

Summary
Activation of the hexosamine pathway (HP) through gain-of-function mutations in its rate-limiting enzyme glutamine fructose-6-phosphate amidotransferase (GFAT-1) ameliorates proteotoxicity and increases lifespan in Caenorhabditis elegans. Here, we investigate the role of the HP in mammalian protein quality control. In mouse neuronal cells, elevation of HP activity led to phosphorylation of both PERK and eIF2α as well as downstream ATF4 activation, identifying the HP as a modulator of the integrated stress response (ISR). Increasing uridine 5′-diphospho-N-acetyl-D-glucosamine (UDP-GlcNAc) levels through GFAT1 gain-of-function mutations or supplementation with the precursor GlcNAc reduces aggregation of the polyglutamine (polyQ) protein Ataxin-3. Blocking PERK signaling or autophagy suppresses this effect. In C. elegans, overexpression of gfat-1 likewise activates the ISR. Consistently, co-overexpression of gfat-1 and proteotoxic polyQ peptides in muscles reveals a strong protective cell-autonomous role of the HP. Thus, the HP has a conserved role in improving protein quality control through modulation of the ISR.
Subject Areas: Biological Sciences, Cell Biology, Functional Aspects of Cell Biology
Graphical Abstract
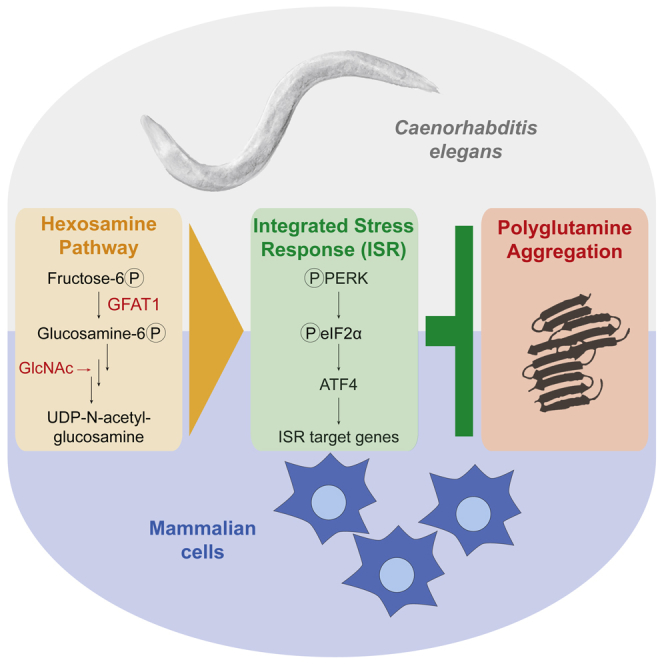
Highlights
-
•
Hexosamine pathway (HP) activation induces the integrated stress response (ISR)
-
•
HP activation ameliorates poly-glutamine aggregation via the ISR and autophagy
-
•
In C. elegans, the HP/ISR axis improves cell autonomous protein homeostasis
-
•
The proteoprotective role of longevity-associated HP is evolutionarily conserved
Biological Sciences; Cell Biology; Functional Aspects of Cell Biology
Introduction
During aging, organisms undergo a progressive decline in physiological function that results in an elevated risk for numerous diseases (Niccoli and Partridge, 2012). A limited number of conserved molecular pathways control life- and healthspan across species and represent potential entry points for treatments of age-related diseases (Kenyon, 2010). However, only few potential drug targets have so far been identified and their role in human aging remains poorly understood. Considering the burden of age-related diseases, it is paramount to identify and understand conserved mechanisms that extend healthspan and promote longevity.
Among the pathways controlling the rate of aging, the upstream signaling nodes including insulin/IGF-1 signaling and the mTOR pathway are well understood, and downstream effectors are partially known. These include the protein homeostasis network, controlling protein synthesis and degradation as key processes. With age, the fidelity of the protein homeostasis network declines (Labbadia and Morimoto, 2015) and by enhancing protein homeostasis, e.g., by boosting autophagy, lifespan can be extended in mice (Pyo et al., 2013). In turn, autophagy is required for lifespan extension in numerous paradigms ranging from caloric restriction to insulin signaling and rapamycin treatment (Madeo et al., 2015). However, there is a gap in knowledge regarding the links between upstream signaling pathways and outputs that orchestrate the coordinated and diverse changes required for extended health of an organism.
The protein homeostasis network comprises all processes that define the dynamics of the proteome: protein synthesis, protein folding, and recycling by the proteasome or through autophagy (Labbadia and Morimoto, 2015). Protein homeostasis is required under normal conditions but is particularly important to be maintained under proteotoxic stress. To detect challenges to proteome stability, cellular compartments possess specialized protein quality surveillance mechanisms that trigger protective responses upon proteotoxic stress. The endoplasmic reticulum (ER) has an unfolded protein response (UPR) with three parallel pathways that are activated by luminal misfolded proteins (UPRER). First, the IRE1 sensor is an endonuclease that specifically triggers mRNA splicing of transcription factor XBP1, permitting expression of downstream stress response genes such as the HSP4/BiP chaperone (Calfon et al., 2002). Second, activated ATF6 is cleaved upon transport to the Golgi, which enables its translocation into the nucleus to act as a transcription factor (Haze et al., 1999). Both pathways coordinate expression of genes that reinstate ER homeostasis. The third UPRER pathway is controlled by PRKR-like endoplasmic reticulum kinase (PERK), which is an ER membrane-localized stress sensor as well. Upon activation, PERK phosphorylates its sole known client, the α subunit of eukaryotic initiation factor-2 (eIF2α), which is a key regulator of protein synthesis (Harding et al., 1999). While PERK activation thus suppresses overall mRNA translation to reduce the burden on the protein folding machinery, specific transcripts that are regulated by upstream open reading frames (uORFs) become induced (Harding et al., 2000, Hinnebusch, 1993, Hinnebusch et al., 1988). These include the stress response gene activating transcription factor (ATF4), which is a key regulator of autophagy (Rzymski et al., 2009). Together, PERK phosphorylation, as an output of cellular stress, thus triggers a coordinated program termed the integrated stress response (ISR).
The protein homeostasis network is challenged not only by external stimuli such as heat shock but also by endogenous metastable proteins. Although these undergo proper protein folding or degradation under normal conditions, they tend to aggregate when the protein homeostasis network becomes insufficient. This not only leads to loss of function of the affected protein but can also result in a gain of toxic function that is often associated with aggregation (Dragatsis et al., 2000, Duyao et al., 1995, Oikawa et al., 2016). Metastable proteins include Aβ amyloid protofibrils, superoxide dismutase, and polyglutamine (polyQ) tracts that form in some proteins such as Huntingtin due to unstable CAG repeats (Harper et al., 1997, Khare et al., 2004, MacDonald et al., 1993). These aggregation-prone proteins are causally linked to Alzheimer's disease, amyotrophic lateral sclerosis, and Huntington's disease, respectively (Glenner and Wong, 1984, Hazeki et al., 1999, Rosen et al., 1993, Wang et al., 2008). Furthermore, the expansion of CAG repeats in the coding region of Ataxin-3 (ATX3) leads to its aggregation, which is causative of Machado-Joseph disease (MJD) (also known as spinocerebellar ataxia-3) (Bevivino and Loll, 2001, Kawaguchi et al., 1994). MJD is a progressive autosomal dominant neurodegenerative disease that causes cerebellar ataxia, and currently there is no effective treatment available (Coutinho and Andrade, 1978, Duarte-Silva and Maciel, 2018).
In a genetic screen for proteotoxic stress resistance in C. elegans we have previously identified the metabolic hexosamine pathway (HP) as a regulator of aging (Denzel et al., 2014). Specifically, we showed that single amino acid substitutions in the pathway's key enzyme glutamine fructose-6-phosphate amidotransferase (GFAT-1) result in gain of function and elevated cellular levels of the HP's product UDP-GlcNAc. This leads to increased activity of protein degradation processes such as ER-associated degradation, proteasome activity, and autophagy. Although these processes are induced and required for the longevity of GFAT-1 gain-of-function mutants, how UDP-GlcNAc triggers the coordinated response of the protein homeostasis network remained unknown (Denzel et al., 2014). Moreover, it was unclear if the HP has a conserved role in mammalian protein homeostasis. Here, we show that HP activation triggers an ER stress response in mammalian cells that results in a significant reduction of aggregated polyQ expanded ATX3 through PERK signaling and the ISR. Using the nematode C. elegans we demonstrate that HP activation modulates the ISR and ameliorates polyQ toxicity in a conserved cell-autonomous manner.
Results
HP activation through specific gain-of-function mutations in GFAT-1 (such as the G451E substitution) as well as GlcNAc supplementation was previously shown to increase lifespan and counter proteotoxicity in the nematode C. elegans (Denzel et al., 2014). To test the impact of HP activation on toxic protein aggregation in mammalian cells, we first established strategies to increase HP flux in mammalian systems (Figure 1A). GFAT1 is highly conserved, and we engineered the G451E point mutation in N2a cells using Crispr/Cas9 (Figure 1B). This gain-of-function substitution increases levels of the HP product UDP-GlcNAc by 4- to 5-fold in mouse N2a cells (Ruegenberg et al., 2020). Supplementation with 10 mM GlcNAc likewise increased the cellular UDP-GlcNAc concentration (Figure 1C). Notably, the two interventions were additive. Consistent with our previous work in the nematode, increased HP flux conferred resistance to the drug tunicamycin in N2a cells (Figures 1D and 1E). Tunicamycin is an inhibitor of UDP-GlcNAc:Dolichylphosphate GlcNAc-1-Phosphotransferase, which catalyzes the first step of N-glycan synthesis utilizing UDP-GlcNAc as substrate (Heifetz et al., 1979). Presumably, elevated UDP-GlcNAc levels “outcompete” tunicamycin and counter the inhibitory effect. Importantly, GlcNAc supplementation also increased UDP-GlcNAc levels in other mammalian systems including mouse primary keratinocytes and multiple human cell lines (Figures 1F and S1A). Moreover, we generated GFAT1 overexpression mice and tested HP activation in primary keratinocytes. Like GlcNAc supplementation, GFAT1 overexpression led to elevated UDP-GlcNAc levels (Figure 1F).
Figure 1.

Hexosamine Pathway Activation in Mammalian Cells
(A) Schematic representation of the hexosamine pathway (HP).
(B) Multiple sequence alignment of a segment of C. elegans GFAT-1 compared with mouse and human GFAT1 (aka GFPT1).
(C) Relative UDP-HexNAc levels (combination of the epimers UDP-GlcNAc and UDP-GalNAc) of WT and GFAT1 G451E engineered N2a cells, and both lines treated with 10 mM GlcNAc for 24 h. Mean + SEM (n ≥ 5), ***p < 0.001(ANOVA).
(D) Representative cell viability (XTT assay) of WT, GFAT1 G451E engineered N2a cells, and both lines supplemented with 10 mM GlcNAc after a 48-h treatment with tunicamycin (TM) doses as indicated.
(E) Cell viability (XTT assay) of WT, GFAT1 G451E engineered N2a cells, and both lines supplemented with 10 mM GlcNAc after a 48-h treatment with 0.5 μg/mL tunicamycin. Mean + SEM (n = 3), **p < 0.01 (ANOVA).
(F) UDP-HexNAc levels in primary keratinocytes isolated from the indicated mouse lines. Prior to sample collection, 10 mM GlcNAc treatment was performed for 24 h. Mean + SEM (n ≥ 5), **p < 0.01, ***p < 0.001 (ANOVA).
See also Figure S1.
Having established independent routes of HP activation we asked whether this activation could alleviate the aggregation of metastable proteins. To this end, we established two independent ATX3-PolyQ expression systems that carry a C-terminal fragment (amino acids 257–360) of ATX3 with a polyQ stretch. First, we assessed the amount of insoluble ATX3-polyQ71 in an inducible Tet-Off expression system in mouse N2a cells (Figure 2A). Upon activation of ATX3-polyQ71 expression by removal of doxycycline, the aggregation-prone fragment was detected in the SDS-insoluble formic acid (FA) fraction (Figure 2B). Treatment with 10 mM GlcNAc, however, fully prevented aggregation of ATX3-polyQ71, whereas supplementation of the negative control D-Arg had no effect (Figure 2B). Second, we transiently expressed tagged ATX3-PolyQ80 in N2a WT and GFAT1 G451E gain-of-function mutant cells (Figure 2C). In line with the results from the inducible system, HP activation by GlcNAc supplementation or genetic GFAT1 gain of function significantly reduced the amount of SDS-insoluble ATX3-PolyQ80 (Figures 2D and 2E). These data demonstrate a conserved role of the HP in improving protein homeostasis and counteracting protein aggregation in mammalian cells.
Figure 2.

HP Activation Ameliorates Protein Aggregation in Mammalian PolyQ Models
(A) Schematic representation of a vector expressing an aggregation-prone ATX3 fragment. A C-terminal fragment of ATX3 cDNA corresponding to amino acid residues 257–360 in the full-length protein was cloned under the control of a tetracycline responsive element (TRE) and tagged with a C-terminal c-myc epitope as described elsewhere (Haacke et al., 2006).
(B) N2a cells stably expressing a Tet-Off operator (tTA) were transiently transfected with the construct described above and treated as indicated (10 mM D-Arg, 10 mM GlcNAc, 1 μg/mL doxycycline). Cells were lysed and processed for fractionation 24 h after treatment and transfection. * Partially formic acid-insoluble material detected by the ATX3 antibody; 1, endogenous ATX3; 2, exogenous ATX3 fragment.
(C) Schematic representation of a vector expressing an aggregation-prone ATX3 fragment including an internal ribosomal entry site (IRES) followed by GFP.
(D) Fractionation of cell lysates from WT, GFAT1 G451E engineered N2a cells, and both treated with 10 mM GlcNAc 48 h after transfection with the ATX3-PolyQ construct (Figure 2C). SOL, SDS-soluble fraction; SDS, SDS wash fraction; FA, SDS-insoluble formic acid fraction.
(E) Quantification of fractionation experiments from Figure 2D. Insoluble fraction was normalized to GFP expression. Mean + SD (n ≥ 4), *p < 0.05, **p < 0.01 (ANOVA).
To understand the cellular and molecular changes that signal activation of the HP to improve protein homeostasis, we performed RNA sequencing in N2a WT and N2a GFAT1 G451E mutant cells. We focused our analysis primarily on regulatory circuits within the protein homeostasis network. Analysis of previously described ATF6, XBP1, and ATF4 target genes (https://chip-atlas.org/target_genes) revealed differential expression of UPRER target genes in GFAT1 G451E mutant cells versus WT controls, which we selectively validated by qPCR (Figure S2A). About 30% of the 177 detected ATF4 target genes were changed in the GFAT1 gain-of-function cells, significantly more than the regulated fraction of all detected genes (23% of 12,626 genes, Figure 3A). About two-thirds of the regulated ATF4 targets were significantly upregulated indicating an ISR induction (Figure S2B). Furthermore, a subset of XBP1 and ATF6 target genes were differentially regulated as well. However, the transcriptional outputs of these other two UPRER branches were slightly underrepresented compared with all regulated genes (20% of XBP1 target genes and 19% of all ATF6 target genes were regulated, Figure 3A).
Figure 3.

HP Activation Induces the Integrated Stress Response
(A) Analysis of differentially expressed genes (p value <0.05) from RNA-seq of WT and GFAT1 G451E N2a cells (n = 3). Total expression changes were compared with expression changes in target genes of the UPRER downstream transcription factors, XBP1, ATF4, and ATF6, respectively. p Values are indicated (Fisher's exact test).
(B) WT and GFAT1 G451E cells were treated with 10 mM GlcNAc for 16 h as indicated and processed for western blotting. A representative set of n ≥ 4 experiments is shown.
(C) Quantification of western blot analysis of Figure 3B. Mean + SD (n ≥ 4), *p < 0.05, **p < 0.01, ***p < 0.001 (ANOVA).
Consistent with an activation of the ISR, HP activation through GFAT1 gain of function and GlcNAc supplementation elevated phosphorylation of the ER-stress sensor PERK and the mRNA translation regulator eIF2α (Figures 3B and 3C). Accordingly, ATF4 protein levels were strongly increased, whereas mRNA levels remained largely unaffected, indicating activation of uORF-regulated translation (Figures 3B, 3C, and S2A). Notably, overall translation rates as assessed by puromycin incorporation were not altered upon any of the treatments (Figures S2C and S2D). We also investigated the status of ATF6 and XBP1 to address the other UPR branches. Upon GlcNAc supplementation or GFAT1 activation, we detected only minor or no effects regarding ATF6 activation and XBP1 splicing (Figures S2E–S2G). Differential regulation in a subset of the respective target genes might be explained by overlapping regulation of UPRER target genes. Taken together, HP activation induces the PERK/eIF2α arm of the UPRER resulting in a transcriptional response.
As HP activation triggered the ISR, we investigated whether this stress response was required for the amelioration of PolyQ aggregation. Again, we first used the ATX3-polyQ71 expression system in mouse N2a cells and found that the GlcNAc-mediated reduction of aggregation was reversed by inhibition of PERK with the specific inhibitor GSK2606414 or by blocking autophagic flux with bafilomycin A (Figures 4A and S3A). Next, we tested polyQ aggregation using both the Flag-HA tagged ATX3-polyQ80 construct containing a GFP internal control and the ATX3-polyQ71 expression system in GFAT1 G451E N2a cells during PERK and autophagy inhibition. In all these approaches, PERK inhibition and reducing autophagy restored polyQ aggregation demonstrating that the ISR-autophagy axis mediates HP-induced benefits (Figures 4A, 4B, and S3B). To rule out possible effects of GSK2606414 or bafilomycin A on the HP, we tested whether the PERK and autophagy responses were downstream of HP flux. Liquid chromatography-mass spectrometry analysis showed that UDP-GlcNAc levels in GFAT1 G451E cells remain significantly elevated after GSK2606414 or bafilomycin A treatment compared with WT N2a cells (Figure S3C).
Figure 4.

HP-Mediated Reduction of polyQ Aggregates Depends on PERK Signaling and Autophagy
(A) WT N2a cells transiently expressing the C-terminal ATX3 fragment described in Figure 2A were treated with the indicated compounds (10 mM GlcNAc, 160 nM BafA, 10 nM PERK inhibitor GSK2606414) and processed for fractionation and western blotting. * Partially formic acid soluble material; 1, endogenous ATX3; 2, exogenous ATX3 fragment.
(B) WT and GFAT1 G451E N2a cells transiently expressing the C-terminal ATX3 fragment as described in Figure 2C were treated with the indicated compounds (160 nM BafA, 10 nM PERK inhibitor GSK2606414) and processed for fractionation and western blotting.
See also Figure S3.
Having uncovered the link between HP flux and protein quality control in a mammalian system we wondered whether the beneficial effects are cell-autonomous or transmitted in a cell non-autonomous manner. Given that protein quality control mechanisms including the ISR are evolutionarily conserved across species from C. elegans to humans, we established a system to test the tissue-specific role of the HP in protein homeostasis in the nematode. First, we assessed ISR activation upon HP activation by either GFAT-1 overexpression under its endogenous promoter (Figure 5A) or by GlcNAc supplementation and found increased phosphorylation of eIF2α in both cases (Figures 5B–5E). Consistently, we found elevated ATF-5 levels (homolog of ATF4) in GFAT-1 overexpressing worms using a GFP reporter strain (Figures 5F and 5G).
Figure 5.

Increased eIF2α Phosphorylation and ATF-5 Expression by gfat-1 Overexpression in C. elegans
(A) Representative image of gfat-1 OE strain (gfat-1P::CFP::gfat-1) at late L4 larval stage with cloning scheme of gfat-1 OE construct.
(B) Representative western blot showing phospho-eIF2α levels in gfat-1P::gfat-1 OE strain relative to wild-type controls at day 1 adult stage.
(C) Quantification of western blots. Mean + SD (n = 5), *p < 0.05 (t test).
(D) Western blot showing phospho-eIF2α levels in wild-type animals treated with 10 mM GlcNAc or D-Arg control for 12 h at day 1 adult stage.
(E) Quantification of western blot in (D). Mean + SD (n = 3), *p < 0.05 (t test).
(F) Representative images of atf-5P::gfp reporter strain in gfat-1P::gfat-1 OE background and WT control at day 1 adult stage. Area marked by red dashed line indicates quantified worm area, excluded is the signal from pharyngeal co-injection marker myo-2::mcherry.
(G) Quantification of GFP expression controlled by atf-5 promoter from microscopy pictures at day 1 adult stage (Figure 4F). Mean + SEM (n = 3, 10 animals per condition and experiment), *p < 0.05 (t test).
We then generated C. elegans overexpressing GFAT-1 specifically in body wall muscle or in neurons (Figures 6A and S4A) and crossed them to Q35::YFP transgenic worms that drive the expression of metastable polyQ proteins solely in the body wall muscle (Morley et al., 2002). We found that polyQ aggregation was significantly reduced by local HP activation (Figures 6B and 6C). This was not the case when GFAT-1 was overexpressed in neurons under the rgef-1 promoter or under its endogenous promoter, which shows only minor activity in the body wall muscle (Figures 5A, 6A, and 6C). Importantly, aggregate reduction occurred without a decrease in total polyQ protein in gfat-1 transgenic animals (Figure S4B). Q35::YFP transgenic animals are well known to show a progressive paralysis phenotype that can be assessed by motility assays (Morley et al., 2002). We found that muscle overexpression of GFAT-1 preserved motility, whereas neuronal GFAT-1 overexpression did not (Figure 6D). Overexpression of GFAT-1 under its endogenous promoter slightly improved motility over control conditions, consistent with a mild induction of HP activity in muscle tissue. To test whether this beneficial effect was linked to GFAT-1 and HP activity, we used RNAi to downregulate gfat-1 and gna-2 expression. GNA-2 is the enzyme downstream of GFAT-1 in the HP. Both interventions largely prevented the motility improvements (Figures 6E and S4C–S4E). Finally, we tested if enhanced motility during GFAT-1 overexpression was dependent on atf-5. Knockdown of atf-5 indeed suppressed the motility phenotype in GFAT-1 transgenic animals (Figure 6F). Together, these findings suggest that cell autonomous HP activation provokes the ISR, reduces polyQ aggregation, and improves motility.
Figure 6.

HP Activation in C. elegans Ameliorates polyQ Toxicity in a Cell-Autonomous Manner
(A) Representative images of tissue-specific gfat-1 OE strains at late L4 larval stage with cloning schemes, muscle-specific gfat-1 OE (myo-3P::CFP::gfat-1, left panel) and pan-neuronal gfat-1 OE (rgef-1P::CFP::gfat-1, right panel).
(B) Representative images of polyQ aggregates in muscle tissue of myo-3P::CFP::gfat-1; unc-54P::Q35::YFP animals relative to controls at day 4 of adulthood. Higher magnification of indicated area in lower panel.
(C) Quantification of Q35::YFP aggregates at day 4 of adulthood from microscopy images. Mean + SD (n = 10 animals per condition) ***p < 0.001 (ANOVA).
(D) Motility assay of gfat-1 OE strains in unc-54P::Q35::YFP background at day 8 of adulthood. Dots are means of individual experiments (≥10 worms per condition). The averages (+SEM, n ≥ 3) are shown as bar graphs. **p < 0.01, ***p < 0.001 (ANOVA).
(E) Motility of control and muscle-specific gfat-1 OE in unc-54P::Q35::YFP background at day 8 of adulthood when grown on control luciferase or gfat-1 RNAi. Means of individual experiments are displayed as dots (≥10 worms per condition). The averages (+SEM, n = 3) are shown as bar graphs. ***p < 0.001 (ANOVA).
(F) Motility of control and muscle-specific gfat-1 OE in unc-54P::Q35::YFP background at day 8 of adulthood when grown on control luciferase or atf-5 RNAi. A representative experiment from two repeats is displayed (n = 30 worms), ***p < 0.001 (ANOVA).
See also Figure S4.
Discussion
Here, we examined the role of the HP in protein homeostasis in nematodes and mammalian cells. Previously, we showed that activating point mutations in GFAT-1 elevate UDP-GlcNAc levels, suppress proteotoxicity, and enhance lifespan in the nematode C. elegans. However, it remained unclear how elevated levels of the metabolite UDP-GlcNAc modulate protein homeostasis and if this function is conserved across species. Therefore, we investigated the consequences of HP activation in mammalian cells. We show that gain-of-function mutations causing specific substitutions of conserved amino acids elevate the HP product UDP-GlcNAc in mouse cells. In cultured neuronal cells, we found that SDS-insoluble polyQ-fused ATX3 was drastically reduced upon elevation of HP activity. Transcriptomics pointed toward an activation of the UPRER, and our biochemical analysis showed strong PERK phosphorylation, whereas XBP1 and ATF6 remained largely unaffected. Downstream of PERK, we noted an induction of the ISR in GFAT1 gain-of-function cells, which was required for the suppression of ATX3-polyQ aggregation. In a C. elegans model of polyQ proteotoxicity in the body wall muscle, we showed that specifically muscle-targeted overexpression of GFAT-1 suppressed toxicity indicating a cell-autonomous mode of action. These results reveal that the HP modulates the ISR to locally improve protein quality control.
Our data establish a link between the metabolic status of the HP and the ISR. It remains unclear, however, how elevated HP metabolites might modulate the key upstream ISR regulators PERK and eIF2α. UDP-GlcNAc is a precursor for O-GlcNAc modifications, but a knockdown of O-GlcNAc transferase OGT, which is essential for this posttranslational modification, did not reduce the capacity of HP activation to clear polyQ aggregates (data not shown). Previous work has shown that glucosamine treatment can lead to ER stress (Lombardi et al., 2012). Moreover, glucosamine supplementation leads to PERK activation and eIF2α phosphorylation (Kline et al., 2006). Together with our work, this suggests a mild HP-induced ER stress that results in PERK activation and eIF2α phosphorylation. Using N2a cells, we indeed observed significant downstream ATF4 activation; however, we did not observe a reduction in bulk protein synthesis or cell growth. This is consistent with a mild effect of HP activation on ER stress that does not achieve a level of full UPR activation.
ATF4 is a key uORF-regulated downstream target of the ISR. ATF4 regulated processes include amino acid metabolism, response to oxidative stress, and apoptosis. GCN4, the yeast ortholog of ATF4, was previously shown to mediate lifespan extension under ribosome subunit depletion (Steffen et al., 2008). In addition, ATF4 has been implicated in mammalian longevity as it is upregulated in distinct types of slow-aging mutant mice (Li et al., 2014, Li and Miller, 2015) and it stimulates production of the pro-survival hormone FGF21 (Salminen et al., 2017). Furthermore, ATF4 is a regulator of a number of autophagy genes (B'Chir et al., 2013, Notte et al., 2015). Consistent with an activation of autophagy downstream of the ISR, the elimination of polyQ aggregates upon HP activation was suppressed by treatment with the autophagy inhibitor bafilomycin A. Supporting this model, a previous study has demonstrated that ER stress modulates autophagy through eIF2α phosphorylation to degrade polyQ aggregates (Kouroku et al., 2007). Furthermore, enhancing eIF2α phosphorylation pharmacologically reduces SDS-insoluble Huntingtin aggregates in a mouse model of Huntington's disease (Krzyzosiak et al., 2018).
Why might enhanced metabolite flux in the HP lead to improved protein quality control? The HP is optimally positioned to reflect the availability of numerous metabolites of key energy pathways. HP flux is only maximized under conditions of sufficient energy and macronutrient availability and is then limited through UDP-GlcNAc-mediated feedback regulation of GFAT1. Protein synthesis and degradation are very energy-consuming pathways, and it is plausible that the HP might signal energy availability. ISR induction typically reduces protein synthesis, whereas this was not the case when the HP was activated. Rather, our data point to enhanced protein degradation and thus, potentially, turnover. HP activation might thus signal conditions supporting proteome maintenance under which toxic protein aggregates are degraded or fail to form toxic conformations in the first place.
Finally, to test our cellular data in vivo, we asked if HP activation can activate the ISR to counter protein aggregation in C. elegans. Previously, we had shown that activating point mutations in GFAT-1 can counter proteotoxicity by inducing protein quality control. We had not detected major transcriptomic changes in gfat-1 gain-of-function worms (unpublished data). As GFAT-1 is most strongly expressed in the pharynx and seam cells of the worm, we might not have detected a UPRER response from these tissues in whole animal lysates. Genetic gfat-1 activation rescued polyQ-mediated toxicity in neurons and in muscle tissue. It was therefore unclear if the GFAT-1 activation had cell non-autonomous effects on protein homeostasis. In the current study we show that GFAT-1 overexpression or GlcNAc treatment indeed increased eIF2α phosphorylation in the nematode, consistent with our mammalian data. Moreover, co-overexpression of polyQ peptides and GFAT-1 in muscle tissue resulted in clear improvements in motility and aggregate clearance, whereas overexpression in neurons did not protect from muscle-specific polyQ proteotoxicity, consistent with a cell autonomous effect.
In all, our data demonstrate a key conserved role of the HP in protein homeostasis through induction of the ISR. Thus, pharmacological activation of the HP might hold potential in protecting from age-related proteotoxic diseases.
Limitations of the Study
First, although we show that the HP affects the UPRER, we do not mechanistically understand how elevated UDP-GlcNAc triggers PERK phosphorylation. This might be due to mild ER stress caused by changes in N-glycosylation or through a more specific mechanism. Second, although our conclusions are based on experiments in tissue culture and in C. elegans, the question remains whether HP activation might have proteoprotective effects in higher mammals. Studies in mice will address this point. Finally, we have investigated only polyQ toxicity in this study. It will be of interest to study the HP in the context of other toxic protein species.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
We thank all M.S.D. and A.A. laboratory members for lively and helpful discussions. We thank Franziska Metge and all members of the bioinformatics core facility and the comparative biology facility at MPI AGE. We thank Thomas Klockgether (DZNE, Bonn) for helpful discussions. We thank the Caenorhabditis Genetics Center (CGC) and Dr. T. Keith Blackwell for worm strains. M.S.D., P.B, and A.A. were supported by the German Federal Ministry of Education and Research (joint research grant EndoProtect 01GQ1423A). A.A. was also supported through the Max Planck Society. M.S.D. was supported by ERC-StG 640254, by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) - Projektnummer 73111208 - SFB 829, and by the Max Planck Society.
Author Contributions
M.H., S.I.D., A.A., and M.S.D. designed the research and wrote the manuscript. M.H., S.I.D., I.S., K.A., B.S., V.K., S.M., and P.B. performed the experiments. M.H., S.I.D., B.S., and P.B. performed the data analysis.
Declaration of Interests
The authors declare no competing interests.
Published: March 27, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2020.100887.
Contributor Information
Peter Breuer, Email: peter.breuer@ukb.uni-bonn.de.
Adam Antebi, Email: antebi@age.mpg.de.
Martin S. Denzel, Email: denzel@age.mpg.de.
Data and Code Availability
Raw sequencing data were deposited to the NCBI Gene Expression Omnibus (GEO) under the accession number GSE140357.
Supplemental Information
References
- B'Chir W., Maurin A.C., Carraro V., Averous J., Jousse C., Muranishi Y., Parry L., Stepien G., Fafournoux P., Bruhat A. The eIF2alpha/ATF4 pathway is essential for stress-induced autophagy gene expression. Nucleic Acids Res. 2013;41:7683–7699. doi: 10.1093/nar/gkt563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bevivino A.E., Loll P.J. An expanded glutamine repeat destabilizes native ataxin-3 structure and mediates formation of parallel beta -fibrils. Proc. Natl. Acad. Sci. U S A. 2001;98:11955–11960. doi: 10.1073/pnas.211305198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calfon M., Zeng H., Urano F., Till J.H., Hubbard S.R., Harding H.P., Clark S.G., Ron D. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature. 2002;415:92–96. doi: 10.1038/415092a. [DOI] [PubMed] [Google Scholar]
- Coutinho P., Andrade C. Autosomal dominant system degeneration in Portuguese families of the Azores Islands. A new genetic disorder involving cerebellar, pyramidal, extrapyramidal and spinal cord motor functions. Neurology. 1978;28:703–709. doi: 10.1212/wnl.28.7.703. [DOI] [PubMed] [Google Scholar]
- Denzel M.S., Storm N.J., Gutschmidt A., Baddi R., Hinze Y., Jarosch E., Sommer T., Hoppe T., Antebi A. Hexosamine pathway metabolites enhance protein quality control and prolong life. Cell. 2014;156:1167–1178. doi: 10.1016/j.cell.2014.01.061. [DOI] [PubMed] [Google Scholar]
- Dragatsis I., Levine M.S., Zeitlin S. Inactivation of Hdh in the brain and testis results in progressive neurodegeneration and sterility in mice. Nat. Genet. 2000;26:300–306. doi: 10.1038/81593. [DOI] [PubMed] [Google Scholar]
- Duarte-Silva S., Maciel P. Pharmacological therapies for Machado-Joseph disease. Adv. Exp. Med. Biol. 2018;1049:369–394. doi: 10.1007/978-3-319-71779-1_19. [DOI] [PubMed] [Google Scholar]
- Duyao M.P., Auerbach A.B., Ryan A., Persichetti F., Barnes G.T., McNeil S.M., Ge P., Vonsattel J.P., Gusella J.F., Joyner A.L. Inactivation of the mouse Huntington's disease gene homolog Hdh. Science. 1995;269:407–410. doi: 10.1126/science.7618107. [DOI] [PubMed] [Google Scholar]
- Glenner G.G., Wong C.W. Alzheimer's disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem. Biophys. Res. Commun. 1984;120:885–890. doi: 10.1016/s0006-291x(84)80190-4. [DOI] [PubMed] [Google Scholar]
- Haacke A., Broadley S.A., Boteva R., Tzvetkov N., Hartl F.U., Breuer P. Proteolytic cleavage of polyglutamine-expanded ataxin-3 is critical for aggregation and sequestration of non-expanded ataxin-3. Hum. Mol. Genet. 2006;15:555–568. doi: 10.1093/hmg/ddi472. [DOI] [PubMed] [Google Scholar]
- Harding H.P., Novoa I., Zhang Y., Zeng H., Wek R., Schapira M., Ron D. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol. Cell. 2000;6:1099–1108. doi: 10.1016/s1097-2765(00)00108-8. [DOI] [PubMed] [Google Scholar]
- Harding H.P., Zhang Y., Ron D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature. 1999;397:271–274. doi: 10.1038/16729. [DOI] [PubMed] [Google Scholar]
- Harper J.D., Wong S.S., Lieber C.M., Lansbury P.T. Observation of metastable Abeta amyloid protofibrils by atomic force microscopy. Chem. Biol. 1997;4:119–125. doi: 10.1016/s1074-5521(97)90255-6. [DOI] [PubMed] [Google Scholar]
- Haze K., Yoshida H., Yanagi H., Yura T., Mori K. Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol. Biol. Cell. 1999;10:3787–3799. doi: 10.1091/mbc.10.11.3787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazeki N., Nakamura K., Goto J., Kanazawa I. Rapid aggregate formation of the huntingtin N-terminal fragment carrying an expanded polyglutamine tract. Biochem. Biophys. Res. Commun. 1999;256:361–366. doi: 10.1006/bbrc.1999.0337. [DOI] [PubMed] [Google Scholar]
- Heifetz A., Keenan R.W., Elbein A.D. Mechanism of action of tunicamycin on the UDP-GlcNAc:dolichyl-phosphate Glc-NAc-1-phosphate transferase. Biochemistry. 1979;18:2186–2192. doi: 10.1021/bi00578a008. [DOI] [PubMed] [Google Scholar]
- Hinnebusch A.G. Gene-specific translational control of the yeast GCN4 gene by phosphorylation of eukaryotic initiation factor 2. Mol. Microbiol. 1993;10:215–223. doi: 10.1111/j.1365-2958.1993.tb01947.x. [DOI] [PubMed] [Google Scholar]
- Hinnebusch A.G., Jackson B.M., Mueller P.P. Evidence for regulation of reinitiation in translational control of GCN4 mRNA. Proc. Natl. Acad. Sci. U S A. 1988;85:7279–7283. doi: 10.1073/pnas.85.19.7279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaguchi Y., Okamoto T., Taniwaki M., Aizawa M., Inoue M., Katayama S., Kawakami H., Nakamura S., Nishimura M., Akiguchi I. CAG expansions in a novel gene for Machado-Joseph disease at chromosome 14q32.1. Nat. Genet. 1994;8:221–228. doi: 10.1038/ng1194-221. [DOI] [PubMed] [Google Scholar]
- Kenyon C.J. The genetics of ageing. Nature. 2010;464:504–512. doi: 10.1038/nature08980. [DOI] [PubMed] [Google Scholar]
- Khare S.D., Caplow M., Dokholyan N.V. The rate and equilibrium constants for a multistep reaction sequence for the aggregation of superoxide dismutase in amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. U S A. 2004;101:15094–15099. doi: 10.1073/pnas.0406650101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kline C.L., Schrufer T.L., Jefferson L.S., Kimball S.R. Glucosamine-induced phosphorylation of the alpha-subunit of eukaryotic initiation factor 2 is mediated by the protein kinase R-like endoplasmic-reticulum associated kinase. Int. J. Biochem. Cell Biol. 2006;38:1004–1014. doi: 10.1016/j.biocel.2005.10.015. [DOI] [PubMed] [Google Scholar]
- Kouroku Y., Fujita E., Tanida I., Ueno T., Isoai A., Kumagai H., Ogawa S., Kaufman R.J., Kominami E., Momoi T. ER stress (PERK/eIF2alpha phosphorylation) mediates the polyglutamine-induced LC3 conversion, an essential step for autophagy formation. Cell Death Differ. 2007;14:230–239. doi: 10.1038/sj.cdd.4401984. [DOI] [PubMed] [Google Scholar]
- Krzyzosiak A., Sigurdardottir A., Luh L., Carrara M., Das I., Schneider K., Bertolotti A. Target-based discovery of an inhibitor of the regulatory phosphatase PPP1R15B. Cell. 2018;174:1216–1228.e9. doi: 10.1016/j.cell.2018.06.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labbadia J., Morimoto R.I. The biology of proteostasis in aging and disease. Annu. Rev. Biochem. 2015;84:435–464. doi: 10.1146/annurev-biochem-060614-033955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W., Li X., Miller R.A. ATF4 activity: a common feature shared by many kinds of slow-aging mice. Aging Cell. 2014;13:1012–1018. doi: 10.1111/acel.12264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W., Miller R.A. Elevated ATF4 function in fibroblasts and liver of slow-aging mutant mice. J. Gerontol. A Biol. Sci. Med. Sci. 2015;70:263–272. doi: 10.1093/gerona/glu040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lombardi A., Ulianich L., Treglia A.S., Nigro C., Parrillo L., Lofrumento D.D., Nicolardi G., Garbi C., Beguinot F., Miele C. Increased hexosamine biosynthetic pathway flux dedifferentiates INS-1E cells and murine islets by an extracellular signal-regulated kinase (ERK)1/2-mediated signal transmission pathway. Diabetologia. 2012;55:141–153. doi: 10.1007/s00125-011-2315-1. [DOI] [PubMed] [Google Scholar]
- MacDonald M.E., Ambrose C.M., Duyao M.P., Myers R.H., Srinidhi C.L.L., Barnes G., Taylor S.A., James M., Groot N., MacFarlane H. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. The Huntington's Disease Collaborative Research Group. Cell. 1993;72:971–983. doi: 10.1016/0092-8674(93)90585-e. [DOI] [PubMed] [Google Scholar]
- Madeo F., Zimmermann A., Maiuri M.C., Kroemer G. Essential role for autophagy in life span extension. J. Clin. Invest. 2015;125:85–93. doi: 10.1172/JCI73946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morley J.F., Brignull H.R., Weyers J.J., Morimoto R.I. The threshold for polyglutamine-expansion protein aggregation and cellular toxicity is dynamic and influenced by aging in Caenorhabditis elegans. Proc. Natl. Acad. Sci. U S A. 2002;99:10417–10422. doi: 10.1073/pnas.152161099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niccoli T., Partridge L. Ageing as a risk factor for disease. Curr. Biol. 2012;22:R741–R752. doi: 10.1016/j.cub.2012.07.024. [DOI] [PubMed] [Google Scholar]
- Notte A., Rebucci M., Fransolet M., Roegiers E., Genin M., Tellier C., Watillon K., Fattaccioli A., Arnould T., Michiels C. Taxol-induced unfolded protein response activation in breast cancer cells exposed to hypoxia: ATF4 activation regulates autophagy and inhibits apoptosis. Int. J. Biochem. Cell Biol. 2015;62:1–14. doi: 10.1016/j.biocel.2015.02.010. [DOI] [PubMed] [Google Scholar]
- Oikawa T., Nonaka T., Terada M., Tamaoka A., Hisanaga S., Hasegawa M. alpha-synuclein fibrils exhibit gain of toxic function, promoting tau aggregation and inhibiting microtubule assembly. J. Biol. Chem. 2016;291:15046–15056. doi: 10.1074/jbc.M116.736355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyo J.O., Yoo S.M., Ahn H.H., Nah J., Hong S.H., Kam T.I., Jung S., Jung Y.K. Overexpression of Atg5 in mice activates autophagy and extends lifespan. Nat. Commun. 2013;4:2300. doi: 10.1038/ncomms3300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen D.R., Siddique T., Patterson D., Figlewicz D.A., Sapp P., Hentati A., Donaldson D., Goto J., O'Regan J.P., Deng H.X. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362:59–62. doi: 10.1038/362059a0. [DOI] [PubMed] [Google Scholar]
- Ruegenberg S., Horn M., Pichlo C., Allmeroth K., Baumann U., Denzel M.S. Loss of GFAT-1 feedback regulation activates the hexosamine pathway that modulates protein homeostasis. Nat. Commun. 2020;11:687. doi: 10.1038/s41467-020-14524-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rzymski T., Milani M., Singleton D.C., Harris A.L. Role of ATF4 in regulation of autophagy and resistance to drugs and hypoxia. Cell Cycle. 2009;8:3838–3847. doi: 10.4161/cc.8.23.10086. [DOI] [PubMed] [Google Scholar]
- Salminen A., Kaarniranta K., Kauppinen A. Integrated stress response stimulates FGF21 expression: Systemic enhancer of longevity. Cell Signal. 2017;40:10–21. doi: 10.1016/j.cellsig.2017.08.009. [DOI] [PubMed] [Google Scholar]
- Steffen K.K., MacKay V.L., Kerr E.O., Tsuchiya M., Hu D., Fox L.A., Dang N., Johnston E.D., Oakes J.A., Tchao B.N. Yeast life span extension by depletion of 60s ribosomal subunits is mediated by Gcn4. Cell. 2008;133:292–302. doi: 10.1016/j.cell.2008.02.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C.E., Tydlacka S., Orr A.L., Yang S.H., Graham R.K., Hayden M.R., Li S., Chan A.W., Li X.J. Accumulation of N-terminal mutant huntingtin in mouse and monkey models implicated as a pathogenic mechanism in Huntington's disease. Hum. Mol. Genet. 2008;17:2738–2751. doi: 10.1093/hmg/ddn175. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw sequencing data were deposited to the NCBI Gene Expression Omnibus (GEO) under the accession number GSE140357.