

Abstract
Low blood phosphate (Pi) reduces muscle function in hypophosphatemic disorders. Which Pi transporters are required and whether hormonal changes due to hypophosphatemia contribute to muscle function is unknown. To address these questions we generated a series of conditional knockout mice lacking one or both house-keeping Pi transporters Pit1 and Pit2 in skeletal muscle (sm), using the postnatally expressed human skeletal actin-cre. Simultaneous conditional deletion of both transporters caused skeletal muscle atrophy, resulting in death by postnatal day P13. smPit1−/−, smPit2−/− and three allele mutants are fertile and have normal body weights, suggesting a high degree of redundance for the two transporters in skeletal muscle. However, these mice show a gene-dose dependent reduction in running activity also seen in another hypophosphatemic model (Hyp mice). In contrast to Hyp mice, grip strength is preserved. Further evaluation of the mechanism shows reduced ERK1/2 activation and stimulation of AMP kinase in skeletal muscle from smPit1−/−; smPit2−/− mice consistent with energy-stress. Similarly, C2C12 myoblasts show a reduced oxygen consumption rate mediated by Pi transport-dependent and ERK1/2-dependent metabolic Pi sensing pathways. In conclusion, we here show that Pit1 and Pit2 are essential for normal myofiber function and survival, insights which may improve management of hypophosphatemic myopathy.
Subject terms: Endocrine system and metabolic diseases, Phosphorus metabolism disorders
Introduction
Inorganic phosphate (Pi) is involved in various cellular processes including DNA and cell membrane synthesis, signal transduction, ATP production, and bone mineralization. Serum Pi is regulated by a hormonal bone-parathyroid-kidney axis consisting of fibroblast growth factor 23 (FGF23), parathyroid hormone (PTH), and 1,25(OH)2-D (calcitriol)1. Familial disorders of Pi homeostasis are caused by mutations in components of this axis that either directly or indirectly (via homeostatic mechanisms) lower serum Pi levels. Furthermore, chronic hypophosphatemia is observed in the often vitamin D-, and therefore Pi-, deficient elderly population2.
How muscle weakness develops in these hypophosphatemic conditions, which Pi transporters are involved and whether the homeostatic hormonal changes, which develop as a result of hypophosphatemia, contribute to reduced muscle function is poorly understood. Previous studies suggest that decreased ATP3,4 and phosphodiesters5 may in part explain the muscle weakness seen in hypophosphatemia. We recently reported that hypophosphatemic myopathy goes along with reduced ATP flux (VATP) and intracellular Pi in an individual with hereditary hypophosphatemic rickets with hypercalciuria (HHRH) and in the sodium-Pi co-transporter Npt2a null mouse model of this disorder6. Basal and insulin-stimulated muscle VATP and Pi uptake have furthermore been shown to be decreased in the offspring of patients with type 2 diabetes7.
While these studies support a direct role for Pi as a substrate for mitochondrial ATP production, it is not clear whether extracellular Pi needs to enter skeletal muscle to improve VATP and muscle function, and which Pi transporters and intracellular signaling pathways are involved. Pit1 and Pit2 are type III sodium-Pi co-transporters encoded by Slc20a1 and Slc20a28. Initially discovered as retroviral receptors9,10, they since have been shown to be ubiquitously expressed Pi-transporters in mammals. Pi-uptake via Pit1 has been shown to maintain ATP levels in pre-osteoblasts11 and chondrocytes12, and it is possible that Pit1 has similar functions in other tissues, including skeletal muscle. In support of a role for Pit1 in skeletal muscle ATP production, Pit1 and Pi levels appear to correlate with fiber-type, with slow twitch having greater levels of Pit1 and Pi than fast twitch fibers13,14, perhaps because slow twitch fibers require more Pi transport to maintain their high oxidative capacity. However, no skeletal muscle phenotype has been described thus far in mice with global ablation of Slc20a1/Pit1, which causes embryonic lethality due to liver and hematopoetic abnormalities15. Likewise, mice with global ablation of Slc20a2/Pit2 are fertile, appear to thrive normally and have comparable body weights without reported skeletal muscle phenotypes16–19. Interestingly, recent evidence suggests that Pit1 can stimulate cell proliferation20, gene expression11, apoptosis21 and activation of the mitogen-activated kinases ERK1/2 in many cells including myocytes11,22 independently of its transport activity. MAPK phosphatases (MKPs) can both positively and negatively regulate myogenesis through coordinate MAPK dephosphorylation23–26. For example, p38 MAPK is believed to be a promyogenic MAPK, while ERK1/2 has been shown to exhibit both positive and negative regulatory roles in myogenesis, and c-Jun NH2 terminus kinases (JNK) appears in some cases to be either dispensable or negative in myogenesis. Furthermore, muscle ablation of ERK1/2 isoforms using HSA-Cre results in severe atrophy of the soleus muscles, revealing their important role for the maintenance of myofibers and neuromuscular synapses in adult mice, particularly in type 1 slow twitch fibers27,28.
Also, it is not clear whether FGF23, PTH, and 1,25(OH)2-D can contribute to hypophosphatemic myopathy by directly affecting muscle function in a Pi transport-independent fashion. For instance, loss-of-function mutations in the sodium-Pi transport protein 2c (SLC34A3) gene have been associated with hypophosphatemic rickets with hypercalciuria (HHRH)29, a disorder that causes renal Pi wasting, rickets and kidney stones, but muscle weakness is less pronounced in these individuals when compared to the muscle weakness seen in X-linked hypophosphatemia (XLH)30 or tumor-induced osteomalacia (TIO)31. FGF23, which is low in HHRH, but elevated in XLH and TIO, was shown to induce left ventricular hypertrophy via activation of the calcineurin-NFAT signaling pathway32, but whether FGF23 impacts skeletal muscle function is unclear33. Initial clinical observations from the use of a novel FGF23 neutralizing therapy34,35 in XLH have shown marked improvement in fatigue, muscle weakness and activity, suggesting that this therapy offers an advantage over standard therapy (oral Pi and calcitriol). Likewise, the Hyp mouse model for XLH has reduced running activity and grip strength, and anti-FGF23 antibodies restore normophosphatemia, endurance and grip strength36. Although no short-term effect of FGF23 on muscle function was reported in ex vivo settings37, chronic exposure to this hormone may be negative for muscle strength as shown in Dmp1 null mice38, which like Hyp mice have FGF23-dependent hypophosphatemia. In addition, in vitamin D-deficient individuals, cholecalciferol therapy was shown to improve mitochondrial activity and restore muscle function39,40. High levels of PTH have been shown to cause myopathy in mice independent of blood Pi, but the mechanism is not known41,42.
To determine the function of Pit1 and Pit2 in skeletal muscle independent of hypophosphatemia and the homeostatic endocrine changes resulting from hypophosphatemia, we generated a series of conditional knockout mice lacking one or two copies of Slc20a1 and Slc20a2 (smPit1−/−; smPit2−/− mice), using the postnatally expressed human skeletal actin (HSA)-cre. These mice show a gene-dose dependent reduction in running activity resembling impaired endurance seen with hypophosphatemia, while interestingly grip strength in these mice is preserved. Furthermore, simultaneous deletion of both transporters in skeletal muscle in smPit1−/−; smPit2−/− mice is lethal postnatally due to skeletal muscle hypoplasia/atrophy with myofiber degeneration, which appears to be caused by energy-stress induced myofiber necrosis.
Results
Generation of skeletal muscle-specific Pit1/2 double knockout mice
We used semi-quantitative RT-PCR to determine that Pit1 and Pit2 are the dominant Pi importers expressed in skeletal muscle (Fig. S1A). In addition to these two Pi importers, the putative Pi exporter Xenotropic And Polytropic Retrovirus Receptor (Xpr1) and the mitochondrial Pi importer PIC/Slc25a3 are expressed at high levels. When compared to Pit1, Pit2 mRNA levels are one order of magnitude higher in most tissues when estimated using this approach (Fig. S1B), which was verified with multiple primer sets.
To avoid the embryonic lethality observed in Pit1 knockout animals15 and to avoid possible systemic changes of Pi homeostasis due to global ablation of Pit216 we employed a conditional approach using cre-recombinase expressed under the human skeletal muscle actin promotor postnatally43. Mutant and WT littermates were generated from a single mating strategy depicted in (Fig. 1A). We confirmed inheritance of the floxed-Pit1, -Pit2 and Cre alleles by PCR from genomic DNA prepared from tail clippings (Fig. 1B). Recombination resulting in excision of exons 3 and 4 of Pit1 and of exon 4 of Pit2 in quadriceps, but not in kidney, gut, brain, lung and liver was confirmed by genomic PCR (Fig. 1C) and consistent with previously reported expression of HSA-Cre using Rosa26tomato reporter mice44. Low level recombination of Pit1fl/fl was detected in heart (Fig. 1D and Fig. S1C), but Pit1 and Pit2 transcript levels were significantly decreased only in the skeletal muscle of smPit1−/−; smPit2−/− mice (Fig. 1E), whereas mRNA levels of both transporters in heart were indistinguishable from WT (Fig. S1D).
Figure 1.

Generation of skeletal muscle-specific smPit1−/−; smPit2−/− double knockout mice. (A) Mating scheme used to target ablation of Pit1 and Pit2 in postnatal skeletal muscle with the human skeletal actin (HSA) promoter Cre. (B) Genotyping PCR for Pit1fl, HSA-Cre, and Pit1wt. (C) Genotyping PCR for the recombinant Pit1delta3,4 and Pit2delta4 in various tissues of a smPit1−/−; smPit2−/− mouse at P10. For raw images see supplement. (D) Semi-quantitative qenomic PCR showed 20% reduced expression of the recombinant Pit1delta3,4 in heart when compared to quadriceps. For raw images see supplement. (E) Semi-quantitative RT-PCR showed reduced expression of Pit1 and Pit2 in quadriceps muscle of smPit1−/− and smPit2−/− mice at P80, respectively. Means ± SEM, n = 5–8, ***p = 0.0002, **p = 0.002, *p = 0.03 vs. WT.
smPit1−/−; smPit2−/− mice develop a severe myopathy and die before two weeks of life
Life birth frequency was close to expected based on Mendelian inheritance with occurrence of a few stillbirths as reflected in the dead/ungenotyped/missing pup number in (Table S3). Growth retardation of smPit1−/−; smPit2−/− mice was evident by P2 (Fig. 2A,B), accompanied by low radiographic bone density (Fig. 2C) and by P10 they had severely impaired mobility (Video V1), inconsistent gastric filling (Fig. 2B) and depleted white adipose stores (Fig. S2D), suggesting that competition for food became progressively challenging for mutants. As a result, all smPit1−/−; smPit2−/− mice died due to hyponutrition by P13. Failure-to-thrive developed likely secondary to myopathy, since myopathy preceded weight loss, was evident at birth and progressed to marked atrophy and myofiber degeneration by P10 (Fig. 2D–F, Fig S2A,B). At P1–4, compared to the WT animal, myofibers in smPit1−/−; smPit2−/− mice were poorly organized, thinner and displayed indistinct and irregularly distributed myofiber nuclei with increased overall cellularity. Less robust but structurally comparable sarcomere striation was evident in smPit1−/−; smPit2−/− mice by light microscopy. At P10, myofiber disintegration was evident, and fibers contained multiple subsarcolemmal nuclei indicative of regenerative attempts. Cleaved caspase-3 cytoplasmic immunostaining (Fig. 2E, inset) was noted in mutant muscle but absent in WT muscle. The diaphragm (Fig. S2B) was likewise affected, while cardiac muscle was histologically normal (Fig. S2E). Skeletal and organ development was qualitatively comparable to WT littermates but quantitatively delayed (Fig. 2D, Fig. S2E). Growth retardation and hyponutrition were supported by histologic evidence of overall smaller size of every structure, atrophic intestinal villi, reduced subcutaneous adipose stores (Fig. S2D, E), and delayed appearance of ossification centers in various bones (Fig. S2C). smPit1−/−; smPit2−/− mice were hypophosphatemic and showed a trend toward hyperphosphaturia at P10 (Fig. 3A, B) along with appropriately suppressed intact FGF23 levels (Fig. 3C). Despite failure to thrive, mutant animals were normoglycemic (Fig. 3D). Absolute serum BUN levels were comparable in smPit1−/−; smPit2−/− and WT mice (Fig. 3E) arguing against significantly impaired renal function of smPit1−/−; smPit2−/− mice.
Figure 2.

smPit1−/−; smPit2−/− mice die before two weeks of life due to starvation. (A) Growth retardation in smPit1−/−; smPit2−/− mice was evident by P2. No difference was observed attributable to presence of HSA-Cre, Pit1fl or Pit2fl (See also Fig. S6B). Means ± SEM, n = indicated, ****p < 0.00002, ***p = 0.0002, **p = 0.002, *p = 0.03 vs. WT. (B) Gross phenotype (P10) shows that smPit1−/−; smPit2−/− mice were much smaller but retain normal proportions, their stomachs were variably filled with milk, and their livers were atrophic (white arrow). (C) Radiographs, at P1, P4 and P10. Smaller size was evident in smPit1−/−; smPit2−/− mice at P1, with reduced growth rate evident by P4, and markedly so by P10. Generalized retardation of mineralization of both axial and appendicular skeleton was noted in mutant animals. smPit1−/−; smPit2−/− mice were proportionately comparable to WT littermates, indicating generalized delay in growth. (D) Hind limb bone, muscle and fat development at P1, P4 and P10. a, c, e: WT; b, d, f: smPit1−/−; smPit2−/−. At P1, smPit1−/−; smPit2−/− mice were slightly smaller (b), and both genotypes had comparable minimal fat stores (asterisk, popliteal subcutaneous fat pad). At P4 and P10, smPit1−/−; smPit2−/− mice exhibited generalized delayed growth, with reduced accumulation of skeletal muscle mass. Delayed ossification of the distal femoral epiphysis at P10 is indicated (black arrows, e, f). Subcutaneous white adipose tissue accumulated in both genotypes through P4, however by P10, this is almost entirely lost in smPit1−/−; smPit2−/− mice (asterisk, popliteal subcutaneous fat pad, c-f). H&E, bar = 500 µm. (E) Muscle phenotype, anterior tibialis muscle, at P1, P4 and P10. a, b: P1. Compared to WT (a), myofibers in smPit1−/−; smPit2−/− mice (b) were poorly organized, thinner, and display indistinct and irregularly distributed myofiber nuclei with increased overall cellularity. c, d: P4. Myofibers in smPit1−/−; smPit2−/− mice (d) were slightly thinner than in WT (c) and had less robust but structurally comparable sarcomeric striation by light microscopy. e, f: P10. Compared to WT (e), myofibers in smPit1−/−; smPit2−/− mice (f) were thinner, with abundant interstitial cellularity (white arrows), and frank myofiber disintegration (black arrows). Sarcomeric striation was retained but was less robust. Cleaved caspase-3 cytoplasmic immunostaining was noted in mutant muscle (inset). H&E, bar = 50 µm (a–f), 20 µm (Inset, f) (F) Muscle phenotype, quadriceps femoris muscle, at P1, P4 and P10. a, b: P1. Compared to WT (a), myofibers in the mutant (b) were thinner with increased overall cellularity. c, d: P4. Myofibers in smPit1−/−; smPit2−/− mice (d) were thinner than in WT (c), with less evident sarcomeric striation by light microscopy. Interstitial regions are expanded in mutant muscle. e, f: P10. Compared to WT (e), myofibers in smPit1−/−; smPit2−/− mice (f) were thinner, with scattered aggregates of increased interstitial cellularity (white arrows). H&E, bar = 50 µm.
Figure 3.

Skeletal muscle ablation of Pit1 and Pit2 results in a mild renal Pi leak at P10. P10 smPit1−/−; smPit2−/− mice were hypophosphatemic (A) and hyperphosphaturic (B) in an FGF23-independent fashion (C). Blood glucose (D) and BUN were within normal limits (E). Means ± SEM, n = 5–10, ***p = 0.0002, **p = 0.002, *p = 0.03 vs. WT.
Energy-stress in smPit1−/−; smPit2−/− skeletal muscle leads to myofiber necrosis
Histological changes observed in all limb, epaxial, and diaphragm muscles from smPit1−/−; smPit2−/− mice include reduction of fiber area starting at P1, which worsened by P10 (Fig. 4A). Overt fiber necrosis seen at P10 under light microscopy were accompanied by a shift to thicker myofibrils (sarcomeres) seen on TEM (Fig. 4B), by reduced myoblast marker expression, and by increased type II Myh4 fast twitch muscle myosin expression in quadriceps muscle (Fig. 4C). There was no evidence for diffuse rhabdomyolyis upon Evans Blue exclusion staining (Fig. S3A) or endoplasmic reticulum (ER) stress based on normal Xbp1 splicing (Fig. S3B) and Chop expression (Fig. S3C) and normal appearance of the rough ER in TEM (Fig. S3G, arrows), which was recently reported in mice lacking Pit1 in growth plate chondrocytes45. There were likewise no signs for proteasomal activation based on normal Fbxo32 expression (Fig. S3D) or for autophagy based on absence of autophagosomes in TEM (Fig. S3G and Fig. S4C,D). Conversely, ERK1/2 activation was reduced in P12 quadriceps muscle obtained from smPit1−/−; smPit2−/− mice, and AMPK phosphorylation was increased (Fig. 4D), consistent with increased energy-stress and reduced muscle ATP production previously described by us6 in the setting of hypophosphatemia. Evaluation by TEM showed reduced muscle mitochondrial area (Fig. 4E), distended mitochondrial cristae and irregular mitochondrial matrix (Fig. S4C, D). Furthermore, a small, non-significant decrease of mitochondrial DNA was seen by semi-quantitative genomic PCR (Fig. S4B). Semi-quantitative RT-PCR analysis (Fig. 4F), furthermore, showed decreased expression of the Pi transporter Slc25a3, the mitochondrial mitofusin 2, the glycolytic enzymes phosphoglycerate kinase (Pgk1) and pyruvate kinase (Pkm1/2), while putative Pi exporter Xpr1, peroxisome proliferator-activated receptor gamma coactivator 1 alpha (Pgc1a) and Fis1 expression was unchanged and mitochondrial transcription factor A (Tfam) expression was increased.
Figure 4.

Energy stress results in reduced muscle fiber and increased myofibril (sarcomere) area in P10 smPit−/−; smPit/2−/− mice. (A) smPit1−/−; smPit2−/− mice showed a reduction of quadriceps fiber area at P10 in paraffin sections stained with H&E. (B) smPit1−/−; smPit2−/− mice showed an increased gastrocnemius myofibril (sarcomere) area at P10 in TEM. A and B show arbitrary units following ImageJ analysis of these sections on the y-axis. (C) Semi-quantitative RT-PCR showed reduced expression of myoblast markers myogenin, Pax7 and fast twitch Myh4 myosin in P10 quadriceps muscle of smPit1−/−; smPit2−/− mice (D) Densitometric analysis of an immunoblot (Fig. S4A) showing increased pAMPK/tAMPK ratio and reduced pERK1/2/tERK1/2 ratio in quadriceps of four different smPit1−/−; smPit2−/− mice at P10. (E) Mitochondrial area and size distribution in TEM of GC at P10. (F) Semi-quantitative RT-PCR to determine gene expression of Xpr1 and PiC transcription factors and metabolic markers in quadriceps muscle of smPit1−/−; smPit2−/− mice at P10. Shown are means ± SEM, n = 3–10, ****p < 0.00002, ***p = 0.0002, **p = 0.002, *p = 0.03 vs. WT.
Reduced oxygen consumption rate (OCR) in the absence of Pi is consistent with energy-stress in C2C12 myoblasts
We next modeled Pi effects using a Seahorse Analyzer in murine C2C12 myoblast cells. Consistent with our observation of energy-stress following Pit1 and Pit2 transporter ablation in vivo, acute response and maximum respiration in C2C12 myoblasts were reduced in the absence of Pi (Fig. 5A). Using permeabilized C2C12 myoblast and selective substrate/inhibitor conditions we were able to test individual mitochondrial complexes. OCR increased with addition of Pi to the mitochondrial stimulation buffer (Fig. 5B). The largest Pi effect was seen when testing complex III dependent respiration following addition of glucose-3-phosphate (G3P, Fig. 5B). Stimulation of OCR by Pi requires activation of ERK1/2, since it can be blocked by pretreatment with the MEK inhibitor UO126 (Fig. 5C). Activation of ERK1/2 by Pi was previously reported for many other cell lines11 and is blocked in C2C12 cells by ablation of Pit1 and Pit2 (Fig. S5A,B), or by pre-treatment with phosphonoformic acid (PFA), an inhibitor of Pi transport46, while PFA does not block insulin-mediated activation of ERK1/2 (Fig. 5D).
Figure 5.

Respiratory chain activity of C2C12 cells. (A) Individual tracing of a coupling assay using intact C2C12 cells and a Seahorse XFe24 analyzer showed reduced oxygen consumption rate (OCR) in cells deprived of Pi (P0) when compared to cells exposed to 10 mM Pi (P10). (B) Individual tracing of a mitochondrial electron-flow assay in permeabilized C2C12 cells in the absence (P0) and in the presence of 10 mM Pi (P10). To stimulate individual complexes, 1 nM Plasma Membrane Permeabilizer (Seahorse) and 100 nM dinitrophenol (DNP) was used along with 10 mM pyruvate/1 mM malate (complex C-I), 1 uM rotenone/10 mM glycerol-3-phosphate (complex C-III) or 10 mM ascorbate/100 nM N,N,N',N'-tetramethyl-1,4-phenylenediamine (TMPD) (complex C-IV), respectively. (C) Summary of several coupling assays as described in A. Pre-treatment with phosphonoformic acid (5 mM PFA), an inhibitor of Pit1, and U0126 (30 uM), an inhibitor of MEK1/2, the kinase upstream of ERK1/2, attenuated the Pi-dependent increase in maximal respiration (D) Summary of several immunoblots of C2C12 lysates following pre-treatment with 5 mM PFA which inhibited stimulation of pERK1/2 by 10 mM Pi (P10), but not by 25 uM human insulin (Ins25). Densitometric results were averaged following z-score transformation of the of individual blots. Shown are means ± SEM, n = 3–5, ****p < 0.00002, ***p = 0.0002, **p = 0.002, *p = 0.03 vs. pre-P0/P0. #p = 0.03 vs. pre-P0/P10 (C) and vs. pre-P0/P10 (D).
Loss of three alleles of Pit1 and Pit2 in skeletal muscle impairs endurance but not grip strength
Different from smPit1−/−; smPit2−/− mice, single and three allele conditional mutants were fertile, thrived normally and had body weights comparable to wildtype animals (Fig. S6D). However, voluntary wheel running (VWR) activity was reduced at P80 for smPit2−/− and smPit1−/−; smPit2−/+ females and for smPit2−/−, smPit1−/+; smPit2−/−, and smPit1−/−; smPit2−/+ males (Fig. 6A–D). The animals took more frequent breaks, which was particularly noticeable around midnight in males (Fig. 6A). VWR activity tests endurance of predominantly slow type I muscles of the lower extremities. Interestingly, a small, but significant improvement of VWR activity was seen in smPit1−/− males. No difference was observed in grip strength (GS) between Pi transporter mutant mice and littermate controls (Fig. S6C). GS tests impact of Pi transporter ablation on fast twitch type II forearm muscles, increased significantly with age and males were stronger than females (Fig. S6A). GS was unchanged even when corrected separately for WT1 and WT2 of each breeding cohorts. No weight difference was detected for the HSA-Cre, Pit1fl or Pit2fl alleles (Fig. S6B,D).
Figure 6.

Loss of three alleles of skeletal muscle Pit1 and Pit2 impairs muscle function. Voluntary hourly wheel-running activity on day 6 of a daily wheel-running study of P80 females (A, C) and males (B, D) was reduced in a gene-dose dependent fashion. Means ± SEM, n = 5–36, ***p = 0.0002, **p = 0.002, *p = 0.03 vs. WT.
Discussion
We here show that smPit1−/−; smPit2−/− mice develop a severe myopathy and die soon after birth. This at first was unexpected since no apparent skeletal muscle phenotype has thus far been described in the respective single knockout mice15–19 and suggests that Pit1 and Pit2 compensate for each other in skeletal muscle. Our findings in smPit1−/−; smPit2−/− mice therefore, for the first time, prove that Pit1 and Pit2 are required for normal myocyte function and survival. Furthermore, subtle deficiencies in running wheel endurance were observed in smPit1−/− and smPit2−/− mice, which might have previously been overlooked in the single global knockout mice.
Pathological evaluation showed features of skeletal myopathy at birth, when smPit1−/−; smPit2−/− mice still appeared otherwise normal compared to their WT littermates, suggesting that this myopathy causes the subsequent failure-to-thrive phenotype of these mice, possibly because myopathy reduces their ability to suckle. As a result smPit1−/−; smPit2−/− mice died by P13, to which respiratory failure may contribute since their diaphragms showed similar myopathic changes. Conversely, heart failure was likely not an important contributing factor to death in these mice, since HSA-cre did not reduce transporter mRNA levels in this organ. Furthermore, cardiac muscle appeared histologically normal at the time of death of smPit1−/−; smPit2−/− mice when compared to WT littermates.
Previous studies suggested that decreased ATP3,4 and phosphodiesters5 may in part explain the muscle weakness seen in hypophosphatemia. We recently reported that hypophosphatemic myopathy goes along with reduced ATP flux (VATP) and IC Pi in an individual with hereditary hypophosphatemic rickets with hypercalciuria (HHRH) and in the sodium-Pi co-transporter Npt2a null mouse6. Consistent with reduced ATP production we observed activation of the energy sensor AMPK, increased Tfam expression and abnormal mitochondrial morphology in skeletal muscle of smPit1−/−; smPit2−/− mice, which may be a result of energy stress47 and provide an explanation for the myodegeneration and muscle necrosis seen in these mice (Fig. 7). pAMPK activates PGC1a which induces nuclear transcription of Tfam and along with p53 translocates to the mitochondria where PGC1a and p53 interact with Tfam to induce mitochondrial biogenesis, to improve respiratory chain function and to decrease Hif1a activity as reviewed in48. Mice lacking skeletal muscle Tfam exhibit exercise intolerance and myopathy49 resembling the one observed in smPit1−/−; smPit2−/− mice.
Figure 7.
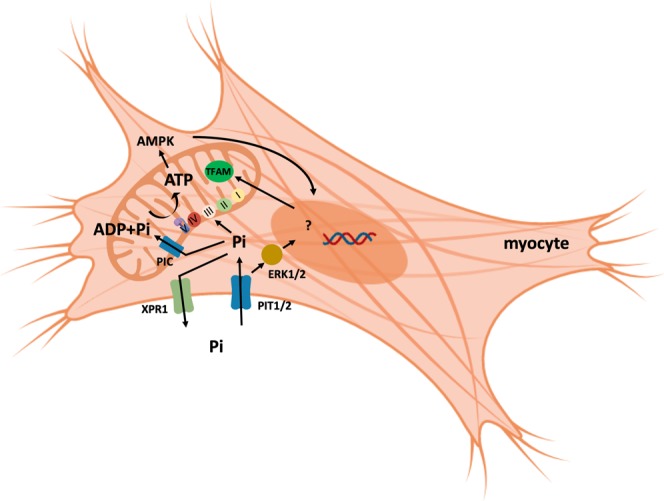
Metabolic Pi-signaling in myocytes. We here provide evidence for an important function of Pit1 and Pit2 to increase intracellular Pi as a substrate for mitochondrial ATP synthesis and/or as a signal to stimulate mitochondrial respiration. Pit1 and Pit2 may also serve as “transceptors” upon binding of blood Pi to activate ERK1/2 in a transport-independent fashion to stimulate myocyte proliferation and differentiation and mitochondrial respiration.
In addition to its function as a substrate for ATP production, Pi activates ERK1/2 in myocytes like in many other cells22 (Fig. 7). Consequently, ERK1/2 phosphorylation was reduced in quadriceps muscle from smPit1−/−; smPit2−/− mice. Our initial observations in murine C2C12 myoblasts suggest that both intracellular and membrane signaling pathways appear to be required for stimulation of mitochondrial respiration by Pi. Furthermore, expression of the muscle stem cell (MSCs) markers Pax7 and myogenin, which are known to be regulated by ERK1/250, was suppressed, and may provide a mechanism, whereby muscle stem cells (MSCs) and regeneration is impaired by ablation of Pit1 and Pit2. However, postnatal worsening of muscle degeneration and presence of multiple subsarcolemmal nuclei indicative of regenerative attempts despite low levels of these two transcription factors suggests that Pit1 and Pit2 are primarily important for muscle fiber survival.
Muscle-specific loss of ERK1/2 was also found to cause loss of neuromuscular integrity in mice28, which may contribute to the myopathy in smPit1−/−; smPit2−/− mice. Finally, Pit1 promotes other transport-independent processes such as proliferation20 and TNF-induced apoptosis in HeLa cells21 and insulin resistance in hepatocytes51. These pathways may be relevant in skeletal muscle as well and may be of interest for further investigation.
Compensation for loss of Pit1 and Pit2 in skeletal muscle may occur on the fiber, tissue and organism level and aim at reducing Pi consumption and keeping IC Pi stable. On the fiber level we observed down-regulation of the mitochondrial Pi importer Slc25A3 (PIC), increased Tfam, decreased Mfn2 levels and abnormal mitochondrial matrix, which may be aimed at improving mitochondrial function and resemble adaptive changes during exercise52.
Mutations in human SLC25A3/PIC cause a severe myopathy affecting skeletal and cardiac muscle in humans53,54 and in mice55. Thus, down-regulation of this transporter may preserve cytosolic Pi, but since mitochondrial uptake of Pi is important for ATP synthesis, this in turn could further worsen muscular energy stress.
On the tissue level we observed a shift to a slow twitch fiber type, which may reflect a compensatory process in response to muscle energy-stress in these fibers which have greater levels of Pit1 and Pi than fast twitch fibers13,14. AMPK plays an important role in regulating muscle mass and regeneration and must be kept within relatively tight boundaries (not too high or too low) for optimal muscle regeneration47. AMPK subunit α1 stimulates anabolism and regulates satellite cell dynamics during regeneration, whereas AMPK subunit α2 regulates muscle degradation during atrophy. Energy stress predominantly activates AMPK subunit α2 via liver kinase B1 (LKB1), while AMPK subunit α1 is only activated by highly‐intense or prolonged exercise, perhaps via Calcium/calmodulin‐dependent protein kinase (CamKK) action, or other AMPK kinases. AMPK subunit α2 stimulates catabolic processes by increasing Foxo3a, Atrogin‐1 and MuRF‐1 expression/activity and increasing autophagy, leading to muscle atrophy under certain circumstances, but has little effect on protein synthesis. AMPK subunit α1 impairs mTOR signaling, slows protein synthesis, and blocks hypertrophy. It will be of interest to determine, which AMPK isoforms contribute to the myopathy in smPit1−/−; smPit2−/− mice and to see, whether expression patterns likewise resemble physiological adaptation to chronic endurance exercise56,57.
On the systemic level we found that smPit1−/−; smPit2−/− mice have significantly lower blood Pi levels, which appears to be a result of renal Pi excretion independent of FGF23. Since HSA-cre is not expressed in the kidneys, we speculate that skeletal muscle of smPit1−/−; smPit2−/− mice secretes a myocyte factor that increases renal excretion of Pi. Candidate muscle-derived factors include IGF-1, FGF-2, IL-15, irisin myostatin and L-BAIBA33, which will be subject of future investigation. Bone loss observed in these mice may be secondary to this renal Pi leak, due to direct action of one of the above myokines on bone metabolism, and/or due to failure to thrive of these mice.
Three allele mutants display reduced VWR activity which further supports the hypothesis that Pit1 and Pit2 are important predominantly for slow twitch fibers4–6,58 as already suggested by the possible shift from fast Myh4 positive fibers to slow Myh7 positive fibers in quadriceps muscle of smPit1−/−; smPit2−/− mice. Furthermore, subtle deficiencies in running wheel endurance were also observed in the single conditional mutant smPit1−/− and smPit2−/− mice, which may have been previously missed during evaluation of the global knockouts for these genes15–19. Interestingly, GS is not affected in mice lacking two or three transporter alleles, which is different from observations in the Hyp mouse model for XLH that displays reduced GS along with reduced VWR activity, and which both are restored by treatment of Hyp mice with anti-FGF23 antibodies36. Likewise, ex-vivo tetanic strength was impaired in Dmp1 null mice38, which like Hyp have elevated FGF23 levels. It is thus conceivable that FGF23 impacts skeletal muscle function33 similar to its effect on the cardiac muscle32. Our Pit1 and Pit2 three allele knockout mice may be a good model to separate effects of hypophosphatemia from effects caused by adaptive hormonal changes as a result of hypophosphatemia and improve our understanding of the pathophysiology of the myopathy seen in hypophosphatemic disorders such as HHRH or XLH59.
Our observations are currently limited by the early failure-to-thrive phenotype of the smPit1−/−; smPit2−/− mice. To further evaluate the role of Pit1 and Pit2 for muscle function and survival in adult mice, we next plan to use an inducible muscle-specific Cre, for example HSA-CrertTA60. Also oral and parenteral Pi supplementation to normalize blood Pi in smPit1−/−; smPit2−/− and three allele knockout mice could be attempted, however, placental Pi transport appears to isolate pups from dietary changes of Pi in the mother61 and thus such pharmacological rescue experiments are likely only feasible postnatally. Of interest will also be genetic rescue experiments using a cross of smPit1−/−; smPit2−/− mice with transgenic mice62 or adenoviral constructs expressing WT and transport-deficient [K71]hPIT1, to test whether substrate-uptake is required to rescue the myopathy of smPit1−/−; smPit2−/− mice. It may also be of interest to test whether ablation of Xpr1 can rescue the myopathy. Xpr1 showed a trend toward what may be interpreted as compensatory down regulation in Fig. 4F and was recently shown to be a widely expressed putative Pi exporter63,64, however, its functions in skeletal muscle are completely unknown. Xpr1 contains an SPX motif, which binds inositol-pyrophosphate (5-InsP7), a compound shown in yeast to communicate IC Pi levels and to regulate the Pi starvation response65. Human mutations in XPR1 cause primary familial brain calcification63,64. Kidney-specific deletion of Xpr1 leads to a decrease in the NAD + /NADH ratio with reduced availability of NAD + for synthetic processes and development of Fanconi syndrome66. As a result, down-regulation of this transporter may preserve cellular Pi and reduce Pi consumption through synthetic processes.
In conclusion, we here show for the first time that Pit1 and Pit2 are essential for normal myofiber function and survival. Hypophosphatemic myopathy is currently treated with Pi supplementation30,59. However, Pi supplements are poorly tolerated and can cause intestinal symptoms, renal calcifications and secondary hyperparathyroidism67. Insights gained from smPit1−/−; smPit2−/− and three allele knockout mice suggest, that optimizing AMPK and ERK1/2 signaling and modification of XPR1 and PIC/SLC25A3 activity may offer alternative strategies for the treatment of hypophosphatemic myopathy.
Methods
Mice and diets
smPit1−/− and smPit2−/− mice, which selectively lack the Pi transporters Pit1(Slc20a1) and Pit2 (Slc20a2), respectively, in skeletal muscle were generated using Pit1fl/fl mice obtained from Dr. Giachelli68, and HSA-Cre mice obtained from Jackson Laboratory (human skeletal actin-Cre, B6.Cg-Tg(ACTA1-cre)79Jme/J; Jackson stock No: 006149)43. Embryonic stem cell clone EPD0243_1_C06 with loxP sites flanking exon-4 of the Slc20a2/Pit2 gene were purchased from the International Knockout Mouse Consortium (IKMC), European Conditional Mouse Mutagenesis Program (EUCOMM). Exon-4, which contains 89 bp, encodes for amino acids 144-172 and its Cre-mediated removal leads to a frame shift and an early stop codon 340 bp downstream. Conditional-ready Pit2 mice were generated by morula aggregation in the Labortierkunde (LTK) of the University of Zurich and mated with Flp-deleter mice (63Flp3B6) purchased from the Jackson Laboratory to generate Pit2fl/fl mice (Pastor-Arroyo et al, in preparation)69.
All mice were generated from a C57Bl/6 background and experimental mice were generated using the mating strategy of Pit1fl/+; Pit2fl/+ females x Pit1fl/+; Pit2fl/+; HSA-Cretg/+ males and Cre-negative littermates were used as controls to minimize bias related to a specific genetic background. Mice were weaned at 3 weeks of age and allowed free access to water and standard chow (1.0% calcium, 0.7% Pi (0.3% bioaccessible Pi; TD.2018S; Harlan Teklad, Madison, WI, USA).
Genotyping at weaning was performed by PCR amplification of genomic DNA extracted from tail clippings using the HotSHOT method70 and amplified by polymerase chain reaction (PCR) as previously described71 using the primers shown in Table S1. Briefly, 75uL alkaline lysis buffer (25mN NaOH, 0.2 mM EDTA) was aliquoted to 1.5 mL tubes containing tissue. Lysates were incubated for 30 min at 95 °C, then neutralized with 75uL neutralization buffer (40 mM Tris-HCl). Genomic DNA was amplified by PCR to genotype mice and determine skeletal muscle-specific ablation of Pit1. Primer pairs used were: wild-type (Pit1wt) and Pit1-floxed (Pit1fl) (F) 819 and (R) 820; wild-type (Pit1wt) and delta allele (Pit1Δe3,4): (F) 819 and (R) 822; ACTA1-cre (HSA-Cre): (F) 823 and (R) 82468 and wild-type (Pit2wt), Pit2-floxed (Pit2fl) (F) 954 and (R) 955, and delta allele (Pit2Δe4) (F) 995 and (R) 996 (Table S1).
Animal research for this study was first approved on October 22, 2014 by the Yale Institutional Animal Care and Use Committee (protocol 2014–11635), was renewed on September 7, 2016, and is valid through Sept. 30, 2020. Yale University has an approved Animal Welfare Assurance (#A3230–01) on file with the NIH Office of Laboratory Animal Welfare. The Assurance was approved on May 5, 2015.
Anatomic phenotyping
A total of 34 animals (Table S2) across 5 litters (P1, P4, P8 and P10) were examined grossly. These included 13 smPit1−/−; smPit2−/− animals with wild-type animals comprising the balance. Animal histopathology was performed on 21 animals (indicated by histopathology numbers in Table S2). Of these, images from animals at P1, P4, and P10 were used to illustrate pathologic findings. Animals were evenly split by sex and genotype. All the methods for the study were carried out in accordance with relevant guidelines and according to the 2013 AVMA guidelines for the euthanasia of animals. Following gross photography, radiography and collection of blood for glucose measurement, soft and bony tissues from all animals were immersed in 10% (w/v) neutral buffered formalin or Bouin’s solution respectively for one week. Cassetted tissues were embedded in paraffin, sectioned at 5 μm, and mounted on glass slides. Sections were stained with hematoxylin and eosin (H&E), examined using a Zeiss Axioskop light microscope and imaged using a Zeiss AxioCam MrC camera.
Transmission electron microscopy
For transmission electron microscopy a 1 mm3 block of the left kidney was fixed in 2.5% glutaraldehyde and 2% paraformaldehyde in 0.1 M Cacodylate buffer for 2 hrs., followed by post-fixation in 1% osmium liquid for 2 hours. Following dehydration using a series of ethanol concentrations (50% to 100%), tissue was embedded in epoxy resin, and polymerization was carried out at 60 °C for overnight as previously described62. After preparing a thin section (50 nm), the tissues were double stained with uranium and lead and observed using a Tecnai Biotwin (LaB6, 80 kV) (FEI, Thermo Fisher, Hillsboro, OR) at the Yale Center for Cellular and Molecular Imaging (YCCMI).
Evan’s Blue staining
To evaluate muscle fiber integrity, 1% Evans blue dye dissolved in sterile saline was administered by intraperitoneal injection at a dose of 1% of body weight to WT and smPit1−/−; smPit2−/− mice at age P10, followed by sacrifice after 24 hrs. and dissection of muscles that were snap-frozen in liquid nitrogen. Muscle cryosections of 10-μm thickness were cut at −21 °C on a Leica (CM3050) cryostat, dipped in cold acetone (−20 °C) for 1 min and then air-dried at room temperature (20–22 °C) (RT). Sections were dipped in xylene (Merck) and mounted with DPx (BDH, Poole, UK) and a glass coverslip. Evans blue was viewed by confocal fluorescent microscopy SP8 LIGHTNING confocal microscope (Leica, Buffalo Grove, IL, USA) at 620 nm excitation and 680 nm emission as described72.
Radiology
Following euthanasia, animals were radiographed in dorsovental and lateral recumbancy using a Universal radiology unit and AGFA Small Animal Digital Radiography system (Agfa HealthCare, Mortsel, Belgium).
Blood and urine parameters
Blood and spot urines were collected as previously described71. Glucose measurements as a measure of recent feeding were taken immediately after removal of the pups from the mother at euthanasia using a handheld glucometer (Accu-Chek, Roche, Switzerland). Creatinine levels were measured by HPLC/MS/MS commercially from serum samples collected at euthanasia (Antech, Fountain Valley, CA, USA). BUN was measured from serum samples using kit 0580 (Stanbio, Boerne, TX, USA). The COBAS Mira Plus automated chemistry analyzer (Roche Diagnostics, Pleasanton, CA, USA) was used to determine plasma and urine inorganic phosphorus. Urinary creatinine was measured by colorimetric assay (DICT-500; BioAssay Systems, Hayward, CA, USA). Concentrations of serum intact fibroblast growth factor (FGF)-23 protein were determined using mouse FGF-23 ELISA assays (60–6500; Immutopics, San Clemente, CA, USA).
Muscle physiology
Forelimb grip strength was measured with the Mouse Grip Strength Meter (1027SM; Columbus Instruments, Columbus, OH, USA) at ages 30, 60, and 120 days. The average grip strength was calculated from three trials conducted in series. Voluntary wheel-running (VWR) was assessed using home cages equipped with running wheels (0297–8; Columbus Instruments) using experimental procedures previously described by other groups at the Yale School of Medicine73,74. Briefly, mice were individually housed for one week and maintained on a 12 h light/dark cycle. Food and water were provided ad libitum. The number of wheel-turns was continuously recorded by ClockLab software (Actimetrics, Wilmette, IL, USA). Data files were exported to MATLAB and converted to Excel spreadsheets using Clocklab Analysis (Actimetrics, Wilmette, IL, USA).
Genomic DNA and RNA extraction and allele-specific PCR from various mouse tissues and C2C12 cells
Gastrocnemius and quadriceps muscle tissue were dissected immediately following sacrifice, snap-frozen (by compression between aluminum clamps pre-chilled in liquid nitrogen), and stored at −70 oC. Genomic DNA was extracted from tissues using the HotSHOT method70 described above. Total RNA from tissues was extracted using TRIzol (15596026; Invitrogen, Carlsbad, CA, USA). Total RNA from C2C12 cells was prepared using RNeasy columns (Qiagen, Valencia, CA). 0.5–2 ug total RNA were reverse-transcribed using the Omniscript RT Kit (205113; Qiagen, Valencia, CA, USA). Semi-quantitative genomic or reverse transcription (RT) PCR was performed with SYBR Green (204143; Quantitect, Qiagen) using the ABI-Step One Plus Cycler (Fisher, Life Technologies, Waltham, MA, USA) and analyzed using the 2-ΔΔCt method. Primer sequences were designed using PrimerBank75 and are shown Table S1. b-actin and 18 S RNA gave comparable results as internal controls (see Fig. S3E,F) and ABI Cycler efficiency measures were all acceptable.
Protein extraction and immunoblot analysis from various mouse tissues and C2C12 cells
Skeletal muscle tissue was homogenized as previously described62 in ice-cold RIPA buffer (50 mM Tris HCL (pH 6.8), 150 mM NaCl, 1% Triton X-100, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate, Protease inhibitor without EDTA (Roche, Mannheim, Germany), 0.4 mM Na-orthovanadate, 50 mM beta-GP, 5 mM NaF). Lysates were incubated on an orbital shaker for 2 hours then centrifuged for 20 min at 12,000 g at 4 °C. Protein amount was measured using a BCA assay (Thermo Fisher Scientific, Waltham, MA, USA). 50 ug of protein were run on a PAGE gel electrophoresis on 10% Tris-HCl/Glycine SDS-polyacrylamide (456–1034, BioRad, CA), electro-transfer to PVDF membranes (162–0218, BioRad, CA, USA) and hybridized with anti-phospho-ERK1/2 (9106, Cell Signaling Technology, MA, or 4730, Cell Signaling Technology, MA, USA) or anti-total-ERK1/2 antibody (4695, Cell Signaling Technology, MA, USA) 1:1000 in phosphate buffered saline containing 0.1% Tween 20 (PBST) and 5% non-fat dry milk at 4 °C overnight, or hybridized with pAMPK rabbit antibody (2535, Cell Signaling Technology, MA, USA, the antibody detects both α1 and α2 isoforms of the catalytic subunit, but does not detect the regulatory β or γ subunits) 1:1000 or tAMPK rabbit antibody (2603, Cell Signaling Technology, MA, USA) 1:1000 in tris buffered saline containing 0.1% Tween 20 (TBST) and 5% BSA (A7888, Sigma, MA, USA) at 4 °C overnight, and detected with horseradish-peroxidase conjugated anti-rabbit IgG (7074 S, Cell Signaling Technology, MA) in PBST + 5% non-fat dry milk at room temperature for 60 min using chemiluminescence/autoradiography (Supersignal 34580, Thermo Fisher Scientific, Waltham, MA, USA).
For C2C12 cells following pretreatment and stimulation the 48-well plate was placed on ice and the culture medium was aspirated. Cells were then lysed with 50 ul lysis buffer (62.5 mM Tris HCL (pH 6.8), 1% SDS, 1 mM EDTA, 1 mM EGTA, 0.05 TiU/ml aprotonin, 1 M PMSF, 100 mM Na-orthovanadate, 0.8% SDS, 3.2% glycerol, 2% beta mercaptoethanol, 0.0015% bromophenolblue). 15 ul cell lysates were then separated, electro-transferred to PVDF membranes and hybridized with anti-phospho-ERK1/2 (9106, Cell Signaling Technology, MA, USA) or (4730, Cell Signaling Technology, MA, USA) or total-ERK1/2 antibody (4695, Cell Signaling Technology, MA, USA) in phosphate buffered saline containing 0.1% Tween 20 (PBST) and 5% non-fat dry milk at 4 °C overnight and detected as described for skeletal muscle lysates above. Following densitometric analysis of the autoradiograms, phospho/total ERK ratios were converted into z-scores to permit statistical evaluation of pooled Western blot experiments.
C2C12 myoblast cell culture
C2C12 myoblasts were cultured at 37 oC under 5% CO2 for up to 8 weeks in high glucose DMEM (pH 7.4; 11965–092; Gibco, Gaitherburg, MD, USA) supplemented with 10% fetal bovine serum (F4135; Sigma, St. Louis, MO, USA). Medium was changed every 3–4 days and cells were split (1:10) once a week with 0.25% trypsin/EDTA. Pi-free DMEM (pH 7.4; 11971–025; Gibco, Gaitherburg, MD, USA) was substituted for complete DMEM when indicated. Human insulin (91077 C, Sigma, St. Louis, MO, USA) was used at a final concentration of 25 ug/ml. The MAP kinase inhibitor UO126 (19–147; Sigma, St. Louis, MO, USA) and the PI-3 kinase inhibitor Ly294002 (L9908; Sigma, St. Louis, MO, USA) were suspended in DMSO and dosed at 10 uM and 50 uM, respectively. Phosphonoformic acid (PFA; P6801; Sigma, St. Louis, MO, USA), a weak competitive inhibitor of Pit1 transport activity, was used at a final concentration of 5 mM.
RNAi ablation studies
C2C12 cells were seeded at 20,000/well and transfected with 20 nM of ON-TARGETplus siRNA (Slc20a1 (Pit1, L-045288–01–0005); GE Dharmacon, Lafayette, CO, USA) using RNAiMAX (13778075; Lipofectamine, Thermo Fisher Scientific, Waltham, MA, USA) diluted in Opti-MEM (31985070; Gibco, Gaithersburg, MD, USA) for delivery. Transfected cells were cultured in Pi-free DMEM for 3 days before further analysis.
Extracellular flux analysis
Cellular respiratory activity was measured using a Seahorse XFe24 Analyzer (Agilent Technologies, Santa Clara, CA, USA). For transfection and non-transfection assays, C2C12 myoblasts were seeded at 40,000 cells/well in Pi-free DMEM (pH 7.4; with 10% FBS and 10 mM HEPES) and cultured for 3 days. Cells were re-fed the day before the assay.
For intact cell assays, microplates were placed in a CO2 free incubator 45 min before the start of the assay to permit evaporation of CO2. Immediately prior to the assay, Pi-free medium was replaced with assay medium (pH 7.4; 8.3 g/l DMEM base (CM038305; Invitrogen, Carlsbad, CA, USA) with 12.5 mM glucose, 31 mM NaCl, 100 mM pyruvate, 2 mM Glutamax-I (35050–061; Gibco, Gaithersburg, MD, USA), and no Pi). Oxygen consumption rate (OCR) was measured in 8.5 min intervals. Mitochondrial respiratory activity was manipulated by serial injection of 10 mM sodium Pi, 1.25 uM oligomycin (an ATP synthase inhibitor), 100 uM 2, 4-dinitrophenol (a mitochondrial uncoupler), and 1 uM antimycin/1 uM rotenone (complex I and III inhibitors). The effect of Pi on intact cell OCR is termed as “acute response”, and the following injection of oligomycin and DNP as “maximal respiration”.
Coupling and electron flow assay protocols were adapted from Divakaruni et al.76, Salabei et al.77, and the manufacturer’s protocols. C2C12 cells were permeabilized with Seahorse Plasma Membrane Permeabilizer (PMP, 102504–100; Agilent Technologies, Santa Clara, CA, USA). Briefly, immediately before each assay, cells were washed once with mannitol/sucrose (MAS) buffer (pH 7.2; 220 mM mannitol, 70 mM sucrose, 10 mM KH2PO4, 5 mM MgCl2, 2 mM HEPES, 1 mM EGTA, and 4 mg/mL fatty acid free-bovine serum albumin) then immersed in MAS buffer containing 1 nM PMP. OCR was measured in 3 min intervals. Mitochondrial respiratory state 3 (ADP + /− Pi + substrate) for coupling assays was induced by injection of the following: 4 mM ADP, 10 mM sodium Pi, 10 mM pyruvate and 1 mM malate (CI), 10 mM succinate (CII), 10 mM glycerol-3-phosphate (G3P, CIII), 100 uM TMPD and 10 mM ascorbic acid (CIV). State 4o (oligomycin-dependent), maximal, and non-mitochondrial respiration were assessed by injection of chemicals mentioned above for intact cells. Electron flow assays were begun with cells immersed in MAS buffer containing PMP, DNP, and complex I-specific substrates, and additional complex-specific substrates were injected in serial order at the concentrations stated above.
Statistical and image analysis
Data are expressed as means ± SEM and were analyzed as previously described62 in Prism 8.0 (GraphPad Software, Inc., La Jolla, CA). Differences between groups were considered significant if p-values obtained with Student’s t-test were < 0.05. The Mann-Whitney U test was used for comparisons when there was evidence by the Shapiro-Wilk normality test that the data were not normally distributed. Two-way ANOVA and Tukey’s test for multiple comparisons was used to determine significant differences between more than two treatment groups with a significance threshold of p < 0.05. Light and electron microscopic images were analysed using ImageJ (version2.0.0-rc-69/1.52p) to determine myofiber, myofibril (sarcomere) and mitochondrial areas.
Supplementary information
Acknowledgements
We are grateful to the Cell Biology Core of the Yale Diabetes Research Center (NIH/NIDDK P30 DK045735) for help with fluorescent microscopic analysis and access to their ABI qPCR instrument, to the Yale O’Brien Center NIH (NIH/NIDDK P30DK079310) for training and access to VWR devices, and biochemical testing of mouse serum and urine samples, to the Mouse Research Pathology Core for histologic slide preparation, to the Yale Mouse Metabolic Phenotyping Core (NIH/NIDDK U24 DK-059635 and NIH/NIDDK R01 DK116774 (G.I.S)) for access to their grip strength device and Seahorse analyzer, and to the Yale Center for Cellular and Molecular Imaging (YCCMI) for help with electron microscopic analysis. N.H. and CAW were supported by the Swiss National Science Foundation (grant no 31003A_176125). D.P. was supported by the Austrian Science Fund (FWF), project no. J 3267.
Author contributions
S.C., B.B.H., J.F. J.S. and D.C. performed all experiments. D.P., A.N. and G.I.S. provided training and devices for grip strength studies. C.Z. performed necropsies and pathological analysis of smPit1−/−; smPit2−/− mice. M.J. and G.I.S. provided the HSA-Cre mice, N.W.C. and C.M.G. provided the Pit1fl/fl mice, N.H. and C.A.W. provided the Pit2fl/fl mice, C.B. conceived the work and together with S.C. and D.C. processed all results, performed statistical analysis and wrote the manuscript.
Competing interests
Sampada Chande, Daniel Caballero, Bryan B. Ho, Jonathan Fetene, Juan Serna, Dominik Pesta, Ali Nasiri, Michael Jurczak, Nicholas W. Chavkin, Nati Hernando, Cecilia M. Giachelli and Caroline Zeiss declare that they have no conflict of interest. Carsten A. Wagner has served on advisory boards to Bayer and Medice and received honoraria from Medice. Gerald I. Shulman serves on the advisory boards for Merck, Novo Nordisk, AstraZeneca, Gilead Sciences and Janseen Research and Development and he receives investigator-initiated support from AstraZeneca, Gilead Sciences and Merck. Clemens Bergwitz has received research support from Nutricia North America, Bethesda, MD, USA, for an unrelated project. He serves on the advisory board for Bayer.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
is available for this paper at 10.1038/s41598-020-59430-4.
References
- 1.Bergwitz C, Jüppner H. Regulation of phosphate homeostasis by PTH, vitamin D, and FGF23. Annu. Rev. Med. 2010;61:91–104. doi: 10.1146/annurev.med.051308.111339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sumukadas D, Jenkinson F, Witham MD. Associations and consequences of hypophosphataemia in older hospitalised women. Age Ageing. 2009;38:112–115. doi: 10.1093/ageing/afn244. [DOI] [PubMed] [Google Scholar]
- 3.Knochel JP. Neuromuscular manifestations of electrolyte disorders. Am. J. Med. 1982;72:521–535. doi: 10.1016/0002-9343(82)90522-8. [DOI] [PubMed] [Google Scholar]
- 4.Hettleman BD, Sabina RL, Drezner MK, Holmes EW, Swain JL. Defective adenosine triphosphate synthesis. An explanation for skeletal muscle dysfunction in phosphate-deficient mice. J. Clin. Invest. 1983;72:582–589. doi: 10.1172/jci111006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ambuhl PM, et al. Metabolic aspects of phosphate replacement therapy for hypophosphatemia after renal transplantation: impact on muscular phosphate content, mineral metabolism, and acid/base homeostasis. Am. J. Kidney Dis. 1999;34:875–883. doi: 10.1016/S0272-6386(99)70045-4. [DOI] [PubMed] [Google Scholar]
- 6.Pesta DH, et al. Hypophosphatemia promotes lower rates of muscle ATP synthesis. FASEB J. 2016;30:3378–3387. doi: 10.1096/fj.201600473R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Petersen KF, Dufour S, Shulman GI. Decreased insulin-stimulated ATP synthesis and phosphate transport in muscle of insulin-resistant offspring of type 2 diabetic parents. PLoS Med. 2005;2:e233. doi: 10.1371/journal.pmed.0020233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Forster IC, Hernando N, Biber J, Murer H. Phosphate transporters of the SLC20 and SLC34 families. Mol. Asp. Med. 2013;34:386–395. doi: 10.1016/j.mam.2012.07.007. [DOI] [PubMed] [Google Scholar]
- 9.Kavanaugh MP, Kabat D. Identification and characterization of a widely expressed phosphate transporter/retrovirus receptor family. Kidney Int. 1996;49:959–963. doi: 10.1038/ki.1996.135. [DOI] [PubMed] [Google Scholar]
- 10.Olah Z, Lehel C, Anderson WB, Eiden MV, Wilson CA. The cellular receptor for gibbon ape leukemia virus is a novel high affinity sodium-dependent phosphate transporter. J. Biol. Chem. 1994;269:25426–25431. [PubMed] [Google Scholar]
- 11.Camalier CE, et al. An integrated understanding of the physiological response to elevated extracellular phosphate. J. Cell Physiol. 2013;228:1536–1550. doi: 10.1002/jcp.24312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sugita Atsushi, Kawai Shinji, Hayashibara Tetsuyuki, Amano Atsuo, Ooshima Takashi, Michigami Toshimi, Yoshikawa Hideki, Yoneda Toshiyuki. Cellular ATP Synthesis Mediated by Type III Sodium-dependent Phosphate TransporterPit-1Is Critical to Chondrogenesis. Journal of Biological Chemistry. 2010;286(4):3094–3103. doi: 10.1074/jbc.M110.148403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Scheibye-Knudsen M, Quistorff B. Regulation of mitochondrial respiration by inorganic phosphate; comparing permeabilized muscle fibers and isolated mitochondria prepared from type-1 and type-2 rat skeletal muscle. Eur. J. Appl. Physiol. 2009;105:279–287. doi: 10.1007/s00421-008-0901-9. [DOI] [PubMed] [Google Scholar]
- 14.Abraham KA, Brault JJ, Terjung RL. Phosphate uptake and PiT-1 protein expression in rat skeletal muscle. Am. J. Physiol. Cell Physiol. 2004;287:C73–78. doi: 10.1152/ajpcell.00510.2003. [DOI] [PubMed] [Google Scholar]
- 15.Beck L, et al. The phosphate transporter PiT1 (Slc20a1) revealed as a new essential gene for mouse liver development. PLoS One. 2010;5:e9148. doi: 10.1371/journal.pone.0009148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bon N, et al. Phosphate-dependent FGF23 secretion is modulated by PiT2/Slc20a2. Mol. Metab. 2018;11:197–204. doi: 10.1016/j.molmet.2018.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bourgine A, et al. Mice with hypomorphic expression of the sodium-phosphate cotransporter PiT1/Slc20a1 have an unexpected normal bone mineralization. PLoS One. 2013;8:e65979. doi: 10.1371/journal.pone.0065979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Crouthamel MH, et al. Sodium-dependent phosphate cotransporters and phosphate-induced calcification of vascular smooth muscle cells: redundant roles for PiT-1 and PiT-2. Arterioscler. Thromb. Vasc. Biol. 2013;33:2625–2632. doi: 10.1161/ATVBAHA.113.302249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jensen N, et al. Loss of function of Slc20a2 associated with familial idiopathic Basal Ganglia calcification in humans causes brain calcifications in mice. J. Mol. Neurosci. 2013;51:994–999. doi: 10.1007/s12031-013-0085-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Beck L, et al. Identification of a Novel Function of PiT1 Critical for Cell Proliferation and Independent of Its Phosphate Transport Activity. J. Biol. Chem. 2009;284:e99959. doi: 10.1074/jbc.M109.053132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Salaun, C. et al. Identification of a novel transport-independent function of PiT1/SLC20A1 in the regulation of TNF-induced apoptosis. J. Biol. Chem. (2010). [DOI] [PMC free article] [PubMed]
- 22.Chande S, Bergwitz C. Role of phosphate sensing in bone and mineral metabolism. Nat. Rev. Endocrinol. 2018;14:637–655. doi: 10.1038/s41574-018-0076-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shi H, et al. Mice lacking MKP-1 and MKP-5 Reveal Hierarchical Regulation of Regenerative Myogenesis. J. Stem Cell Regen. Biol. 2015;1:1–7. doi: 10.15436/2741-0598.15.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shi H, et al. Improved regenerative myogenesis and muscular dystrophy in mice lacking Mkp5. J. Clin. Invest. 2013;123:2064–2077. doi: 10.1172/JCI64375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shi H, et al. MAP kinase phosphatase-1 deficiency impairs skeletal muscle regeneration and exacerbates muscular dystrophy. FASEB J. 2010;24:2985–2997. doi: 10.1096/fj.09-150045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bennett AM, Tonks NK. Regulation of distinct stages of skeletal muscle differentiation by mitogen-activated protein kinases. Sci. 1997;278:1288–1291. doi: 10.1126/science.278.5341.1288. [DOI] [PubMed] [Google Scholar]
- 27.Wang S, Seaberg B, Paez-Colasante X, Rimer M. Defective Acetylcholine Receptor Subunit Switch Precedes Atrophy of Slow-Twitch Skeletal Muscle Fibers Lacking ERK1/2 Kinases in Soleus Muscle. Sci. Rep. 2016;6:38745. doi: 10.1038/srep38745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Seaberg B, et al. Muscle-derived extracellular signal-regulated kinases 1 and 2 are required for the maintenance of adult myofibers and their neuromuscular junctions. Mol. Cell Biol. 2015;35:1238–1253. doi: 10.1128/MCB.01071-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bergwitz C, et al. SLC34A3 mutations in patients with hereditary hypophosphatemic rickets with hypercalciuria predict a key role for the sodium-phosphate cotransporter NaPi-IIc in maintaining phosphate homeostasis. Am. J. Hum. Genet. 2006;78:179–192. doi: 10.1086/499409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Carpenter TO, Imel EA, Holm IA, Jan de Beur SM, Insogna KL. A clinician’s guide to X-linked hypophosphatemia. J. Bone Min. Res. 2011;26:1381–1388. doi: 10.1002/jbmr.340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Drezner MK. Tumor-induced osteomalacia. Rev. Endocr. Metab. Disord. 2001;2:175–186. doi: 10.1023/A:1010006811394. [DOI] [PubMed] [Google Scholar]
- 32.Faul C, et al. FGF23 induces left ventricular hypertrophy. J. Clin. Invest. 2011;121:4393–4408. doi: 10.1172/JCI46122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Brotto M, Bonewald L. Bone and muscle: Interactions beyond mechanical. Bone. 2015;80:109–114. doi: 10.1016/j.bone.2015.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ruppe MD, et al. Effect of fourmonthly doses of a human monoclonal anti-FGF23 antibody (KRN23) on quality of life in X-linked hypophosphatemia. Bone. Rep. 2016;5:158–162. doi: 10.1016/j.bonr.2016.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Suzanne et al, Thomas Carpenter. in ASBMR (Denver, Colorado, 2017).
- 36.Aono Y, et al. Anti-FGF-23 neutralizing antibodies ameliorate muscle weakness and decreased spontaneous movement of Hyp mice. J. Bone Min. Res. 2011;26:803–810. doi: 10.1002/jbmr.275. [DOI] [PubMed] [Google Scholar]
- 37.Avin KG, et al. Fibroblast growth factor 23 does not directly influence skeletal muscle cell proliferation and differentiation or ex vivo muscle contractility. Am. J. Physiol. Endocrinol. Metab. 2018;315:E594–E604. doi: 10.1152/ajpendo.00343.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wacker MJ, et al. Skeletal Muscle, but not Cardiovascular Function, Is Altered in a Mouse Model of Autosomal Recessive Hypophosphatemic Rickets. Front. Physiol. 2016;7:173. doi: 10.3389/fphys.2016.00173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sinha A, Hollingsworth KG, Ball S, Cheetham T. Improving the vitamin D status of vitamin D deficient adults is associated with improved mitochondrial oxidative function in skeletal muscle. J. Clin. Endocrinol. Metab. 2013;98:E509–513. doi: 10.1210/jc.2012-3592. [DOI] [PubMed] [Google Scholar]
- 40.Schubert L, DeLuca HF. Hypophosphatemia is responsible for skeletal muscle weakness of vitamin D deficiency. Arch. Biochem. Biophys. 2010;500:157–161. doi: 10.1016/j.abb.2010.05.029. [DOI] [PubMed] [Google Scholar]
- 41.Baczynski R, et al. Effect of parathyroid hormone on energy metabolism of skeletal muscle. Kidney Int. 1985;28:722–727. doi: 10.1038/ki.1985.190. [DOI] [PubMed] [Google Scholar]
- 42.Smogorzewski M, Piskorska G, Borum PR, Massry SG. Chronic renal failure, parathyroid hormone and fatty acids oxidation in skeletal muscle. Kidney Int. 1988;33:555–560. doi: 10.1038/ki.1988.33. [DOI] [PubMed] [Google Scholar]
- 43.Miniou P. Gene targeting restricted to mouse striated muscle lineage. Nucleic Acids Research. 1999;27(19):27e–27. doi: 10.1093/nar/27.19.e27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Leu M, et al. Erbb2 regulates neuromuscular synapse formation and is essential for muscle spindle development. Dev. 2003;130:2291–2301. doi: 10.1242/dev.00447. [DOI] [PubMed] [Google Scholar]
- 45.Couasnay G, et al. PiT1/Slc20a1 Is Required for Endoplasmic Reticulum Homeostasis, Chondrocyte Survival, and Skeletal Development. J. Bone Min. Res. 2019;34:387–398. doi: 10.1002/jbmr.3609. [DOI] [PubMed] [Google Scholar]
- 46.Collins JF, Bai L, Ghishan FK. The SLC20 family of proteins: dual functions as sodium-phosphate cotransporters and viral receptors. Pflug. Arch. 2004;447:647–652. doi: 10.1007/s00424-003-1088-x. [DOI] [PubMed] [Google Scholar]
- 47.Thomson David. The Role of AMPK in the Regulation of Skeletal Muscle Size, Hypertrophy, and Regeneration. International Journal of Molecular Sciences. 2018;19(10):3125. doi: 10.3390/ijms19103125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Drake JC, Wilson RJ, Yan Z. Molecular mechanisms for mitochondrial adaptation to exercise training in skeletal muscle. FASEB J. 2016;30:13–22. doi: 10.1096/fj.15-276337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Handschin C, et al. Abnormal glucose homeostasis in skeletal muscle-specific PGC-1alpha knockout mice reveals skeletal muscle-pancreatic beta cell crosstalk. J. Clin. Invest. 2007;117:3463–3474. doi: 10.1172/JCI31785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Olguin HC, Pisconti A. Marking the tempo for myogenesis: Pax7 and the regulation of muscle stem cell fate decisions. J. Cell Mol. Med. 2012;16:1013–1025. doi: 10.1111/j.1582-4934.2011.01348.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Forand A, et al. Disruption of the Phosphate Transporter Pit1 in Hepatocytes Improves Glucose Metabolism and Insulin Signaling by Modulating the USP7/IRS1 Interaction. Cell Rep. 2016;16:2736–2748. doi: 10.1016/j.celrep.2016.08.012. [DOI] [PubMed] [Google Scholar]
- 52.Theilen NT, Kunkel GH, Tyagi SC. The Role of Exercise and TFAM in Preventing Skeletal Muscle Atrophy. J. Cell Physiol. 2017;232:2348–2358. doi: 10.1002/jcp.25737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mayr JA, et al. Deficiency of the mitochondrial phosphate carrier presenting as myopathy and cardiomyopathy in a family with three affected children. Neuromuscul. Disord. 2011;21:803–808. doi: 10.1016/j.nmd.2011.06.005. [DOI] [PubMed] [Google Scholar]
- 54.Bhoj EJ, et al. Pathologic Variants of the Mitochondrial Phosphate Carrier SLC25A3: Two New Patients and Expansion of the Cardiomyopathy/Skeletal Myopathy Phenotype With and Without Lactic Acidosis. JIMD Rep. 2015;19:59–66. doi: 10.1007/8904_2014_364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kwong JQ, et al. Genetic deletion of the mitochondrial phosphate carrier desensitizes the mitochondrial permeability transition pore and causes cardiomyopathy. Cell Death Differ. 2014;21:1209–1217. doi: 10.1038/cdd.2014.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Allen DL, et al. Cardiac and skeletal muscle adaptations. voluntary Wheel. running mouse. J. Appl. Physiol. 2001;90:1900–1908. doi: 10.1152/jappl.2001.90.5.1900. [DOI] [PubMed] [Google Scholar]
- 57.Egan B, Zierath JR. Exercise metabolism and the molecular regulation of skeletal muscle adaptation. Cell Metab. 2013;17:162–184. doi: 10.1016/j.cmet.2012.12.012. [DOI] [PubMed] [Google Scholar]
- 58.Knochel JP. Models of hypophosphatemia and phosphate depletion. Adv. Exp. Med. Biol. 1982;151:191–198. doi: 10.1007/978-1-4684-4259-5_25. [DOI] [PubMed] [Google Scholar]
- 59.Bergwitz Clemens, Miyamoto Ken-Ichi. Hereditary hypophosphatemic rickets with hypercalciuria: pathophysiology, clinical presentation, diagnosis and therapy. Pflügers Archiv - European Journal of Physiology. 2018;471(1):149–163. doi: 10.1007/s00424-018-2184-2. [DOI] [PubMed] [Google Scholar]
- 60.Iwata M, et al. A novel tetracycline-responsive transgenic mouse strain for skeletal muscle-specific gene expression. Skelet. Muscle. 2018;8:33. doi: 10.1186/s13395-018-0181-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ma Y, et al. FGF23 Is Not Required to Regulate Fetal Phosphorus Metabolism but Exerts Effects Within 12 Hours After Birth. Endocrinol. 2017;158:252–263. doi: 10.1210/en.2016-1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chande S, Ho B, Fetene J, Bergwitz C. Transgenic mouse model for conditional expression of influenza hemagglutinin-tagged human SLC20A1/PIT1. PLoS One. 2019;14:e0223052. doi: 10.1371/journal.pone.0223052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Giovannini D, Touhami J, Charnet P, Sitbon M, Battini JL. Inorganic phosphate export by the retrovirus receptor XPR1 in metazoans. Cell Rep. 2013;3:1866–1873. doi: 10.1016/j.celrep.2013.05.035. [DOI] [PubMed] [Google Scholar]
- 64.Legati A, et al. Mutations in XPR1 cause primary familial brain calcification associated with altered phosphate export. Nat. Genet. 2015;47:579. doi: 10.1038/ng.3289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wild R, et al. Control of eukaryotic phosphate homeostasis by inositol polyphosphate sensor domains. Sci. 2016;352:986–990. doi: 10.1126/science.aad9858. [DOI] [PubMed] [Google Scholar]
- 66.Ansermet C, et al. Renal Fanconi Syndrome and Hypophosphatemic Rickets in the Absence of Xenotropic and Polytropic Retroviral Receptor in the Nephron. J. Am. Soc. Nephrol. 2017;28:1073–1078. doi: 10.1681/ASN.2016070726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ovejero D, Gafni RI, Collins MT. 1,25-Dihydroxyvitamin D as Monotherapy for XLH: Back to the Future? J. Bone Min. Res. 2016;31:925–928. doi: 10.1002/jbmr.2858. [DOI] [PubMed] [Google Scholar]
- 68.Festing MH, Speer MY, Yang HY, Giachelli CM. Generation of mouse conditional and null alleles of the type III sodium-dependent phosphate cotransporter PiT-1. Genes. 2009;47:858–863. doi: 10.1002/dvg.20577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wagner Carsten A., Rubio-Aliaga Isabel, Hernando Nati. Renal phosphate handling and inherited disorders of phosphate reabsorption: an update. Pediatric Nephrology. 2017;34(4):549–559. doi: 10.1007/s00467-017-3873-3. [DOI] [PubMed] [Google Scholar]
- 70.Truett GE, et al. Preparation of PCR-quality mouse genomic DNA with hot sodium hydroxide and tris (HotSHOT) Biotechniques. 2000;29:52–54. doi: 10.2144/00291bm09. [DOI] [PubMed] [Google Scholar]
- 71.Li Y, et al. Response of Npt2a knockout mice to dietary calcium and phosphorus. PLOS ONE. 2017;12:e0176232. doi: 10.1371/journal.pone.0176232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Saria A, Lundberg JM. Evans blue fluorescence: quantitative and morphological evaluation of vascular permeability in animal tissues. J. Neurosci. Methods. 1983;8:41–49. doi: 10.1016/0165-0270(83)90050-x. [DOI] [PubMed] [Google Scholar]
- 73.Sahay A, et al. Increasing adult hippocampal neurogenesis is sufficient to improve pattern separation. Nat. 2011;472:466–470. doi: 10.1038/nature09817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Duman CH, Schlesinger L, Russell DS, Duman RS. Voluntary exercise produces antidepressant and anxiolytic behavioral effects in mice. Brain Res. 2008;1199:148–158. doi: 10.1016/j.brainres.2007.12.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wang X, Seed B. A PCR primer bank for quantitative gene expression analysis. Nucl. Acid. Res. 2003;31:1–8. doi: 10.1093/nar/gkg120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Divakaruni AS, Rogers GW, Murphy AN. Measuring Mitochondrial Function in Permeabilized Cells Using the Seahorse XF Analyzer or a Clark-Type Oxygen Electrode. Current protocols in toxicology. 2014;60:25.22.21–16. doi: 10.1002/0471140856.tx2502s60. [DOI] [PubMed] [Google Scholar]
- 77.Salabei JK, Gibb AA, Hill BG. Comprehensive measurement of respiratory activity in permeabilized cells using extracellular flux analysis. Nat. Protoc. 2014;9:421–438. doi: 10.1038/nprot.2014.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.