

Abstract
The aim of this study was to investigate the effects of an extract of Qiweibaizhu powder combined with Debaryomyces hansenii on the gut microbiota of antibiotic-treated mice with diarrhea. Mice were gavaged with a mixture of gentamycin sulfate and cefradine to induce diarrhea. After diarrhea was observed, 25% dose of ultra-micro Qiweibaizhu powder extract combined with 25% dose of Debaryomyces hansenii (QCD) was gavaged to mice with diarrhea. DNA of intestinal contents in mice was extracted for 16S rRNA gene sequence analysis by high-throughput sequencing following treatment finished. The results showed that the QCD increased the species richness and diversity, but did not recover the diversity to the original level. Antibiotics and QCD significantly altered the composition of gut microbiota at different taxonomic levels. At the genus level, the relative abundance of Bacteroidales S24-7 group_unidentified and Bacteroides returned to baseline after QCD treatment. Additionally, QCD suppressed the growth of Oscillospira and Ruminococcus, and promoted the proliferation of Erysipelotrichaceae_norank and Blautia compared with the healthy and diarrheal mice. Our results indicated that QCD modulated the diversity and composition of the gut microbiota in antibiotic-treated mice with diarrhea. The synergistic effect between Qiweibaizhu powder extract and Debaryomyces hansenii may be related to Bifidobacterium and Bacteroidales S24-7 group_unidentified.
Keywords: Qiweibaizhu powder, Debaryomyces hansenii, Gut microbiota, Antibiotic-associated diarrhea, Diversity
Introduction
Antibiotic-associated diarrhea (AAD) is a drug-induced disease commonly regarded as an adverse effect of antibiotics. It has been reported that 5–25% of patients who use antibiotics experience diarrhea (Lichtman et al. 2016). Gut microbiota dysbiosis is responsible for the development of AAD (Hogenauer et al. 1998; Anand et al. 2017). Therefore, the key aspect of AAD treatment is re-establishing the balance of gut microbiota.
Qiweibaizhu powder (QWBZP), a traditional Chinese herbal formula in “Xiaoer Yaozheng Zhijue”, has been used to treat infantile diarrhea in China for thousands of years. Our studies have supported that QWBZP is a valuable therapeutic for AAD, and cures diarrhea by adjusting the intestinal microecology and re-establishing the gut microbiota balance (Tan et al. 2012; Zhang et al. 2014). Superfine pulverization technology can enhance the dissolution of effective components in traditional Chinese medicine (TCM) and notably decrease the dosage of TCM (Deng et al. 2011). Our previous studies confirmed that 50% dose of ultra-micro QWBZP extract is equivalent to the 100% dose of traditional QWBZP extract when treating AAD. Additionally, 50% dose of ultra-micro QWBZP extract is superior to the 100% dose of traditional QWBZP extract in regulating the metabolic diversity of gut microbiota, repairing intestinal mucosa, and promoting the proliferation of yeast and Bifidobacterium (Zhang et al. 2014; Deng et al. 2011).
Probiotics are live microorganisms that confer a health benefit on the host when administered in adequate amounts (Silverman et al. 2017). Usually, Lactobacillus, Bifidobacterium, yeast, Enterococcus and Bacillus are used to treat AAD (Goldenberg et al. 2015; Szajewska et al. 2016). In addition to Saccharomyces boulardii, several yeast strains, such as Debaryomyces hansenii (D. hansenii), Torulaspora delbrueckii, and Kluyveromyces lactis, also possess antibacterial and probiotic effects (Hatoum et al. 2012). D. hansenii, isolated from natural habitats, food and animal intestines, has been found to have anti-inflammatory effects on human dendritic cells (Ochangco et al. 2016). Furthermore, D. hansenii can stimulate innate immunity, antioxidant parameters, and immune-related gene expression in newborn goats (Angulo et al. 2019). We isolated D. hansenii from the gut of experimental mice and revealed its potency in modulating the diversity of gut bacteria and stimulating the density of Lactobacillus and Bifidobacterium (Guo et al. 2015; He et al. 2017).
Clinically, the combination of TCM and probiotics effectively alleviates diarrhea and shortens the clinical treatment time (Lei et al. 2018). Importantly, the dosages of TCM and probiotics were notably reduced, which is cost saving. Based on both QWBZP extract and D. hansenii can modulate the gut microbiota and cure AAD, we wanted to know whether the combination of the two has synergistic effect in AAD treatment. Our previous studies showed that 25% dose of ultra-micro QWBZP extract combined with 25% dose of D. hansenii (QCD) exhibited synergistic effects in recovering the density of total intestinal bacteria and Escherichia coli, and in increasing the diversity of Lactobacillus. The therapeutic effect of QCD is equivalent to the 50% dose of ultra-micro QWBZP extract and 100% dose of traditional QWBZP extract (Guo et al. 2015; Long et al. 2018; Liu et al. 2016). In this study, we further discussed the synergistic mechanism of QCD by high-throughput sequencing and provided support for the clinical application of QCD.
Materials and methods
Preparation of 100% dose of ultra-micro QWBZP extract
QWBZP is composed of the slices of seven Chinese herbs: Ginseng Radix et Rhizoma (6 g), Aucklandiae Radix (6 g), Poria (10 g), Atractylodis Macrocephalae Rhizoma Tostum (10 g), Pogostemonis Herba (10 g), Puerariae Lobatae Radix (10 g), and Glycyrrhizae Radix et Rhizoma (3 g). All Chinese herb slices were purchased from The First Hospital of Hunan University of Chinese Medicine. The slices of the seven Chinese herbs were processed into ultra-micro powder, and subsequently brewed in boiling water. The solution was centrifuged at 4000 × g, and the supernatant was collected. The concentration of the supernatant was 2 g mL−1, and was subsequently diluted to 25% dose. The diluted supernatant was stored at 4 °C and reheated to 25–30 °C before use.
Preparation of 100% dose of D. hansenii
D. hansenii was activated according to our previous study (Guo et al. 2015). In short, D. hansenii was inoculated into liquid potato sucrose medium and shaken at 28 °C for 36 h. The suspension was centrifuged at 2000 × g for 4 min. The sediment was washed 1–2 times using sterile stroke-physiological saline solution and then transferred to a centrifuge tube. After counting with a hemocytometer, the number of D. hansenii was 1010 cells mL−1, and was subsequently diluted to 25% dose for further use.
Reagents
The antibiotic mixture was composed of gentamicin sulfate (No. 5120106, Yichang Renfu Pharmaceutical Co., Ltd.) and cefradine (No. 110804, Suzhou Zhonghua Pharmaceutical Industry Co., Ltd.). The concentration of the antibiotic mixture was 62.5 g L−1 (Zhang et al. 2014). Tris-saturated phenol–chloroform–isoamyl alcohol (25:24:1), protease K, TE buffer, lysozyme, and acetone were purchased from Beijing Dingguo Changsheng Biotechnology Co., Ltd. and were used to extract DNA.
Animal ethics statement
Eighteen specific-pathogen-free (SPF) Kunming (KM) mice (nine males and nine females, weight 18–22 g, license number [SCXK (Xiang) 2013-0004]) were purchased from Hunan Slaccas Jingda Laboratory Animal Co., Ltd. Fodder was provided by the Laboratory Animal Center of Hunan University of Chinese Medicine. The study was approved by the Animal Care and Use Committee of Hunan University of Chinese Medicine.
Experimental design
After adaptive feeding (3 days) under controlled conditions (23–25 °C, 50–70% humidity and a 12 h light/12 h dark cycle), the mice were randomly divided into three groups with three males and three females in each group: healthy group (qck), AAD group (qm), and QCD treatment group (qjq). The mice in the AAD group and QCD treatment group were first gavaged with a mixture of gentamycin sulfate and cefradine (0.35 mL) twice per day for 5 days, while the mice in the healthy group were gavaged with the same amount of sterile water. After diarrhea was observed, the mice in the healthy group and AAD group were gavaged with sterile water, and the mice in the QCD treatment group were administered QCD (0.35 mL), twice per day for 4 days. After 4 days of treatment, all mice were euthanized by cervical dislocation, and the intestinal contents were collected immediately. The intestinal contents of one male and one female in the same group were mixed and then stored at 4 °C for further use.
16S rRNA gene sequence analysis
Microbial genomic DNA was extracted from each sample according to the protocol in our previous report (Wu et al. 2012). The variable region of bacterial 16S rRNA V4 was amplified using the primers 520F (5′-AYTGGGYDTAAAGNG-3′) and 802R (5′-TACNVGGGTATCTAATCC-3′). The PCR amplification system included 2.0 µL of dNTPs (2.5 mmol L−1), 5.0 µL of 5 × Q5 reaction buffer, 5.0 µL of 5 × Q5 high enhancer, 1.0 µL of forward primer (10 µmol L−1), 1.0 µL of reverse primer (10 µmol L−1), 0.25 µL of Q5 polymerase (5 U µL−1), 2.0 µL of template DNA (0.2 ng µL−1), and 8.75 µL of sterilized ddH2O. The PCR conditions were as follows: 98 °C for 30 s; 25 cycles of 98 °C for 30 s, 50 °C for 30 s and 72 °C for 30 s; and 72 °C for 5 min. The PCR products were examined by 2% agarose gel electrophoresis. Sequencing was performed by Shanghai Personal Biotechnology Co., Ltd.
Bioinformatic analyses
Quantitative insights into microbial ecology (QIIME, version 1.7.0, https://qiime.org/) was used to analyze raw DNA sequences, operational taxonomic units (OTUs), rank abundance curves, community structures, and beta diversity (Caporaso et al. 2010; Edgar 2010; Oberauner et al. 2013). Alpha diversity and species abundance were analyzed by MOTHUR (version 1.35.1, https://www.mothur.org/) (White et al. 2009). Information on species evolution and abundance was provided by MEGAN (https://ab.inf.uni-tuebingen.de/software/megan/) (Huson et al. 2011). The Greengene (Release 13.8, https://greengenes.secondgenome.com/) database was used to annotate taxonomic information. SPSS 21.0 (IBM Corp., Armonk, NY, USA) was used to compare the differences among the three groups. Analyses of significant differences were performed by an independent one-way analysis of variance (ANOVA) in terms of the least significant difference (LSD) multiple comparison test.
Results
DNA sequences of the intestinal bacteria in AAD mice
All the raw data were submitted to NCBI (Accession number: SRP245727). A total of 1,221,890 high-quality sequences were detected in all samples. The average proportions of high-quality sequences in the healthy group, AAD group and QCD treatment group were 92.63%, 92.23%, and 93.84%, respectively (Fig. 1). The QCD treatment group possessed more DNA sequences than the other two groups. The results indicated that QCD treatment increased the number of gut bacteria species in mice with AAD.
Fig. 1.
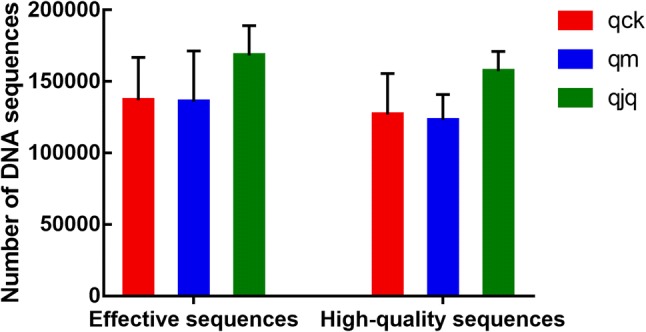
DNA sequences of the intestinal bacteria in different groups. qck, healthy group; qm, antibiotic-associated diarrhea (AAD) group; qjq, Qiweibaizhu powder extract combined with Debaryomyces hansenii (QCD) treatment group
OTU number of the intestinal bacteria in AAD mice
High-quality sequences were clustered at 97% similarity by QIIME. The unique and shared OTUs in different groups are presented in Fig. 2. The Venn diagram shows that 1451 OTUs were identified in total, and 576 of these OTUs were shared by the three groups. In addition, the numbers of OTUs identified in the healthy group, AAD group, and QCD treatment group were 944, 1001, and 1140, respectively. Briefly, QCD treatment increased the OTU number, which implied that QCD treatment increased the number of intestinal bacteria.
Fig. 2.
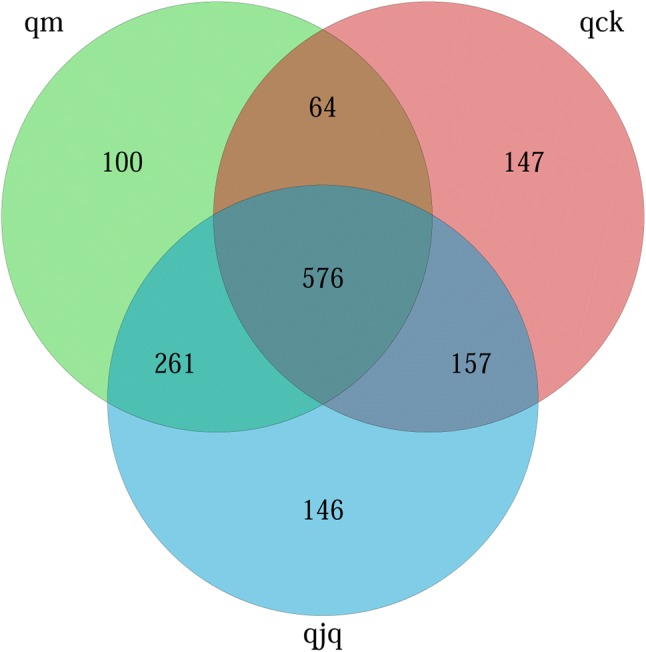
Shared operational taxonomic unit (OTU) analysis of the different groups. Venn diagram of OTUs based on the sequences with a threshold similarity of 97%. qck, healthy group; qm, AAD group; qjq, QCD treatment group
Alpha diversity of the intestinal bacteria in AAD mice
Alpha diversity indices (Chao, ACE, Shannon and Simpson) reflect community richness and diversity. Commonly, Chao and ACE are related to community richness, while Shannon and Simpson are associated with the diversity of species. In this study, the highest community richness was observed in the QCD treatment group, but the differences among the three groups were not significant. On the other hand, significant differences in the Shannon index were found between the control group and the other two groups. QCD treatment increased the species diversity, which was reduced by antibiotics. However, the species diversity did not fully recover to normal levels (Fig. 3). These results were also confirmed by the rank abundance curve (Fig. 4).
Fig. 3.
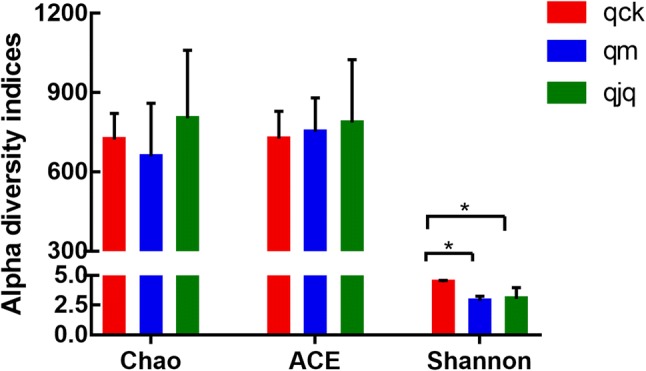
Alpha diversity analysis of the gut microbiota in different groups based on the Chao, ACE, and Shannon indices. qck, healthy group; qm, AAD group; qjq, QCD treatment group. Data are the mean ± SE, n = 3. *p < 0.05 vs. the healthy group
Fig. 4.

The OTU abundance of each sample in different groups. Each polyline represents one sample. The larger the curve span, the richer the species. The flatter the curve, the more homogeneous the species. qck 1–3, healthy group; qm1-3, AAD group; qjq1-3, QCD treatment group
Beta diversity of the intestinal bacteria in AAD mice
Principal component analysis (PCA) was used to compare the structure of the intestinal bacteria among different groups. The variation in PC1 and PC2 represented 70.98% of the total difference among the samples in this study (Fig. 5). After weighted UniFrac analysis, the nonmetric multidimensional scaling (NMDS) results showed that the samples in the healthy group and QCD treatment group were relatively concentrated. However, the distance between the healthy group and the other two groups was large (Fig. 6). This means that the microbial composition of the AAD group and QCD treatment group was dramatically different from that of the healthy group.
Fig. 5.

Bacterial similarity among different groups as represented by principal component analysis (PCA). Each point in the figure represents a sample. qck, healthy group; qm, AAD group; qjq, QCD treatment group
Fig. 6.

Bacterial similarity among different groups as represented by nonmetric multidimensional scaling (NMDS). The NMDS score plot is based on weighted UniFrac analysis. Each point in the figure represents a sample. qck, healthy group; qm, AAD group; qjq, QCD treatment group
The composition of gut bacteria at different taxonomic levels
Figure 7 shows the number of taxa at different taxonomic levels. Compared with the healthy group and AAD group, the QCD treatment group possessed the largest number of species, but no significant differences were detected among the three groups at any taxonomic level.
Fig. 7.
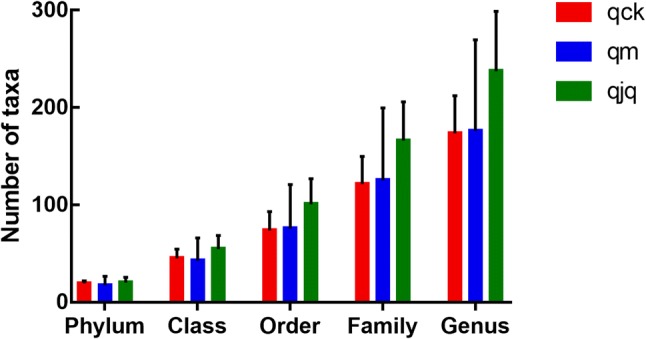
Number of taxa at different taxonomic levels in different groups. qck, healthy group; qm. AAD group; qjq, QCD treatment group. Data are the mean ± SE, n = 3
Thirty-one phyla were identified in this study. Firmicutes and Bacteroidetes were the two predominant phyla in the three groups. The relative abundance of Firmicutes in the healthy group, AAD group, and QCD treatment group was 49.6%, 57.5%, and 59.1%, respectively. Bacteroidetes accounted for 44.9%, 37.0%, and 26.9% in the healthy group, AAD group and QCD treatment group, respectively (Fig. 8). Antibiotics use increased the relative abundance of Firmicutes while reducing the relative abundance of Bacteroidetes. Although QCD treatment enhanced the increasing and decreasing tendencies of Firmicutes and Bacteroidetes, respectively, the relative abundances of Firmicutes and Bacteroidetes among the three groups were not statistically significant (p > 0.05). Euryarchaeota was found in the healthy group and QCD treatment group but not in the AAD group. TM7 and Tenericutes were detected in only the healthy group. BRC1, OD1, and Synergistetes were found in only the AAD group, whereas Chlamydiae was found in only the QCD treatment group.
Fig. 8.

Histogram of microbiota community structures at the phylum level. qck, healthy group; qm, AAD group; qjq, QCD treatment group
Obvious changes in the microbial composition at the genus level were observed. Figure 9 shows the abundance and evolutionary relationship of the bacterial species. The healthy group was predominantly composed of Bacteroidales S24-7 group_unidentified, Clostridiales_norank, and Lactobacillus, which accounted for 30.9%, 20.8%, and 8.6% of the total microbial population, respectively. In contrast, antibiotics use resulted in Bacteroides (22.3%), Eubacterium (12.9%), and Parabacteroides (9.5%) becoming the predominant genera. Moreover, Bacteroidales S24-7 group_unidentified, Blautia, and Erysipelotrichaceae_norank were the predominant genera in the QCD treatment group, accounting for 23.9%, 15.1%, and 13.1%, respectively. In summary, antibiotics and QCD treatment significantly altered the microbial composition.
Fig. 9.

The abundances and evolutionary relationships of intestinal bacteria in different groups. qck, healthy group; qm, AAD group; qjq, QCD treatment group
Antibiotics use diminished the relative abundance of Bacteroidales S24-7 group_unidentified (p = 0.01), Clostridiales_ norank (p = 0.003), Lactobacillus, Oscillospira (p = 0.014), Ruminococcus (p = 0.045), Rikenellaceae_norank (p = 0.003), Bifidobacterium, and Sutterella, while increasing the relative abundance of Bacteroides (p = 0.00), Parabacteroides, Erysipelotrichaceae_norank, Eubacterium (p = 0.037), Blautia, Akkermansia, and Peptostreptococcaceae_norank (p = 0.031) (Table 1). Notably, the relative abundances of Bacteroidales S24-7 group_unidentified and Bacteroides recovered to baseline after QCD treatment. Blautia (p = 0.04) and Erysipelotrichaceae_norank (p = 0.004) increased notably after QCD treatment.
Table1.
Relative abundance of the intestinal bacteria in different groups at the genus level
| Genus | qck | qm | qjq |
|---|---|---|---|
| Bacteroidales S24-7 group_unidentified | 0.3090 ± 0.0355 | 0.0467 ± 0.0201** | 0.2389 ± 0.1441# |
| Clostridiales_norank | 0.2080 ± 0.8933 | 0.0079 ± 0.0112** | 0.0037 ± 0.0034** |
| Lactobacillus | 0.0862 ± 0.0485 | 0.0335 ± 0.0533 | 0.0088 ± 0.0097 |
| Bacteroides | 0.0605 ± 0.0238 | 0.2228 ± 0.0275** | 0.0235 ± 0.0227## |
| Oscillospira | 0.0527 ± 0.0235 | 0.0120 ± 0.0061* | 0.0064 ± 0.0060** |
| Parabacteroides | 0.0221 ± 0.0082 | 0.0953 ± 0.0942 | 0.0024 ± 0.0030 |
| Ruminococcus | 0.0142 ± 0.0083 | 0.0039 ± 0.0024* | 0.0000 ± 0.0000* |
| Rikenellaceae_norank | 0.0106 ± 0.0031 | 0.0013 ± 0.0020** | 0.0013 ± 0.0017** |
| Erysipelotrichaceae_norank | 0.0055 ± 0.0009 | 0.0431 ± 0.0238 | 0.1309 ± 0.0549**# |
| Eubacterium | 0.0032 ± 0.0023 | 0.1293 ± 0.0349* | 0.1009 ± 0.0936 |
| Blautia | 0.0028 ± 0.0006 | 0.0828 ± 0.0512 | 0.1505 ± 0.1090* |
| Akkermansia | 0.0008 ± 0.0007 | 0.0187 ± 0.0203 | 0.0010 ± 0.0008 |
| Bifidobacterium | 0.0011 ± 0.0012 | 0.0007 ± 0.0007 | 0.0476 ± 0.0750 |
| Sutterella | 0.0032 ± 0.0010 | 0.0006 ± 0.0006 | 0.0345 ± 0.0304 |
| Peptostreptococcaceae_norank | 0.0000 ± 0.0000 | 0.0134 ± 0.0086* | 0.0045 ± 0.0053 |
Data are the mean ± SD, n = 3
qck, healthy group; qm, AAD group; qjq, QCD treatment group
Compared with the healthy group, *p < 0.05, **p < 0.01. Compared with the AAD group, #p < 0.05
Discussion
It is well known that antibiotics alter the diversity and composition of gut microbiota, a condition known as dysbiosis, which allows for infection, diarrhea, and inflammation. Probiotics play a vital role in maintaining the balance of intestinal microecology. However, probiotics did not shift the overall diversity of the gut microbiota (Grazul et al. 2016). In fact, probiotics remodel the intestinal microecology of an individual recovering from antibiotic treatment by changing the composition of intestinal bacteria (Grazul et al. 2016). In accordance with Grazul et al., we found that although QWBZP extract and D. hansenii did cause an increase in microbial diversity when used alone or in combination, the diversity of gut microbiota was not restored to its original levels. This result indicated that QCD treatment helped to restore microbial diversity during the recovery period, but a long time was still required for the gut microbiota become rebalanced after antibiotics use.
Notably, antibiotics use and QCD treatment led to a drastic change in the gut microbiota composition. The Bacteroides population is commonly believed to be beneficial to the host’s nutrition, mucosa, and immunity. On the other hand, Bacteroides can escape into the sterile peritoneum when colonic integrity is disrupted and act as opportunistic pathogens (Wick and Sears 2010). Enterotoxigenic Bacteroides fragilis (ETBF) is the only strain of Bacteroides associated with diarrhea (Wick and Sears 2010). In this study, antibiotics induced the growth of Bacteroides. In contrast, QCD treatment efficiently eliminated Bacteroides to the original level. This result indicated that the diarrhea induced by gentamicin sulfate and cefradine may be due to ETBF infection, and that QCD treatment can relieve ETBF infection.
Hernández et al. (2019) evaluated the association of the gut microbiota in patients with Clostridioides difficile (C. difficile) infection (CDI) and CDI risk factors. It was found that Peptostreptococcaceae was positively correlated with Akkermansia, which may predict the presence of CDI (Hernández et al. 2019). Yutin and Galperin (2013), suggested that C. difficile should be reclassified within Peptostreptococcaceae in the order Clostridiales. Hence, this phylogenetic relationship could explain why Peptostreptococcaceae is of prime importance in CDI. In our study, the relative abundance of Peptostreptococcaceae was increased by antibiotics, which indicated that the diarrhea induced by gentamicin sulfate and cefradine may also be due to C. difficile infection.
Sangster et al. (2016) also observed an increase in Akkermansia in CDI patients. Over the excessive growth of Akkermansia was also found in the gut of individuals who received special antibiotic therapy (Derrien et al. 2017). Akkermansia is a mucin-degrading bacterium that was isolated from human fecal samples. There has been a growing interest in Akkermansia because of its correlation to health and because of its potential as a biomarker for disease (Geerlings et al. 2018). Furthermore, Akkermansia was found to increase the expression of immune response genes and strengthen the mucosal barrier in mice (Everard et al. 2013; Derrien et al. 2011; Fujio-Vejar et al. 2017). This study revealed that the number of Akkermansia was increased by antibiotics. The immune responses triggered by Akkermansia may be regarded as a self-recovery mechanism and be responsible for this change. Furthermore, QCD treatment decreased the relative abundance of Akkermansia.
In this study, the relative abundance of Ruminococcus, Oscillospira, and Blautia also changed notably after QCD treatment. The dysbiosis of feedlot cattle with hemorrhagic diarrhea was characterized by increasing Blautia and decreasing Ruminococcus and Oscillospira, while dogs with acute hemorrhagic diarrhea were characterized by decreasing Ruminococcus and Blautia (Zeineldin et al. 2018; Suchodolski et al. 2012). Although the changes in Ruminococcus, Oscillospira, and Blautia in different animals and diarrhea were debatable, our study was in line with the observations of Zeineldin. However, QCD treatment enhanced the tendency of the change in the three genera.
Our previous study revealed that a low concentration of QWBZP extract promoted the proliferation of D. hansenii in vitro (Guo et al. 2013). A recent study confirmed that yeast promoted the growth and survival rate of other coexisting probiotics by increasing the biofilm formation capacity and hydrophobicity of probiotics, increasing lactic acid levels, reducing pH values, and decreasing the reducing sugars and free amino nitrogen levels of probiotics (Zoumpourtikoudi et al. 2018). In this study, QCD treatment accelerated the growth of Bifidobacterium. Bifidobacterium can regulate intestinal immune homeostasis by inhibiting the infection of pathogenic bacteria, improving the function of the mucosal barrier, suppressing proinflammatory cytokines, and altering the function of dendritic cells (Azad et al. 2018). Therefore, the synergistic effect of QWBZP extract and D. hansenii may be associated with increased Bifidobacterium. According to previous studies, QWBZP extract and D. hansenii, when used alone or in combination, promoted the proliferation of Lactobacillus in vitro. In contrast, QCD treatment reduced the relative abundance of Lactobacillus in vivo. QCD treatment showed different effects on Lactobacillus in vitro and in vivo.
Bacteroidales family S24-7 is highly localized to the intestinal tracts of homeothermic animals and is recognized to degrade fiber and polysaccharide (Ormerod et al. 2016; Garcia-Mazcorro et al. 2018). Considering the function of S24-7 family, it seemed that S24-7 family members promoted the efficiency of carbohydrate metabolism in the QCD treatment group. In particular, S24-7 was found to be notably higher in mice that efficiently metabolized Panax ginseng (Dong et al. 2017). Interestingly, Bacteroidales S24-7 group_unidentified increased in the gut of mice with AAD, but did not recover to baseline after D. hansenii treatment (He et al. 2017). However, the abundance of Bacteroidales S24-7 group_unidentified returned to normal levels after treatment with QCD. The results implied that Bacteroidales S24-7 group_unidentified may be the key genus that responded to QWBZP extract. Moreover, Bacteroidales S24-7 group_unidentified may be regarded as evidence to suggest the synergistic effect between QWBZP extract and D. hansenii. In summary, D. hansenii increased the relative abundance of Bacteroidales S24-7 group_unidentified, which was drastically reduced by antibiotics. Moreover, increasing Bacteroidales S24-7 group_unidentified helped to metabolize Panax ginseng, which is an important ingredient of QWBZP, and promoted the efficacy of QWBZP.
Conclusions
Taken together, QCD treatment dramatically changed the structure of gut microbiota, and these changes were benefit to cure AAD. Bifidobacterium and Bacteroidales S24-7 group_unidentified may be regarded as the key genera to show the synergistic effect between QWBZP extract and D. hansenii. The exact synergistic mechanism of QCD in treating AAD requires further researches.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (81573951) and Innovation Project for Graduate Students of Hunan University of Chinese Medicine (2019CX08).
Author contributions
ZT designed the study. Material preparation, data collection, and analysis were performed by GX, YW, TZ, and KS. The first draft of the manuscript was written by GX. The decision to submit the manuscript for publication was made by all the authors.
Compliance with ethical standards
Conflict of interest
The authors declare that there is no conflict of interests regarding the publication of this paper.
Institutional animal care and use committee statement
The study was approved by the Animal Ethics and Welfare Committee of Hunan University of Chinese Medicine.
References
- Anand A, Bharadwaj R, Pol S. Antibiotic associated diarrhea with special reference to Clostridium difficile. IJTDH. 2017;24(4):1–10. doi: 10.9734/IJTDH/2017/34541. [DOI] [Google Scholar]
- Angulo M, Reyes-Becerril M, Cepeda-Palacios R, Tovar-Ramírez D, Esteban MÁ, Angulo C. Probiotic effects of marine Debaryomyces hansenii CBS 8339 on innate immune and antioxidant parameters in newborn goats. Appl Microbiol Biotechnol. 2019;103(5):2339–2352. doi: 10.1007/s00253-019-09621-5. [DOI] [PubMed] [Google Scholar]
- Azad MAK, Sarker M, Li T, Yin J. Probiotic species in the modulation of gut microbiota: an overview. Biomed Res Int. 2018;8:9478630. doi: 10.1155/2018/9478630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010;7(5):335–336. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng HJ, Wu H, Peng XS, Cai GX, Zhang X, Tan ZJ, et al. Effects of ultra-micro powder of Qiweibaizhu on dysbacteriosis in mice with diarrhea. Progress Modern Biomed. 2011;11(19):3635–3636. doi: 10.13241/j.cnki.pmb.2011.19.008. [DOI] [Google Scholar]
- Derrien M, Belzer C, de Vos WM. Akkermansia muciniphila and its role in regulating host functions. Microb Pathog. 2017;106:171–181. doi: 10.1016/j.micpath.2016.02.005. [DOI] [PubMed] [Google Scholar]
- Derrien M, Van Baarlen P, Hooiveld G, Norin E, Müller M, de Vos WM. Modulation of mucosal immune response, tolerance, and proliferation in mice colonized by the mucin-degrader Akkermansia muciniphila. Front Microbiol. 2011;2:166. doi: 10.3389/fmicb.2011.00166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong WW, Xuan FL, Zhong FL, Jiang J, Wu S, Li D, et al. Comparative analysis of the rats' gut microbiota composition in animals with different ginsenosides metabolizing activity. J Agric Food Chem. 2017;65(2):327–337. doi: 10.1021/acs.jafc.6b04848. [DOI] [PubMed] [Google Scholar]
- Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 2010;26(19):2460–2461. doi: 10.1093/bioinformatics/btq461. [DOI] [PubMed] [Google Scholar]
- Everard A, Belzer C, Geurts L, Ouwerkerk JP, Druart C, Bindels LB, et al. Cross-talk between Akkermansia muciniphila and intestinal epithelium controls diet-induced obesity. Proc Natl Acad Sci USA. 2013;110:9066–9071. doi: 10.1073/pnas.1219451110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujio-Vejar S, Vasquez Y, Morales P, Morales P, Magne F, Vera-Wolf P, et al. The gut microbiota of healthy chilean subjects reveals a high abundance of the phylum Verrucomicrobia. Front Microbiol. 2017;8:1221. doi: 10.3389/fmicb.2017.01221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Mazcorro JF, Mills DA, Murphy K, Noratto G. Effect of barley supplementation on the fecal microbiota, caecal biochemistry, and key biomarkers of obesity and inflammation in obese db/db mice. Eur J Nutr. 2018;57(7):2513–2528. doi: 10.1007/s00394-017-1523-y. [DOI] [PubMed] [Google Scholar]
- Geerlings SY, Kostopoulos I, de Vos WM, Belzer C. Akkermansia muciniphila in the human gastrointestinal tract: when, where, and how? Microorganisms. 2018;6(3):E75. doi: 10.3390/microorganisms6030075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldenberg JZ, Lytvyn L, Steurich J, Parkin P, Mahant S, Johnston BC. Probiotics for the prevention of pediatric antibiotic-associated diarrhea. Cochrane Database Syst Rev. 2015;12:CD004827. doi: 10.1002/14651858.CD004827.pub4. [DOI] [PubMed] [Google Scholar]
- Grazul H, Kanda LL, Gondek D. Impact of probiotic supplements on microbiome diversity following antibiotic treatment of mice. Gut Microbes. 2016;7(2):101–114. doi: 10.1080/19490976.2016.1138197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo KX, Tan ZJ, Xie MZ, She Y, Wang XH. The synergic effect of ultra-micro powder Qiweibaizhu combined with yeast on dysbacteriotic diarrhea mice. Chin J Appl Environ Biol. 2015;21(1):61–67. doi: 10.3724/SP.J.1145.2013.10002. [DOI] [Google Scholar]
- Guo KX, Zhou SN, Tan ZJ, Cai Y, She Y, Cai GX. Study on inhibition and boosting action of Qiweibaizhusan to intestinal yeast. Progress Modern Biomed. 2013;13(27):5259–5263. doi: 10.13241/j.cnki.pmb.2013.27.010. [DOI] [Google Scholar]
- Hatoum R, Labrie S, Fliss I. Antimicrobial and probiotic properties of yeasts: from fundamental to novel applications. Front Microbiol. 2012;3:421. doi: 10.3389/fmicb.2012.00421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He L, Long CX, Liu YW, Guo YF, Xiao NQ, Zhou JT (2017) Effects of Debaryomyces hansenii treatment on intestinal microorganisms in mice with antibiotics-induced diarrhea. 3 Biotech 7:347. Doi: 10.1007/s13205-017-0953-9. [DOI] [PMC free article] [PubMed]
- Hernández M, de Frutos M, Rodríguez-Lázaro D, Lôpez-Urrutia L, Quijada NM, Eiros JM. Fecal microbiota of toxigenic Clostridioides difficile-associated diarrhea. Front Microbiol. 2019;9:3331. doi: 10.3389/fmicb.2018.03331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogenauer C, Hammer HF, Krejs GJ, Reisinger EC. Mechanisms and management of antibiotic-associated diarrhea. Clin Infect Dis. 1998;27(4):702–710. doi: 10.1086/514958. [DOI] [PubMed] [Google Scholar]
- Huson DH, Mitra S, Ruscheweyh HJ, Weber N, Schuster SC. Integrative analysis of environmental sequences using MEGAN4. Genome Res. 2011;21(9):1552–1560. doi: 10.1101/gr.120618.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei JF, Yu LP, Zhang N. Study of Shenling-Baizhu granule combined with Saccharomyces boulardii treatment for pediatric antibiotic associated diarrhea. Int J Trad Chin Med. 2018;40(7):613–615. doi: 10.3760/cma.j.issn.1673-4246.2018.07.009. [DOI] [Google Scholar]
- Lichtman JS, Ferreyra JA, Ng KM, Smits SA, Sonnenburg JL, Elias JE. Host-microbiota interactions in the pathogenesis of antibiotic-associated diseases. Cell Rep. 2016;14(5):1049–1061. doi: 10.1016/j.celrep.2016.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu YJ, Xiao XY, Deng YL, Guo KX, She Y, Tan ZJ. Effect of Qiweibaizhusan combined with yeast on intestinal Lactobacillus diversity in dysbacteriotic diarrhea mice. Space Med Med Eng. 2016;29(3):175–180. doi: 10.16289/j.cnki.1002-0837.2016.03.004. [DOI] [Google Scholar]
- Long CX, He L, Guo KX, Tan ZJ, Yin KK. Effect of Qiwei Baizhu powder combined with yeast on the intestinal bacteria diversity in dysbacteriotic diarrhea. CJITWM. 2018;38(1):66–70. doi: 10.7661/j.cjim.20171207.299. [DOI] [Google Scholar]
- Oberauner L, Zachow C, Lackner S, Hogenauer C, Smolle KH, Berg G. The ignored diversity: complex bacterial communities in intensive care units revealed by 16S pyrosequencing. Sci Rep. 2013;3:1413. doi: 10.1038/srep01413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochangco HS, Gamero A, Smith IM, Christensen JE, Jespersen L, Arneborg N. In vitro investigation of Debaryomyces hansenii strains for potential probiotic properties. World J Microbiol Biotechnol. 2016;32(9):141. doi: 10.1007/s11274-016-2109-1. [DOI] [PubMed] [Google Scholar]
- Ormerod KL, Wood DL, Lachner N, Gellatly SL, Daly JN, Parsons JD, et al. Genomic characterization of the uncultured Bacteroidales family S24-7 inhabiting the guts of homeothermic animals. Microbiome. 2016;4(1):36. doi: 10.1186/s40168-016-0181-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sangster W, Hegarty JP, Schieffer KM, Wright JR, Hackman J, Toole DR, et al. Bacterial and fungal microbiota changes distinguish C. difficile infection from other forms of diarrhea: results of a prospective inpatient study. Front Microbiol. 2016;7:789. doi: 10.3389/fmicb.2016.00789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverman MA, Konnikova L, Gerber JS. Impact of antibiotics on necrotizing enterocolitis and antibiotic-associated diarrhea. Gastroenterol Clin North Am. 2017;46(1):61–76. doi: 10.1016/j.gtc.2016.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suchodolski JS, Markel ME, Garcia-Mazcorro JF, Unterer S, Heilmann RM, Dowd SE, et al. The fecal microbiome in dogs with acute diarrhea and idiopathic inflammatory bowel disease. PLoS ONE. 2012;7(12):e51907. doi: 10.1371/journal.pone.0051907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szajewska H, Canani RB, Guarino A, Hojsak I, Indrio F, Kolacek S, et al. Probiotics for the prevention of antibiotic-associated diarrhea in children. J Pediatr Gastroenterol Nutr. 2016;62(3):495–506. doi: 10.1097/MPG.0000000000001081. [DOI] [PubMed] [Google Scholar]
- Tan ZJ, Wu H, Liu FL, Cai Y, Cai GX, Zhang HL, et al. Effect of ultra-micro powder qiweibaishusan on the intestinal microbiota and enzyme activities in mice. Acta Ecol Sin. 2012;32(21):6856–6863. doi: 10.5846/stxb201109271422. [DOI] [Google Scholar]
- White JR, Nagarajan N, Pop M. Statistical methods for detecting differentially abundant features in clinical metagenomic samples. PLoS Comput Biol. 2009;5(4):e1000352. doi: 10.1371/journal.pcbi.1000352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wick EC, Sears CL. Bacteroides spp. and diarrhea. Curr Opin Infect Dis. 2010;23(5):470–474. doi: 10.1097/QCO.0b013e32833da1eb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, Zhou SN, Guo C, Tan ZJ, Cai GX, Zeng A, et al. A metagenome DNA extracting method of intestinal flora in mice for molecular diversity analysis based on PCR technology. Chin J Microecol. 2012;24(7):648–651. doi: 10.13381/j.cnki.cjm.2012.07.003. [DOI] [Google Scholar]
- Yutin N, Galperin MY. A genomic update on clostridial phylogeny: gram-negative spore formers and other misplaced clostridia. Environ Microbiol. 2013;15:2631–2641. doi: 10.1111/1462-2920.12173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeineldin M, Aldridge B, Lowe J. Dysbiosis of the fecal microbiota in feedlot cattle with hemorrhagic diarrhea. Microb Pathog. 2018;115:123–130. doi: 10.1016/j.micpath.2017.12.059. [DOI] [PubMed] [Google Scholar]
- Zhang HL, Cai Y, Tan ZJ, Zhou SN, Guo KX, She Y. Effects of ultra-micro powder Qiweibaizhu on metabolism diversity of intestinal microflora in diarrhea mice with dysbacteriosis. Chin J Appl Environ Biol. 2014;20(1):93–100. doi: 10.3724/SP.J.1145.2014.00093. [DOI] [Google Scholar]
- Zoumpourtikoudi V, Pyrgelis N, Chatzigrigoriou M, Tasakis RN, Touraki M. Interactions among yeast and probiotic bacteria enhance probiotic properties and metabolism offering augmented protection to Artemia franciscana against Vibrio anguillarum. Microb Pathog. 2018;125:497–506. doi: 10.1016/j.micpath.2018.10.022. [DOI] [PubMed] [Google Scholar]