

Abstract
Throughout the central nervous system extracellular adenosine serves important neuroprotective and neuromodulatory functions. However, current understanding of the in vivo regulation and effects of adenosine is limited by the spatial and temporal resolution of available measurement techniques. Here, we describe an enzyme-linked microelectrode array (MEA) with high spatial (7500 μm2) and temporal (4 Hz) resolution that can selectively measure extracellular adenosine through the use of self-referenced coating scheme that accounts for interfering substances and the enzymatic breakdown products of adenosine. In vitro, the MEAs selectively measured adenosine in a linear fashion (r2=0.98±0.01, concentration range=0–15 μM, limit of detection = 0.96±0.5 μM). In vivo the limit of detection was 0.04±0.02 μM, which permitted real-time monitoring of the basal extracellular concentration in rat cerebral cortex (4.3±1.5 μM). Local cortical injection of adenosine through a micropipette produced dose-dependent transient increases in the measured extracellular concentration (200 nL: 6.8±1.8 μM; 400 nL: 19.4±5.3 μM) [P<0.001]. Lastly, local injection of dipyridamole, which inhibits transport of adenosine through equilibrative nucleoside transporter, raised the measured extracellular concentration of adenosine by 120% (5.6→12.3 μM) [P<0.001]. These studies demonstrate that MEAs can selectively measure adenosine on temporal and spatial scales relevant to adenosine signaling and regulation in normal and pathologic states.
Keywords: Amperometry, Voltammetry, Microdialysis
1. Introduction
Extracellular adenosine is an important neuromodulator of the central nervous system affecting heart rate (Drury and Szent-Gyorgyi, 1929; Tupone et al., 2013), sleep (Bjorness et al., 2009; Huang et al., 2014), and breathing (Spyer and Thomas, 2000). Accumulations of extracellular adenosine are also a vital part of a neuroprotective negative feedback loop to reduce synaptic activity and increase delivery of energy substrates (Dunwiddie and Masino, 2001). During periods of high energy demand, the extracellular concentration rises through dephosphorylation of adenine nucleotides by ecto-nucleotidases and transport from the intracellular environment (Illes and Zimmermann, 1999). In the extracellular space, activation of A1 receptors suppresses synaptic transmission, while activation of A2A receptors increases delivery of metabolic substrates (Dunwiddie, 1980; Pascual et al., 2005; Pedata et al., 2014). As a result, adenosine has exhibited neuroprotective effects in a variety of pathologic situations including: ischemia (Chen et al., 2014; Cui et al., 2013; Laghi Pasini et al., 2000; Weigand et al., 1999), trauma (Burnstock, 2015; Clark et al., 1997; Robertson et al., 2001), epilepsy (Dunwiddie and Worth, 1982; Masino et al., 2014; Zhang et al., 1990), cortical spreading depolarization (Lindquist and Shuttleworth, 2012, 2014), and seizure (During and Spencer, 1992; Miranda et al., 2014; Van Gompel et al., 2014).
Insights into the regulation of extracellular adenosine have been limited by a lack of selective in vivo techniques that are capable of quantifying the extracellular concentration with sufficient spatial/temporal resolution. Microdialysis (MD), a semi-permeable membrane that permits passive diffusion of neurochemicals from the extracellular space into the collection fluid for analysis, has been used over the past 30 years to measure extracellular adenosine (Hagberg et al., 1987). While MD is capable of selectively quantifying the extracellular concentration, the poor temporal (min–hrs) and spatial (mm) resolution of the technique limits the ability to discern changes on the temporal and spatial scales of physiologic and pathologic processes (Park and Gidday, 1990; Pedata et al., 1993). To overcome these methodological limitations, electrochemical detection methods have been developed that use a combination of three enzymes – nucleoside phosphorylase (NP), xanthine oxidase (XO), and adenosine deaminase (ADA) – to convert adenosine into the reporter molecule, H2O2 (Dale, 1998). The first generation probe, which consisted of two similar sensors one sensitive to adenosine and the other sensitive to the enzymatic breakdown products, provided a means to selectively quantify the extracellular concentration with improved spatial (surface area ~200 mm2) and temporal (1–2 min) resolution (Dale et al., 2000). However, the relatively large size of the probe limited the response time and produced considerable damage to the parenchyma, which may confound the physiological measurements (Llaudet et al., 2003). As such, second generation sensors were developed with improved spatial (surface area ~25 mm2) and temporal (< 5 s) resolution (Llaudet et al., 2003), but suffered from reduced selectivity as they were also sensitive to the adenosine enzymatic breakdown products that inhibited quantification of the extracellular concentration (Llaudet et al., 2003). Recently, a new class of adenosine electrodes has been developed that utilize the specific oxidation properties of adenosine combined with fast scan cyclic voltammetry (FSCV) to produce a selective sensor with excellent spatial (surface area ~1.5 mm2) and temporal (< 1 s) resolution (Nguyen et al., 2014; Swamy and Venton, 2007). While this technique enables rapid monitoring of extracellular adenosine in discrete functional circuits, methodological limitations of FSCV preclude quantification of the basal concentration (Nguyen and Venton, 2015).
Here, we describe a novel technique using enzyme-linked microelectrode arrays (MEA) and a self-referencing methodology to selectively quantify the extracellular adenosine concentration with excellent temporal (4 Hz) and spatial (surface area 7.5 mm2) resolution that is capable of monitoring discrete functional brain circuits, such as sub-regions of the hippocampus (Hinzman et al., 2010a; Stephens et al., 2011), prefrontal cortex (Mattinson et al., 2011; Miller et al., 2015), and whisker-barrel cortex (Thomas et al., 2012). Our initial results suggest MEAs are capable of selectively monitoring the extracellular adenosine concentration with sufficient temporal resolution to study the intrinsic regulatory mechanisms.
2. Methods
2.1. Animals
Six male Sprague-Dawley rats weighing 325–400 g (Harlan Laboratories, Inc.) were used in the experiments. Animals were exposed to a 12 h light/dark cycle, with food and water available ad libitum in accordance with the standards of the Association for Assessment and Accreditation of Laboratory Animal Care International. All procedures were performed during the light cycle, were approved by the University of Cincinnati Institutional Animal Care and Use Committee, and conformed to the Animal Welfare Act and the National Institutes of Health Guide for the Care and Use of Laboratory Animals (National Research Council).
2.2. Microelectrode Array Preparation
MEAs consisted of four platinum (Pt) recording sites (50 × 150 μm2) arranged vertically on the tip of the electrode with 50 μm spacing between each site (Quanteon, LLC, Lexington, KY). The MEAs were configured in a self-referenced manner, where the two distal electrodes were sensitive to adenosine and the two proximal electrodes served as sentinel sites that were sensitive to both interfering molecules and the enzymatic breakdown products of extracellular adenosine, but not adenosine (Fig. 1). This provided a more selective measure and permitted determination of basal extracellular adenosine concentrations (Burmeister and Gerhardt, 2001; Burmeister et al., 2002). The distal recording sites were coated with an ADA enzyme solution that consisted of 1% bovine serum albumin (BSA) (Sigma-Aldrich, St. Louis, MO), 0.125% glutaraldehyde (Sigma-Aldrich), 0.5 units of xanthine oxidase (XO) (Sigma-Aldrich) and 0.5 units of nucleoside phosphorylase (NP) (Sigma-Aldrich), and 2 units of adenosine deaminase (ADA) (Worthington Biochem, Lakewood, NJ). A small drop, approximately 0.1 μL, of the ADA containing solution was applied to the distal pair of Pt recording sites manually using a dissecting microscope and a microsyringe. The solution was allowed to dry for one minute, and repeated two more times for a total of three coats. For the sentinel sites a similar solution was prepared including all of the above components except for ADA. This solution was used to coat the two proximal sites of the MEA in the same fashion. The MEAs were then cured for 24 h at 25 °C. To restrict interference from large molecules in vivo (e.g., ascorbic acid), a size-exclusion layer of 1,3-phenylenediamine (mPD) was electroplated onto the Pt recording sites prior to in vitro calibration. The electroplating procedure was as follows: MEAs were connected to the FAST-16 mkI system (Fast Analytical Sensor Technology Mark I; Quanteon, L.L.C., Nicholasville, KY) and the Pt recording site area (tip) of the MEA was placed in a 5 mM mPD solution (Acros Organics, Morris Plains, NJ). A triangular potential wave with ±0.25 V peak amplitude, offset of −0.5 V, and frequency of 0.05 Hz was then applied for 20 min (Hinzman et al., 2010b, 2012).
Fig. 1.
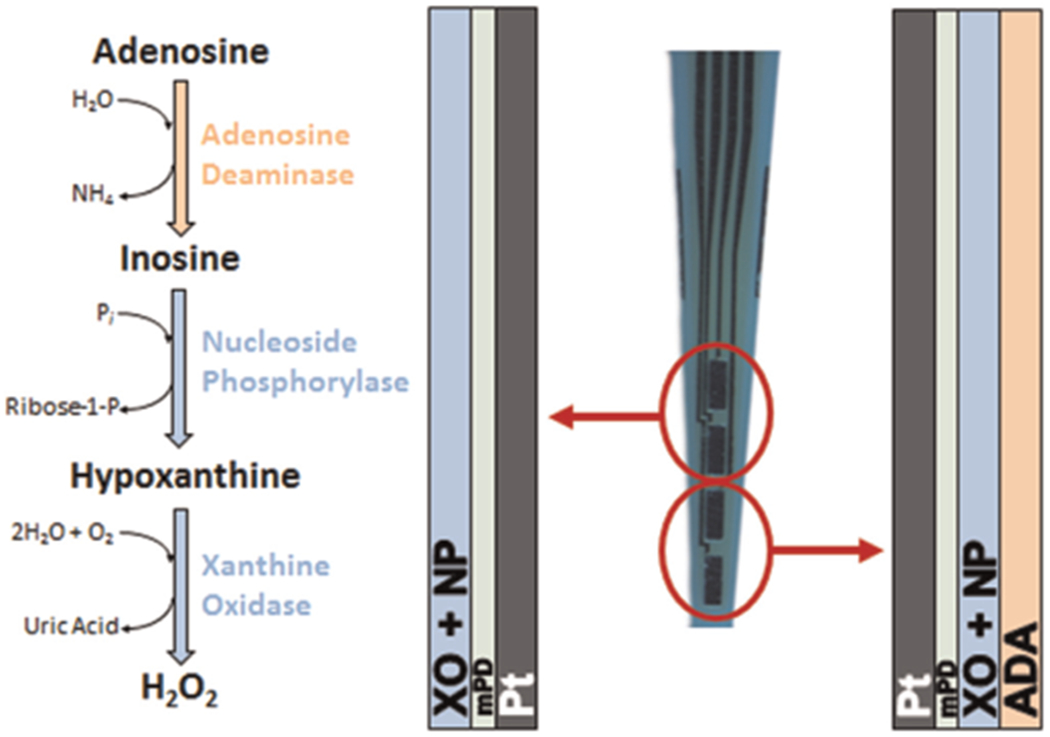
Microelectrode array coating scheme for adenosine measures. Diagram depicts chemical reactions and enzymatic coating scheme applied to the platinum (Pt) recording sites of each microelectrode array (MEA). Generation of the reporter molecule, H2O2, from extracellular adenosine required three enzymes (ADA, NP, XO). The distal recording sites were coated with ADA, XO, and NP, while the proximal sites were coated with only XO and NP. An exclusion layer (mPD) prevented large-molecule interferents (ascorbic acid) from reaching the Pt surface, while still allowing passage of H2O2 for oxidation. Pt recording sites 50 × 150 μm2.
2.3. MEA calibration
In vitro calibrations were conducted prior to in vivo experimentation to assess the capability of the MEAs to selectively measure adenosine. Constant potential amperometry, +0.7 V versus an Ag/AgCl reference electrode, was performed to oxidize the reporter molecule H2O2. This two-electron oxidation reaction, which occurs at the Pt recording sites, generated a current which was then amplified and digitized by the FAST-16 mkI system (Quanteon, LLC). During in vitro calibration, the MEAs were placed in a continuously-stirred 40 mL solution of 0.05 M phosphate-buffered saline maintained at 37 °C with a recirculating water bath (Gaymar Industries Inc., Orchard Park, NY).
2.4. In vivo drug delivery
For local application of solutions into the rat brain, a glass micropipette (1.5 mm outside diameter, 0.86 mm inside diameter; Sutter Instruments, Novato, CA) was pulled and bumped to form an outer tip diameter of ~10 μM (P-97, Sutter Instruments). The micropipette was fixed to the shank of the MEA recording site using sticky wax, with the tip of the pipette secured ~300 μm above the most distal recording site. The micropipette was filled with a solution of either 200 μM adenosine (Sigma-Aldrich) or 200 μM dipyridamole (DPR) (Sigma-Aldrich), a competitive equilibrative nucleoside transporter (ENT) inhibitor that produces an increase in extracellular adenosine levels in vivo (Park and Gidday, 1990). All drugs were dissolved to their final concentrations in physiological saline, sterile filtered (0.20 μm), and adjusted to a pH of 7.4. The micropipette used for drug delivery was connected via tubing to a Picospritzer III (Parker-Hannifin, Cleveland, OH) with settings adjusted to consistently deliver volumes between 200 and 400 nL. Pressure was applied from 10–30 psi for 0.3–3 s and volume displacement was monitored via stereomicroscope.
2.5. In vivo recordings
Animals were anesthetized throughout the duration of the procedures. Induction was achieved with 5% isoflurane for five minutes, after which the surgical site was prepared by shaving and applying 70% alcohol and betadine solution. The rat’s eyes were then treated with artificial tears, a rectal thermometer was inserted, and an Ag/AgCl reference electrode was placed subcutaneously in the neck. During recordings, anesthesia was maintained at 2–3% isoflurane (30% oxygen, 70% nitrous oxide) and body temperature was maintained at 37 °C with a homeothermic heading pad (Harvard Apparatus, Holliston, MA). Rats were mounted in a stereotaxic frame and the skull was exposed using a midline scalp incision with retraction of the right temporalis muscle. A craniotomy was made over parietal cortex (~2 mm2; ML: + 2→+4 mm and AP: −1→−2 mm from bregma). The dura was incised with a 30 gauge needle and the exposed brain was kept moist with physiologic saline irrigation. The MEA and attached micropipette were implanted into the cortex avoiding major vasculature so that the most proximal recording site resided just below the pial surface. At the conclusion of the experiment, rats were euthanized by overdose of anesthesia and decapitation.
3. Experimental design and data analysis
All recordings were performed at a frequency of 4 Hz using constant potential amperometry. After the MEA reached a stable baseline (10–15 min), the basal extracellular adenosine concentration was calculated as a 10 s average prior to the first drug application. The concentration was calculated by subtracting the background current from the sentinel sites (−ADA) from the current of the adenosine sensitive sites (+ADA) and then converting the resulting current (pA) into concentration by dividing by the sensitivity (pA/μM) obtained during the calibration. The smallest amount the adenosine-sensitive sensors could detect (limit of detection, LOD) was calculated as 3 times the standard deviation of the baseline noise over a 10 s period. To examine the effect of locally applied adenosine on the extracellular adenosine concentration, adenosine was injected through the micropipette every five minutes and the corresponding signals were analyzed by the following parameters: peak amplitude (μM); Trise (s), the time for the signal to reach the peak amplitude; T80 (s), the time for the signal to decay by 80% from the peak amplitude; duration (s); and uptake rate (μM/s) (s−1), which is based on the slope of the linear regression of the natural log transformation of the decay over time (T80) (Hinzman et al., 2010b; Nickell et al., 2007; Thomas et al., 2009). To examine the effect of the competitive uptake inhibitor, DPR, on the extracellular adenosine concentration, the drug was locally applied every 5 min and the maximum increase in extracellular adenosine and total duration were calculated for each signal. Amperometric data were analyzed using custom MATLAB® program. All data were analyzed using a two-tailed t-test. Data are presented as mean ±SD, and statistical significance was defined as P<0.05.
4. Results
4.1. In vitro detection of adenosine
Calibrations were conducted prior to in vivo use to test the capability of the MEAs to selectively measure adenosine and to generate a standard curve for the conversion of current to adenosine concentration (Fig. 2). After the sensor reached a stable baseline (~10–15 min), the LOD of the sensors was calculated as 3 times the standard deviation of the baseline noise in the 10 s prior to the addition of ascorbic acid. Then, MEAs were exposed to ascorbic acid (250 μM), the major interfering substance in the brain, to ensure the mPD exclusion layer prevented ascorbic acid from reaching the Pt surface. Then, MEAs were exposed to three sequential concentrations of adenosine (5, 10, 15 μM) to ensure a selective and linear enzymatic (ADA, NP, XO) production of H2O2 (Fig. 2). Next, single additions of inosine (5 μM) and xanthine (5 μM) allowed verification of proper enzymatic performance of NP and XO to produce H2O2. In addition, two sensors were exposed to other possible interfering substances (dopamine and norepinephrine, 2 μM) to ensure the mPD size-exclusion layer prevented these compounds from reaching the platinum surface. Lastly, the MEAs were exposed to 8.8 μM H2O2 as a positive control to confirm MEA performance.
Fig. 2.
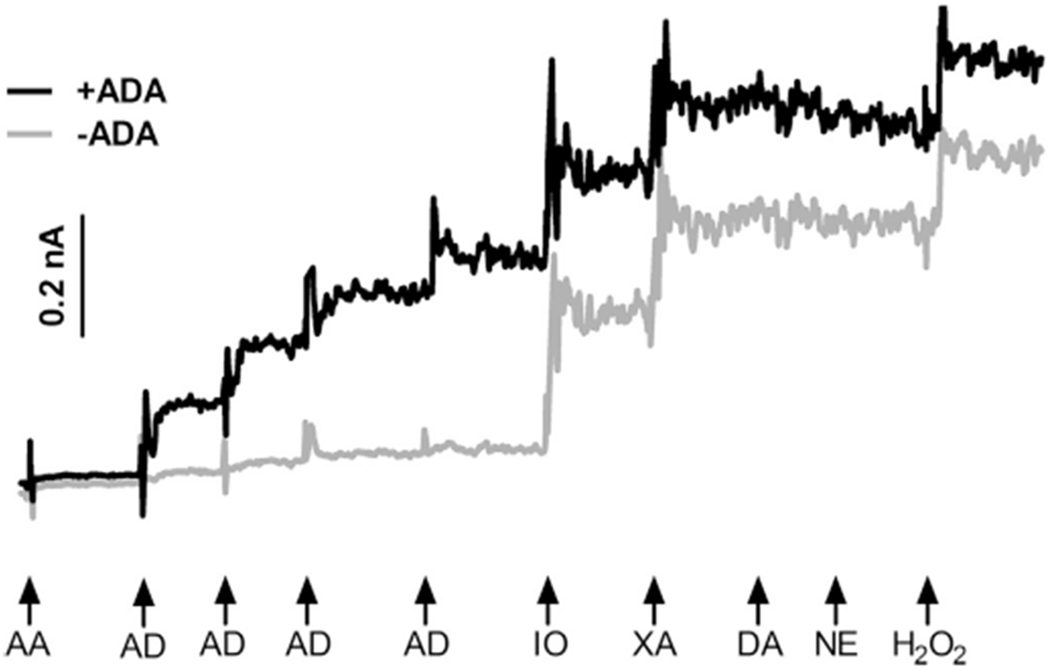
In vitro calibration. Representative amperometric current tracings of the ADA-sensitive recording site (+ADA) and a sentinel recording site (−ADA). Addition of the major interferent ascorbic acid (AA) produced a minor increase in current as the mPD exclusion layer prevented oxidation at the Pt surface. Three sequential additions of adenosine (AD) produced a step-wise increase in current on the +ADA sites. Additions of of inosine (IO) and xanthine (XA) produced similar increases in current on both sites, ensuring enzymatic performance. Sensors were insensitive to additions of other potential interfering substances, dopamine (DA) and norepinephrine (NE). Lastly, addition of H2O2 confirmed similar electrode performance.
The in vitro performance of the sensors was sufficient for in vivo detection of extracellular adenosine. The LOD of the sensors was 0.96±0.5 μM and the selectivity of the sensors to measure adenosine compared to the major interfering substance in the brain (the number of molecules of adenosine detected for each molecule of ascorbic acid) was 106±96:1.The MEAs responded to adenosine in a linear fashion (r2=0.98±0.01) with an average slope of 40.2±2.8 pA/μM (n=14 sites). Thus, in vitro, the MEAs selectively measured adenosine in a linear fashion.
4.2. In vivo detection of exogenously applied adenosine
To examine MEA performance in vivo, the MEA with an attached micropipette filled with 200 μM adenosine was stereotactically implanted into the rat cortex. Using the self-referencing method, subtraction of the current generated upon enzymatic breakdown of both inosine and xanthine (−ADA) from the adenosine signal (+ADA) provided a selective measure of extracellular adenosine. After the MEA reached a stable baseline (~10 min), the basal extracellular adenosine concentration (4.3±1.5 μM, n=6) was averaged over a 10 s period prior to the first drug application. The in vivo LOD of the MEAs (0.04±0.02 μM) was also calculated during this period as 3 times the standard deviation of the baseline noise. Local application of 200 nL into the cortex transiently and reproducibly increased the current on the sensor with ADA and to a lesser extent on the sensor without ADA (Fig. 3). The average amplitude of the adenosine signal (n=9) was 6.8±1.8 μM with a rise time of 5.7±2.1 s. The average time for signal to decay by 50% (T50) was 17.7±8.8 s and by 80% (T80) was 32.9±12.2 s producing an uptake rate of 0.028±0.020 μM/s. Local application of 400 nL of adenosine produced significantly larger measured concentrations (19.4±5.3 μM) [t(13)=6.6, P<0.001]. Thus, in vivo the MEAs could measure increases in extracellular adenosine from exogenous application in a dose-dependent manner.
Fig. 3.
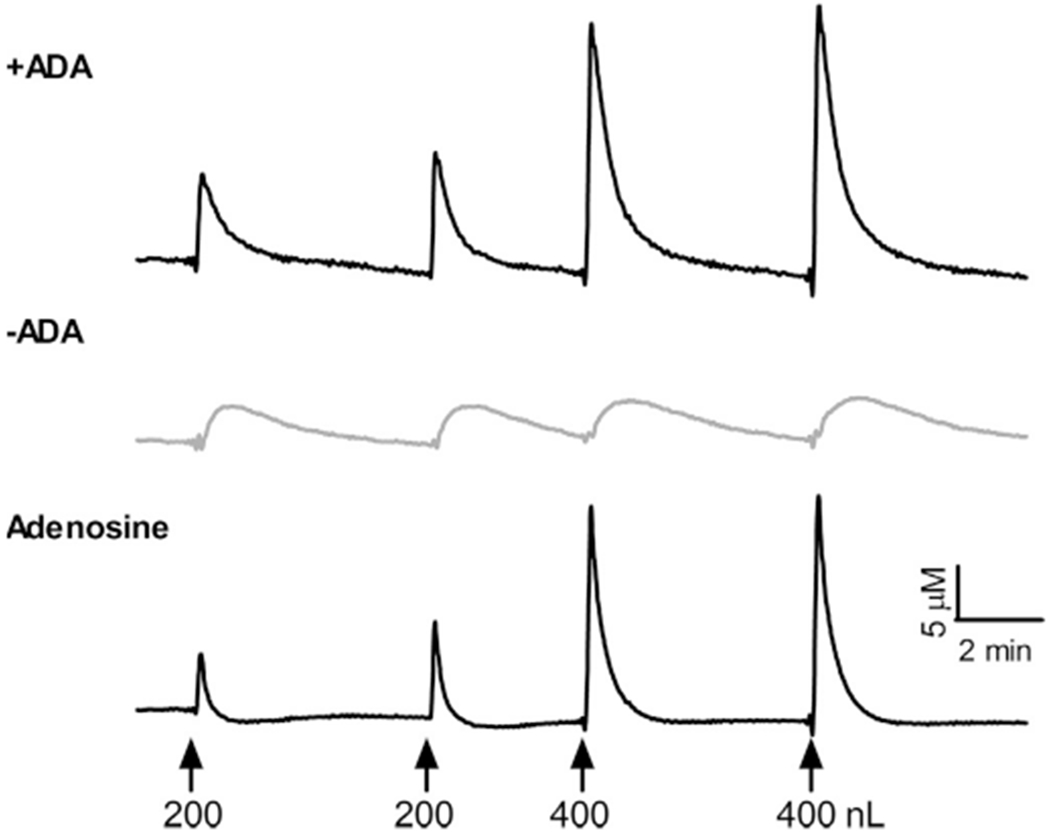
In vivo detection of exogenously applied adenosine. Representative in vivo recording of adenosine-sensitive recording site (+ADA), sentinel recording sites (−ADA), and subtracted signal depicting increases in extracellular adenosine during local injection 200 μM adenosine. Exogenous application of adenosine produced transient increases in extracellular adenosine in a dose-dependent manner (↑ time of injection).
4.3. In vivo detection of endogenous accumulations of adenosine
Next, we examined the capability of the sensors to monitor endogenous increases in extracellular adenosine. We inhibited reuptake of adenosine through the ENT, the predominant method of extracellular adenosine clearance, by locally applying an ENT inhibitor (DPR). Local application of 200 nL of 200 μM DPR significantly raised the extracellular concentration of adenosine by 120% (5.6→12.3 μM) [t(16)=8.0, P<0.001] (Fig. 4). The endogenous increase in extracellular adenosine was transient with an average duration of 84.0±28.0 s and was reproducible with subsequent injections (Fig. 2). Thus, MEAs are capable of examining the endogenous mechanisms regulating the extracellular adenosine concentration.
Fig. 4.
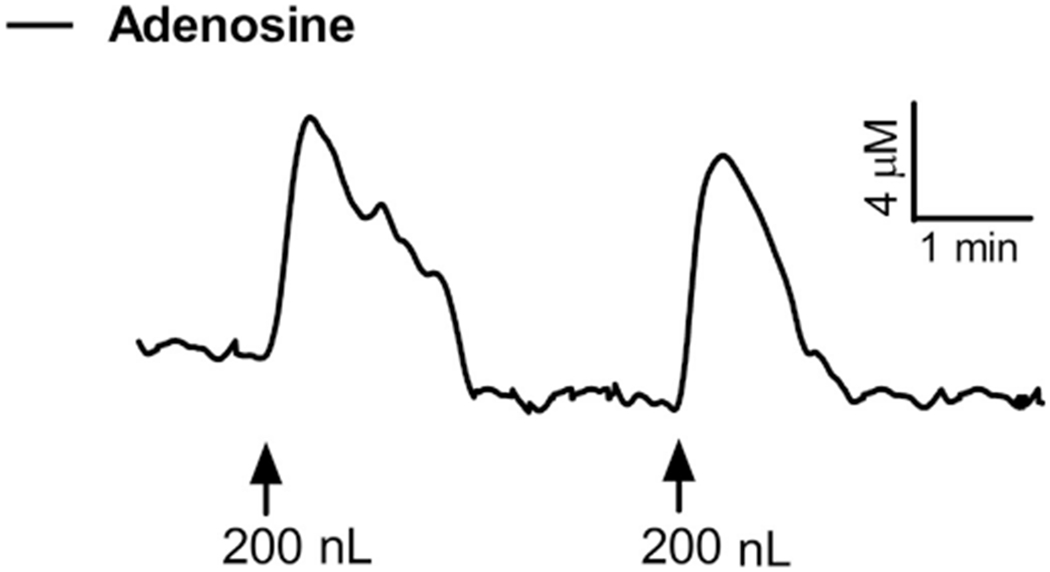
In vivo MEA response to endogenous adenosine. Representative in vivo recording depicting the change in extracellular adenosine (+ADA site – sentinel site) during local application of 200 nL of 200 μM dipyridamole (DPR), a competitive inhibitor of the equilibrative nucleoside transporter (ENT). Local injection produced a transient 120% increase in extracellular adenosine that was reproducible (↑ time of injection).
5. Discussion
5.1. Summary
Here, we have developed a novel self-referenced enzyme-linked MEA to monitor extracellular adenosine in vivo that accounts for any potential false signals from major interferents (i.e. ascorbic acid) and the adenosine enzymatic breakdown products (inosine and xanthine). In vitro, the sensors could selectively measure adenosine in a linear fashion. In vivo, the LOD of the sensors was sufficient for quantification of the basal adenosine concentration. In addition, the sensors measured increases in extracellular adenosine from exogenous application in a dose-dependent manner with sufficient temporal resolution to quantify signal decay. Lastly, the MEAs could monitor endogenous increases in the extracellular concentration, via inhibition of adenosine reuptake, demonstrating the ability of the sensors to examine the intrinsic regulatory mechanisms. Thus, we have developed a novel sensor with excellent spatial and temporal resolution that can be used to selectively monitor extracellular adenosine and further understanding of the role of adenosine in physiologic and pathologic states.
5.2. Advantages of in vivo monitoring with MEAs
Our approach improves on the spatial resolution of the first generation electrochemical probes that were also configured in a self-referenced manner (Dale et al., 2000). As a result, we have provided a novel sensor that selectively measures extracellular adenosine and produces minimal damage to the brain tissue (Hinzman et al., 2015; Rutherford et al., 2007) permitting real-time monitoring in discrete brain regions. The major advantage of the self-referenced design is the ability to subtract out false signals from major interferents and the enzymatic break-down products (inosine and xanthine) that a single electrode may fail to distinguish (Llaudet et al., 2003), which require additional experimental controls to identify the source of signal (Lindquist and Shuttleworth, 2012). The self-referenced configuration of the MEA and low LOD (0.04 μM) allow measurement of the basal adenosine concentration, which has been previously found to range 0.04–10 μM in the rat brain (Latini and Pedata, 2001). We found a basal concentration of 4.3 μM in the rat somatosensory cortex that is consistent with this range. Another advantage of the MEA is the improved temporal resolution that permits detailed analysis of the decay of the adenosine signal. Overall, these methodological improvements permit the study of the intrinsic mechanisms regulating the extracellular adenosine concentration. Here, inhibition of adenosine re-uptake by the ENT significantly increased the extracellular adenosine concentration by 120%, which is similar prior results showing a 70–200% increase (Ballarin et al., 1991; Hagberg et al., 1987).
5.3. Limitations of in vivo monitoring with MEAs
The major limitation of the MEA configuration used in this study is the inability to simultaneously monitor multiple neurochemicals of interest. While MD has limited spatial and temporal resolution, analysis of the dialysate can provide simultaneous quantification of many neurochemicals. Thus, MD can provide an extensive neurochemical profile of the extracellular space during the sampling period. However, recent advances in electrochemical sensors have shown that MEAs can selectively monitor either directly or indirectly (via enzyme-based generation of reporter molecule) many important neurochemicals such as: glucose (Burmeister et al., 2004), lactate (Burmeister et al., 2005), nitric oxide, (Barbosa et al., 2008) and the neurotransmitters glutamate (Burmeister et al., 2002; Hinzman et al., 2010b), choline (Burmeister et al., 2008), acetylcholine (Burmeister et al., 2008), and dopamine (Burmeister et al., 2000). Thus, combinations of these configurations on a single MEA could in principle provide a “lab on a chip” that is capable of simultaneous real-time monitoring of multiple neurochemical species.
6. Conclusions
Here, we have developed a MEA with excellent spatial and temporal resolution to selectively monitor extracellular adenosine in discrete functional brain circuits. Furthermore, this technique could be combined with selective measures of other important neurochemicals and neurotransmitters on a single device, a real-time “lab on a chip”, with the resolution to advance understanding of cerebral physiology and pathophysiology.
Acknowledgments
Support provided by Mayfield Education and Research Foundation (JMH, RDT, JAH) and by Alpha Omega Alpha Carolyn L. Kuckein Student Research Fellowship (JLG).
Abbreviations:
- AA
Ascorbic Acid
- ADA
Adenosine Deaminase
- ATP
Adenosine-Triphosphate
- BSA
Bovine Serum Albumin
- DPR
Dipyridamole
- ENT
Equilabrative Nucleoside Transporter
- FSCV
Fast Scan Cyclic Voltammetry
- IO
Inosine
- LOD
Limit of Detection
- MD
Microdialysis
- MEA
Microelectrode Array
- mPD
1,3-phenylenediamine
- NP
Nucleoside Phosphorylase
- Pt
Platinum
- XA
Xanthine
- XO
Xanthine Oxidase
Footnotes
Conflict of interest
Greg Gerhardt is the sole proprietor of Quanteon, LLC, which makes the electrochemistry hardware used for the adenosine measurements. JEQ has served as a consultant to Quanteon, LLC. No other authors have a conflict of interest.
References
- Ballarin M, Fredholm BB, Ambrosio S, Mahy N, 1991. Acta Physiol. Scand 142 (1), 97–103. [DOI] [PubMed] [Google Scholar]
- Barbosa RM, Lourenco CF, Santos RM, Pomerleau F, Huettl P, Gerhardt GA, Laranjinha J, 2008. Methods Enzymol 441, 351–367. [DOI] [PubMed] [Google Scholar]
- Bjorness TE, Kelly CL, Gao T, Poffenberger V, Greene RW, 2009. J. Neurosci 29 (5), 1267–1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burmeister JJ, Coates TD, Gerhardt GA, 2004. Conf. Proc. IEEE. Eng. Med. Biol. Soc 7, 5348–5351. [DOI] [PubMed] [Google Scholar]
- Burmeister JJ, Gerhardt GA, 2001. Anal. Chem 73 (5), 1037–1042. [DOI] [PubMed] [Google Scholar]
- Burmeister JJ, Moxon K, Gerhardt GA, 2000. Anal. Chem 72 (1), 187–192. [DOI] [PubMed] [Google Scholar]
- Burmeister JJ, Palmer M, Gerhardt GA, 2005. Biosens. Bioelectron 20 (9), 1772–1779. [DOI] [PubMed] [Google Scholar]
- Burmeister JJ, Pomerleau F, Huettl P, Gash CR, Werner CE, Bruno JP, Gerhardt GA, 2008. Biosens. Bioelectron 23 (9), 1382–1389. [DOI] [PubMed] [Google Scholar]
- Burmeister JJ, Pomerleau F, Palmer M, Day BK, Huettl P, Gerhardt GA, 2002. J. Neurosci. Methods 119 (2), 163–171. [DOI] [PubMed] [Google Scholar]
- Burnstock G, 2015. Neuropharmacology, 212–219.26518371 [Google Scholar]
- Chen F, Qi Z, Luo Y, Hinchliffe T, Ding G, Xia Y, Ji X, 2014. Prog. Neurobiol 115, 246–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark RS, Carcillo JA, Kochanek PM, Obrist WD, Jackson EK, Mi Z, Wisneiwski SR, Bell MJ, Marion DW, 1997. Neurosurgery 41 (6), 1284–1292, discussion 1292–1283. [DOI] [PubMed] [Google Scholar]
- Cui M, Bai X, Li T, Chen F, Dong Q, Zhao Y, Liu X, 2013. PLoS One 8 (2), e57065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dale N, 1998. J. Physiol 511, 265–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dale N, Pearson T, Frenguelli BG, 2000J. Physiol 526 (Pt 1), 143–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drury AN, Szent-Gyorgyi A, 1929. J. Physiol 68 (3), 213–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunwiddie TV, 1980. Epilepsia 21 (5), 541–548. [DOI] [PubMed] [Google Scholar]
- Dunwiddie TV, Masino SA, 2001. Annu. Rev. Neurosci 24, 31 −55. [DOI] [PubMed] [Google Scholar]
- Dunwiddie TV, Worth T, 1982. J. Pharmacol. Exp. Ther 220 (1), 70–76. [PubMed] [Google Scholar]
- During MJ, Spencer DD, 1992. Ann. Neurol 32 (5), 618–624. [DOI] [PubMed] [Google Scholar]
- Hagberg H, Andersson P, Lacarewicz J, Jacobson I, Butcher S, Sandberg M, 1987. J. Neurochem 49 (1), 227–231. [DOI] [PubMed] [Google Scholar]
- Hinzman JM, DiNapoli VA, Mahoney EJ, Gerhardt GA, Hartings JA, 2015. Exp. Neurol 267, 243–253. [DOI] [PubMed] [Google Scholar]
- Hinzman JM, Thomas TC, Burmeister JJ, Quintero JE, Huettl P, Pomerleau F, Gerhardt GA, Lifshitz J, 2010a. J. Neurotrauma 27 (5), 889–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinzman JM, Thomas TC, Burmeister JJ, Quintero JE, Huettl P, Pomerleau F, Gerhardt GA, Lifshitz J, 2010b. J. Neurotrauma 27 (5), 889–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinzman JM, Thomas TC, Quintero JE, Gerhardt GA, Lifshitz J, 2012. J. Neurotrauma 29 (6), 1197–1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang ZL, Zhang Z, Qu WM, 2014. Int. Rev. Neurobiol 119, 349–371. [DOI] [PubMed] [Google Scholar]
- Illes P, Zimmermann H, 1999. Nucleotides and their Receptors in the Nervous System, 1st ed Science Elsevier, Amsterdam, New York. [Google Scholar]
- Laghi Pasini F, Guideri F, Picano E, Parenti G, Petersen C, Varga A, Di Perri T, 2000. Brain Res. Bull 51 (4), 327–330. [DOI] [PubMed] [Google Scholar]
- Latini S, Pedata F, 2001. J. Neurochem 79 (3), 463–484. [DOI] [PubMed] [Google Scholar]
- Lindquist BE, Shuttleworth CW, 2012. Neuroscience 223, 365–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindquist BE, Shuttleworth CW, 2014. J. Cereb. Blood Flow Metab 34 (11), 1779–1790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llaudet E, Botting NP, Crayston JA, Dale N, 2003. Biosens. Bioelectron 18 (1), 43–52. [DOI] [PubMed] [Google Scholar]
- Masino SA, Kawamura M Jr., Ruskin DN, 2014. Int. Rev. Neurobiol 119, 233–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattinson CE, Burmeister JJ, Quintero JE, Pomerleau F, Huettl P, Gerhardt GA, 2011. J. Neurosci. Methods 202 (2), 199–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller EM, Quintero JE, Pomerleau F, Huettl P, Gerhardt GA, Glaser PE, 2015. J. Neurosci. Methods [DOI] [PubMed] [Google Scholar]
- Miranda MF, Hamani C, de Almeida AC, Amorim BO, Macedo CE, Fernandes MJ, Nobrega JN, Aarao MC, Madureira AP, Rodrigues AM, Andersen ML, Tufik S, Mello LE, Covolan L, 2014. Front. Cell Neurosci 8, 312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen MD, Lee ST, Ross AE, Ryals M, Choudhry VI, Venton BJ, 2014. PLoS One 9 (1), e87165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen MB, Venton J, 2015. Comput. Struct. Biotechnol. J 13, 47–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nickell J, Salvatore MF, Pomerleau F, Apparsundaram S, Gerhardt GA, 2007. Neurobiol. Aging 28 (11), 1737–1748. [DOI] [PubMed] [Google Scholar]
- Park TS, Gidday JM, 1990. J. Cereb. Blood Flow Metab 10 (3), 424–427. [DOI] [PubMed] [Google Scholar]
- Pascual O, Casper KB, Kubera C, Zhang J, Revilla-Sanchez R, Sul JY, Takano H, Moss SJ, McCarthy K, Haydon PG, 2005. Science 310 (5745), 113–116. [DOI] [PubMed] [Google Scholar]
- Pedata F, Latini S, Pugliese AM, Pepeu G, 1993. J. Neurochem 61 (1), 284–289. [DOI] [PubMed] [Google Scholar]
- Pedata F, Pugliese AM, Coppi E, Dettori I, Maraula G, Cellai L, Melani A, 2014. Mediat. Inflamm 2014, 805198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson CL, Bell MJ, Kochanek PM, Adelson PD, Ruppel RA, Carcillo JA, Wisniewski SR, Mi Z, Janesko KL, Clark RS, Marion DW, Graham SH, Jackson EK, 2001. Crit. Care Med 29 (12), 2287–2293. [DOI] [PubMed] [Google Scholar]
- Rutherford EC, Pomerleau F, Huettl P, Stromberg I, Gerhardt GA, 2007. J. Neurochem 102 (3), 712–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spyer KM, Thomas T, 2000. Brain Res. Bull 53 (1), 121–124. [DOI] [PubMed] [Google Scholar]
- Stephens ML, Quintero JE, Pomerleau F, Huettl P, Gerhardt GA, 2011. Neurobiol. Aging 32 (5), 811–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swamy BE, Venton BJ, 2007. Anal. Chem 79 (2), 744–750. [DOI] [PubMed] [Google Scholar]
- Thomas TC, Grandy DK, Gerhardt GA, Glaser PE, 2009. Neuropsychopharmacology 34 (2), 436–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas TC, Hinzman JM, Gerhardt GA, Lifshitz J, 2012. J. Neurotrauma 29 (2), 187–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tupone D, Madden CJ, Morrison SF, 2013. J. Neurosci 33 (36), 14512–14525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Gompel JJ, Bower MR, Worrell GA, Stead M, Chang SY, Goerss SJ, Kim I, Bennet KE, Meyer FB, Marsh WR, Blaha CD, Lee KH, 2014. Epilepsia 55 (2), 233–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weigand MA, Michel A, Eckstein HH, Martin E, Bardenheuer HJ, 1999. Anesthesiology 91 (2), 414–421. [DOI] [PubMed] [Google Scholar]
- Zhang G, Franklin PH, Murray TF, 1990. Neurosci. Lett 114 (3), 345–350. [DOI] [PubMed] [Google Scholar]