome, and rates of adverse effects similar to those who did continue to have their data recorded.
3. Continuous In the case where attrition for a continuous outcome is between 0 and 30% and completer‐only data were reported, we have reproduced these.
Assessment of heterogeneity
1. Clinical heterogeneity We considered all included studies without any comparison to judge clinical heterogeneity.
2. Statistical 2.1 Visual inspection We visually inspected graphs to investigate the possibility of statistical heterogeneity.
2.2 Employing the I‐squared statistic This provided an estimate of the percentage of inconsistency thought to be due to chance. I‐squared estimate greater than or equal to 50% was interpreted as evidence of high levels of heterogeneity (Higgins 2003).
Assessment of reporting biases
Reporting biases arise when the dissemination of research findings is influenced by the nature and direction of results (Egger 1997). These are described in section 10.1 of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2008). We are aware that funnel plots may be useful in investigating reporting biases but are of limited power to detect small‐study effects. We did not use funnel plots for outcomes where there were ten or fewer studies, or where all studies were of similar sizes. In other cases, where funnel plots were possible, we sought statistical advice in their interpretation.
Data synthesis
Where possible we employed a fixed‐effect model for analyses. We understand that there is no closed argument for preference for use of fixed or random‐effects models. The random‐effects method incorporates an assumption that the different studies are estimating different, yet related, intervention effects. This does seem true to us, however, random‐effects does put added weight onto the smaller of the studies ‐ those trials that are most vulnerable to bias. For this reason we favour using fixed‐effect models employing random‐effects only when investigating heterogeneity.
Subgroup analysis and investigation of heterogeneity
If data are clearly heterogeneous we checked that data are correctly extracted and entered and that we had made no unit of analysis errors. If the high levels of heterogeneity remained we did not undertake a meta‐analysis at this point for if there is considerable variation in results, and particularly if there is inconsistency in the direction of effect, it may be misleading to quote an average value for the intervention effect. We would have wanted to explore heterogeneity. We pre‐specified no characteristics of studies that may be associated with heterogeneity except quality of trial method. If no clear association could be shown by sorting studies by quality of methods a random‐effects meta‐analysis was performed. Should another characteristic of the studies be highlighted by the investigation of heterogeneity, perhaps some clinical heterogeneity not hitherto predicted but plausible causes of heterogeneity, these post‐hoc reasons will be discussed and the data analysed and presented. However, should the heterogeneity be substantially unaffected by use of random‐effects meta‐analysis and no other reasons for the heterogeneity be clear, the final data were presented without a meta‐analysis.
Sensitivity analysis
If necessary, we analysed the effect of including studies with high attrition rates in a sensitivity analysis. We aimed to include trials in a sensitivity analysis if they are described as 'double‐blind' but only implied randomisation. If we found no substantive differences within primary outcome when these high attrition and 'implied randomisation' studies were added to the overall results, we included them in the final analysis. However, if there was a substantive difference we only used clearly randomised trials and those with attrition lower than 30%.
Results
Description of studies
For substantive descriptions of studies please see Characteristics of included studies and Characteristics of excluded studies tables.
Results of the search
The original search in 1998 produced a list of 16 references. Further inspection of abstracts and collation of references into individual studies reduced the list to seven, of which only two could be included. A new search, using a new electronic search term (see search strategy in Appendix 2), was carried out in 2002; this produced a list of 35 possible new references. From this list, we found eight new studies to add to the review. We eventually excluded four of these, making the total number of excluded studies nine. Three studies were added to the included studies table. The 2005 search found six new references; two were additional references for Fenton 2001 ‐ a study already included, and two were not relevant to the review. The final two references were for Emsley 2002 a study previously classed as 'ongoing'. Our 2008 update found 151 references from 42 studies, 21 of these references relating to nine studies were relevant to the review. One of these studies (Peet 2001a) was already included in the review, five others (Bentsen 2007, Blanchet 2008, Korbanic 2005, Murck 2001 and Richtand 2007) are ongoing. One further study (Amminger 2007 ) was excluded leaving two new trials (Berger 2007 and Emsley 2006) for inclusion. This review now includes eight studies.
Included studies
For the 2008 update we included two new studies, Berger 2007 and Emsley 2006. We now have eight included studies in this review with a total of 517 people randomised (further details can be found in the Characteristics of included studies table).
1. Length of studies Study duration ranged from six (Wolkin 1986) to 16 weeks (Fenton 2001).
2. Setting Studies take place in a mixture of hospital and community settings.
3. Participants All included studies involved people with DSM‐IV schizophrenia although Wolkin 1986 (n=16) does include one person with bipolar affective disorder and does not use any diagnostic criteria. Fenton 2001 also includes people with schizo‐affective disorder. Most participants are chronically ill, and are still symptomatic despite exposure to antipsychotic drugs. Peet 2001b, however, specifically includes only people who are recently ill and have no previous exposure to neuroleptics. Berger 2007 also only randomises relatively young people experiencing their first psychotic episode. All participants in Emsley 2006 have established neuroleptic induced tardive dyskinesia. All studies are mixed sex apart from Wolkin 1986. Wolkin 1986 includes men suffering not only from serious mental illness but also neuroleptic‐induced mild tardive dyskinesia. Emsley 2002 does not state the sex of participants. The overall age range of participants is 18‐65 years.
4. Study size Overall, sample size is small. Peet 2002, the largest study includes 122 people. The size of the other trials ranges from 16 (Wolkin 1986) to 90 (Fenton 2001).
5. Interventions All included studies give people polyunsaturated fatty acids not synthesised by the body. The majority of trials compare EFA supplementation with placebo supplementation and use omega‐3 EFAs, either eicosapentaenoic acid (EPA), its ester, ethyl‐eicosapentaenoic acid (E‐EPA) or docosahexanoic acid (DHA). Peet 2001a also compares EPA supplementation with DHA supplementation. Wolkin 1986 is the only trial to compare an omega‐6 EFA, gamma‐linolenic acid (GLA) with placebo. Peet 2001b is the only trial not to supplement neuroleptic treatment. In this study participants are allocated eicosapentaenoic acid or placebo as sole treatment unless it becomes necessary to prescribe standard antipsychotic drugs during the trial. Some trials use specific doses of E‐EPA supplementation. Fenton 2001 compares 2 g/day E‐EPA with placebo but also give all participants an additional supplement of Vitamin E. Peet 2002 compares different doses (1 g/day, 2 g/day or 4 g/day) of E‐EPA with placebo. Berger 2007 and Emsley 2006 compare a single dose of 2 g/day E‐EPA with placebo while Emsley 2002 compares 3 g/day E‐EPA with placebo.
6. Outcomes 6.1 Missing outcomes Only four main treatment effects are evaluated: global state; mental state; adverse events and leaving the study early. None of the studies reported on mortality, direct measures of compliance, relapse, patient and carer satisfaction, social functioning or cost of treatment.
6.2 Scales Nine rating scales are used to collect scale data, only four of these provide useful, non skewed continuous data ‐ the Positive and Negative Syndrome Scale (PANSS), the Abnormal Involuntary Movement Scale (AIMS), the Simpson and Angus Scale (SAS) and the MADRS. All are validated and peer‐reviewed. Scale details are provided below:
6.2.1. Abnormal Involuntary Movement Scale ‐ AIMS (Guy 1976). A 12‐item scale designed to record the occurrence of dyskinetic movements. Each item is rated from zero (none) to four (severe). Low score indicates low levels of dyskinetic movements. This scale was used in Wolkin 1986.
6.2.2 Positive and Negative Syndrome Scale ‐ PANSS (Kay 1986). A brief clinician‐rated scale used to assess mental state and severity of psychopathology. There are 30 items, each item is scored on a scale of one (absent) to seven (extreme). The PANSS can be divided into three sub‐scales measuring severity of (i) general psychopathology, (ii) positive symptoms (PANSS‐P), and (iii) negative symptoms (PANSS‐N). A low score indicates low levels of psychopathology. This scale was used in Peet 2001a.
6.2.3 Montgomery Asberg Depression Rating Scale ‐ MADRS (Montgomery 1979) A 65‐item comprehensive psychopathology scale was used to identify the 17 most commonly occurring symptoms in primary depressive illness. Ratings are based on ten items, with higher scores indicating more symptoms.
6.2.4 Simpson Angus Scale ‐ SAS (Simpson 1970) This 10‐item scale, with a scoring system of zero to four for each item, measures drug‐induced parkinsonism, a short‐term drug‐induced movement disorder. A low score indicates low levels of parkinsonism.
Excluded studies
The review now contains ten excluded studies. Only one, Peet 1996, was not randomised. Heresco‐Levy 1996, Maurer 2002, Silbergeld 1973 and Vaddadi 1986 are randomised but all use interventions not specifically relevant to this review. Heresco‐Levy 1996 uses glycine (an amino acid) to supplement standard antipsychotic treatments, Maurer 2002 compares olanzapine with haloperidol and Silbergeld 1973 compares dexamethasone with placebo. Vaddadi 1986 investigates whether depot antipsychotic withdrawal could take place while all study participants took EFA supplementation. Glen 1996, Holman 1983 and Vaddadi 1989, are relevant trials of EFA supplementation they do not report any usable data. In addition, Holman 1983 is not explicit about allocation; it is, however, a double‐blind trial. We have contacted the authors of these trials and if the required data is supplied, we will include these trials in a future update. Amminger 2007 compares E‐EPA with placebo but randomised people at high risk of psychosis. Puri 2001 does randomise people with schizophrenia but those with Huntington's disease. Details of all excluded trials can be found in the 'Characteristics of excluded studies' table.
Awaiting assessment
No studies are awaiting assessment.
Ongoing studies
There are now five ongoing studies in this review (Bentsen 2007, Blanchet 2008, Korbanic 2005, Murck 2001 and Richtand 2007 ).
Risk of bias in included studies
A summary of risk of bias can be found in Figure 1 and Figure 2.
1.

Methodological quality graph: review authors' judgments about each methodological quality item presented as percentages across all included studies.
2.

Methodological quality summary: review authors' judgments about each methodological quality item for each included study.
Allocation
All studies stated they were randomised but Emsley 2002, Fenton 2001 and Wolkin 1986 did not describe the randomisation techniques and we have to classify them as unclear quality with moderate risk of selection bias. Berger 2007, Emsley 2006, Peet 2001a, Peet 2001b and Peet 2002 all describe how allocation is concealed via pre‐coded packages, each with a unique randomisation number generated by a company independent to the trial. These trials therefore are classified as clear quality with a low risk of selection bias and low risk of overestimate of positive effect.
Blinding
All trials stated that they were double‐blind and all but Emsley 2002 and Wolkin 1986 described how this was attempted. Berger 2007, Emsley 2006, Fenton 2001, Peet 2001a, Peet 2001b and Peet 2002 used capsules that were identical in both appearance and taste. No trial, however, tested whether their attempts at blinding were successful. We have to rate the risk of observer bias in Emsley 2002 and Wolkin 1986 as unclear, which adds to their potential for overestimate of positive effects. The other trials all have a low risk of observer bias.
Incomplete outcome data
All of the studies that had people leaving early (Peet 2001a and Peet 2002 had no loss) present data on loss and give descriptions. Wolkin 1986, however, has 37% loss to follow up for both groups. This is greater than the pre‐stated limit of 30% and the data reported in this study for mental state could not be presented in this review as bias could be introduced to the final analysis if only conducted on those completing the study. Of the other five studies that had low attrition rates, one (Peet 2001b) did not describe how they dealt with missing data, three (Emsley 2002; Emsley 2006 and Fenton 2001) undertook an 'intention‐to‐treat' (ITT) analysis and one trial (Berger 2007) uses multiple imputation to deal with lost data. Based on this information we assigned most trials a low risk of attrition bias but had to rate Wolkin 1986 as a high risk trial.
Selective reporting
Compared to the trials used in the first version of this review, the quality of data reporting has fallen. This is mainly due to the larger new trials Berger 2007,Emsley 2006, and Peet 2002 who report more outcomes, and in particular use more scales to collect continuous data but do not present data useful to this review, and for some outcomes no data are presented. These trials had a high risk of selective reporting bias. The other trials present data for all outcomes and for most outcomes the data reporting is above average with use of use valid scales and presentation of variances with mean total scores.
Other potential sources of bias
In half of the trials there is a high risk of other sources of bias, mainly author subjectivity. For four trials (Peet 2001a; Peet 2001b, Peet 2002 and Wolkin 1986) funding comes from the drug companies supplying trial medication and in Peet 2002 one of the main authors, Professor Horrobin, was an employee of the sponsoring drug company at the time of the trial. Another trial with a potential risk of bias is Berger 2007. This is a grant sponsored study, the main author however, is a consultant for several pharmaceutical companies. These sources of bias in these trials combined with their tendency to selectively report outcomes gave us reason to judge the risk of bias in the studies to be high, with these authors likely to be overestimating any true positive effect, and underestimating negative effects.
Effects of interventions
1. COMPARISON 1. ANY DOSE OMEGA‐3 (E‐EPA OR EPA) versus PLACEBO 1.1 Global State Peet 2001b (short term, up to 12 weeks) and Fenton 2001 (medium term, 13‐26 weeks) report data for global state. Peet 2001b is an unusual study as people are unmedicated prior to randomisation. The need for neuroleptics during the trial appears to favour omega‐3 supplementation with fewer people requiring neuroleptic drugs in this group but the result is not quite statistically significant (n=30, 1 RCT, RR 0.73 CI 0.54 to 1.00). Peet 2001b also reports on the number of days that people in the study are free of standard antipsychotics. This is considerably less for those allocated to the omega‐3 group, but data are skewed so are presented in a table. Fenton 2001 reported scale data from Clinical Global Impression endpoint scores. There is no difference between groups (n=87, 1 RCT, MD 0.00 CI ‐0.29 to 0.29). Berger 2007 presents numbers achieving a symptomatic response by 12 weeks and there are no significant differences between groups for this outcome for all participants (n=69, 1 RCT, RR 0.90 CI 0.5 to 1.63) or for nonaffective participants (n=53, 1 RCT, RR 0.56 CI 0.25 to 1.25).
1.2 Mental State Peet 2001b judges that an improvement of greater than 25% on the PANSS scale is clinically meaningful for this group of patients. Results show a significant improvement in PANSS scores, favouring the omega‐3 EFA group for people who are unmedicated when included in the study (n=30, 1 RCT, RR 0.54 CI 0.30 to 0.96, NNT3 CI 2 to 29). In Peet 2001a, there is a suggestion that people already on antipsychotic drugs when randomised to receive omega‐3 EFA, did show higher level of improvement compared with those receiving placebo but the statistical significance is borderline (n=29, 1 RCT, RR 0.62 CI 0.37 to 1.05). Similar results are found for the average endpoint scores on the PANSS scale. Peet 2001b found unmedicated participants receiving omega‐3 have significantly lower endpoint scores than those receiving placebo for the short term (n=30, 1 RCT, MD ‐12.5 CI ‐22.38 to ‐2.62). Medicated participants receiving omega‐3 supplementation in Peet 2001a also have significantly lower endpoint scores, again for the short term, but the difference is borderline (n=29, 1 RCT, MD ‐10.4 CI ‐20.35 to ‐0.45). Medium‐term data, reported by Fenton 2001, does not favour either group (n=87, 1 RCT, MD ‐1.0 CI ‐8.15 to 6.15). Fenton 2001 reports data for depression as measured on the MADRS. Skewed data, presented in this review in 'Additional tables' suggests no effect for the medium term.
1.3 Adverse effects Fenton 2001 reports usable data for movement disorders as measured by the AIMS and Simpson and Angus scales. Their skewed data are presented in a table but do not suggest any effect of omega‐3 supplementation.
1.4 Leaving the study early Seven studies have low or no attrition (<10% total). Combining data from those with low attrition, Berger 2007, Emsley 2006, Fenton 2001, Peet 2001a, Peet 2001b and Peet 2002, and suggests that there are no differences in numbers between groups leaving the study early (n=679, 6 RCTs, RR 0.85 CI 0.51 to 1.40).
2. COMPARISON 2. ANY DOSE OMEGA‐6 (GLA) versus PLACEBO 2.1 Tardive dyskinesia One small study (Wolkin 1986) finds no significant difference between groups for the average short‐term endpoint AIMS score by the end of trial (n=16, 1 RCT, MD 1.30 CI ‐1.96 to 4.56).
2.2 Leaving the study early Three people in each of the groups left one small study early (n=16, 1 RCT, RR 1.0 CI 0.28 to 3.54).
3. COMPARISON 3. ANY DOSE OMEGA 3 (E‐EPA or EPA) versus ANY DOSE OMEGA‐3 (DHA) 3.1 Mental state One small (n=31) study compares the effects of different omega‐3 EFAs (Peet 2001a). For the outcome of no clinically important improvement in mental state in the short term (>25% improvement in PANSS scores), E‐EPA is no different to DHA (n=31, 1 RCT, RR 0.66 CI 0.39 to 1.11). Measuring improvement in mental state by the difference in change of PANSS scores also shows no difference between groups (n=31, WMD ‐9.80 CI ‐20.97 to 1.37).
3.2 Leaving the study early There is no loss from either group for this comparison (n=31, study duration 12 weeks).
4. COMPARISON 4. SPECIFIC DOSE OMEGA‐3 (E‐EPA) versus PLACEBO 4.1 Global state Fenton 2001 compares supplementation with less than 1 g/day E‐EPA with antipsychotic supplementation with placebo and finds no difference between groups on the CGI scale in the medium term (n=87, 1 RCT, MD 0.00 CI ‐0.29 to 0.29).
4.2 Mental State Again Fenton 2001 reports usable data for medium‐term endpoint PANSS scores. There is no difference between people allocated less than 1 g/day E‐EPA supplementation and those receiving placebo (n=78, 1 RCT, MD ‐1.00 CI ‐8.15 to 6.15). Emsley 2002 presents skewed data for percentage change in PANSS scores from baseline and results suggest a positive effect for those receiving 3 g/day E‐EPA supplementation compared with those receiving placebo; this data can be found in 'Additional tables'.
4.3 Adverse effects Unless stated, all data for adverse effects are provided by Peet 2002, a short‐term study. For the outcome of 'experiencing at least one adverse effect' no differences between groups are found for any dose (1 g/day E‐EPA vs placebo n=63 , 1 RCT, RR 0.97 CI 0.60 to 1.56; 2 g/day E‐EPA vs placebo n=63, 1 RCT, RR 0.67 CI 0.37 to 1.20; 4 g/day E‐EPA vs placebo n=58, 1 RCT, RR 1.15 CI 0.72 to 1.82).
Short‐term use of E‐EPA supplementation is not clearly associated with gastrointestinal problems such as diarrhoea and nausea but most data comes from one small study with few events (Peet 2002). Data from two studies, the short‐term Peet 2002 and medium‐term Fenton 2001 do not show that people receiving less than 1 g/day of E‐EPA are more likely to experience diarrhoea than those on placebo (n=150, 2 RCTs, RR 2.04 CI 0.91 to 4.54). However, these data are heterogeneous and the results of the medium‐term Fenton 2001 do suggest that there may be a link with the use of low dose E‐EPA supplementation and diarrhoea (n=87, 1 RCT, RR 17.39 CI 1.03 to 292).
Peet 2002 records how many people have experienced liver and bilary tract problems, metabolic and nutritional difficulties (for example weight gain), and musculoskeletal adverse effects. No differences are apparent for any dose of E‐EPA supplementation compared with placebo. The same study looked for any psychiatric sequelae of giving different doses of E‐EPA supplementation, psychosexual difficulties and adverse effects such as infections. Again although it appears as though there is an effect, no statistically significant differences are apparent. Finally, rashes, urinary problems and 'any other' adverse effects are rare and not different for any doses.
4.4 Leaving the study early Overall there are no significant differences in loss between groups. Fenton 2001 (medium term) and Peet 2002 (short term) report data for <1 g/day E‐EPA compared with placebo supplementation. No differences in numbers leaving the study early are found between groups (n=150, 2 RCTs, RR 1.61 CI 0.71 to 3.67). Results from data provided by Berger 2007, Emsley 2006 and Peet 2002 also show no difference between groups for the 2 g/day E‐EPA comparison (n=307, 3 RCTs, RR 1.17 CI 0.54 to 2.55) and the 4 g/day E‐EPA comparison with placebo (n=58, 1 RCT, RR 2.30 CI 0.22 to 23.94). Emsley 2002 also finds no short‐term differences in loss between groups for a 3 g/day dose of E‐EPA compared with placebo (n=40, 1 RCT, RR 3.00 CI 0.13 to 69.52)
5. Heterogeneity and publication bias Both the possibility of heterogeneity and publication bias are impossible to test on such a small number of studies.
Discussion
Summary of main results
1. COMPARISON 1: ANY DOSE OMEGA‐3 (E‐EPA or EPA) versus PLACEBO 1.1 Global state Peet 2001b investigates the short‐term effects of omega‐3 compared with placebo for people with schizophrenia not medicated at the start of the study. Global outcomes tend to favour the EPA group although none are statistically significant and expecting a clear result from a study of only 30 people is ambitious. The Peet 2001b trial generates important hypotheses for future studies. Fenton 2001, a generally less favourable study for EPA supplementation of antipsychotic drugs, at least in the medium term, uses the CGI to measure global state and also finds no effect. Berger 2007 found no effect for numbers achieving symptomatic response when assessing all participants. When the nonaffective participants were separated out a positive affect of EPA supplementation seems to appear but this difference fails to make significance. The numbers in this study, again are small. No firm conclusions can be made from any of these results but they certainly do not support the use of EPA outside of the experimental situation.
1.2 Mental state Overall the mental state of both medicated and unmedicated people, if measured by a 25% improvement on the PANSS scale, is significantly better for those receiving omega‐3 EFA compared with placebo (NNT 3 CI 2 to 8). The trialist judged that an improvement of greater than 25% on the PANSS scale is clinically meaningful for this group of patients. We feel this to be unlikely, and are in keeping with other reviewers in this field (Hunter 2003), but leave the final judgement to the reader. Short‐term PANSS scores from Peet 2001a and Peet 2001b concur and are favourable for the EPA group but again the mean differences may not be very clinically meaningful and the longer‐term study (Fenton 2001) finds no effect. Whether clinically significant or not, omega‐3 EPAs may be having an effect worthy of more research. These are drugs working in quite a different way to standard antipsychotics and, therefore, may teach us much about the pathology of schizophrenia.
1.3 Adverse effects In keeping with the complete lack of a suggestion of a global clinical effect in Fenton 2001, this study finds that EPA supplementation does not result in any movement disorders as measured by the AIMS and Simpson and Angus scales. If EPA did have some clinical effect, an issue that is still debatable, this apparent lack of adverse effects would become more important.
1.4 Leaving the study early Considering that some drug treatments gain a licence using data from studies with over 50% loss to follow up over similar time periods, it is good to see such low attrition (<10% total) in these trials. There is nothing to suggest that EPAs encourage loss to follow up.
2. COMPARISON 2: ANY DOSE OMEGA‐6 (GLA) versus PLACEBO 2.1 Tardive dyskinesia Wolkin 1986 investigates omega‐6 EFA supplementation for the chronic condition of tardive dyskinesia, a complication of long‐term use of antipsychotic drugs, and records AIMS ratings on 16 people after six weeks of treatment. No differences are found between treatment groups. This same small (n=16), short study finds no difference between groups for leaving before the end of trial. Nothing is proved or disproved by investigation in such a small short trial. The effects of omega‐6 EPAs for people with tardive dyskinesia are unproven.
3. COMPARISON 3: ANY DOSE OMEGA ‐3 (E‐EPA or EPA) versus ANY DOSE OMEGA ‐3 (DHA) 3.1 Mental state When comparing different types of omega‐3 supplementation it does appear as though E‐EPA is more effective at producing a greater than 25% change on the PANSS compared to DHA. This result, however, comes from one small (n=31) study (Peet 2001a) and therefore no firm conclusions should be made.
4. COMPARISON 4: SPECIFIC DOSES of OMEGA‐3 (E‐EPA) versus PLACEBO 4.1 Global and mental state Very low dose E‐EPA (<1 g/day) does not seem to have any clear effect on global or mental state when compared with placebo over 16 weeks, but numbers are very small and, as for all results in this review, inconclusive.
4.2 Adverse effects Most adverse effect data was derived from Peet 2002 (n=122, short term), and all events are rare and results are inconclusive. Any doses of E‐EPA, when compared with placebo, are not associated with frequent adverse effects. Adding the few data from the one medium‐term study (Fenton 2001) results in the synthesised results becoming heterogeneous, and this one study does suggest that there may be a link with the use of low dose E‐EPA supplementation and diarrhoea (n=87, 1 RCT, RR 17.39 CI 1.03 to 292).
4.3 Leaving the study early Within these studies, all doses of E‐EPA seem acceptable to people with schizophrenia.
Overall completeness and applicability of evidence
Randomised studies investigating the effects of polyunsaturated acids for those with schizophrenia have been carried out for over 20 years now, but the majority randomise relatively small numbers of people and none so far have been longer than 16 weeks. While assessing the short‐term effects of supplementation is important for the acute symptoms of schizophrenia such as mental state, the chronic symptoms of schizophrenia such as impaired global and social functioning may take longer to improve and such short‐term studies can not adequately address these problems.
Another issue with applicability is that all trials used manufactured enriched oils as supplements to antipsychotic medication rather than dietary supplementation with foods rich in EFAs. It was not possible to assess the effects of manufactured oils compared to oils obtained naturally from the diet. Access to these specific enriched supplements may be too difficult and expensive for many people.
Outcome reporting was mainly symptom and physician‐oriented. Patient‐oriented global and functional outcomes, such as social functioning, ability to work, patient and carer satisfaction and family burden were not reported. There is clearly a need for studies focusing not only on symptoms, but also on general and social functioning, family burden and patient acceptability.
The setting of trials were a mixture of hospital and community, reflecting the situation most people with schizophrenia experience.
Overall the completeness and applicability of evidence for those suffering from schizophrenia is poor. Despite realistic settings, many useful outcomes were not investigated and the important long‐term effects were also overlooked. It is also disappointing that recent trials are concentrating on investigating the effects of highly‐refined enriched oils rather than the possible effects simple dietary supplementation may have.
Quality of the evidence
Overall, the methodological quality of the eight included studies is good. The process of randomisation is, in the main, described and blindness is also described but remained untested. The studies are, however, small and short. Only Peet 2002 randomises over 100 people and only Fenton 2001 is over three months in duration. Despite good methodological quality, data reporting is poor, and the quality of evidence suffers. It is unfortunate that the largest study (Peet 2002) fails to report variances with the average scores thus rendering most of the scale‐derived data useless. It is also disappointing that the more recent studies, Berger 2007, Emsley 2002 and Emsley 2006, have no overall effect on the results of this review. Their main effect is to lower the quality of evidence and increase risk of bias with higher rates of selective outcome reporting and poor presentation of unusable data. With so few good quality data, all findings can still only be considered as hypothesis generating.
Potential biases in the review process
We have only worked with published reports and by doing this we may be perpetuating a reporting and publishing bias.
Agreements and disagreements with other studies or reviews
This review substantially updates past work in that two new studies are included. The findings of this review are similar to previous versions, but this is due to a lack of new data rather than new data confirming previous results. Although the conclusions remain the same, the positive findings of previous versions are now more questionable because of the new Risk of Bias table function (Figure 1 and Figure 2) of this version of RevMan. This table highlights more potential bias in the included trials and their likelihood of overestimating positive effects.
Authors' conclusions
Implications for practice.
1. For people with schizophrenia Those suffering from serious mental illnesses should find data on the effects of EFA supplementation inconclusive. Volunteering to be involved in ongoing studies may help resolve the issue, but it is important that the researchers record and release all clinically relevant data.
2. For clinicians Despite two new studies there is very little useful new data and there is currently still no reason for clinicians to either encourage or discourage the use of polyunsaturated fatty acids. If a person with schizophrenia wishes to use one then, perhaps, an omega‐3 preparation should be the preferred option. Clinicians should be able to prescribe an omega‐3 preparation as part of a well‐designed randomised trial.
3. For policy makers This review provides no evidence to support change in policy.
4. For funders The value of polyunsaturated fatty acids for treating schizophrenia is still unproven. The intriguing theory behind their use and even the possibility of a clinical effect could create a whole new path of research. Support of a substantial study would seem warranted.
Implications for research.
1. General By not fully implementing the CONSORT recommendations (Begg 1996, Moher 2001), trialists and editors have done a disservice to people with schizophrenia.
2. Specific There are still too few data on the role that essential fatty acid supplementation plays in the treatment of people with schizophrenia. Its use is based on relatively new theoretical hypotheses. The data in this review neither refute nor strongly support the need for further trials. Certainly future trials should be large (probably >150 per treatment arm), and their duration over one year (please see suggested study design Table 5). In addition, unless clinically relevant outcomes are measured, the real value of omega‐3 or omega‐6 EFA supplementation will remain in doubt.
1. Suggested design of study.
| Type of study | Patient | Intervention | Outcomes | Notes |
| Allocation: randomised, with sequence generation and concealment of allocation clearly described. Blindness: double, tested. Design: parallel group. Duration: 2, 6 and 12 months. | Diagnosis: any person with schizophrenia History: stable on antipsychotic medication. N=300.* Age: any. Sex: both. | 1. Neuroleptic medication + ethyl‐eicosapentaenoic acid: dose 3 g/day. N=150. 2. Neuroleptic medication alone. N=150. | Service use: readmission/relapse**, time in hospital, use of other psychotropic medications. Global response: CGI (binary).** Mental state: CGI (binary). Serious adverse effects: any**, list. Acceptability of treatment to patient, staff and carers (binary outcome). Economic outcomes. | * Size of study with sufficient power to highlight ˜10% difference between groups for primary outcome. ** Primary outcome. |
What's new
| Date | Event | Description |
|---|---|---|
| 5 October 2011 | Amended | Contact details updated. |
History
Protocol first published: Issue 4, 1998 Review first published: Issue 1, 1999
| Date | Event | Description |
|---|---|---|
| 4 August 2010 | Amended | Contact details updated. |
| 11 November 2009 | Amended | Plain language summary revised |
| 23 February 2009 | New search has been performed | New search carried out and two new included studies added. |
| 23 April 2008 | Amended | Converted to new review format. |
| 24 May 2006 | New citation required and conclusions have changed | Substantive amendment |
Notes
Previously published under the title of 'Polyunsaturated fatty acid (fish or evening primrose oil) for schizophrenia'.
Acknowledgements
The reviewers would like to thank Clive Adams and the Cochrane Schizophrenia Group editorial base staff past and present for advice and administrative support, Crispin Bennett for list of current research and clarification of types of EFA's, David Horrobin for advice and information about types of EFA's and Oliver Gillie for pointing out the lack of clarity in description of types of EFA's used in studies.
Appendices
Appendix 1. Original search
1. Search for the original review 1.1 Biological Abstracts on Silver Platter (1985 to February 1998) We used the Cochrane Schizophrenia Group's terms for randomised controlled trials and for schizophrenia combined with the phrase:
[and (phospholip* or EFA or EPA or MaxEPA or DHA or ALA or omega*) or (acid near1 (arachi* or docosahex* or fatty)) or (fish near3 oil*) or (fatty near3 acid* near3 (n‐3 or n‐6)) or (membran* near2 (hypothes* or neuronal*)) or (evening near3 primrose) or (oil near3 (flax or linseed))]
1.2 CINAHL on Silver Platter (1982 to February 1998) We used the Cochrane Schizophrenia Group's terms for randomised controlled trials and for schizophrenia combined with the phrase:
[and (phospholip* or EFA or EPA or MaxEPA or DHA or ALA or omega*) or (acid near1 (arachi* or docosahex* or fatty)) or (fish near3 oil*) or (fatty near3 acid* near3 (n‐3 or n‐6)) or (membran* near2 (hypothes* or neuronal*)) or (evening near3 primrose) or (oil near3 (flax or linseed)) or explode "FISH‐OILS"/ all topical subheadings / all age subheadings or explode "FATTY‐ACIDS"/ all topical subheadings / all age subheadings or explode "FATTY‐ACIDS,‐ESSENTIAL"/ all topical subheadings / all age subheadings or explode "FATTY‐ACIDS,‐OMEGA‐3"/ all topical subheadings / all age subheadings or explode "ARACHIDONIC‐ACIDS"/ all topical subheadings / all age subheadings or explode "PLANT‐OILS"/ all topical subheadings / all age subheadings or explode "PHOSPHOLIPIDS"/ all topical subheadings / all age subheadings]
1.3 The Cochrane Library (Issue 4, 1999) We used the Cochrane Schizophrenia Group's terms for randomised controlled trials and for schizophrenia combined with the phrase:
[and (phospholip* or EFA or EPA or MaxEPA or DHA or ALA or omega*) or (acid near1 (arachi* or docosahex* or fatty)) or (fish near3 oil*) or (fatty near3 acid* near3 (n‐3 or n‐6)) or (membran* near2 (hypothes* or neuronal*)) or (evening near3 primrose) or (oil near3 (flax or linseed)) or LIPIDS‐AND‐ANTILIPEMIC‐AGENTS*:ME or FATTY‐ACIDS*:ME or FATTY‐ACIDS‐OMEGA‐3*:ME or PHOSPHOLIPIDS*:ME or NEUROTRANSMITTERS‐AND‐NEUROTRANSMITTER‐AGENT*:ME or MEMBRANE‐LIPIDS*:ME]
1.4 The Cochrane Schizophrenia Group's Register (June 1998) We used the phrase:
[(phospholip* or EFA or EPA or MaxEPA or DHA or ALA or omega*) or (acid and (arachi* or docosahex* or fatty)) or (fish and oil*) or (fatty and acid* and (n‐3 or n‐6)) or (membran* and (hypothes* or neuronal*)) or (evening and primrose) or (oil and (flax or linseed))]
1.5 EMBASE (January 1980 to February 1998) We used the Cochrane Schizophrenia Group's terms for randomised controlled trials and for schizophrenia combined with the phrase:
[and (phospholip* or EFA or EPA or MaxEPA or DHA or ALA or omega*) or (acid near1 (arachi* or docosahex* or fatty)) or (fish near3 oil*) or (fatty near3 acid* near3 (n‐3 or n‐6)) or (membran* near2 (hypothes* or neuronal*)) or (evening near3 primrose) or (oil near3 (flax or linseed)) or explode "FISH‐OIL"/ all subheadings or explode "LINSEED‐OIL"/ all subheadings or explode "PRIMROSE‐OIL"/ all subheadings or explode "ARACHIDONIC‐ACID"/ all subheadings or explode "DOCOSAHEXAENOIC‐ACID"/ all subheadings or explode "OMEGA‐3‐FATTY‐ACID"/ all subheadings or explode "LIPID‐MEMBRANE"/ all subheadings or explode "NERVE‐CELL‐MEMBRANE"/ all subheadings or explode "FATTY‐ACID"/ all subheadings or explode "ESSENTIAL‐FATTY‐ACID"/ all subheadings or explode "MEMBRANE‐PHOSPHOLIPID"/ all subheadings or explode "PHOSPHOLIPID"/ all subheadings]
1.6 MEDLINE on Silver Platter (January 1966 to February 1998) We used the Cochrane Schizophrenia Group's terms for randomised controlled trials and for schizophrenia combined with the phrase:
[and (phospholip* or EFA or EPA or MaxEPA or DHA or ALA or omega*) or (acid near1 (arachi* or docosahex* or fatty)) or (fish near3 oil*) or (fatty near3 acid* near3 (n‐3 or n‐6)) or (membran* near2 (hypothes* or neuronal*)) or (evening near3 primrose) or (oil near3 (flax or linseed)) or explode "FISH‐OILS"/ all subheadings or explode "LINSEED‐OIL"/ all subheadings or explode "FATTY‐ACIDS"/ all subheadings or explode "FATTY‐ACIDS,‐ESSENTIAL"/ all subheadings or explode "FATTY‐ACIDS,‐OMEGA‐3"/ all subheadings or explode "DOCOSAHEXAENOIC‐ACIDS"/ all subheadings or explode "ARACHIDONIC‐ACIDS"/ all subheadings or explode "ARACHIDONIC‐ACID"/ all subheadings or explode "MEMBRANE‐LIPIDS"/ all subheadings or explode "CELL‐MEMBRANE"/ all subheadings or explode "SYNAPTIC‐MEMBRANES"/ all subheadings or explode "PHOSPHOLIPIDS"/ all subheadings]
1.7 PsycLIT on Silver Platter (January 1974 to February 1998) We used the Cochrane Schizophrenia Group's terms for randomised controlled trials and for schizophrenia combined with the phrase:
and (phospholip* or EFA or EPA or MaxEPA or DHA or ALA or omega*) or (acid near1 (arachi* or docosahex* or fatty)) or (fish near3 oil*) or (fatty near3 acid* near3 (n‐3 or n‐6)) or (membran* near2 (hypothes* or neuronal*)) or (evening near3 primrose) or (oil near3 (flax or linseed)) or explode "FATTY‐ACIDS"or explode "PHOSPHATIDES" or explode "LIPIDS" or explode "MEMBRANES" or explode NEUROTRANSMITTERS"]
We also inspected citations for additional terms, and if found we added these to the above searches and repeated the process.
1.8 Cited reference searching 1.8.1 ISI database ‐ Science Citation Index and Social Science Citation Index We sought each of the included studies as a cited reference on the above databases. We then inspected reports of articles that had cited these studies in order to identify further trials.
1.8.2 Reference lists We examined the references cited in all included trials in order to identify any missing studies.
1.9 Personal contact We contacted the authors of all studies initially selected for inclusion in order to identify further relevant trials.
Appendix 2. Previous update searches
1. The update of 2002 and 2005 1.1 Electronic searches We searched the Cochrane Schizophrenia Group's Register (July 2002 and July 2005) with the phrase:
[(*phospholip* or * EFA* or * EPA* or * MaxEPA* or * DHA* or * ALA* or * omega* or *arachi* or *docosahex* or *fish oil* or *fatty acid* or *evening primrose* or *flax* or *linseed* or *eicosapentaenoic* in title, abstract, index terms of REFERENCE)]
The Schizophrenia Groups trials register is based on regular searches of BIOSIS Inside; CENTRAL; CINAHL; EMBASE; MEDLINE and PsycINFO; the hand searching of relevant journals and conference proceedings, and searches of several key grey literature sources. A full description is given in the Group's module.
Appendix 3. Cochrane Schizophrenia Group trials register search strategy
[(*phospholip* or * EFA* or * EPA* or * MaxEPA* or * DHA* or * ALA* or *omega* or *arachi* or *docosahex* or *fish oil* or *fatty acid* or *evening primr* or *flax* or *linseed* or *eicosapentaenoic* or *polyunsaturat* in title, abstract, index terms of REFERENCE) or (*phospholip* or *docosahex* or *fish o* or *fatty a* or *evening p* or *eicosap* or *polyu* in intervention field in STUDY)]
This register is compiled by systematic searches of major databases, hand searches and conference proceedings (see Group Module). The Cochrane Schizophrenia Group Trials Register is maintained on Meerkat 1.5.1. This version of Meerkat stores references as studies. When an individual reference is selected through a search, all references which have been identified as the same study are also selected.
Data and analyses
Comparison 1. ANY DOSE OMEGA‐3 (E‐EPA or EPA ) versus PLACEBO.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Global state: 1. Needing standard neuroleptics by end of trial (short term) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 untreated when included in study | 1 | 30 | Risk Ratio (M‐H, Random, 95% CI) | 0.74 [0.54, 1.02] |
| 2 Global state: 2. Average scale score by end of trial (CGI, medium term, high=poor) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 3 Global state: 3. Average number of days on neuroleptics during trial (short term, skewed data) | Other data | No numeric data | ||
| 4 Global state: 4. Not achieving symptomatic response by 12 weeks ‐ all participants | 1 | 69 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.90 [0.50, 1.63] |
| 5 Global state: 5. Not achieving symptomatic response by 12 weeks ‐ nonaffective participants | 1 | 53 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.56 [0.25, 1.24] |
| 6 Mental state: 1. Not improved by end of trial (<25% improvement on PANSS scale, short term) | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 6.1 untreated when included in study | 1 | 30 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.54 [0.30, 0.96] |
| 6.2 receiving neuroleptics when included in study | 1 | 29 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.62 [0.37, 1.05] |
| 7 Mental state: 2. Average scale score by end of trial (PANSS, high=poor) | 3 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 7.1 untreated when included in study (short term) | 1 | 26 | Mean Difference (IV, Fixed, 95% CI) | ‐12.5 [‐22.38, ‐2.62] |
| 7.2 receiving neuroleptics when included in study (short term) | 1 | 29 | Mean Difference (IV, Fixed, 95% CI) | ‐10.40 [‐20.35, ‐0.45] |
| 7.3 receiving neuroleptics when included in study (medium term) | 1 | 87 | Mean Difference (IV, Fixed, 95% CI) | ‐1.0 [‐8.15, 6.15] |
| 8 Mental state: 3. Average scale score by end of trial (MADRS, medium term, high=poor, skewed data) | Other data | No numeric data | ||
| 9 Adverse events: Average scale score by end of trial (medium term, high=poor, skewed data) | Other data | No numeric data | ||
| 10 Adverse events: Average time to first tardive dyskinesia response | 1 | 77 | Mean Difference (IV, Fixed, 95% CI) | ‐0.40 [‐1.94, 1.14] |
| 11 Leaving the study early | 6 | 679 | Risk Ratio (M‐H, Random, 95% CI) | 0.85 [0.51, 1.40] |
| 11.1 short term | 5 | 345 | Risk Ratio (M‐H, Random, 95% CI) | 0.71 [0.31, 1.59] |
| 11.2 medium term | 3 | 334 | Risk Ratio (M‐H, Random, 95% CI) | 0.95 [0.50, 1.81] |
| 12 Adverse events: Scale change score by end of trial (ESRS) (skewed data) | Other data | No numeric data |
1.1. Analysis.

Comparison 1 ANY DOSE OMEGA‐3 (E‐EPA or EPA ) versus PLACEBO, Outcome 1 Global state: 1. Needing standard neuroleptics by end of trial (short term).
1.2. Analysis.

Comparison 1 ANY DOSE OMEGA‐3 (E‐EPA or EPA ) versus PLACEBO, Outcome 2 Global state: 2. Average scale score by end of trial (CGI, medium term, high=poor).
1.3. Analysis.
Comparison 1 ANY DOSE OMEGA‐3 (E‐EPA or EPA ) versus PLACEBO, Outcome 3 Global state: 3. Average number of days on neuroleptics during trial (short term, skewed data).
| Global state: 3. Average number of days on neuroleptics during trial (short term, skewed data) | ||||
|---|---|---|---|---|
| Study | Interventions | N | Mean | SD |
| Peet 2001b | EPA | 15 | 35.1 | 34.7 |
| Peet 2001b | Placebo | 15 | 65.3 | 18.9 |
1.4. Analysis.

Comparison 1 ANY DOSE OMEGA‐3 (E‐EPA or EPA ) versus PLACEBO, Outcome 4 Global state: 4. Not achieving symptomatic response by 12 weeks ‐ all participants.
1.5. Analysis.

Comparison 1 ANY DOSE OMEGA‐3 (E‐EPA or EPA ) versus PLACEBO, Outcome 5 Global state: 5. Not achieving symptomatic response by 12 weeks ‐ nonaffective participants.
1.6. Analysis.

Comparison 1 ANY DOSE OMEGA‐3 (E‐EPA or EPA ) versus PLACEBO, Outcome 6 Mental state: 1. Not improved by end of trial (<25% improvement on PANSS scale, short term).
1.7. Analysis.

Comparison 1 ANY DOSE OMEGA‐3 (E‐EPA or EPA ) versus PLACEBO, Outcome 7 Mental state: 2. Average scale score by end of trial (PANSS, high=poor).
1.8. Analysis.
Comparison 1 ANY DOSE OMEGA‐3 (E‐EPA or EPA ) versus PLACEBO, Outcome 8 Mental state: 3. Average scale score by end of trial (MADRS, medium term, high=poor, skewed data).
| Mental state: 3. Average scale score by end of trial (MADRS, medium term, high=poor, skewed data) | ||||
|---|---|---|---|---|
| Study | Interventions | N | Mean | SD |
| Fenton 2001 | EPA | 43 | 6.2 | 4.2 |
| Fenton 2001 | Placebo | 44 | 6.6 | 4.7 |
1.9. Analysis.
Comparison 1 ANY DOSE OMEGA‐3 (E‐EPA or EPA ) versus PLACEBO, Outcome 9 Adverse events: Average scale score by end of trial (medium term, high=poor, skewed data).
| Adverse events: Average scale score by end of trial (medium term, high=poor, skewed data) | |||||
|---|---|---|---|---|---|
| Study | Scale | Interventions | N | Mean | SD |
| Fenton 2001 | AIMS | EPA | 43 | 2.7 | 5.3 |
| Fenton 2001 | Placebo | 44 | 3.2 | 5.4 | |
| Fenton 2001 | SAS | EPA | 43 | 2.4 | 3.1 |
| Fenton 2001 | Placebo | 44 | 2.7 | 3.8 | |
1.10. Analysis.

Comparison 1 ANY DOSE OMEGA‐3 (E‐EPA or EPA ) versus PLACEBO, Outcome 10 Adverse events: Average time to first tardive dyskinesia response.
1.11. Analysis.

Comparison 1 ANY DOSE OMEGA‐3 (E‐EPA or EPA ) versus PLACEBO, Outcome 11 Leaving the study early.
1.12. Analysis.
Comparison 1 ANY DOSE OMEGA‐3 (E‐EPA or EPA ) versus PLACEBO, Outcome 12 Adverse events: Scale change score by end of trial (ESRS) (skewed data).
| Adverse events: Scale change score by end of trial (ESRS) (skewed data) | |||||
|---|---|---|---|---|---|
| Study | Subscale | Intervention | Mean score change | SD | Total |
| Emsley 2006 | Parkinson | E‐EPA | ‐0.8 | 3.2 | 39 |
| Emsley 2006 | Placebo | ‐1.1 | 3.3 | 38 | |
| Emsley 2006 | Dystonia | E‐EPA | 0.05 | 0.5 | 39 |
| Emsley 2006 | Placebo | 0.4 | 0.5 | 38 | |
| Emsley 2006 | Akathisia | E‐EPA | ‐0.1 | 0.4 | 39 |
| Emsley 2006 | Placebo | ‐0.06 | 0.7 | 38 | |
Comparison 2. ANY DOSE OMEGA‐6 (GLA) versus PLACEBO.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Tardive dyskinesia: Average scale score by end of trial (AIMS, short term, high=poor) | 1 | 16 | Mean Difference (IV, Fixed, 95% CI) | 1.30 [‐1.96, 4.56] |
| 2 Leaving the study early (short term) | 1 | 16 | Risk Ratio (M‐H, Random, 95% CI) | 1.0 [0.28, 3.54] |
2.1. Analysis.

Comparison 2 ANY DOSE OMEGA‐6 (GLA) versus PLACEBO, Outcome 1 Tardive dyskinesia: Average scale score by end of trial (AIMS, short term, high=poor).
2.2. Analysis.

Comparison 2 ANY DOSE OMEGA‐6 (GLA) versus PLACEBO, Outcome 2 Leaving the study early (short term).
Comparison 3. ANY DOSE OMEGA‐3 (EPA or E‐EPA) versus ANY DOSE OMEGA‐3 (DHA).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Mental state: 1. Not improved by end of trial (<25% improvement on PANSS scale, short term) | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 2 Mental state: 2. Percentage change in scale score by end of trial (PANSS, short term, high=good) | 1 | 31 | Mean Difference (IV, Fixed, 95% CI) | ‐9.80 [‐20.97, 1.37] |
| 3 Leaving the study early (short term) | 1 | 29 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
3.1. Analysis.

Comparison 3 ANY DOSE OMEGA‐3 (EPA or E‐EPA) versus ANY DOSE OMEGA‐3 (DHA), Outcome 1 Mental state: 1. Not improved by end of trial (<25% improvement on PANSS scale, short term).
3.2. Analysis.
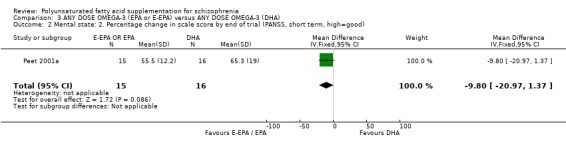
Comparison 3 ANY DOSE OMEGA‐3 (EPA or E‐EPA) versus ANY DOSE OMEGA‐3 (DHA), Outcome 2 Mental state: 2. Percentage change in scale score by end of trial (PANSS, short term, high=good).
3.3. Analysis.

Comparison 3 ANY DOSE OMEGA‐3 (EPA or E‐EPA) versus ANY DOSE OMEGA‐3 (DHA), Outcome 3 Leaving the study early (short term).
Comparison 4. SPECIFIC DOSES OF E‐EPA versus PLACEBO.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Global state: Average scale score by end of trial (CGI, medium term, high=poor) | 1 | 87 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [‐0.29, 0.29] |
| 1.1 E‐EPA (<1g/day) | 1 | 87 | Mean Difference (IV, Fixed, 95% CI) | 0.0 [‐0.29, 0.29] |
| 2 Mental state: 1. Average scale score by end of trial (PANSS, medium term, high=poor) | 1 | 87 | Mean Difference (IV, Fixed, 95% CI) | ‐1.0 [‐8.15, 6.15] |
| 2.1 E‐EPA (<1g/day) | 1 | 87 | Mean Difference (IV, Fixed, 95% CI) | ‐1.0 [‐8.15, 6.15] |
| 3 Mental state: 2. Percentage change in total scores by end of trial (PANSS, medium term, high =good, skewed) | Other data | No numeric data | ||
| 4 Adverse events: General 1. Experiencing at least one adverse effect (short term) | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 4.1 E‐EPA (<1g/day) | 1 | 63 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.97 [0.60, 1.58] |
| 4.2 E‐EPA (2g/day) | 1 | 63 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.67 [0.37, 1.20] |
| 4.3 E‐EPA (4g/day) | 1 | 58 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.15 [0.72, 1.82] |
| 5 Adverse events: Specific 1a. Gastrointestinal (diarrhoea) | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 5.1 E‐EPA (<1g/day) | 2 | 150 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.04 [0.91, 4.54] |
| 5.2 E‐EPA (2g/day) | 1 | 63 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.14 [0.02, 1.06] |
| 5.3 E‐EPA 4g/day | 1 | 58 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.49 [0.14, 1.72] |
| 6 Adverse events: Specific 1b. Gastrointestinal (nausea, short term) | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 6.1 E‐EPA (<1g/day) | 1 | 63 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.91 [0.12, 68.81] |
| 6.2 E‐EPA (2g/day) | 1 | 63 | Risk Ratio (M‐H, Fixed, 95% CI) | 6.79 [0.36, 126.24] |
| 6.3 E‐EPA (4g/day) | 1 | 56 | Risk Ratio (M‐H, Fixed, 95% CI) | 8.62 [0.47, 159.37] |
| 7 Adverse events: Specific 2. Liver and biliary tract (short term) | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 7.1 E‐EPA (<1g/day) | 1 | 63 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.91 [0.12, 68.81] |
| 7.2 E‐EPA (2g/day) | 1 | 63 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 7.3 E‐EPA (4g/day) | 1 | 63 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 8 Adverse events: Specific 4. Metabolic and nutritional (short term) | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 8.1 E‐EPA (<1g/day) | 1 | 63 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.97 [0.06, 14.82] |
| 8.2 E‐EPA (2g/day) | 1 | 63 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.97 [0.06, 14.82] |
| 8.3 E‐EPA (4g/day) | 1 | 58 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.44 [0.38, 31.20] |
| 9 Adverse events: Specific 5. Musculoskeletal (short term) | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 9.1 E‐EPA (<1g/day) | 1 | 63 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.32 [0.01, 7.65] |
| 9.2 E‐EPA (2g/day) | 1 | 63 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.32 [0.01, 7.65] |
| 9.3 E‐EPA (4g/day) | 1 | 58 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.38 [0.02, 8.98] |
| 10 Adverse events: Specific 6. Psychiatric (short term) | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 10.1 E‐EPA (<1g/day) | 1 | 63 | Risk Ratio (M‐H, Fixed, 95% CI) | 8.73 [0.49, 155.62] |
| 10.2 E‐EPA (2g/day) | 1 | 63 | Risk Ratio (M‐H, Fixed, 95% CI) | 4.85 [0.24, 97.11] |
| 10.3 E‐EPA (4g/day) | 1 | 58 | Risk Ratio (M‐H, Fixed, 95% CI) | 14.86 [0.88, 252.11] |
| 11 Adverse events: Specific 7. Reproductive (short term) | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 11.1 E‐EPA (<1g/day) | 1 | 63 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.32 [0.01, 7.65] |
| 11.2 E‐EPA (2g/day) | 1 | 63 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.32 [0.01, 7.65] |
| 11.3 E‐EPA (4g/day) | 1 | 58 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.30 [0.22, 23.94] |
| 12 Adverse events: Specific 8. Infections and respiratory system | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 12.1 E‐EPA (<1g/day) | 2 | 150 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.42 [0.95, 6.19] |
| 12.2 E‐EPA (2g/day) | 1 | 63 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.19 [0.02, 1.57] |
| 12.3 E‐EPA (4g/day) | 1 | 58 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.69 [0.18, 2.62] |
| 13 Adverse events: Specific 9. Skin (short term) | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 13.1 E‐EPA (<1g/day) | 1 | 63 | Risk Ratio (M‐H, Fixed, 95% CI) | 4.85 [0.24, 97.11] |
| 13.2 E‐EPA (2g/day) | 1 | 63 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.91 [0.12, 68.81] |
| 13.3 E‐EPA (4g/day) | 1 | 58 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.43 [0.15, 80.83] |
| 14 Adverse events: Specific 10. Urinary (short term) | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 14.1 E‐EPA (<1g/day) | 1 | 63 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.94 [0.18, 20.30] |
| 14.2 E‐EPA (2g/day) | 1 | 63 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.32 [0.01, 7.65] |
| 14.3 E‐EPA (4g/day) | 1 | 58 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.38 [0.02, 8.98] |
| 15 Adverse events: Specific 11. Vision (short term) | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 15.1 E‐EPA (<1g/day) | 1 | 63 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.97 [0.06, 14.82] |
| 15.2 E‐EPA (2g/day) | 1 | 63 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.32 [0.01, 7.65] |
| 15.3 E‐EPA (4g/day) | 1 | 58 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.38 [0.02, 8.98] |
| 16 Adverse events: Specific 12. Other (short term) | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 16.1 E‐EPA (<1g/day) | 1 | 63 | Risk Ratio (M‐H, Fixed, 95% CI) | 4.85 [0.24, 97.11] |
| 16.2 E‐EPA (2g/day) | 1 | 63 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.91 [0.12, 68.81] |
| 16.3 E‐EPA (4g/day) | 1 | 58 | Risk Ratio (M‐H, Fixed, 95% CI) | 8.0 [0.43, 148.25] |
| 17 Leaving the study early | 5 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 17.1 E‐EPA (<1g/day) | 2 | 153 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.61 [0.71, 3.67] |
| 17.2 E‐EPA (2g/day) | 3 | 307 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.17 [0.54, 2.55] |
| 17.3 E‐EPA (3g/day) | 1 | 40 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.0 [0.13, 69.52] |
| 17.4 E‐EPA (4g/day) | 1 | 58 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.30 [0.22, 23.94] |
4.1. Analysis.
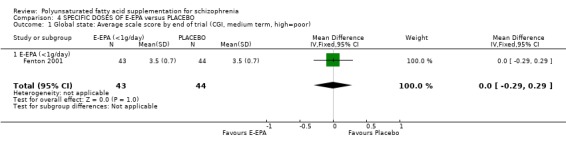
Comparison 4 SPECIFIC DOSES OF E‐EPA versus PLACEBO, Outcome 1 Global state: Average scale score by end of trial (CGI, medium term, high=poor).
4.2. Analysis.

Comparison 4 SPECIFIC DOSES OF E‐EPA versus PLACEBO, Outcome 2 Mental state: 1. Average scale score by end of trial (PANSS, medium term, high=poor).
4.3. Analysis.
Comparison 4 SPECIFIC DOSES OF E‐EPA versus PLACEBO, Outcome 3 Mental state: 2. Percentage change in total scores by end of trial (PANSS, medium term, high =good, skewed).
| Mental state: 2. Percentage change in total scores by end of trial (PANSS, medium term, high =good, skewed) | ||||
|---|---|---|---|---|
| Study | Interventions | N | Mean | SD |
| Emsley 2002 | E‐EPA (3g/day) | 20 | 12.6 | 12.6 |
| Emsley 2002 | Placebo | 20 | 3.1 | 13.3 |
4.4. Analysis.

Comparison 4 SPECIFIC DOSES OF E‐EPA versus PLACEBO, Outcome 4 Adverse events: General 1. Experiencing at least one adverse effect (short term).
4.5. Analysis.

Comparison 4 SPECIFIC DOSES OF E‐EPA versus PLACEBO, Outcome 5 Adverse events: Specific 1a. Gastrointestinal (diarrhoea).
4.6. Analysis.

Comparison 4 SPECIFIC DOSES OF E‐EPA versus PLACEBO, Outcome 6 Adverse events: Specific 1b. Gastrointestinal (nausea, short term).
4.7. Analysis.

Comparison 4 SPECIFIC DOSES OF E‐EPA versus PLACEBO, Outcome 7 Adverse events: Specific 2. Liver and biliary tract (short term).
4.8. Analysis.

Comparison 4 SPECIFIC DOSES OF E‐EPA versus PLACEBO, Outcome 8 Adverse events: Specific 4. Metabolic and nutritional (short term).
4.9. Analysis.

Comparison 4 SPECIFIC DOSES OF E‐EPA versus PLACEBO, Outcome 9 Adverse events: Specific 5. Musculoskeletal (short term).
4.10. Analysis.

Comparison 4 SPECIFIC DOSES OF E‐EPA versus PLACEBO, Outcome 10 Adverse events: Specific 6. Psychiatric (short term).
4.11. Analysis.

Comparison 4 SPECIFIC DOSES OF E‐EPA versus PLACEBO, Outcome 11 Adverse events: Specific 7. Reproductive (short term).
4.12. Analysis.

Comparison 4 SPECIFIC DOSES OF E‐EPA versus PLACEBO, Outcome 12 Adverse events: Specific 8. Infections and respiratory system.
4.13. Analysis.

Comparison 4 SPECIFIC DOSES OF E‐EPA versus PLACEBO, Outcome 13 Adverse events: Specific 9. Skin (short term).
4.14. Analysis.

Comparison 4 SPECIFIC DOSES OF E‐EPA versus PLACEBO, Outcome 14 Adverse events: Specific 10. Urinary (short term).
4.15. Analysis.

Comparison 4 SPECIFIC DOSES OF E‐EPA versus PLACEBO, Outcome 15 Adverse events: Specific 11. Vision (short term).
4.16. Analysis.

Comparison 4 SPECIFIC DOSES OF E‐EPA versus PLACEBO, Outcome 16 Adverse events: Specific 12. Other (short term).
4.17. Analysis.

Comparison 4 SPECIFIC DOSES OF E‐EPA versus PLACEBO, Outcome 17 Leaving the study early.
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Berger 2007.
| Methods | Allocation: randomised. Blindness: double. Duration: 12 weeks. Setting: inpatient and outpatient. | |
| Participants | Diagnosis: DSM‐IV first episode psychosis. N=80. Sex: 54M, 15F (NOTE only 69 participants analysed ‐ see risk of bias table). Age: 15‐29 years. Exclusions: drug induced psychosis, other organic brain disorders, history of intellectual disability, history of head injury. History: first episode psychosis, currently psychotic. | |
| Interventions | 1. 2 g ( 2 X 500 mg capsules, twice a day) purified E‐EPA (ethyl eicosapentaenoic acid) plus current antipsychotic medication. N=35.
2. 2 g (2 X 500 mg capsules, twice a day) placebo oil. N=34. Benzodiazepines, chlorpromazine, zuclopenthixol acetate (allowed for behaviour control) Zolpidem tartrate (allowed for insomnia) Selective serotonin reuptake inhibitors (allowed for depression) |
|
| Outcomes | Leaving the study early. Unable to use: Global state: time to first response, CGI, GAF scores (no mean, SD presented). Mental state: BPRS, SANS, CDSS scores (no usable data presented). Social functioning: SOFAS score (no usable data presented). Adverse effects: SAS, EPS, BAS UKU scores (no usable data presented). |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Adequate sequence generation? | Low risk | Randomised code list. |
| Allocation concealment? | Low risk | Sealed envelopes. |
| Blinding? All outcomes | Low risk | Both interventions in identical tasteless capsules. Key to allocation code kept from participants and researchers until end of data entry. |
| Incomplete outcome data addressed? All outcomes | Low risk | Numbers leaving study early described and explained ‐ all left before first analysis and code breaking. Analysis available for all remaining participants. Missing data from those who left early dealt with by multiple imputation. |
| Free of selective reporting? | High risk | Several outcomes not reported. |
| Free of other bias? | High risk | Main author consultant for several major antipsychotic drug companies. |
Emsley 2002.
| Methods | Allocation: randomised. Blindness: double. Duration: 12 weeks. Setting: unclear. Consent: written. | |
| Participants | Diagnosis: schizophrenia (DSM‐IV). N=40. Sex: not given. Age: 18 ‐ 55 years, mean ˜ 46 years. Exclusions: substance abuse, significant medical conditions. History: chronic, mean duration of illness˜23 years, Positive and Negative Syndrome Scale total score >50. | |
| Interventions | 1. Ethyl‐eicosapentaenoic acid: 3 g/day (three 500 mg capsules twice daily). N=20.
2. Placebo (liquid paraffin). N=20. Previous neuroleptic treatment remained constant throughout the trial. No other medication apart from analgesics for headaches and lorazepam for insomnia if required. |
|
| Outcomes | Mental state: PANSS scores.
Leaving the study early. Unable to use: Mental state: PANSS subscale scores (no mean, no SD). Adverse events: EPRS scores (no mean, no SD). |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Adequate sequence generation? | Unclear risk | No description given. |
| Allocation concealment? | Unclear risk | No description given. |
| Blinding? All outcomes | Unclear risk | No description given. |
| Incomplete outcome data addressed? All outcomes | Low risk | LOCF, ITT. |
| Free of selective reporting? | Low risk | All outcomes reported. |
| Free of other bias? | Unclear risk | Grant sponsored, medication supplied by Laxdale Ltd. |
Emsley 2006.
| Methods | Allocation: randomised. Blindness: double. Duration: 12 weeks. Setting: inpatient and outpatient from single site. Consent: written. | |
| Participants | Diagnosis: DSM‐IV schizophrenia or schizoaffective disorder with established TD. N= 84. Age: 18‐60 years. Sex: 51M, 26F* Exclusions: substance abuse, significant other medical or neurological illness, pregnancy, breastfeeding, receiving clozapine. History: stable, received fixed dose antipsychotic medication for at least 6 weeks prior to trial entry. | |
| Interventions | 1. 2 g/day E‐EPA: 2 X 500 mg capsules twice a day plus current antipsychotic medication. N=39.
2. Placebo: 2 X 500 mg capsules twice a day plus current antipsychotic medication. N=38. Allowed anticholinergic medication for treatment emergent EPS, anxiolytic or hypnotic medication for insomnia or anxiety, medication for any physical conditions. |
|
| Outcomes | Adverse effects: time to first response (TD).
Leaving the study early. Unable to use: Mental state: numbers with significant improvement, change in score on PANSS (no usable data presented). Adverse effects: numbers with significant improvement on CGI subscore for TD (no usable data presented), change in scores on ESRS (skewed data). |
|
| Notes | 84 randomised, 77 analysed* | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Adequate sequence generation? | Unclear risk | Assignment of numbers by independent clinical trials company but not described how achieved. |
| Allocation concealment? | Low risk | Identical sealed packs. |
| Blinding? All outcomes | Low risk | Identical sealed packs, identical capsules, code not broken until after trial. |
| Incomplete outcome data addressed? All outcomes | Low risk | LOCF, ITT. |
| Free of selective reporting? | Low risk | All outcomes reported but most data presentation unusable. |
| Free of other bias? | Unclear risk | Supported by grant from Stanley Medical/Alliance Research Institute, medication supplied by Amarin Neuroscience Ltd. |
Fenton 2001.
| Methods | Allocation: randomised. Blindness: double. Duration: 16 weeks. Setting: outpatient clinic. Consent: given. | |
| Participants | Diagnosis: schizophrenia or schizoaffective disorder (DSM‐IV). N=90. History: residual symptoms despite neuroleptic treatment. Sex: 70 men, 20 women. Age: 18‐65 years, mean˜40 years. Exclusions: substance abuse, bleeding disorder, mental deficiency, already taking fish oil supplements, anticoagulants, cholestyramine or clofibrate antilipaemic agents. | |
| Interventions | 1. Ethyl‐eicosapentaenoic acid: dose 500 mg/day + vitamin E. N=43.
2. Mineral oil placebo + vitamin E. N=44.* Previous neuroleptic treatment remained constant throughout trial. |
|
| Outcomes | Global state: CGI score. Mental state: PANSS, MADRS scores. Adverse events: AIMS, SAS scores, other observed events. Leaving the study early. | |
| Notes | * data for 87 participants only. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Adequate sequence generation? | Unclear risk | No description given. |
| Allocation concealment? | Unclear risk | No description given. |
| Blinding? All outcomes | Low risk | Identical capsules used, raters blind to treatment. |
| Incomplete outcome data addressed? All outcomes | Low risk | LOCF, ITT. |
| Free of selective reporting? | Low risk | All outcomes reported and presented with means and SD. |
| Free of other bias? | Unclear risk | Supported by grant from Stanley Medical/Alliance Research Institute. |
Peet 2001a.
| Methods | Allocation: randomised. Blindness: double. Duration: 12 weeks. Setting: outpatient clinic. Consent: given. | |
| Participants | Diagnosis: schizophrenia (DSM‐IV). N=55. History: symptomatic despite neuroleptic treatment. Sex: 30 men, 15 women*. Age:˜30‐56 years. Exclusions: other significant physical or psychiatric illness. | |
| Interventions | 1. Eicosapentaenoic acid enriched oil (containing 2 g of EPA). N=15.
2. Docosahexanoic acid oil (containing DHA)*. N=16.
3. Placebo (corn oil) N=14. Previous neuroleptic treatment remained constant throughout trail. |
|
| Outcomes | Mental state: PANSS improved, not improved. Leaving the study early. | |
| Notes | * only 45 participants analysed. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Adequate sequence generation? | Low risk | Unique randomisation number generated by a company independent to the trial. |
| Allocation concealment? | Low risk | Pre‐coded packages, code kept from researchers. |
| Blinding? All outcomes | Low risk | Identical capsules in appearance and taste, raters blind to allocation. |
| Incomplete outcome data addressed? All outcomes | Unclear risk | Loss to follow up described but how this data was dealt with not described. |
| Free of selective reporting? | Low risk | All outcomes reported. |
| Free of other bias? | High risk | Laxdale Ltd supplied trial medication and some financial support. |
Peet 2001b.
| Methods | Allocation: random assignment. Blindness: double. Duration: 12 weeks. Setting: psychiatric clinic. Consent: given. | |
| Participants | Diagnosis: schizophrenia (DSM‐IV ). N=30. History: either first episode or newly relapsed, all currently unmedicated. Sex: 18 men, 12 women. Age: ˜25‐45 years. Exclusions: not described. | |
| Interventions | 1. Eicosapentaenoic acid enriched oil (oil containing 2 g of EPA). N=15.
2. Placebo (corn oil). N=15. Aim of study was to keep participants unmedicated but if neuroleptics were required after randomisation, they were given. |
|
| Outcomes | Global state: need for neuroleptics. Mental state: PANSS score. Leaving the study early. | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Adequate sequence generation? | Low risk | Unique randomisation number generated by a company independent to the trial. |
| Allocation concealment? | Low risk | Pre‐coded packages, code kept from researchers. |
| Blinding? All outcomes | Low risk | Identical capsules in appearance and taste, raters blind to allocation. |
| Incomplete outcome data addressed? All outcomes | Unclear risk | Loss to follow up described but how this data was dealt with not described. |
| Free of selective reporting? | Low risk | All outcomes reported. |
| Free of other bias? | High risk | Laxdale Ltd supplied trial medication and some financial support. |
Peet 2002.
| Methods | Allocation: random assignment. Blindness: double. Duration: 12 weeks. Setting: outpatient clinic. Consent: given. | |
| Participants | Diagnosis: schizophrenia (DSM‐IV). N=122. History: symptomatic despite neuroleptic treatment. Sex: 66M, 39F*. Age: ˜18 ‐ 65 years. Exclusions: suffered from schizophrenia for over 20 years, other significant medical or neurological disorder, substance abuse, risk of self harm, taking of concurrent co comitant medication, pregnancy, participation in other study within 90 days of trial, abnormal lab results. | |
| Interventions | 1. Ethyl‐eicosapentenoic acid: dose 1 g/day. N=32.
2. Ethyl‐eicosapentaenoic acid: dose 2 g/day. N=32.
3. Ethyl‐eicosapentaenoic acid: dose 4 g/day. N=27.
4. Placebo. N=31. Previous neuroleptic treatment remained constant throughout trial. |
|
| Outcomes | Adverse effects: observed events.
Leaving the study early. Unable to use: Global state: CGI (no data). Mental state: MADRS, PANSS, (no SD). Compliance with medication (no data). Adverse effects: AIMS, BAS, LUNSERS, SAS, (no SD). |
|
| Notes | * only 115 participants analysed. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Adequate sequence generation? | Low risk | Unique randomisation number generated by a company independent to the trial. |
| Allocation concealment? | Low risk | Pre‐coded packages, code kept from researchers. |
| Blinding? All outcomes | Low risk | Identical capsules in appearance and taste, raters blind to allocation. |
| Incomplete outcome data addressed? All outcomes | Unclear risk | Loss to follow up described but how this data was dealt with not described. |
| Free of selective reporting? | High risk | Some outcomes had no data presentation in results. |
| Free of other bias? | High risk | Main author Dr. Malcolm Peet received research funding from Laxdale, Prof. Horrobin was an employee of Laxdale at the time and other members of the E‐E Multicentre Study Group received remuneration from Laxdale for the time spent on the study. Laxdale supplied medication for trial. |
Wolkin 1986.
| Methods | Allocation: random assignment. Blindness: double. Duration: 6 weeks. Setting: hospital and community. | |
| Participants | Diagnosis: schizophrenia (15), bipolar affective disorder (1). N=16. History: chronically ill, long exposure to antipsychotic drugs, mild TD over 1 yr. Sex: men. Age: average˜55 years. | |
| Interventions | 1. Omega‐6 EFA (gamma‐linolenic acid): dose 600 mg/day. N=8.
2. Placebo. N=8. Previous neuroleptic treatment remained constant throughout trial. |
|
| Outcomes | Adverse effects: AIMS. Unable to use: Leaving the study early (loss > 30%) Mental state: BPRS score (loss > 30%). |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Adequate sequence generation? | Unclear risk | No description given. |
| Allocation concealment? | Unclear risk | No description given. |
| Blinding? All outcomes | Unclear risk | No description given. |
| Incomplete outcome data addressed? All outcomes | High risk | Significant loss occurred but not addressed in text. |
| Free of selective reporting? | Low risk | All outcomes reported. |
| Free of other bias? | Unclear risk | Supported in part by Efamol ltd. |
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Amminger 2007 | Allocation: randomised. Participants: young adolescents at risk of schizophrenia or psychosis. |
| Glen 1996 | Allocation: randomised. Participants: people with DSM‐III‐R schizophrenia. Intervention: 500 mg/day oil of evening primrose vs placebo. Outcomes: all data unusable. |
| Heresco‐Levy 1996 | Allocation: randomised. Participants: people with treatment‐resistant chronic schizophrenia. Intervention: continued antipsychotic drugs + glycine vs continued antipsychotic drugs + placebo, no EFA. |
| Holman 1983 | Allocation: not stated, double‐blind. Participants: people with chronic schizophrenia. Intervention: continued antipsychotic drugs + EFA vs continued antipsychotic drugs + placebo. Outcomes: unable to use ‐ loss to follow up (> 50% & original group not reported); mental state (BPRS ‐ EFA group data only); and behaviour (MACC ‐ EFA group data only). |
| Maurer 2002 | Allocation: randomised. Participants: people with DSM‐IV schizophrenia. Intervention: haloperidol vs olanzapine. |
| Peet 1996 | Allocation: not randomised. |
| Puri 2001 | Allocation: randomised. Participants: people with Huntington's disease. |
| Silbergeld 1973 | Allocation: randomised. Participants: people with schizophrenia, depression, anxiety. Intervention: dexamethasone (1 mg/day) vs placebo. |
| Vaddadi 1986 | Two studies. Allocation: randomised. Participants: people with treatment‐resistant chronic schizophrenia. Intervention: Study 1. Continued antipsychotic drugs + EFA vs placebo depot injections + EFA. Study 2. Continued antipsychotic drugs + EFA vs placebo depot injections + placebo, not evaluating EFA supplementation. |
| Vaddadi 1989 | Allocation: randomised. Participants: people with schizophrenia, mild neuroleptic‐induced TD. Intervention: continued antipsychotic drugs + EFA + vitamin E vs continued antipsychotic drugs + placebo + vitamin E. Outcomes: unable to use ‐ loss to follow up (numbers not reported by group); mental state (CPRS ‐ no SD); movement disorders (AIMS ‐ no SD); and cognitive function (WMS ‐ no SD). |
Clinical Scales AIMS ‐ Abnormal Involuntary Movements Scale BPRS ‐ British Psychiatric Rating Scale CPRS ‐ Comprehensive Psychopathological Rating Scale MACC ‐ Motility Affect Cooperation Communication Scale WMS ‐ Wechsler Memory Scale
Miscellaneous EFA ‐ Essential fatty acid/s SD ‐ Standard deviation
Characteristics of ongoing studies [ordered by study ID]
Bentsen 2007.
| Trial name or title | A multicentre, placebo‐controlled trial of eicosapentaenoic acid (EPA) and antioxidant supplementation in the treatment of schizophrenia and related disorders. |
| Methods | Allocation: randomised. Blindness: double. Duration: 16 weeks. |
| Participants | Diagnosis: schizophrenia or related psychoses. N=99. Age: 18‐40 years. Sex: male and female. History: admitted within 21 days of trial, less than 15 years since first diagnosis. Exclusions: substance abuse, allergy to medication. |
| Interventions | 1. E‐EPA and/or Vitamins E+C plus regular medication. 2. Placebo. |
| Outcomes | Mental state: PANSS. Adverse effects: UKURS, lab results. |
| Starting date | 2007 |
| Contact information | |
| Notes | Has been finished, no data yet. |
Blanchet 2008.
| Trial name or title | Efficacy of docosahexaenoic acid on tardive dyskinesia. |
| Methods | Allocation: randomised. Blindness: double. |
| Participants | Diagnosis: people with tardive dyskinesia. |
| Interventions | 1. Omega‐3 fish oil capsules (including DHA). 2. Placebo. |
| Outcomes | Unknown. |
| Starting date | February 2008. |
| Contact information | |
| Notes |
Korbanic 2005.
| Trial name or title | CAD (coronary artery disease) risk in schizophrenia: effect of omega‐3 fatty acid supplementation. |
| Methods | Unknown. |
| Participants | Diagnosis: people with clinically stable schizophrenia. |
| Interventions | 1. Omega‐3 polyunsaturated fatty acid administration, lipid lowering drug administration. |
| Outcomes | Mental state: reduction of illness severity. Physical: reduction of coronary artery disease. |
| Starting date | Unknown. |
| Contact information | |
| Notes |
Murck 2001.
| Trial name or title | A multicentre, double‐blind, randomised, parallel group, placebo‐controlled, dose‐ranging pilot study of ethyl‐eicosapentaenoate (ethyl‐EPA) in patients with schizophrenia. |
| Methods | Allocation: randomised. Blindness: double. |
| Participants | Diagnosis: schizophrenia. |
| Interventions | 1. Varied doses of E‐EPA. 2. Placebo. |
| Outcomes | |
| Starting date | |
| Contact information | |
| Notes |
Richtand 2007.
| Trial name or title | Omega‐3 fatty acid deficiency replacement in early schizophrenia. |
| Methods | Allocation: randomised. Blindness: double. |
| Participants | Diagnosis: schizophrenia. |
| Interventions | 1. Dietary supplementation with omega‐3 fatty acids. 2. Placebo. |
| Outcomes | |
| Starting date | January 2008. |
| Contact information | |
| Notes |
EPA: Eicosapentaenoic acid. PANSS: Positive and negative syndrome scale.
Differences between protocol and review
We have substantially reformatted this review in light of changes to software (RevMan 5).
Contributions of authors
Claire Irving: protocol writing, searching, trial selection, data extraction, completion of report. Completion of 2002, 2005, 2008 update. Roger Mumby‐Croft: protocol writing, trial agreement, data agreement, help with completion of report. Advice and confirmation of trial status for the 2002 update. Tony Joy: protocol writing, trial agreement, help (as lay reader) with completion of report. Advice and confirmation of trial status for the 2002 update.
Sources of support
Internal sources
Cochrane Schizophrenia Group General Fund, UK.
School of Business, Oxford Brookes University, UK.
External sources
No sources of support supplied
Declarations of interest
None known.
Edited (no change to conclusions)
References
References to studies included in this review
Berger 2007 {published data only}
- Berger G, Wood S, Proffitt T, McConchie M, Khan A, O'Donnell C, Yuen H, Smith D, Horrobin D, McGorry P. Ethyl‐eicosapentaenoic acid (E‐EPA) supplementation in early psychosis. A double‐blind add on standard therapy in 80 drug naive or early treated first episode psychosis patients. Schizophrenia Research 2004;70(1):41. [Google Scholar]
- Berger GE, Proffitt TM, McConchie M, Yuen H, Wood SJ, Amminger GP, Brewer W, McGorry PD. Ethyl‐eicosapentaenoic acid in first‐episode psychosis: a randomised placebo‐controlled trial. Journal of Clinical Psychiatry 2007;68(12):1867‐75. [DOI] [PubMed] [Google Scholar]
- Berger GE, Proffitt TM, McConchie MA, Wood SJ, Yuen HP, McGorry PD. Ethyl‐Eicosapentaenoic acid (E‐EPA) supplementation in early psychosis. A double‐blind, randomised, placebo controlled trial (RCT) comparing 2g E‐EPA versus placebo add on therapy in 80 drug naive or early treated first episode psychosis. Schizophrenia Bulletin 2005;31:475. [Google Scholar]
- McConchie MA, Berger GE, Proffitt TM, Yuen HP, Wood S, smith D, Horrobin D, McGorry PD. Effect of diagnostic heterogeneity on response to ethyl‐eicosapentaenoic acid (EPA) in first episode psychosis (FEP). Proceedings of the 12th Biennial Winter Workshop on Schizophrenia; 2004 Feb 7‐13, Davos, Switzerland. Davos, 2004.
- Proffitt T, Wood S, Yuen H, McConchie M, Brewer W, Horrobin D, McGorry P, Berger G. Ethyl‐Eicosapentaenoic acid (E‐EPA) supplementation in first‐episode psychosis: effects on cognition mediated by lipid metabolic status. Schizophrenia Research 2004;70(1):40. [Google Scholar]
Emsley 2002 {published data only}
- Emsley R, Myburgh C, Oosthuizen P, Rensburg SJ. Randomised, placebo‐controlled study of ethyl‐eicosapentaenoic acid as supplemental treatment in schizophrenia. American Journal of Psychiatry 2002;159:1596‐8. [DOI] [PubMed] [Google Scholar]
Emsley 2006 {published data only}
- Emsley R. A double‐blind, randomised, parallel‐group comparison of ethyl‐eicosapentaenoic acid (e‐epa) versus placebo as add‐on medication in patients with established tardive dyskinesia. www.clinicaltrials.gov 2005.
- Emsley R, Niehaus DJH, Koen L, Oosthuizen PP, Turner HJ, Carey P, Rensburg SJ, Maritz JS, Murck H. The effects of eicosapentaenoic acid in tardive dyskinesia: a randomised placebo‐controlled trial. Schizophrenia Research 2006;84(1):112‐20. [DOI] [PubMed] [Google Scholar]
Fenton 2001 {published data only}
- Fenton W, Dickerson F, Boronow JJ, Hibbeln JR, Knable MB. A placebo‐controlled trial of omega‐3 fatty acid (ethyl eicosapentaenoic acid) supplementation for residual symptoms and cognitive impairment in schizophrenia. American Journal of Psychiatry 2001;158(12):2071‐4. [DOI] [PubMed] [Google Scholar]
- Fenton WS, Boronow J, Dickerson F, Hibbein J, Knable MB. Randomised trial of supplemental EPA for residual symptoms of schizophrenia. Biological Psychiatry 2000;47:S159‐60. [Google Scholar]
- Fenton WS, Dickerson F, Boronow JJ, Hibbein JR, Knable MB. Placebo‐controlled trial of omega‐3 fatty acid. Proceedings of the 155th Annual Meeting of the American Psychiatric Association; 2002 May 18‐23; Philadelphia,USA. Philadelphia, 2002.
Peet 2001a {published data only}
- Peet M. International multi‐centre, double‐blind, placebo controlled trial of EPA as an adjunct to existing neuroleptic treated schizophrenic patients. National Research Register 2001.
- Peet M, Brind J, Ramchand CN, Shah S, Vankar GK. Two double‐blind placebo‐controlled pilot studies of eicosapentaenoic acid in the treatment of schizophrenia. Schizophrenia Research 2001;49:243‐51. [DOI] [PubMed] [Google Scholar]
- Peet M, Laugharne J, Mellor J. Double‐blind trial of N3 fatty acid supplementation in the treatment of schizophrenia. Schizophrenia Research 1997;24:209. [Google Scholar]
- Peet M, Mellor J. Double‐blind, placebo controlled trial of N‐3 polyunsaturated fatty acids as an adjunct to neuroleptics. Schizophrenia Research 1998;29:160. [Google Scholar]
- Shah S, Vankar GK, Telang SD, Ramchand CN, Peet M. Eicosapentaenoic acid (EPA) as an adjunct in the treatment of schizophrenia. Schizophrenia Research 1998;29:158. [DOI] [PubMed] [Google Scholar]
Peet 2001b {published and unpublished data}
- Peet M, Brind J, Ramchand CN, Sha S, Vankar GK. Two double‐blind placebo‐controlled pilot studies of eicosapentaenoic acid in the treatment of schizophrenia. Schizophrenia Research 2001;49:243‐51. [DOI] [PubMed] [Google Scholar]
Peet 2002 {published and unpublished data}
- Horrobin DF. The role of phospholipids in schizophrenia. Proceedings of the 7th World Congress of Biological Psychiatry; 2001 July 1‐6; Berlin, Germany. Berlin, 2001.
- Horrobin DF, Bennett CN, Peet M. Correlation between clinical improvement and red cell fatty acid changes when treating schizophrenia with eicosapentaenoic acid. Proceedings of the 8th International Congress on Schizophrenia Research; 2001 April 28‐ May 2; British Columbia, Canada. British Columbia, 2001:232.
- Horrobin Df, Jenkins K, Bennett CN, Christie WW. Eicosapentaenoic acid and arachidonic acid: collaboration and not antagonism is the key to biological understanding. Prostaglandins, Leukotrienes and Essential Fatty Acids 2002;66(1):83‐90. [DOI] [PubMed] [Google Scholar]
- Peet M. A multi‐centre, double‐blind, randomised, parallel group, placebo controlled, dose ranging pilot study of ethyl eicosapentaenoate (ethly EPA) in patients with schizophrenia. National Research Register 2001.
- Peet M. Eicosapentaenoic acid (EPA) is effective in relieving schizophrenic symptoms in patients on clozapine. Proceedings of the 8th International Congress on Schizophrenia Research; 2001 April 28 ‐ May 2; British Columbia, Canada. British Columbia, 2001:241‐2.
- Peet M, Horrobin DF, Rowlands P, Murray R, Wet C, Kendall K, Abed R, Lloyd G, Barber M, Hellewell J, Stirling J, Hay A, Roberts S, Puri B, Lekh S. A dose‐ranging exploratory study of the effects of ethyl‐eicosapentaenoate in patients with persistent schizophrenic symptoms. Journal of Psychiatric Research 2002;36:7‐18. [DOI] [PubMed] [Google Scholar]
- Shah S, Ramchand CN, Peet M. Double blind pilot study of eicosapentaenoic acid (EPA) as the sole treatment for schizophrenia. Schizophrenia Research 2000;41(1):27. [DOI] [PubMed] [Google Scholar]
Wolkin 1986 {published data only}
- Wolkin A, Jordan B, Peselow E, Rubinstein M, Rotrosen J. Essential fatty acid supplementation in tardive dyskinesia. American Journal of Psychiatry 1986;143:912‐4. [DOI] [PubMed] [Google Scholar]
References to studies excluded from this review
Amminger 2007 {published data only}
- Amminger GP. Indicated prevention with omega‐3 fatty acids in adolescents with 'at‐risk‐mental‐state' for psychosis: a randomised, double blind, placebo‐controlled treatment trial. www.clinicaltrials.gov 2006.
- Amminger GP Schafer MR. Is it feasible to conduct a RCT in ultra‐high risk individuals at a child and adolescent psychiatric service?. Proceedings of the 5th International Conference on Early Psychosis; 2006 Oct 4‐6; Birmingham, UK. Birmingham, 2006.
- Amminger GP, Schafer MR. Indicated prevention with omega‐3 fatty acids in adolescents at ultra‐high risk for psychosis ‐ rationale, methods and 3‐months outcome. Proceedings of the 5th International Conference on Early Psychosis; 2006 Oct 4‐6, Birmingham, UK. Birmingham, 2006.
- AmmingerG, Schaefer MR, Papageorgiou K, Becker J, Mossaheb N, Harrigan SM, McGorry PD, Berger GE. Omega‐3 fatty acids reduce the risk of early transition to psychosis in ultra‐high risk individuals: a double‐blind randomised, placebo‐controlled treatment study. Schizophrenia Bulletin 2007;33(2):418‐9. [Google Scholar]
- Klier C, Hollmann M, Schlogelhofer M, Mossaheb N, Friedrich M, Amminger PG. Indicated prevention with omega‐3 fatty acids (epa/dha) in adolescents with "at‐risk‐mental‐state" for psychosis. Proceedings of the XIII World Congress of Psychiatry; 2005 Sept 10‐15; Cairo, Egypt. Cairo, 2005.
Glen 1996 {published data only}
- Glen AIM, Cooper SJ, Rybakowski J, Vaddadi K, Brayshaw N, Horrobin DF. Membrane fatty acids, niacin flushing and clinical parameters. Prostaglandins, Leukotrienes and Essential Fatty Acids 1996;55:9‐15. [DOI] [PubMed] [Google Scholar]
Heresco‐Levy 1996 {published data only}
- Heresco Levy U, Javitt DC, Ermilov M, Mordel C, Horowitz A, Kelly D. Double‐blind, placebo‐controlled, crossover trial of glycine adjuvant therapy for treatment‐resistant schizophrenia. British Journal of Psychiatry 1996;169:610‐7. [DOI] [PubMed] [Google Scholar]
Holman 1983 {published data only}
- Holman CP, Bell AFJ. A trial of evening primrose oil in the treatment of chronic schizophrenia. Journal of Orthomolecular Psychiatry 1983;12:302‐4. [Google Scholar]
Maurer 2002 {published data only}
- Maurer I, Volz HP, Czekalla J, Holstein W, Streck S, Winnefel K. Reduction of arachidonic acid levels by haloperidol in patients with schizophrenia. Schizophrenia Research 2002;53(3 suppl 1):209. [Google Scholar]
Peet 1996 {published data only}
- Laugharne JDE, Mellor JE, Peet M. Fatty acids and schizophrenia. Lipids 1996;31:163‐5. [DOI] [PubMed] [Google Scholar]
- Mellor JE, Laugharne JDE, Peet M. Omega‐3 fatty acid supplementation in schizophrenic patients. Human Psychopharmacology 1996;11:39‐46. [Google Scholar]
- Mellor JE, Laugharne JDE, Peet M. Schizophrenic symptoms and dietary intake of n‐3 fatty acids. Schizophrenia Research 1995;18:85‐6. [DOI] [PubMed] [Google Scholar]
- Peet M, Laugharne JDE, Mellor J, Ramchand CN. Essential fatty acid deficiency in erythrocyte membranes from chronic schizophrenic patients, and the clinical effects of dietary supplementation. Prostaglandins, Leukotrienes and Essential Fatty Acids 1996;55:71‐5. [DOI] [PubMed] [Google Scholar]
Puri 2001 {published data only}
- Puri BK, Bydder GM, Counsell SJ, Corridan BJ, Richardson AJ, Hajnal JV, Appel C, McKee HM, Vaddadi KS, Horrobin DF. MRI and neuropsychological improvement in huntington disease following ethyl‐EPA treatment. Clinical Neuroscience 2002;13(1):123‐6. [DOI] [PubMed] [Google Scholar]
Silbergeld 1973 {published data only}
- Silbergeld S, Noble E. Corticosteroids in psychiatric patients: Subacute and diurnal effects on free fatty acid and catecholamine metabolism. Journal of Psychiatric Research 1973;10:59‐71. [DOI] [PubMed] [Google Scholar]
Vaddadi 1986 {published data only}
- Vaddadi KS, Gilleard CJ, Mindham RH, Butler R. A controlled trial of prostaglandin E1 precursor in chronic neuroleptic resistant schizophrenic patients. Psychopharmacology Berlin 1986;88:362‐7. [DOI] [PubMed] [Google Scholar]
Vaddadi 1989 {published data only}
- Vaddadi KS, Courtney P, Gilleard CJ, Manku MS, Horrobin DF. A double‐blind trial of essential fatty acid supplementation in patients with tardive dyskinesia. Psychiatry Research 1989;27:313‐23. [DOI] [PubMed] [Google Scholar]
- Vaddadi KS, Gilleard CJ. Essential fatty acids, tardive dyskinesia and schizophrenia. Omega‐6 Essential Fatty acids: Pathophysiology and Roles in Clinical Medicine. Alan R, Liss. Inc, 1990:333‐43. [Google Scholar]
References to ongoing studies
Bentsen 2007 {published data only}
- Bentsen H, Lingjaerde O. A multicentre, placebo‐controlled trial of eicosapentaenoic acid (EPA) and antioxidant supplementation in the treatment of schizophrenia and related disorders. www.clinicaltrials.gov 2007.
- Landro NI, Bentsen H. The effects of a fatty acid and antioxidants on neuropsychological functioning in schizophrenia and related psychoses. Schizophrenia Bulletin 2007;33:529‐30. [Google Scholar]
Blanchet 2008 {published data only}
- Blanchet PJ, Stip E. Efficacy of docosahexaenoic acid on tardive dyskinesia. www.clinicaltrials.gov 2008.
Korbanic 2005 {published data only}
- Korbanic CJ, Yao JK. CAD risk in schizophrenia: effect of omega‐3 fatty acid supplementation. www.clinicaltrials.gov 2005.
Murck 2001 {published data only}
- Murck H. A multicentre, double blind, randomised, parallel group, placebo controlled dose ranging pilot study of ethyl‐eicosapentaenoate (ethyl‐epa) in patients with schizophrenia. Current Controlled Trials 2001.
Richtand 2007 {published data only}
- Richtand N. Omega‐3 fatty acid deficiency replacement in early schizophrenia. www.clinicaltrials.gov 2007.
Additional references
Altman 1996
- Altman DG, Bland JM. Detecting skewness from summary information. BMJ 1996;313:1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Begg 1996
- Begg C, Cho M, Eastwood S, Horton R, Moher D, Olkin I, Pitkin R, Rennie D, Schulz KF, Simel D, Stroup DFSO. Improving the quality of reporting of randomised controlled trials. The CONSORT statement [comment]. JAMA 1996;276:637‐9. [DOI] [PubMed] [Google Scholar]
Bland 1997
- Bland JM. Statistics notes. Trials randomised in clusters. BMJ 1997;315:600. [DOI] [PMC free article] [PubMed] [Google Scholar]
Christensen 1998
- Christensen O, Christensen E. Fat consumption and schizophrenia. Acta Psychiatrica Scandinavica 1998;78:587‐91. [DOI] [PubMed] [Google Scholar]
Di Marzo 1992
- Marzo V, Piomelli D. Participation of prostaglandin E2 in dopamine D2 receptor‐dependent potentiation of arachidonic response. Journal of Neurochemisty 1992;59:379‐82. [DOI] [PubMed] [Google Scholar]
Divine 1992
- Divine GW, Brown JT, Frazier LM. The unit of analysis error in studies about physicians' patient care behaviour. Journal of Internal Medicine 1992;7(6):623‐9. [DOI] [PubMed] [Google Scholar]
Donner 2002
- Donner A, Klar N. Issues in the meta‐analysis of cluster randomised trials. Statistics in Medicine 2002;21:2971‐80. [DOI] [PubMed] [Google Scholar]
Egger 1997
- Egger M, Davey‐smith G, Schneider M, Minder C. Bias in meta‐analysis, detected by a simple graphical test. BMJ 1997;315:629‐34. [DOI] [PMC free article] [PubMed] [Google Scholar]
Elbourne 2002
- Elbourne D, Altman DG, Higgins JPT, Curtina F, Worthingtond HV, Vaile A. Meta‐analyses involving cross‐over trials: methodological issues. International Journal of Epidemiology 2002;31(1):140‐9. [DOI] [PubMed] [Google Scholar]
Glen 1994
- Glen AIM, Glen EMT, Horrobin D, Vaddadi KS, Spellman M, Morse‐Fisher N, Ellisk, Skinner FS. A red cell membrane abnormality in a subgroup of schizophrenic patients: evidence for two diseases. Schizophrenia Research 1994;12:53‐61. [DOI] [PubMed] [Google Scholar]
Gulliford 1999
- Gulliford MC, Ukoumunne OC, Chinn S. Components of variance and intraclass correlations for the design of community‐based surveys and intervention studies: data from Health Survey for England 1994. American Journal of Epidemiology 1999;149:876‐83. [DOI] [PubMed] [Google Scholar]
Guy 1976
- Guy Q. ECDEU assessment manual for psychopharmacology. ADM 76‐338. Rockville, MD: US Department of Health, Education and Welfare, 1976. [Google Scholar]
Heresco‐Levy 1998
- Heresco‐Levy U, Javitt DCAD. The role of N‐methyl‐D‐aspartate (NMDA) receptor‐mediated neurotransmission in the pathophysiology and therapeutics of psychiatric syndromes. European Neuropsychopharmacology 1998;8:141‐52. [DOI] [PubMed] [Google Scholar]
Higgins 2003
- Higgins JPT, Thompson SG, Deeks JJ, Altman DG. Measuring inconsistency in meta‐analyses. BMJ 2003;327:557‐60. [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2008
- Higgins, JPT, Green S (editors). Cochrane Handbook for systematic reviews of interventions 5.0.1 [updated September 2008]. The Cochrane Collaboration, 2008. [Google Scholar]
Horrobin 1991
- Horrobin DF, Manku MS, Hillman H, Iain A, Glen M. Fatty acid levels in the brains of schizophrenics and normal controls. Biological Psychiatry 1991;30:795‐805. [DOI] [PubMed] [Google Scholar]
Horrobin 1994
- Horrobin DF, Glen AIM, Vaddadi KSO. The membrane hypothesis of schizophrenia. Schizophrenia Research 1994;13:195‐207. [DOI] [PubMed] [Google Scholar]
Hunter 2003
- Hunter RH, Gilbody SM, Joy CB, Kennedy E, Song F. Risperidone versus typical antipsychotic medication for schizophrenia. Cochrane Database of Systematic Reviews 2003, Issue 1. [DOI: 10.1002/14651858.CD000440] [DOI] [PubMed] [Google Scholar]
Jablensky 1992
- Jablensky A, Sartorius N, Ernberg G, Anker M, Korten A, Cooper JE, Day R, Bertelsen A. Schizophrenia: manifestations, incidence and course in different cultures. A World Health Organization ten‐country study. Psychological Medicine Monograph Supplement 1992;20:1‐97. [DOI] [PubMed] [Google Scholar]
Kay 1986
- Kay SR, Opler LA, Fiszbein A. Positive and Negative Syndrome Scale(PANSS) manual. North Tonawanda (NY): Multi‐Health Systems, 1986. [Google Scholar]
Marshall 2000
- Marshall M, Lockwood A, Bradley C, Adams C, Joy C, Fenton M. Unpublished rating scales: a major source of bias in randomised controlled trials of treatments for schizophrenia. British Journal of Psychiatry 2000;176:249‐52. [DOI] [PubMed] [Google Scholar]
Mellor 1995
- Mellor J, Laugharne JDE, Peet M. Schizophrenic symptoms and dietary intake of n‐3 fatty acids. Schizophrenia Research 1995;18:85‐6. [DOI] [PubMed] [Google Scholar]
Mellor 1996
- Mellor JE, Laugharne JDE, Peet M. Omega‐3 fatty acid supplementation in schizophrenic patients. Human Psychopharmacology 1996;11:39‐46. [Google Scholar]
Moher 2001
- Moher D, Schulz KF, Altman D. The CONSORT statement: revised recommendations for improving the quality of reports of parallel‐group randomised trials. JAMA 2001;285:1987‐91. [DOI] [PubMed] [Google Scholar]
Montgomery 1979
- Montgomery SA, Asberg M. A new depression scale designed to be sensitive to change. British Journal of Psychiatry 1979;134:382‐9. [DOI] [PubMed] [Google Scholar]
Nunez 1993
- Nunez EA, editor. Fatty acids and cell signalling. Prostaglandins Leukotrienes EFAs 1993;12:53‐61. [DOI] [PubMed] [Google Scholar]
Puri 1997
- Puri BK, Easton T, Richardson AJ. Normalisation of positive and negative symptoms of schizophrenia following dietary supplementation with essential fatty acids: a case study. Biological Psychiatry 1997;42:S189. [Google Scholar]
Simpson 1970
- Simpson GM, Angus JWS. A rating scale for extra‐pyramidal side‐effects. Acta Psychiatrica Scandinavica Supplementum 1970;212:11‐9. [DOI] [PubMed] [Google Scholar]
Ukoumunne 1999
- Ukoumunne OC, Gulliford MC, Chinn S, Sterne JAC, Burney PGJ. Methods for evaluating area‐wide and organisation‐based intervention in health and health care: a systematic review. Health Technology Assessment 1999;3(5):1‐75. [PubMed] [Google Scholar]
WHO 1998
- WHO. Global Health Statistics Online. Available: http//www.who.ch/msa/mnh/ems/rates/schizo.htm. 24th July 1998.
Wiersma 1998
- Wiersma D, Nienhuis FJ, Slooff CJ, Giel R. Natural course of schizophrenic disorders: a 15‐year follow‐up of a Dutch incidence cohort. Schizophrenia Bulletin 1998;24:75‐85. [DOI] [PubMed] [Google Scholar]
Xia 2007
- Xia J, Adams CE, Bhagat N, Bhagat V, Bhoopathi P, El‐Sayeh H, Pinfold V, Takriti Y. The Leeds Outcomes Stakeholders Survey (LOSS) Study. Proceedings of the 15th Cochrane Colloquium; 2007 Oct 23‐27; Sao Paulo. 2007.
References to other published versions of this review
Joy 2000
- Joy CB, Mumby‐Croft R, Joy LA. Polyunsaturated fatty acid supplementation (fish or evening primrose oil) for schizophrenia: a Cochrane review. Schizophrenia Research 2000;41(1):27. [DOI] [PubMed] [Google Scholar]