

Abstract
The lymphatic system is an active player in the pathogenesis of several human diseases, including lymphedema and cancer. Relevant models are needed to advance our understanding of lymphatic biology in disease progression to improve therapy and patient outcomes. Currently, there are few 3D in vitro lymphatic models that can recapitulate the physiological structure, function, and interactions of lymphatic vessels in normal and diseased microenvironments. Here, we developed a 3D microscale lymphatic vessel (μLYMPH) system for generating human lymphatic vessels with physiological tubular structure and function. Consistent with characteristics of lymphatic vessels in vivo, the endothelium of cultured vessels was leaky with an average permeability of 1.38 x 10−5 ± 0.29 x 10−5 cm/s as compared to 0.68 x 10−5 ± 0.13 x 10−5 cm/s for blood vessels. This leakiness also resulted in higher uptake of solute by the lymphatic vessels under interstitial flow, demonstrating recapitulation of their natural draining function. The vessels secreted appropriate growth factors and inflammatory mediators. Our system identified the follistatin/activin axis as a novel pathway in lymphatic vessel maintenance and inflammation. Moreover, the μLYMPH system provided a platform for examining crosstalk between lymphatic vessels and tumor microenvironmental components, such as breast cancer-associated fibroblasts (CAFs). In co-culture with CAFs, vessel barrier function was significantly impaired by CAF-secreted IL-6, a possible pro-metastatic mechanism of lymphatic metastasis. Targeted blocking of the IL-6/IL-6R signaling pathway with an IL-6 neutralizing antibody fully rescued the vessels, demonstrating the potential of our system for screening therapeutic targets. These results collectively demonstrate the μLYMPH system as a powerful model for advancing lymphatic biology in health and disease.
Keywords: lymphatic vessel, microfluidic, organotypic, tumor microenvironment
Introduction
The lymphatic system is a crucial component of the circulatory and immune systems. It complements blood circulation, modulating interstitial fluid transport, immune cell trafficking, and lipid absorption [1]. Impairment of lymphatic vessels can result in lymphedema, i.e., the swelling of tissue due to interstitial fluid accumulation [1]. The lymph node microenvironment can also facilitate lymphoma progression and dissemination (e.g. non-Hodgkin’s lymphoma) [2]. Moreover, lymphangiogenesis is a critical step in the metastasis of cancer cells to secondary organs [3]. Despite their importance in normal health and disease, the lymphatics are often understudied, especially in comparison to blood vasculature. However, there has been renewed effort and substantial progress in advancing lymphatic biology in the last two decades, following recognition of the lymphatic system as a key player in the pathogenesis of human diseases [3]. The discovery of protein markers for isolating lymphatic endothelial cells (LECs) in 1995 [4] and 1999 [5–7] has enabled in vitro lymphatic cultures and modeling.
Conventional methods for modeling lymphatic vessels comprise LECs cultured as monolayers on plastic surfaces (2D in vitro), LECs cultured in or on a matrix (3D in vitro), excised vessels from mice cultured in a matrix (3D ex vivo), and mouse models (in vivo) [8]. The usefulness of these models can be measured on a scale of tractability (i.e. repeatability and control over experimental parameters) versus physiological relevance (i.e. recapitulation of in vivo geometry and interactions). In vitro LEC cultures enable robust and repeatable experimentation but have limited capability in recapitulating microenvironmental cues (e.g. cell-cell communication, fluid shear stress, and matrix forces). In contrast, animal models provide relevant in vivo microenvironments; yet, experimental control is challenging in these dynamic systems. Current organotypic approaches, such as the lymphatic ring assay [9], offer a possible bridge between in vitro and in vivo approaches. Nevertheless, their dependence on excised vessels from animal sources reduces repeatability and reproducibility. There is an opportunity, then, to develop organotypic models using human lymphatic tissue that balance tractability and recapitulation of in vivo lymphatic structure, function, and microenvironmental interactions in a reliable and repeatable manner.
Microfluidic organotypic models, also known as microphysiological systems or organs-on-chips, show promise to this end. These models enable 3D culture at the physical length scales of cells and tissue. They can recapitulate the form, function, and biophysical and biochemical microenvironments of various organs [10–12]. In the context of vasculature, organotypic vessels have tubular structure, barrier function, and respond to microenvironmental cues (e.g. growth factors) as similar to in vivo [13–19]. While endothelial monolayers and cells-in-gels in microchannels can mimic vascular physiology [20–23], capturing the tubular structure of vessels is not trivial and has implications in structure-function relationships. Geometry alone can alter endothelial cell signaling and phenotype [24]. To date, organotypic models of the lymphatic system are scarce. Existing models have examined the effect of cyclic adenosine monophosphate on the permeability of the lymphatic endothelium [25], and the interstitial fluid drainage function of artificial lymphatics (i.e. lumens without LECs) [26]. Further development of microfluidic organotypic lymphatic models would accelerate basic and translational lymphatics research.
Here, we developed a microscale lymphatic vessel (μLYMPH) system for culturing human lymphatic endothelial vessels. The system enables several capabilities for mechanistically studying lymphatic vessel biology, including: (1) basic characterization of vessel phenotype, (2) assessment of vessel cytokine secretion and barrier capacity, (3) assessment of vessel response to exogenous stimuli, (4) simulation of diseased microenvironments via co-culture with disease-specific stromal cells, and (5) identification of potential therapeutic targets for lymphatic-tumor microenvironmental interactions. Vessels generated in the μLYMPH system had patent tubular structure with diameters in the range of 200-250 μm and expressed classical endothelial junctional proteins (e.g. CD31, vascular endothelial cadherin - VE-cadherin, and zonula occluens-1 - ZO-1) continuously throughout their endothelium, which are characteristics representative of pre-collecting/collecting lymphatic vessels in vivo [27,28]. In comparison to blood vessels cultured in the same system, the lymphatic vessels had comparatively leakier endothelia allowing significantly more solute drainage into the vessels. Moreover, vessel physiology was substantially altered when they were stimulated with exogenous lymphangiogenic and inflammatory factors, and co-cultured with fibroblasts. Co-culture with breast CAFs increased vessel permeability through IL-6 signaling, a possible mechanism for promoting lymphatic metastasis. Importantly, barrier function was fully recovered by neutralizing excess IL-6 in the co-culture. Collectively, these results demonstrate the utility of the μLYMPH system for generating functional human lymphatic vessels and enabling the study of lymphatic biology in physiological microenvironments.
Materials and Methods
μLYMPH system and vessel culture
The μLYMPH system is polydimethylsiloxane (PDMS)-based, consisting of a luminal rod suspended in an extracellular matrix (ECM) gel chamber (Fig. 1a). Devices are fabricated and assembled based on our previously established LumeNEXT approach [18] (Fig. S1). Briefly, the top and bottom layers of the device were fabricated via standard soft lithography using silicon masters containing SU-8 100 photoresist features (Y13273, MicroChem, Newton, MA). The bottom layer contains the ECM gel chamber and a channel for suspending the lumen rod. The top layer forms the cover for the chamber and contains ports for fluid handling. PDMS with a 1:10 ratio of curing agent to pre-polymer was used for all device layers and lumen rods.
Figure 1.

μLYMPH system concept and vessel culture. a Exploded view of device layers. A bottom layer contains the extracellular matrix gel chamber and a lumen rod suspended across the chamber supported by a channel. A top layer forms the cover for the chamber and contains ports for fluid handling. b Schematic of fully assembled device and cultured vessel. c Representative images of devices and a lymphatic vessel with a diameter of 238 μm, cultured in 3 mg/mL collagen type I gel. d Viability images of vessels cultured over 7 days for gel densities ranging from 3 mg/mL to 6 mg/mL. Live cells are green and dead cells are red. The live fraction percentages for all densities are > 85%, with a maximum of 92% for 3 mg/mL. There are regions of cell detachment for 5 mg/mL and 6 mg/mL (dashed outlines). Live fraction percentage values are the average of n = 3 individual vessels with error bars as one standard deviation. *p ≤ 0.05, **p ≤ 0.01.
Lumen rods were made using 25-gauge hypodermic needles (inner diameter ~250 μm, 14-840-84, Fisher Scientific, Pittsburgh, PA). Uncured PDMS was pushed through the needles using a syringe, and the needles were heated at 80°C for 3 hours. Polymerized rods were removed from the needles and cut to fit across the gel chambers of the devices. Following assembly of the device layers, rods were inserted into the channel with a tweezer. Completed devices were then bonded to glass-bottom culture dishes (P50G-1.5-30-F, MatTek Corporation, Ashland, MA) and stored until use.
Devices were UV sterilized for 15 min prior to cell culture. The main steps of the culture process are illustrated in Fig. S1. To minimize delamination of collagen in the gel chamber, devices were treated with a 2% polyethyleneimine solution (03880, Sigma-Aldrich, St. Louis, MO) at room temperature for 10 min, followed by treatment with a 0.1% glutaraldehyde solution (G6257, Sigma-Aldrich, St. Louis, MO) at room temperature for 30 min. Devices were then washed five times with sterile deionized water. A solution of rat-tail collagen type I (354249, BD Biosciences, San Jose, CA) was prepared on ice and neutralized using 0.5 M NaOH. The recipes for different collagen densities are provided in Table S1. Devices were filled with the prepared collagen solution (6 μL per device), incubated at room temperature for 10 min to initiate polymerization, and then transferred to 37 °C for 1 hour. For the fibroblast co-cultures, a similar collagen gel solution was prepared containing normal or cancer-associated fibroblasts (250 cells/μL). Devices were filled with the cell-laden collagen solution (6 μL per device) and incubated at 37 °C for 1 hour.
After gel polymerization, lumen rods were removed using a tweezer to reveal empty lumens molded in the collagen gel matrices. Prior to cell seeding, lumens were incubated with a fibronectin solution (33 μg/mL, 10 μL per lumen) at room temperature for 30 min to support cell adhesion. HLECs were trypsinized with 0.05% Trypsin-EDTA (25300062, ThermoFisher Scientific, Waltham, MA), suspended in EGM-2 MV medium (~20,000 cell/μL), and seeded into the fibronectin-coated lumens (4 μL per lumen). HLEC-filled lumens were incubated at 37 °C for an initial cell attachment period of 2 hours, where devices were manually flipped every 25 min to ensure homogeneous cell coverage of the lumen wall. Following this process, lumens were supplemented with 10 μL of medium and cultured overnight at 37 °C. Cultured vessels were refreshed with medium twice per day post-seeding. During each feeding, vessels were washed three times with medium to remove dead cells and then maintained with 10 μL of medium. All fluid handling procedures were conducted with standard pipettes.
Vessel culture with and without flow
To assess cell alignment in the lymphatic vessels, vessels were cultured in static and flow conditions, where flow was generated via manual pipetting of media during daily vessel feedings. To achieve static culture, one culture dish of vessels was maintained in 2 mL of media over the entire culture period without daily media exchanges. Vessels cultured in static and flow conditions were fixed after 7 days and stained with phalloidin and DAPI. F-actin images were acquired with the Nikon TI® Eclipse inverted microscope. For each condition, alignment was quantified as the angle of a cell’s major axis for at least 20 cells per vessel and presented as histograms with Gaussian curve fits. All images were analyzed using Fiji[29].
Cell culture
Human lymphatic endothelial cells isolated from the lymph node (HLECs, 2500, ScienCell, Carlsbad, CA) and human umbilical vein endothelial cells (HUVECs, C2517A, Lonza, Allandale, NJ) were cultured separately in standard cell culture flasks coated with fibronectin (5μg/cm2, F1141-5MG, Sigma Aldrich, St. Louis, MO) at a starting cell concentration of 5 x 105 as per supplier instructions. Cultures were maintained with Endothelial Basal Medium-2 (EBM-2, CC-3156) supplemented with EGM-2 MV SingleQuot Kit (CC-4147, Lonza, Allandale, NJ). HLECs and HUVECs were cultured to 95% confluency at passages 4 and 5 for all experiments. Normal mammary fibroblasts (provided by Dr. Lisa Arendt’s lab at UW-Madison) and breast cancer-associated fibroblasts (provided by Dr. Andreas Friedl’s lab at UW-Madison) were cultured separately in standard flasks at a starting cell concentration of 5 x 105. Fibroblasts were maintained with Dulbecco’s Modified Eagle Medium (11965092, ThermoFisher Scientific, Waltham, MA) supplemented with 10% fetal bovine serum. All cultures were kept in a humidified incubator at 37 °C with 5% CO2.
Flow cytometry
HUVECs and HLECs were cultured in standard flasks as described in the ‘Cell culture’ section. Cells were lifted with PBS + 5 mM EDTA and resuspended in PBS at 10 x 106 cells/mL prior to live/dead staining with Ghost Red 780 (13-0865, Tonbo Biosciences, San Diego, CA) following manufacturer’s instructions. 1 x 106 cells were aliquoted per stain or control sample and 5μl of Fc Block (564219, BD Biosciences, San Jose, CA) was added to the resuspended HUVECs and HLECs. Cells were stained with a cocktail of CD31, LYVE1, and podoplanin antibodies using CD31-PE/Cy7 (303117), LYVE-1-A647 (FAB20892R), and Podoplanin-PE (337003), respectively. All antibodies were purchased from BioLegend. Cells were then fixed and permeabilized prior to intracellular staining of PROX1 using the FOXP3 staining kit (A25866A, ThermoFisher Scientific, Waltham, MA) as per manufacturer’s instructions and PROX-1-A488 (NBP1-30045AF488, Novus Biologicals, Centennial, CO). FMO controls were performed for each of the antibodies. Live/dead controls used a mixture of heat killed and live cells stained with ghost red only, for each cell type. Compensation controls used Ultracomp ebeads labelled with the same amount of antibody as the cells (01-2222-41, ThermoFisher Scientific, Waltham, MA). Marker expression was quantified using the Attune™ NxT flow cytometer (ThermoFisher Scientific). Analysis was completed with the FlowJo software (BD Biosciences), gating on size, singlets and live cells.
Live/dead assays
The viability of cultured vessels was quantified using calcein AM (C3100MP, ThermoFisher Scientific, Waltham, MA) and ethidium homodimer-1 (E1169, ThermoFisher Scientific, Waltham, MA). A staining solution was prepared using 1 mL of EBM-2 medium containing 2.5 μL of calcein AM for live cells and 2 μL of ethidium homodimer-1 for dead cells. Prior to staining, cultured vessels were washed three times with EBM-2. Vessels were then incubated with staining solution (10 μL per vessel) at 37 °C for 30 min and imaged immediately afterwards.
Cytokine quantification
Cytokines secreted into the media for vessel mono- and co-cultures were analyzed using a human growth factor panel (HAGP1MAG-12K, EMD Millipore, Billerica, MA). Media was collected from six individual vessels (10 ul per vessel) over two days (culture days 3 and 4) and pooled to generate sufficient volume for four technical replicates per experimental condition. The panel was prepared following manufacturer instructions and measured using the MAPGPIX system (Luminex Corp., Austin, TX). Cytokine concentrations were quantified with the Luminex xPONENT software.
Quantitative reverse transcription PCR (qRT-PCR)
Transcription levels of endothelial junctional proteins, CD31, VE-cadherin, and ZO-1, were measured qRT-PCR using TaqMan probes (ThermoFisher Scientific). mRNA isolation was performed using Dynabeads mRNA direct purification kit (61011, ThermoFisher Scientific, Waltham, MA) and reverse transcription was conducted with the RNA to cDNA kit (4387406, ThermoFisher Scientific, Waltham, MA). The ΔΔCT method was used to assess relative gene expression, where expression of cell junctional protein genes PECAM1 (Hs01065279_m1), CDH5 (Hs00901465_m1), and TJP1 (Hs01551861_m1) were normalized to the housekeeping gene GAPDH (Hs01922876_m1).
Dextran diffusion assays
Solute transport from out of the vessels (i.e. permeability) and into the vessels (i.e. drainage) was measured by dextran diffusion. Texas Red dextran (10 kDa, D1828, and 70kDa, D1830, ThermoFisher Scientific, Waltham, MA) solutions were prepared to working concentrations of 1 μM. For the permeability assays, 3 μL of dextran solution was added to each vessel such that fluid was flush with the lip of the ports to minimize flow from a pressure head. Solute transport was measured over 15 minutes per vessel. Permeability coefficients were calculated using equation 1 [30]:
| Eq. 1 |
where Io is the total initial intensity outside the vessel, If is the total intensity outside the vessel at 15 minutes, to is the initial time point, tf is the final time point of 15 minutes, and D is vessel diameter. For the drainage assays, 3 μL of dextran solution was added to one gel chamber port to create a pressure head for interstitial flow toward the vessel. Solute transport was measured over 30 minutes per vessel. Drainage coefficients were calculated using a modified version of equation 1, equation 2:
| Eq. 2 |
where Iovessel is the total initial intensity inside the vessel, Ifvessel is the total intensity inside the vessel at 30 minutes, to is the initial time point, tf is the final time point of 30 minutes, and D is vessel diameter. Dextran diffusion was imaged with the Nikon TI® Eclipse inverted microscope. All images were analyzed using Fiji.
Blocking and neutralization
MAZ51 (676492, EMD Millipore, Billerica, MA) was used to inhibit VEGF-C/VEGFR-3 signaling. Anti-IL-6R antibody (ab47215, Abcam, Cambridge, UK) and anti-IL-6 antibody (501109, BioLegend, San Diego) were used to inhibit IL-6/IL-6R signaling. Mouse IgG1 (400102, BioLegend, San Diego, CA) and rat IgG1 (400413, BioLegend, San Diego) antibodies were used as isotype controls for the IL-6/IL-6R inhibition experiments. To inhibit VEGFR-3, cultured vessels were primed for 1 hour with media containing MAZ51 (either 1 μM or 5 μM). Subsequently, MAZ51 was added to media containing VEGF-C (50 ng/mL) used to stimulate vessels over culture days 3 and 4. Vessels were washed with fresh media without VEGF-C or MAZ51 on day 5 and used for dextran diffusion analysis. To block vessels stimulated with IL-6 and in co-culture with CAFs, vessels were treated with anti-IL-6R antibody (either 5 μg/mL or 25 μg/mL) overnight from day 2 to day 3. Blocked vessels were then treated with media containing IL-6 (50 ng/mL) or left to co-culture with CAFs over days 3 and 4, and used on day 5 for dextran diffusion analysis. To neutralize IL-6 for the CAF co-cultures, vessels were treated with fresh medium containing 25 μg/mL of anti-IL-6 antibody from day 2 to day 4 and used on day 5 for dextran diffusion analysis.
Immunofluorescent staining and imaging
All vessels prepared for immunofluorescent staining were cultured at least 3 days post-seeding to ensure confluency. Washing buffer (0.1% PBS-Tween 80) and blocking buffer (3% BSA in 0.1% PBS-Tween 80) were made in advance and stored at 4 °C until use. Each vessel was 1) incubated with 5 μL of 4% paraformaldehyde at room temperature for 15 min for fixation, 2) incubated with 5 μL of 0.2% Triton X-100 at room temperature for 30 min for permeabilization, 3) incubated with 10 μL of blocking buffer at 4 °C overnight. All vessels were washed three times with sterile PBS in between each step.
For antibody-based staining of lymphatic-specific markers and cell junctional proteins, primary antibodies were diluted to desired concentrations with staining buffer (blocking buffer plus 1% PBS-Tween 80 at 10:1 v/v) (Table S2). Vessels were washed three times with washing buffer and incubated with primary antibodies at 4 °C overnight. To remove excess primary antibodies from the collagen gel matrix, vessels were washed multiple times for one day (10 min per wash). Secondary antibodies were prepared at desired concentrations using the staining buffer supplemented with 10% goat serum to reduce aspecific binding (Table S2). Vessels were incubated with the prepared antibodies at room temperature for 1 hour. Stained vessels were washed over two days and stored in sterile PBS until imaging. Alexa Fluor 488 phalloidin (A12379, ThermoFisher Scientific, Waltham, MA) and DAPI (D3571, ThermoFisher Scientific, Waltham, MA) were used to stain actin cytoskeleton and nuclei, respectively.
Fluorescent images were acquired using a Nikon TI® Eclipse inverted microscope (Melville, New York) and processed using the National Institutes of Health ImageJ software. Confocal images were acquired using a Leica SP8 3X STED Super-resolution microscope (Wetzlar, Germany) in the UW-Madison Optical Imaging Core.
Statistical analysis
GraphPad Prism 7 (GraphPad Software, La Jolla, CA) was used for statistical analysis. Significance tests were performed using multiple unpaired, two-tailed Student’s t-test with Bonferroni-Dunn multiple comparisons test (Fig. 3c), ordinary one-way ANOVA with Bonferroni’s multiple comparisons test (Fig. 1d, 4c, 5e, 6c–e, 7d, and 8c–d), and unpaired, two-tailed Student’s t-test (Fig. 4d). Tests were considered significant for p ≤ 0.05. The number of replicates ranged from n = 3 to n = 6 for each experimental condition.
Figure 3.
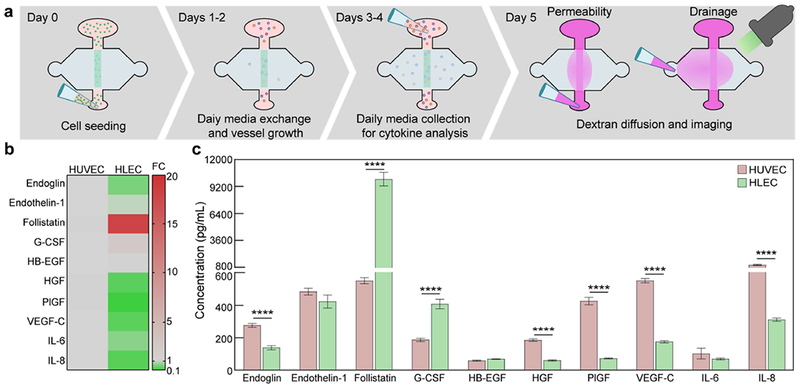
Lymphatic vessel cytokine secretion. a Schematic of experimentation timeline over five days. b Direct comparison of the fold change in secretion levels between blood (HUVEC) vessels and lymphatic (HLEC) vessels, with blood vessel levels taken as baseline. Overall, lymphatic vessels have lower secretion levels of the measured growth and inflammatory factors. However, follistatin expression is 18-fold higher for the lymphatic vessels. c Concentrations of secreted growth and inflammatory factors. Blood and lymphatic vessels have unique secretion profiles. There are significant differences in the secretion of endoglin, follistatin, G-CSF, HGF, PlGF, VEGF-C, and IL-8. Concentration values are the averages of n = 4 technical replicates of media pooled from 6 individual vessels for each condition.
Figure 4.
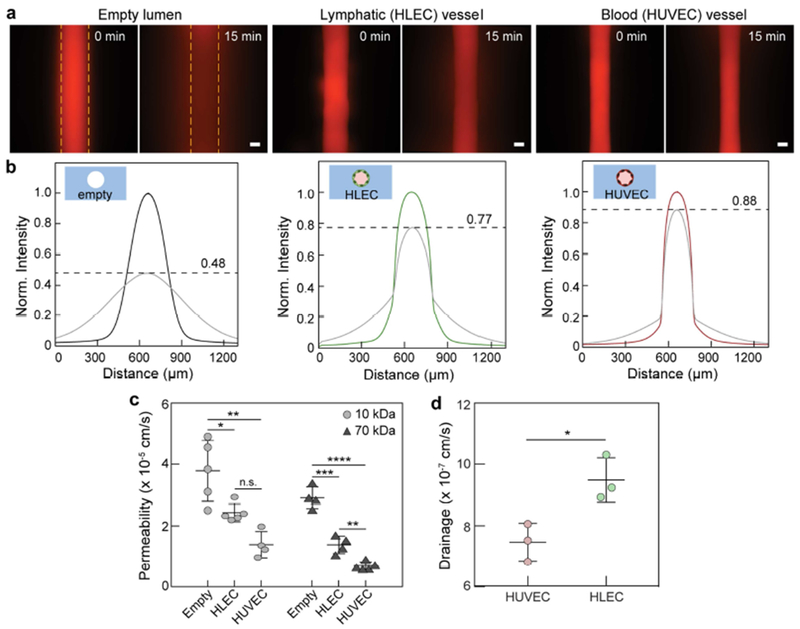
Lymphatic vessel barrier function and drainage capacity. a Representative images of 70kDa dextran diffusion in empty, lymphatic, and blood vessels over 15 min. Dashed lines indicate lumen boundaries. b Normalized intensity profiles of 70 kDa dextran diffusion, where lymphatic vessels have moderate barrier capacity in comparison to blood vessels. The initial concentration decreases by 12%, 23%, and 52% for blood, lymphatic, and empty vessels, respectively. c Quantification of vessel permeability. For both 10 kDa and 70 kDa dextran, lymphatic vessels are ~2-fold leakier than blood vessels. Both endothelial vessels, however, provide barrier function in comparison to empty lumens. d Quantification of drainage capacity. Lymphatic vessels uptake significantly more 70 kDa dextran over 30 min. as compared to blood vessels. Permeability values are the averages of at least n = 4 individual vessels. Drainage values are the averages of n = 3 individual vessels. All error bars are one standard deviation. Scale bars are 100 μm. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001.
Figure 5.
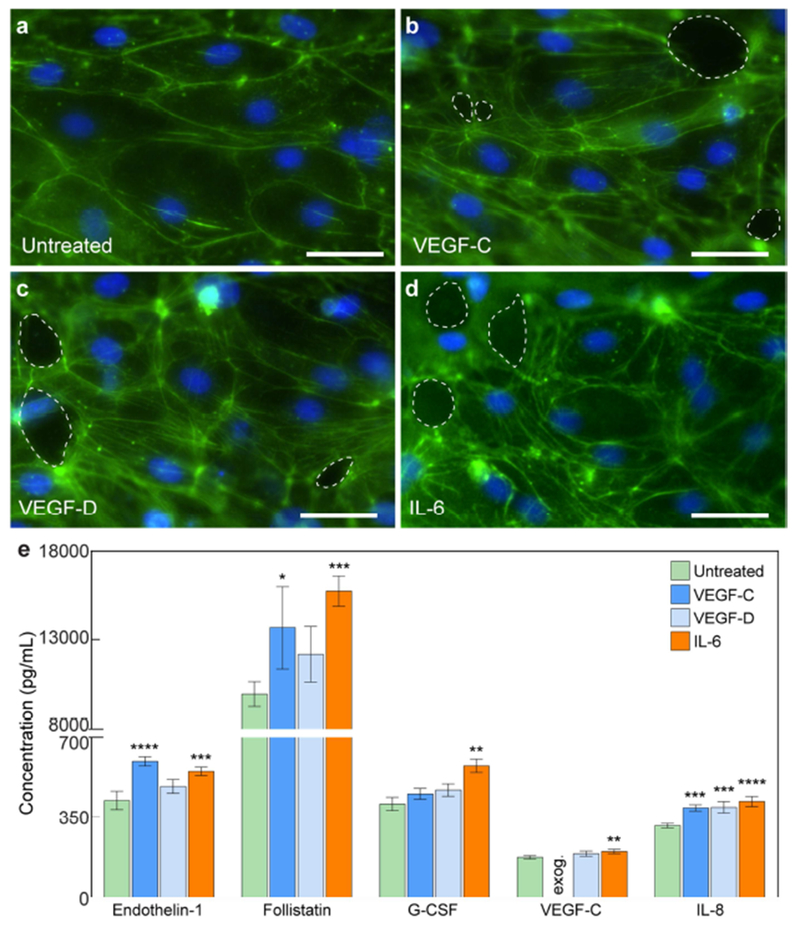
Morphological and secretion responses of lymphatic vessels to exogenous lymphangiogenic and inflammatory stimuli. a-d Images of lymphatic endothelia under VEGF-C, VEGF-D, and IL-6 stimulation (F-actin in green and nuclei in blue). In comparison to untreated vessels, LECs in stimulated vessels express increased actin stress fibers. There are also holes the vessel wall (dashed outlines). e Cytokine concentrations for untreated and stimulated conditions. VEGF-C and IL-6 induce significant changes in all presented cytokines. Concentration values are the averages of n = 4 technical replicates of media pooled from 6 individual vessels for each condition. All error bars are one standard deviation. Scale bars are 10 μm. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001.
Figure 6.
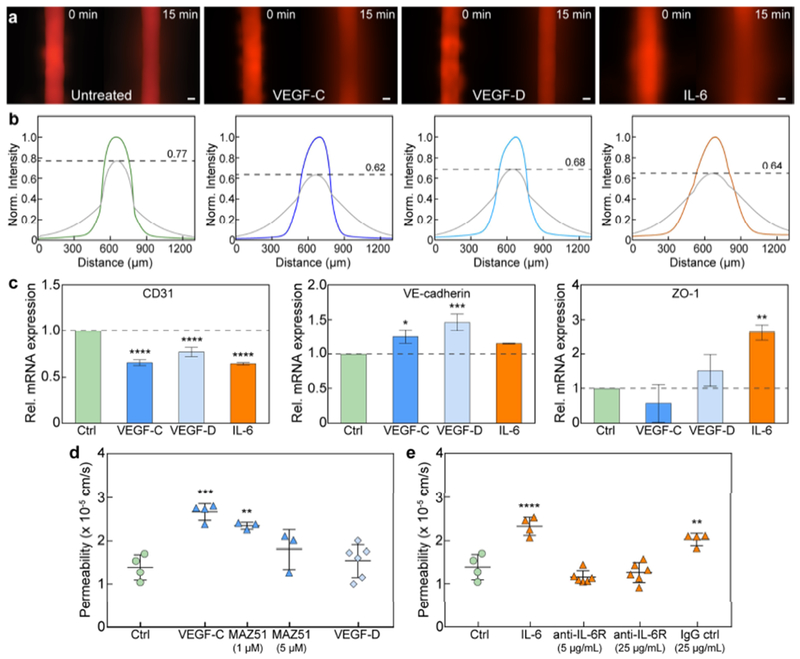
Barrier response of lymphatic vessels to exogenous lymphangiogenic and inflammatory stimuli. a Images of 70 kDa dextran diffusion in untreated vessels and vessels stimulated with VEGF-C, VEGF-D, and IL-6. b Normalized intensity profiles of dextran diffusion. The initial concentration decreases by 38%, 32%, and 36% for VEGF-C, VEGF-D, and IL-6 stimulation, respectively, as compared to 23% for untreated vessels. c Transcriptional expression of intercellular junctions under stimulation. CD31 expression is significantly downregulated for all cases. VE-cadherin expression is upregulated after VEGF-C and VEGF-D treatment, and ZO-1 after IL-6 stimulation. Relative mRNA values are the averages of n = 3 technical replicates with each replicate representing two individual vessels. d Vessel permeability significantly increases after VEGF-C treatment. Barrier function can be rescued by inhibiting the phosphorylation of VEGFR3, the receptor for VEGF-C, with MAZ51 (partially at 1 μM and fully at 5 μM). e IL-6 stimulation significantly reduces barrier capacity, which can be mitigated via antibody-mediated blocking of the IL-6 receptor on the lymphatic vessels. All permeability values are the averages of at least n = 3 individual vessels. All error bars are one standard deviation. Scale bars are 100 μm. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, ****p ≤ 0.0001.
Figure 7.
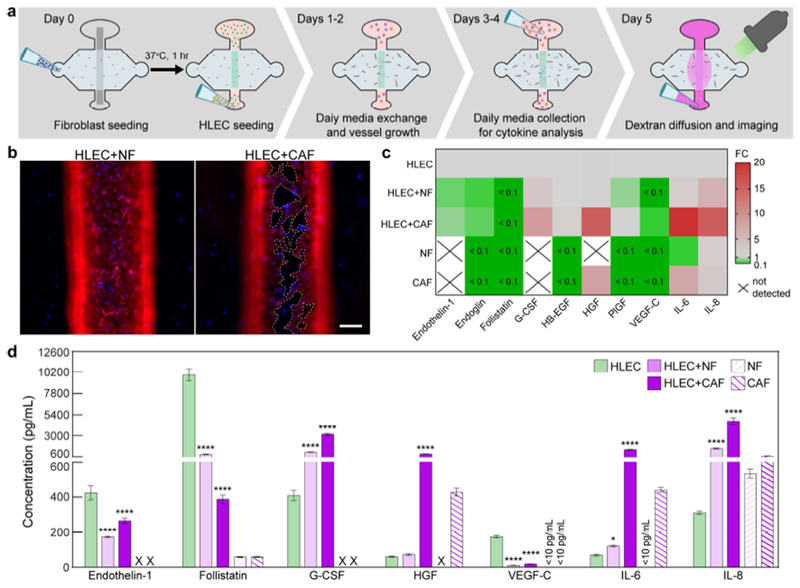
Lymphatic vessel co-culture with normal mammary fibroblasts (NFs) and cancer-associated fibroblasts (CAFs). a Schematic of co-culture experimentation timeline over 5 days. b Images of vessels co-cultured with NFs and CAFs (vimentin in red and nuclei in blue). The lymphatic endothelium is highly destabilized when CAFs are present, where cells have detached from the extracellular matrix (holes indicated by dashed outlines). c Direct comparison of the fold change in secretion levels between fibroblast co-cultures and vessel monoculture, with monoculture levels taken as baseline. CAFs increase the secretion of pro-inflammatory mediators (IL-6 and IL-8) by >15-fold and substantially downregulate the expression of endoglin, follistatin, and VEGF-C. Secretion of G-CSF and HGF is also upregulated by 8-fold and 15-fold, respectively. Secretion levels NF and CAF monocultures are also compared to the HLEC monoculture, where CAFs secrete HGF and IL-6 ~7-fold and 6-fold higher, respectively. d Cytokine concentrations for fibroblast co-cultures. CAFs drive vessels toward a pro-inflammatory phenotype significantly more than NFs and amplify the secretion of key growth factors, e.g. HGF, involved in tumor progression. Concentration values are the averages of n = 4 technical replicates of media pooled from 6 individual vessels for each condition. Error bars are one standard deviation. Scale bar is 100 μm. *p ≤ 0.05, ***p ≤ 0.001, ****p ≤ 0.0001.
Figure 8.
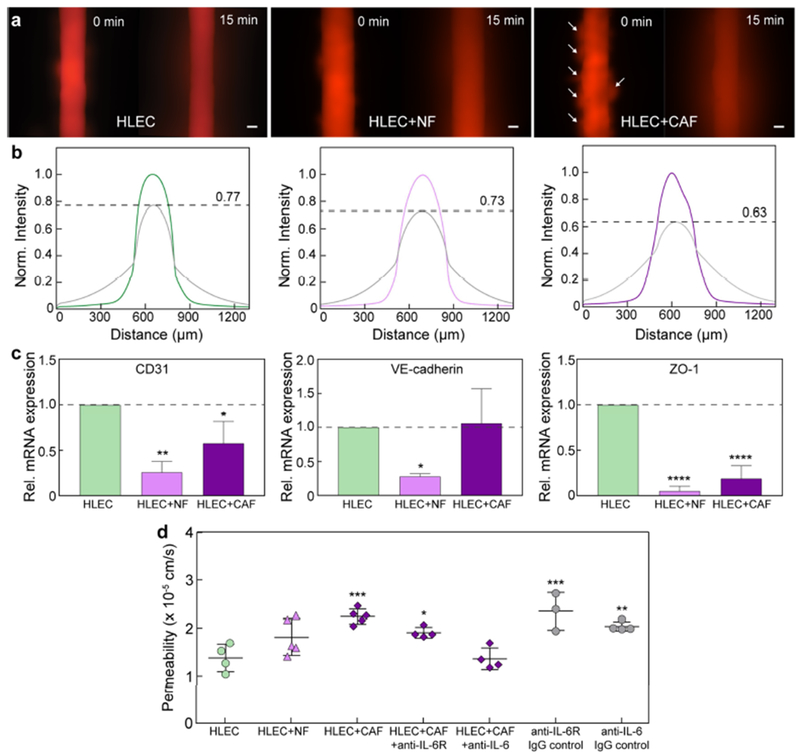
Barrier response of lymphatic vessels co-cultured with normal mammary fibroblasts (NFs) and cancer-associated fibroblasts (CAFs). a Images of 70 kDa dextran diffusion for vessels in monoculture and co-culture. There are multiple holes in the endothelium for the CAF co-culture, as indicated by the focal diffusion regions (arrows). b Normalized intensity profiles of dextran diffusion. The initial concentration decreases by 27% and 37% when co-cultured with NFs and CAFs, respectively, as compared to 23% for vessels in monoculture. c Transcriptional expression of intercellular junctions under co-culture. CD31 and ZO-1 mRNA levels are significantly downregulated for both cases. VE-cadherin expression is downregulated for NFs. Relative mRNA values are the averages of n = 3 technical replicates with each replicate representing two individual vessels. d CAFs significantly increase lymphatic vessel permeability. Barrier function can be rescued, in part, by blocking the IL-6 receptor on the lymphatic vessels. Alternatively, vessel permeability was completely normalized when IL-6 was neutralized with an anti-IL-6 antibody. Permeability values are the averages of at least n = 3 individual vessels. All error bars are one standard deviation. Scale bars are 100 μm. *p ≤ 0.05, **p ≤ 0.01, ****p ≤ 0.0001.
Results
Generation of 3D functional lymphatic endothelial vessels.
We report an organotypic lymphatic vessel model, called the μLYMPH system (Fig. 1), that enables the culture of human lymphatic endothelial vessels and the study of lymphatic vessel biology in tailored microenvironments, such as the tumor microenvironment. The μLYMPH system is a two-layer polydimethylsiloxane (PDMS) device fabricated using standard soft lithography, consisting of a removable PDMS lumen rod suspended across a gel chamber (Fig. 1a). To generate a lymphatic vessel, collagen type I gel is polymerized around the lumen rod, the rod is removed leaving an empty lumen, and the subsequent lumen is seeded with primary human lymphatic endothelial cells (HLECs) isolated from the lymph node (ScienCell) forming vessels with diameters in the range of 200-250 μm (Fig. 1b and c, and Fig. S1). We previously demonstrated this approach for forming HUVEC and iPSC-derived endothelial vessels with high reproducibility[18,19].
Lymphatic vessels were viable for at least seven days for collagen densities ranging from 3 mg/mL to 6 mg/mL; however, softer collagen gels (3 mg/mL and 4 mg/mL) produced significantly more viable vessels with a maximum live fraction of 92% for 3 mg/mL (Fig. 1d). At the higher densities of 5 mg/mL and 6mg/mL, there was cell detachment from the endothelium as indicated by the dashed outlines in Fig. 1d. Matrix stiffening with increasing collagen density has been shown to alter endothelial cell behavior[31], suggesting that the current lymphatic vessels sense and respond to the changing mechanical composition of the surrounding collagen matrix. Importantly, the lymphatic vessels are patent tubular structures (Fig. 2a), allowing perfusion of growth media through the vessels. Lymphatic phenotype of the vessels was characterized by the immunofluorescent staining of LYVE1 (Fig. 2b) and flow cytometry quantification of LYVE1, podoplanin and PROX1 (Fig. 2c). These expression profiles are consistent with the literature[32]. Interestingly, the HUVECs used to generate blood vessels also express LYVE1 and PROX1, which has been shown to occur in vitro[33,34]. Formation of mature lymphatic endothelia was confirmed by the presence of cell-cell junctional proteins, including CD31, VE-cadherin, and ZO-1 (Fig. 2b). Corresponding images of lymphatic markers and junctional proteins for 2D cultured HLEC monolayers are shown in Fig. S2. To transport media through cultured vessels, we leveraged passive pumping[35], where a bolus of media was transported from the small port to the large port due to the difference in Laplace pressures of fluid droplets at the ports. Vessels were perfused with media in this manner at least 2-3 times per day for maintenance. This manner of perfusion mimics the pulsatile nature of lymph flow through lymphatic vessels[1]. In comparison to vessels maintained in the static condition without daily perfusion, we observed endothelial cell alignment in the direction of fluid flow (angle of 90 degrees) as a result of the repeated media exchanges (Fig. 2d and 2e), which is representative of lymphatic endothelial cell behavior in vivo [36].
Figure 2.

Vessel structure and markers. a Confocal image of a lymphatic vessel showing patent tubular structure. b Immunofluorescent of lymphatic marker - LYVE1, endothelial cell marker - CD31, and endothelial cell junctions - CD31, VE-cadherin, and ZO-1. c Flow cytometry quantification of CD31, LYVE1, podoplanin, and PROX1 for both blood (HUVEC) and lymphatic (HLEC) endothelial cells used in the study. HUVECs express CD31, LYVE1, and PROX1, but not podoplanin. HLECs express all four markers. d Cells in the endothelium align in the direction of fluid flow from daily medium exchanges as compared to cells maintained in excess medium without flow (static condition). e Histograms of cell alignment for static and flow conditions. A higher number of cells in the flow condition align in the direction of flow (angle of 90 degrees). Histogram frequency data was generated by combining values of n = 4 individual vessels for each condition.
Cultured lymphatic vessels are functionally different from blood vessels.
The μLYMPH system enables analysis of lymphatic vessel biology, including cytokine secretion and barrier function. These readouts were obtained over a five-day period, which involved culturing the vessels in growth media for two days post-seeding, collecting media for secretion analysis over a subsequent two days, and then assessing vessel permeability at day five with a dextran diffusion assay (Fig. 3a). We measured the secretion levels of a panel of growth factors and inflammatory mediators with a multiplex magnetic bead-based immunoassay (i.e. Luminex MAGPIX) (Fig. 3b and 3c). Both blood and lymphatic vessels produced measurable levels of all cytokines in the panel. In comparison to blood vessels, lymphatic vessels have a unique secretion profile and generally expressed lower levels of the measured cytokines (Fig. 3b). However, there was a particularly striking 18-fold difference in the secretion of follistatin. While blood vessels are known sources of follistatin[37,38], our finding implicates lymphatic vasculature as a major alternative source not previously known. Granulocyte colony stimulating factor (G-CSF) was also produced 2-fold more by the lymphatic vessels. Conversely, the blood vessels expressed significantly higher levels of endoglin (2-fold), hepatocyte growth factor (HGF) (3-fold), placental growth factor (PlGF) (6-fold), vascular endothelial growth factor (VEGF)-C (3-fold), and interleukin (IL)-8 (3-fold).
A hallmark of endothelial vessels is their capacity to regulate the passage of molecules through their endothelium. The permeability of lymphatic vessels cultured in the μLYMPH system was assessed by diffusion assays using 10 kDa and 70 kDa Texas Red dextran. As compared to empty lumens, both blood and lymphatic vessels demonstrated significant barrier function and regulated the passage of the dextran molecules (Fig. 4a and 4b). Lymphatic vessels, however, were leakier than blood vessels with a 23% decrease in the peak intensity of the diffusion profile over 15 minutes versus a 10% decrease for blood vessels (Fig. 4b). We also measured the permeability coefficients of the vessels for both 10 kDa and 70 kDa dextran (Fig. 4c). For the 10 kDa molecular weight, the permeability coefficients of the lymphatic and blood vessels were 2.43 x 10−5 ± 0.29 x 10−5 cm/s and 1.39 x 10−5 ± 0.43 x 10−5 cm/s, respectively. Similarly, for the 70 kDa case, lymphatic vessels were more permeable with a coefficient of 1.38 x 10−5 ± 0.29 x 10−5 cm/s as compared to 0.68 x 10−5 ± 0.13 x 10−5 cm/s for the blood vessels. The barrier capacities of both vessel types were significantly higher for the 70 kDa dextran, indicating tighter regulation of biomolecules in that size range (e.g. serum albumin, ~67 kDa). To further distinguish the physiology of the lymphatic and blood vessels, solute drainage into each vessel type was measured by adding 70 kDa dextran solution to one gel chamber port to generate interstitial flow (Fig. 3a). Drainage into the lymphatic vessels was 1.3-fold higher than for the blood vessels (Fig. 4d), which demonstrates that our model can capture the natural drainage function of the lymphatics. Taken together, these results demonstrate the suitability of the μLYMPH system for assessing lymphatic vessel barrier function, which is distinctly different from blood vessels cultured in the same system.
Lymphangiogenic and inflammatory cytokine stimulation alters vessel function.
To demonstrate the applicability of the μLYMPH system for examining lymphatic biology beyond basic vessel characterization, we assessed the response of cultured vessels in lymphangiogenic and inflammatory microenvironments simulated by exogenous VEGF-C, VEGF-D, and IL-6 cytokine stimulation. Using a similar workflow as depicted in Fig. 3a, VEGF-C (50 ng/mL), VEGF-D (50 ng/mL), and IL-6 (50 ng/mL) were added to the vessels at two days post-seeding and cultured up to five days. For all conditions, there were notable morphological changes to the endothelium in comparison to untreated vessels, namely, increased actin stress fibers and cell detachment (Fig. 5a–d). Cell alignment, however, remained consistent for all stimulated vessels. In Fig. 5e, we limit the results to cytokines with concentration levels greater than 150 pg/mL to better visualize changes in the secretion profile (cytokines with concentration levels less than 150 pg/mL are shown in Fig. S3). Stimulation of the vessels with VEGF-C and IL-6 significantly altered the secretion profile, more so than stimulation with VEGF-D (Fig. 5e). There was, however, a significant increase in IL-8 concentration by VEGF-D stimulation. Specifically, VEGF-C stimulation triggered significant increases in the secretion of endothelin-1, follistatin, and IL-8. IL-6 stimulation significantly increased the secretion levels of all presented factors (endothelin-1, follistatin, G-CSF, VEGF-C, and IL-8).
Similarly, barrier function was significantly altered when the lymphatic vessels were stimulated with VEGF-C, VEGF-D, and IL-6. Solute (70 kDa dextran) diffusion for all three stimulatory conditions was substantially faster in comparison to untreated vessels, where the peak intensity decreased by 38%, 32%, and 36% for VEGF-C, VEGF-D, and IL-6 stimulated vessels, respectively, as compared to 23% for untreated vessels over 15 minutes (Fig. 6a and 6b). Stimulated vessels also responded at the transcriptional level with changes in the mRNA expression of their cell-cell junctional proteins. CD31 transcription was reduced for all stimulatory conditions, whereas VE-cadherin mRNA expression increased for VEGF-C and VEGF-D stimulation, and ZO-1 mRNA expression increased for IL-6 stimulation (Fig. 6c). Moreover, we measured significant increases in permeability for the VEGF-C and IL-6 conditions in comparison to untreated vessels. VEGF-C stimulation increased the permeability of the lymphatic vessels by ~2-fold to 2.67 x 10−5 ± 0.19 x 10−5 cm/s from a baseline value of 1.38 x 10−5 ± 0.29 x 10−5 cm/s (Fig. 6d). However, this increase was mitigated by supplementing MAZ51 to the VEGF-C media used to stimulate the vessels. MAZ51 inhibits VEGF-C signaling by preventing the autophosphorylation of its receptor, VEGFR3[39]. At a dosage of 1 μM MAZ51, the VEGF-C stimulated vessels were still significantly more permeable than the untreated vessels, however, they were less permeable than vessels treated with VEGF-C alone. At a higher dosage of 5 μM MAZ51, VEGF-C stimulated vessels were fully rescued with no measurable difference in permeability as compared to the untreated vessels. Similar results were observed for the IL-6 stimulated vessels, where their permeability was increased by ~2-fold following IL-6 treatment (Fig. 6e). IL-6 induced barrier dysfunction was prevented by treating the lymphatic vessels with anti-IL6R (5 μg/mL and 25 μg/mL), an antibody blocking the binding of IL-6 to its receptor. Collectively, these data demonstrate the utility of the μLYMPH system for simulating different microenvironments by exogenous cytokine stimulation and for targeting molecular pathways that impair barrier function.
Cancer-associated fibroblasts promote a pro-inflammatory lymphatic microenvironment.
We assessed the capacity of the μLYMPH system for examining lymphatic vessel biology in diseased microenvironments, such as the tumor microenvironment (TME), by co-culturing lymphatic vessels with breast cancer-associated fibroblasts (CAFs). CAF-induced responses were compared to co-cultures with normal mammary fibroblasts (NFs). Fibroblast co-cultures were enabled by embedding either CAFs or NFs into the collagen gel prior to forming and seeding the lumen with HLECs (Day 0 in Fig. 7a). The co-cultures were maintained and analyzed using the same workflow as the lymphatic vessel monocultures. A dramatic morphological response was observed for vessels co-cultured with CAFs, where HLECs detached in large areas of the endothelium (dashed outlines in Fig. 7b). There was no observable cell detachment for the NF co-culture controls. Regarding cytokine secretion, co-culture with CAFs induced larger fold changes in cytokine concentration than with NFs, as compared to vessel monocultures (Fig. 7c). Specifically, CAFs upregulated the secretion of pro-tumorigenic growth factors, G-CSF (8-fold) and HGF (15-fold), and pro-inflammatory mediators, IL-6 (20-fold) and IL-8 (15-fold). Interestingly, follistatin secretion, which was at ng/mL levels for the monoculture condition, was reduced to pg/mL levels after co-culture with both CAFs and NFs. Endothelin-1, endoglin, and VEGF-C were also reduced by at least 10-fold for both fibroblast co-culture conditions. PlGF was solely downregulated by NF co-culture.
Notably, cytokine concentrations measured in Fig. 7d indicate that CAFs and NFs either regulate vessel secretion or contribute to the total concentration by their own secretion (cytokines with concentrations <150 pg/mL are shown in Fig. S4). For example, endothelin-1, follistatin, G-CSF, and VEGF-C levels (as well as endoglin, HB-EGF, PlGF levels in Fig. S4) were altered in the fibroblast co-cultures despite negligible secretion of these cytokines by the CAF and NF monocultures, suggesting their associated pathways in the lymphatic vessels are regulated by interactions with the fibroblasts. In the cases of HGF, IL-6, and IL-8, their levels significantly increased in the co-cultures predominantly due to secretion by the CAFs and NFs, as indicated by the accompanying CAF and NF monocultures. Indeed, it is well-known that CAFs are the primary contributors of HGF, IL-6, and IL-8 in the TME[40], demonstrating the capability of our approach to recapitulate pertinent TME stromal interactions.
CAFs also significantly impaired the barrier function of the lymphatic vessels. In agreement with the observed cell detachment in Fig. 7b, vessels co-cultured with CAFs were substantially leakier (leakage from areas of cell detachment indicated by arrows in Fig. 8a), with a 37% decrease in the peak intensity of the diffusion profile over 15 minutes, as compared to 27% and 23% for the NF co-culture and vessel monoculture conditions, respectively (Fig. 8b). However, the impact of CAFs on the transcription of vessel junctional proteins, CD31, VE-cadherin, and ZO-1, was less pronounced in comparison to that of the NFs (Fig. 8c). The downregulation in transcriptional activity measured for the NF co-cultures did not correlate to morphological changes (e.g. cell detachment) or a significant increase in solute diffusion in the vessels. These data suggest that reduction in cell-cell junctional integrity (at the transcription level) may not be the primary mechanism of barrier dysfunction induced by the CAFs. Moreover, vessel permeability significantly increased from 1.38 x 10−5 ± 0.29 x 10−5 cm/s in monoculture to 2.24 x 10−5 ± 0.16 x 10−5 cm/s in co-culture with the CAFs (Fig. 8d). There was a moderate, but insignificant, increase in vessel permeability to 1.81 x 10−5 ± 0.38 x 10−5 cm/s induced by the NFs. Importantly, neutralization of excess IL-6 produced in the co-culture with CAFs, using an anti-IL-6 antibody, fully normalized vessel permeability, while blocking IL-6R on the HLECs partially recovered vessel barrier function (Fig. 8d). Collectively, these results demonstrate the utility of the μLYMPH system for simulating diseased microenvironments, such as the TME. Notably, our co-culture approach enables direct crosstalk between lymphatic vessels and pertinent tumor stromal cells (e.g. CAFs), providing new insight into biological mechanisms of lymphatic dysfunction.
Discussion
Organotypic lymphatic models are few in number, especially in comparison to models of blood vasculature. Models that can recapitulate lymphatic vessel structure and function in relevant microenvironments would advance basic and translational lymphatics research. We developed the μLYMPH system that enables the study of lymphatic vessel biology in normal and diseased microenvironments. To the best of our knowledge, only one other study has demonstrated the culture of lymphatic vessels with in vivo tubular structure [25], which is critical for recapitulating vessel function in vitro [24]. Other microfluidic models have been developed for examining lymphatic vessel permeability and lymphangiogenesis [22,23]; however, they lack in vivo tubular structure. Our system offers capabilities comparable to and beyond existing models including: 1) generation of human lymphatic vessels with physiologically relevant structure and function, 2) characterization of vessel response under cytokine stimulation, 3) co-culture of vessels with stromal components, and 4) assessment of molecular pathways as potential therapeutic targets.
Lymphatic vessels have unique structure-function relationships as compared to their blood vessel counterparts. They are integral components of the blood-lymph loop, specializing in interstitial fluid drainage and recirculation to blood, as well as immunoregulation [1]. The lymphatic vascular tree consists of initial, pre-collecting, and collecting vessels, each having phenotypic differences. For instance, the spatial organization of junctional proteins, such as VE-cadherin, ZO-1, and CD31, differs between the vessel types [27,41]. These proteins form discontinuous button junctions for the initial lymphatic vessels to facilitate fluid drainage and continuous zipper junctions for the pre-collecting and collecting vessels to tightly regulate fluid transport [27,41]. Vessel diameter also changes throughout the lymphatic vascular tree, where diameters range from 10-60 μm for initial vessels [42] and >200 μm for pre-collecting/collecting vessels [28]. Our cultured lymphatic vessels express all three junctional proteins uniformly throughout the endothelium and have diameters in the range of 200-250 μm, suggesting recapitulation of the pre-collecting/collecting phenotype. This is likely the case given the vessels are generated by patterning empty lumens with LECs, rather than grown via lymphvasculogenesis. Nevertheless, the expression of the junctional proteins indicates the vessels have capacity to regulate molecular transport across their endothelia. Indeed, our permeability measurements confirmed moderate barrier function for the lymphatic vessels, situated between little-to-no barrier function for the empty lumens and high barrier function for the blood vessels. This finding is consistent with observations that lymphatic vessels are typically leakier than blood vessels [43]. The permeability of the cultured lymphatic vessels also depended on the size of the diffusing molecule (i.e. 10 kDa versus 70 kDa dextran), demonstrating their capacity to selectively control molecular transport. In comparison to a previous in vitro study on assessing lymphatic vessel permeability [25], our baseline permeability is comparable when adjusted for differences in vessel diameters. Our permeability measurements, however, are typically higher than those quantified for collecting vessels in mouse models [44], a discrepancy likely due to species variance. Furthermore, we showed that solute drainage into the lymphatic vessels was representative of in vivo behavior when compared to the blood vessels. Collectively, our vessel characterization and permeability results demonstrate the capacity of the μLYMPH system for generating physiological and functional lymphatic vessels in vitro.
Lymphatic and blood vessels leverage different molecular pathways for survival and growth, which is evident from their dissimilar cytokine secretion profiles. Previous studies have shown that several angiogenic growth factors, including endoglin, HGF, and VEGF-C measured in our study, can induce lymphangiogenesis [45–48]. Specifically, VEGF-C (and VEGF-D) has been well-characterized as a potent lymphangiogenic factor that activates the VEGFR-3 pathway in LECs [48]. However, our results showed significantly higher VEGF-C secretion by the cultured blood vessels. This finding is consistent with previous work demonstrating higher expression of VEGF-C by blood endothelial cells [49], supporting its role as an angiogenic factor and as a paracrine regulator of lymphatic vessel growth [50,51]. Other than VEGF-C, it is unclear what reciprocal factors are secreted by lymphatic vessels for modulating blood vessel growth. G-CSF, a pro-angiogenic factor [52], is one possible candidate as it was expressed significantly more by the cultured lymphatic vessels. G-CSF also promotes neutrophil differentiation and mobilization to sites of physiological stress via blood circulation [53]. Our results suggest lymphatic vessels may work synergistically with blood vessels in modulating this process. Similarly, IL-8, a well-known neutrophil chemoattractant [54], was measured in both the cultured lymphatic and blood vessels, further suggesting their combined role in regulating the innate immune response. Moreover, the most striking difference in the cytokine secretion profiles, and perhaps the largest differentiator of lymphatic-blood vessel function, was the expression of follistatin (i.e. 18-fold higher for the lymphatic vessels). Follistatin antagonizes activin A, a member of the transforming growth factor-β superfamily involved in cell proliferation, differentiation, and apoptosis depending on the specific microenvironment [55]. In the context of vasculature, the follistatin/activin axis modulates angiogenesis, lymphangiogenesis, and inflammation [38,56,57]. Follistatin is expressed in the circulation, however, there is speculation on its specific source(s). Previous studies have shown that blood endothelial cells and hepatocytes contribute largely to plasma follistatin [37,38,58]. Consistent with these findings, our results confirmed blood endothelial cells as sources of follistatin. However, its significantly higher secretion by LECs suggests lymphatic vessels as a major contributor to plasma follistatin via recirculation of lymph fluid to blood. The follistatin/activin axis may also play an important role in autocrine regulation of lymphatic vessel maintenance, and in lymphatic vessel crosstalk with other tissues. Collectively, the cytokine secretion results demonstrate the utility of the μLYMPH system for culturing functionally distinct lymphatic vessels and identifying specific molecular pathways important to lymphatic biology.
Lymphatic vessels cultured in the μLYMPH system respond to exogenous lymphangiogenic (VEGF-C, VEGF-D) and inflammatory (IL-6) stimuli. As aforementioned, VEGF-C and VEGF-D are key regulators of lymphangiogenesis via the VEGFR-3 pathway [48]. IL-6 is a classic pro-inflammatory cytokine and has been known to induce lymphatic vessel dysfunction in vitro and in vivo [59,60]. Stimulation of lymphatic vessels with all three cytokines resulted in morphological and functional changes, including increased actin stress fibers, holes in the endothelium, altered cytokine secretion, and impaired barrier function. Regarding barrier function, VEGF-C and IL-6 elicited significant increases in vessel permeability, as consistent with similar data for LEC monolayers [59,61]. In the IL-6 study [59], the authors observed a correlation between reduced VE-cadherin protein expression and increased vessel permeability. Our mRNA expression data indicate the opposite, where VE-cadherin mRNA transcription increased following IL-6 stimulation. This discrepancy may be due to differences in approach (i.e. lumens vs monolayers, or gene vs protein expression). Moreover, our mRNA expression and permeability data collectively suggest barrier function may be CD31 mediated, as its transcription was downregulated in all conditions while VE-cadherin and ZO-1 transcription increased or remained near baseline. Furthermore, the increase in vessel permeability is likely compounded by LEC contraction as indicated by the observed increase in actin stress fibers, resulting in localized loss in junctional integrity and holes in the endothelium. Importantly, barrier dysfunction induced by VEGF-C and IL-6 stimulation could be rescued by inhibiting their respective pathways, demonstrating the applicability of the μLYMPH system as a potential therapeutic discovery and screening platform.
In contrast to the established role of blood vasculature in inflammation, the role of lymphatic vasculature remains understudied. Classically, the lymphatic system has been considered a passive player in modulating inflammatory responses, however, emerging data suggests otherwise [62]. Specifically, lymphatic vessels actively remodel (e.g. enlarge) and proliferate to facilitate the clearance of excess interstitial fluid and pro-inflammatory cytokines from the site of inflammation [63,64]. These responses are typically mediated by the increased expression of lymphangiogenic growth factors, such as VEGF-C [63,64]. Our cultured lymphatic vessels exhibited a similar resolution mechanism by releasing more VEGF-C in response to IL-6 treatment. IL-6 stimulation also increased the expression of neutrophil chemotactic factors, G-CSF and IL-8, suggesting neutrophils play a role in modulating lymphatic vascular inflammation. Interestingly, a previous study demonstrated that neutrophils contribute to inflammation-associated lymphangiogenesis by secreting VEGF-D [65]. Furthermore, inflamed vessels released significantly more follistatin, which is a response that typically follows a rise in activin expression during inflammatory insult to neutralize activin-induced inflammatory effects [57]. A similar follistatin response was observed when vessels were stimulated with VEGF-C. These results indicate lymphatic vessels leverage the follistatin/activin axis as an alternate pathway in regulating inflammatory and lymphangiogenic responses, in addition to the IL-6/IL-6R and VEGF-C/VEGFR-3 pathways. Collectively, our vessel stimulation results demonstrate the utility of the μLYMPH system for examining vessel response in tailored microenvironments.
In the context of cancer, lymphatic vessels, like blood vessels, are conduits for the spread of primary cancer cells to secondary sites in the body. Regional nodal metastasis often precedes distant metastasis, and is a prognostic factor for assessing patient survival [66,67]. The interactions between lymphatic vessels and cancer cells that promote cancer progression typically involve lymphatic release of chemokines that enhance cancer cell invasion (e.g. CCL21/CCR7) and tumor lymphangiogenesis via overexpression of VEGF-C by cancer cells [68,69]. There is less known about the interactions between lymphatic vessels and tumor stromal cells that contribute to pro-malignancy and lymphatic metastasis. We examined the impact of breast cancer CAFs on lymphatic vessel cytokine signaling and barrier function to provide initial insight into lymphatic vessel-tumor stroma interactions. Overall, our fibroblast co-culture results indicate that CAFs promote a pro-tumorigenic and pro-inflammatory lymphatic microenvironment. Specifically, CAFs alone contributed to the overexpression of HGF, a pro-tumorigenic factor that enhances cancer cell invasion [70]. They also enhanced lymphatic vessel secretion of G-CSF, which promotes breast cancer metastasis by recruiting tumor-associated neutrophils [71]. There is speculation on the sources of G-CSF in the TME, whether it is largely contributed by tumor cells or tumor stromal cells or both. Our results indicate that LECs are potential stromal sources via CAF stimulation, a mechanism not previously known. Moreover, CAFs induced vessel inflammation by releasing significant levels of pro-inflammatory cytokines, IL-6 and IL-8 [70], and reducing vessel secretion of follistatin. Interestingly, follistatin has been implicated as a prognostic factor in breast cancer, where higher levels correlated with reduced invasion and better patient survival [72]. Taken together, breast CAFs may have the capacity to ‘recognize’ follistatin as an anti-metastatic cytokine and consequently, inhibit its expression. Ultimately, the pro-inflammatory mechanisms induced by the CAFs resulted in significant barrier dysfunction, which has potential implications for lymphatic metastasis. We were able to fully rescue the vessels by neutralizing IL-6 secreted in the CAF co-cultures. Blocking IL-6R on the lymphatic endothelium partially rescued barrier function. This blockade of IL-6 signaling indicates that IL-6 plays a major role in lymphatic vessel dysfunction and may warrant its further investigation as a target to inhibit progression of tumors that commonly rely on lymphatic vessels for spread to regional lymph nodes. Notably, our mRNA expression data showed downregulation of CD31, VE-cadherin, and ZO-1 for both the CAF and NF co-cultures, suggesting that loss of junctional integrity may not be the primary mechanism of CAF-induced barrier dysfunction. Indeed, tumor cells can cause endothelial necroptosis (i.e. inflammatory cell death) as a pro-metastatic mechanism [73], which may extend to CAFs as indicated by the large areas of cell detachment on the endothelium of co-cultured vessels. Collectively, our co-culture results demonstrate the usefulness of the μLYMPH system for examining lymphatic vessel function in diseased microenvironments, such as the TME.
In conclusion, we developed the μLYMPH system to examine lymphatic vessel biology in normal and diseased microenvironments. Our approach enables the generation of 3D human lymphatic vessels with in vivo-like structure-function relationships distinct from blood vessels. The system can be tailored to assess vessel response in different stimulatory conditions. Its capacity for co-culture further extends its modeling capabilities to relevant diseased microenvironments, such as the TME. We identified the follistatin/activin axis as a pathway unique to lymphatic vessels, which has a potential role in modulating lymphatic vascular inflammation and tumor microenvironmental interactions. Importantly, we demonstrated the efficacy of the μLYMPH system as a therapeutic screening platform, being able to mitigate cytokine (VEGF-C and IL-6) and CAF-induced barrier dysfunction by inhibiting relevant molecular pathways. Overall, our system advances the capabilities of existing in vitro lymphatic models, offering a powerful alternative to animal models.
Supplementary Material
Acknowledgements
We acknowledge a Postdoctoral Fellowship from the Natural Sciences and Engineering Research Council of Canada to M.M.G. and a fellowship from the UW-Madison Graduate Engineering Research Scholars program to K.M.L. Research reported in this publication was supported by NIH (R01EB010039) and the Wisconsin Head & Neck Cancer SPORE (P50DE026787). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. Furthermore, we are grateful to Dr. Lisa Arendt’s lab for providing the normal human mammary fibroblasts and Dr. Andreas Friedl’s lab for the breast cancer-associated fibroblasts.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Competing interests
David J. Beebe holds equity in Bellbrook Labs, LLC, Tasso Inc., Salus Discovery LLC, Stacks to the Future, LLC and Onexio Biosystems, LLC.
Data availability
The raw/processed data required to reproduce these findings cannot be shared at this time due to technical or time limitations.
References
- [1].Swartz M, The physiology of the lymphatic system, Adv. Drug Deliv. Rev 50 (2001) 3–20. doi: 10.1016/S0169-409X(01)00150-8. [DOI] [PubMed] [Google Scholar]
- [2].Küppers R, Mechanisms of B-cell lymphoma pathogenesis, Nat. Rev. Cancer 5 (2005) 251 10.1038/nrc1589. [DOI] [PubMed] [Google Scholar]
- [3].Padera TP, Meijer EFJ, Munn LL, The Lymphatic System in Disease Processes and Cancer Progression, Annu. Rev. Biomed. Eng 18 (2016) 125–158. doi: 10.1146/annurev-bioeng-112315-031200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Kaipainen A, Korhonen J, Mustonen T, van Hinsbergh VW, Fang GH, Dumont D, Breitman M, Alitalo K, Expression of the fms-like tyrosine kinase 4 gene becomes restricted to lymphatic endothelium during development., Proc. Natl. Acad. Sci 92 (1995) 3566–3570. doi: 10.1073/pnas.92.8.3566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Wigle JT, Oliver G, Prox1 Function Is Required for the Development of the Murine Lymphatic System, Cell. 98 (1999) 769–778. doi: 10.1016/S0092-8674(00)81511-1. [DOI] [PubMed] [Google Scholar]
- [6].Breiteneder-Geleff S, Soleiman A, Kowalski H, Horvat R, Amann G, Kriehuber E, Diem K, Weninger W, Tschachler E, Alitalo K, Kerjaschki D, Angiosarcomas Express Mixed Endothelial Phenotypes of Blood and Lymphatic Capillaries, Am. J. Pathol 154 (1999) 385–394. doi: 10.1016/S0002-9440(10)65285-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Banerji S, Ni J, Wang S-X, Clasper S, Su J, Tammi R, Jones M, Jackson DG, LYVE-1, a New Homologue of the CD44 Glycoprotein, Is a Lymph-specific Receptor for Hyaluronan, J. Cell Biol 144 (1999) 789–801. doi: 10.1083/jcb.144.4.789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Bruyère F, Noël A, Lymphangiogenesis: in vitro and in vivo models, FASEB J. 24 (2010) 8–21. doi: 10.1096/fj.09-132852. [DOI] [PubMed] [Google Scholar]
- [9].Bruyère F, Melen-Lamalle L, Blacher S, Roland G, Thiry M, Moons L, Frankenne F, Carmeliet P, Alitalo K, Libert C, Sleeman JP, Foidart J-M, Noël A, Modeling lymphangiogenesis in a three-dimensional culture system, Nat. Methods 5 (2008) 431–437. doi: 10.1038/nmeth.1205. [DOI] [PubMed] [Google Scholar]
- [10].Huh D, Torisawa Y, Hamilton GA, Kim HJ, Ingber DE, Microengineered physiological biomimicry: Organs-on-Chips, Lab Chip. 12 (2012) 2156. doi: 10.1039/c2lc40089h. [DOI] [PubMed] [Google Scholar]
- [11].Bhatia SN, Ingber DE, Microfluidic organs-on-chips, Nat. Biotechnol 32 (2014) 760–772. doi: 10.1038/nbt.2989. [DOI] [PubMed] [Google Scholar]
- [12].Zhang B, Korolj A, Lai BFL, Radisic M, Advances in organ-on-a-chip engineering, Nat. Rev. Mater 3 (2018) 257–278. doi: 10.1038/s41578-018-0034-7. [DOI] [Google Scholar]
- [13].Chrobak KM, Potter DR, Tien J, Formation of perfused, functional microvascular tubes in vitro, Microvasc. Res 71 (2006) 185–196. doi: 10.1016/j.mvr.2006.02.005. [DOI] [PubMed] [Google Scholar]
- [14].Raghavan S, Nelson CM, Baranski JD, Lim E, Chen CS, Geometrically Controlled Endothelial Tubulogenesis in Micropatterned Gels, Tissue Eng. Part A 16 (2010) 2255–2263. doi: 10.1089/ten.tea.2009.0584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Zheng Y, Chen J, Craven M, Choi NW, Totorica S, Diaz-Santana A, Kermani P, Hempstead B, Fischbach-Teschl C, Lopez JA, Stroock AD, In vitro microvessels for the study of angiogenesis and thrombosis, Proc. Natl. Acad. Sci 109 (2012) 9342–9347. doi: 10.1073/pnas.1201240109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Bischel LL, Lee S-H, Beebe DJ, A Practical Method for Patterning Lumens through ECM Hydrogels via Viscous Finger Patterning, J. Lab. Autom 17 (2012) 96–103. doi: 10.1177/2211068211426694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Bischel LL, Young EWK, Mader BR, Beebe DJ, Tubeless microfluidic angiogenesis assay with three-dimensional endothelial-lined microvessels, Biomaterials. 34 (2013) 1471–1477. doi: 10.1016/j.biomaterials.2012.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Jiménez-Torres JA, Peery SL, Sung KE, Beebe DJ, LumeNEXT: A Practical Method to Pattern Luminal Structures in ECM Gels, Adv. Healthc. Mater 5 (2016) 198–204. doi: 10.1002/adhm.201500608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Ingram PN, Hind LE, Jiménez-Torres JA, Huttenlocher A, Beebe DJ, An Accessible Organotypic Microvessel Model Using iPSC-Derived Endothelium, Adv. Healthc. Mater 1700497 (2017) 1700497. doi: 10.1002/adhm.201700497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Kim S, Lee H, Chung M, Jeon NL, Engineering of functional, perfusable 3D microvascular networks on a chip, Lab Chip. 13 (2013) 1489. doi: 10.1039/c3lc41320a. [DOI] [PubMed] [Google Scholar]
- [21].Jeon JS, Bersini S, Whisler JA, Chen MB, Dubini G, Charest JL, Moretti M, Kamm RD, Generation of 3D functional microvascular networks with human mesenchymal stem cells in microfluidic systems, Integr. Biol 6 (2014) 555–563. doi: 10.1039/C3IB40267C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Sato M, Sasaki N, Ato M, Hirakawa S, Sato K, Sato K, Microcirculation-on-a-Chip: A Microfluidic Platform for Assaying Blood- and Lymphatic-Vessel Permeability, PLoS One. 10 (2015) e0137301. doi: 10.1371/journal.pone.0137301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Kim S, Chung M, Jeon NL, Three-dimensional biomimetic model to reconstitute sprouting lymphangiogenesis in vitro, Biomaterials. 78 (2016) 115–128. doi : 10.1016/j.biomaterials.2015.11.019. [DOI] [PubMed] [Google Scholar]
- [24].Bischel LL, Sung KE, Jiménez-Torres JA, Mader BR, Keely PJ, Beebe DJ, The importance of being a lumen, FASEB J. 28 (2014) 4583–4590. doi: 10.1096/fj.13-243733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Price GM, Chrobak KM, Tien J, Effect of cyclic AMP on barrier function of human lymphatic microvascular tubes, Microvasc. Res 76 (2008) 46–51. doi: 10.1016/j.mvr.2008.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Wong KHK, Truslow JG, Khankhel AH, Chan KLS, Tien J, Artificial lymphatic drainage systems for vascularized microfluidic scaffolds, J. Biomed. Mater. Res. Part A 101A (2013) 2181–2190. doi: 10.1002/jbm.a.34524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Ulvmar MH, Mäkinen T, Heterogeneity in the lymphatic vascular system and its origin, Cardiovasc. Res 111 (2016) 310–321. doi: 10.1093/cvr/cvw175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Mohanakumar S, Majgaard J, Telinius N, Katballe N, Pahle E, Hjortdal V, Boedtkjer D, Spontaneous and α-adrenoceptor-induced contractility in human collecting lymphatic vessels require chloride, Am. J. Physiol. Circ. Physiol 315 (2018) H389–H401. doi: 10.1152/ajpheart.00551.2017. [DOI] [PubMed] [Google Scholar]
- [29].Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez J-Y, White DJ, Hartenstein V, Eliceiri K, Tomancak P, Cardona A, Fiji: an open-source platform for biological-image analysis, Nat. Methods 9 (2012) 676 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Huxley VH, Curry FE, Adamson RH, Quantitative fluorescence microscopy on single capillaries: alpha-lactalbumin transport., Am. J. Physiol 252 (1987) H188–97. doi: 10.1152/ajpheart.1987.252.1.H188. [DOI] [PubMed] [Google Scholar]
- [31].Byfield FJ, Reen RK, Shentu T-P, Levitan I, Gooch KJ, Endothelial actin and cell stiffness is modulated by substrate stiffness in 2D and 3D, J. Biomech 42 (2009) 1114–1119. doi: 10.1016/j.jbiomech.2009.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Xiong Y, Brinkman CC, Famulski KS, Mongodin EF, Lord CJ, Hippen KL, Blazar BR, Bromberg JS, A robust in vitro model for trans-lymphatic endothelial migration, Sci. Rep 7 (2017) 1633. doi: 10.1038/s41598-017-01575-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Nguyen VA, Fürhapter C, Obexer P, Stössel H, Romani N, Sepp N, Endothelial cells from cord blood CD133+CD34+ progenitors share phenotypic, functional and gene expression profile similarities with lymphatics, J. Cell. Mol. Med 13 (2009) 522–534. doi: 10.1111/j.1582-4934.2008.00340.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Cooley LS, Handsley MM, Zhou Z, Lafleur MA, Pennington CJ, Thompson EW, Pöschl E, Edwards DR, Reversible transdifferentiation of blood vascular endothelial cells to a lymphatic-like phenotype in vitro, J. Cell Sci 123 (2010) 3808–3816. doi: 10.1242/jcs.064279. [DOI] [PubMed] [Google Scholar]
- [35].Walker GM, Beebe DJ, A passive pumping method for microfluidic devices, Lab Chip. 2 (2002) 131. doi: 10.1039/b204381e. [DOI] [PubMed] [Google Scholar]
- [36].Sweet DT, Jiménez JM, Chang J, Hess PR, Mericko-Ishizuka P, Fu J, Xia L, Davies PF, Kahn ML, Lymph flow regulates collecting lymphatic vessel maturation in vivo, J. Clin. Invest 125 (2015) 2995–3007. doi: 10.1172/JCI79386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Michel U, Schneider O, Kirchhof C, Meisel S, Smirnov A, Wiltfang J, Rieckmann P, Production of follistatin in porcine endothelial cells: differential regulation by bacterial compounds and the synthetic glucocorticoid RU 28362., Endocrinology. 137 (1996) 4925–4934. doi: 10.1210/endo.137.11.8895365. [DOI] [PubMed] [Google Scholar]
- [38].Kozian DH, Ziche M, Augustin HG, The activin-binding protein follistatin regulates autocrine endothelial cell activity and induces angiogenesis., Lab. Invest 76 (1997) 267–76. http://www.ncbi.nlm.nih.gov/pubmed/9042163. [PubMed] [Google Scholar]
- [39].Kirkin V, Thiele W, Baumann P, Mazitschek R, Rohde K, Fellbrich G, Weich H, Waltenberger J, Giannis A, Sleeman JP, MAZ51, an indolinone that inhibits endothelial cell and tumor cell growth in vitro, suppresses tumor growth in vivo., Int. J. Cancer 112 (2004) 986–93. doi: 10.1002/ijc.20509. [DOI] [PubMed] [Google Scholar]
- [40].Cirri P, Chiarugi P, Cancer-associated-fibroblasts and tumour cells: a diabolic liaison driving cancer progression, Cancer Metastasis Rev. 31 (2012) 195–208. doi: 10.1007/s10555-011-9340-x. [DOI] [PubMed] [Google Scholar]
- [41].Baluk P, Fuxe J, Hashizume H, Romano T, Lashnits E, Butz S, Vestweber D, Corada M, Molendini C, Dejana E, McDonald DM, Functionally specialized junctions between endothelial cells of lymphatic vessels., J. Exp. Med 204 (2007) 2349–62. doi: 10.1084/jem.20062596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Margaris KN, Black R. a, Modelling the lymphatic system: challenges and opportunities, J R Soc Interface. 9 (2012) 601–612. doi: 10.1098/rsif.2011.0751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].O’Morchoe CC, O’Morchoe PJ, Differences in lymphatic and blood capillary permeability: ultrastructural-functional correlations., Lymphology. 20 (1987) 205–9. http://www.ncbi.nlm.nih.gov/pubmed/3328024. [PubMed] [Google Scholar]
- [44].Scallan JP, Huxley VH, In vivo determination of collecting lymphatic vessel permeability to albumin: A role for lymphatics in exchange, J. Physiol 588 (2010) 243–254. doi: 10.1113/jphysiol.2009.179622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Niessen K, Zhang G, Ridgway JB, Chen H, Yan M, ALK1 signaling regulates early postnatal lymphatic vessel development, Blood. 115 (2010) 1654–1661. doi: 10.1182/blood-2009-07-235655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Kajiya K, Hirakawa S, Ma B, Drinnenberg I, Detmar M, Hepatocyte growth factor promotes lymphatic vessel formation and function, EMBO J. 24 (2005) 2885–2895. doi: 10.1038/sj.emboj.7600763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Cao R, Bjorndahl M. a, Gallego MI, Chen S, Religa P, Hansen a J., Cao Y, Hepatocyte growth factor is a novel lymphangiogenic factor with an indirect mechamism of action, Blood. (2006) 3531–3537. doi: 10.1182/blood-2005-06-2538.Supported. [DOI] [PubMed] [Google Scholar]
- [48].Jussila L, Alitalo K, Vascular growth factors and lymphangiogenesis., Physiol. Rev 82 (2002) 673–700. doi: 10.1152/physrev.00005.2002. [DOI] [PubMed] [Google Scholar]
- [49].Kriehuber E, Breiteneder-Geleff S, Groeger M, Soleiman A, Schoppmann SF, Stingl G, Kerjaschki D, Maurer D, Isolation and Characterization of Dermal Lymphatic and Blood Endothelial Cells Reveal Stable and Functionally Specialized Cell Lineages, J. Exp. Med 194 (2001) 797–808. http://jem.rupress.org/content/194/6/797.abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Cao Y, Linden P, Farnebo J, Cao R, Eriksson A, Kumar V, Qi J-H, Claesson-Welsh L, Alitalo K, Vascular endothelial growth factor C induces angiogenesis in vivo, Proc. Natl. Acad. Sci 95 (1998) 14389–14394. http://www.pnas.org/content/95/24/14389.abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Nakao S, Zandi S, Hata Y, Kawahara S, Arita R, Schering A, Sun D, Melhorn MI, Ito Y, Lara-Castillo N, Ishibashi T, Hafezi-Moghadam A, Blood vessel endothelial VEGFR-2 delays lymphangiogenesis: an endogenous trapping mechanism links lymph- and angiogenesis, Blood. 117 (2011) 1081–1090. http://www.bloodjournal.org/content/117/3/1081.abstract. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Natori T, Sata M, Washida M, Hirata Y, Nagai R, Makuuchi M, G-CSF stimulates angiogenesis and promotes tumor growth: potential contribution of bone marrow-derived endothelial progenitor cells., Biochem. Biophys. Res. Commun 297 (2002) 1058–61. http://www.ncbi.nlm.nih.gov/pubmed/12359263. [DOI] [PubMed] [Google Scholar]
- [53].Roberts AW, G-CSF: A key regulator of neutrophil production, but that’s not all!, Growth Factors. 23 (2005) 33–41. doi: 10.1080/08977190500055836. [DOI] [PubMed] [Google Scholar]
- [54].Hammond ME, Lapointe GR, Feucht PH, Hilt S, Gallegos CA, Gordon CA, Giedlin MA, Mullenbach G, Tekamp-Olson P, IL-8 induces neutrophil chemotaxis predominantly via type I IL-8 receptors., J. Immunol 155 (1995) 1428–33. http://www.ncbi.nlm.nih.gov/pubmed/7636208. [PubMed] [Google Scholar]
- [55].Phillips DJ, De Kretser DM, Follistatin: A multifunctional regulatory protein, Front. Neuroendocrinol 19 (1998) 287–322. doi: 10.1006/frne.1998.0169. [DOI] [PubMed] [Google Scholar]
- [56].Heinz M, Niederleithner HL, Puujalka E, Soler-Cardona A, Grusch M, Pehamberger H, Loewe R, Petzelbauer P, Activin A Is Anti-Lymphangiogenic in a Melanoma Mouse Model, J. Invest. Dermatol 135 (2015) 212–221. doi: 10.1038/jid.2014.328. [DOI] [PubMed] [Google Scholar]
- [57].Jones KL, Mansell A, Patella S, Scott BJ, Hedger MP, de Kretser DM, Phillips DJ, Activin A is a critical component of the inflammatory response, and its binding protein, follistatin, reduces mortality in endotoxemia, Proc. Natl. Acad. Sci 104 (2007) 16239–16244. doi: 10.1073/pnas.0705971104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Hansen JS, Rutti S, Arous C, Clemmesen JO, Secher NH, Drescher A, Gonelle-Gispert C, Halban PA, Pedersen BK, Weigert C, Bouzakri K, Plomgaard P, Circulating Follistatin Is Liver-Derived and Regulated by the Glucagon-to-Insulin Ratio., J. Clin. Endocrinol. Metab 101 (2016) 550–60. doi: 10.1210/jc.2015-3668. [DOI] [PubMed] [Google Scholar]
- [59].Cromer WE, Zawieja SD, Tharakan B, Childs EW, Newell MK, Zawieja DC, The effects of inflammatory cytokines on lymphatic endothelial barrier function., Angiogenesis. 17 (2014) 395–406. doi: 10.1007/s10456-013-9393-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Aldrich MB, Sevick-Muraca EM, Cytokines are systemic effectors of lymphatic function in acute inflammation., Cytokine. 64 (2013) 362–9. doi: 10.1016/j.cyto.2013.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Breslin JW, Yuan SY, Wu MH, VEGF-C alters barrier function of cultured lymphatic endothelial cells through a VEGFR-3-dependent mechanism., Lymphat. Res. Biol 5 (2007) 105–13. doi: 10.1089/lrb.2007.1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Wilting J, Becker J, Buttler K, Weich HA, Lymphatics and inflammation., Curr. Med. Chem 16 (2009) 4581–92. http://www.ncbi.nlm.nih.gov/pubmed/19903150. [DOI] [PubMed] [Google Scholar]
- [63].Huggenberger R, Siddiqui SS, Brander D, Ullmann S, Zimmermann K, Antsiferova M, Werner S, Alitalo K, Detmar M, An important role of lymphatic vessel activation in limiting acute inflammation., Blood. 117 (2011) 4667–78. doi: 10.1182/blood-2010-10-316356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Lachance P-A, Hazen A, Sevick-Muraca EM, Lymphatic Vascular Response to Acute Inflammation, PLoS One. 8 (2013) e76078 10.1371/journal.pone.0076078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Tan KW, Chong SZ, Wong FHS, Evrard M, Tan SM-L, Keeble J, Kemeny DM, Ng LG, Abastado J-P, Angeli V, Neutrophils contribute to inflammatory lymphangiogenesis by increasing VEGF-A bioavailability and secreting VEGF-D, Blood. 122 (2013) 3666–3677. http://www.bloodjournal.org/content/122/22/3666.abstract. [DOI] [PubMed] [Google Scholar]
- [66].McAllaster JD, Cohen MS, Role of the lymphatics in cancer metastasis and chemotherapy applications, Adv. Drug Deliv. Rev 63 (2011) 867–875. doi: 10.1016/j.addr.2011.05.014. [DOI] [PubMed] [Google Scholar]
- [67].Stacker SA, Williams SP, Karnezis T, Shayan R, Fox SB, Achen MG, Lymphangiogenesis and lymphatic vessel remodelling in cancer, Nat. Rev. Cancer 14 (2014) 159–172. doi: 10.1038/nrc3677. [DOI] [PubMed] [Google Scholar]
- [68].Alitalo A, Detmar M, Interaction of tumor cells and lymphatic vessels in cancer progression, Oncogene. 31 (2011) 4499 10.1038/onc.2011.602. [DOI] [PubMed] [Google Scholar]
- [69].Stachura J, Wachowska M, Kilarski WW, Guc E, Golab J, Muchowicz A, The dual role of tumor lymphatic vessels in dissemination of metastases and immune response development., Oncoimmunology. 5 (2016) e1182278. doi: 10.1080/2162402X.2016.1182278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Erdogan B, Webb DJ, Cancer-associated fibroblasts modulate growth factor signaling and extracellular matrix remodeling to regulate tumor metastasis., Biochem. Soc. Trans 45 (2017) 229–236. doi: 10.1042/BST20160387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Mouchemore KA, Anderson RL, Hamilton JA, Neutrophils, G-CSF and their contribution to breast cancer metastasis., FEBS J 285 (2018) 665–679. doi: 10.1111/febs.14206. [DOI] [PubMed] [Google Scholar]
- [72].Zabkiewicz C, Resaul J, Hargest R, Jiang WG, Ye L, Increased Expression of Follistatin in Breast Cancer Reduces Invasiveness and Clinically Correlates with Better Survival, Cancer Genomics Proteomics. 14 (2017) 241–251. doi: 10.21873/cgp.20035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Strilic B, Yang L, Albarrán-Juárez J, Wachsmuth L, Han K, Müller UC, Pasparakis M, Offermanns S, Tumour-cell-induced endothelial cell necroptosis via death receptor 6 promotes metastasis., Nature. 536 (2016) 215–8. doi: 10.1038/nature19076. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.