

Abstract
G-protein-coupled receptors (GPCRs) are seven transmembrane spanning receptors that regulate a wide array of intracellular signaling cascades in response to various stimuli. To do so, they couple to different heterotrimeric G proteins and adaptor proteins, including arrestins. Importantly, arrestins were shown to regulate GPCR signaling through G proteins, as well as promote G protein-independent signaling events. Several research groups have reported successful isolation of exclusively G protein-dependent and arrestin-dependent signaling downstream of GPCR activation using biased agonists or receptor mutants incapable of coupling to either arrestins or G proteins. In the latter category, the DRY mutant of the angiotensin II type 1 receptor was extensively used to characterize the functional selectivity downstream of AT1AR. In an attempt to understand histamine 1 receptor signaling, we characterized the signaling capacity of the H1R DRY mutant in a panel of dynamic, live cell biosensor assays, including arrestin recruitment, heterotrimeric G protein activation, Ca2+ signaling, protein kinase C activity, GTP binding of RhoA, and activation of ERK1/2. Here, we show that both H1R DRY mutant and the AT1AR DRY mutant are capable of efficient activation of G protein-mediated signaling. Therefore, contrary to the common belief, they do not constitute suitable tools for the dissection of the arrestin-mediated, G protein-independent signaling downstream of these receptors.
Introduction
G protein-coupled receptors (GPCRs) constitute the largest family of cell surface proteins involved in signaling in response to various stimuli that underlie many cellular and physiological processes. Agonist binding to GPCRs evokes rearrangements in the intramolecular interactions within the seven transmembrane domain structure of the receptor that result in receptor activation and its coupling to heterotrimeric (Gαßγ) G proteins.1 This leads to G protein activation and switching of the canonical GPCR signaling via second messengers. Subsequent cessation of this G protein-dependent signaling occurs via the recruitment of arrestins to the cytoplasmic surface of the receptor, a process that is enhanced by receptor phosphorylation by G protein-coupled receptor kinases, GRKs.2,3 Of four arrestin isoforms, arrestin1 and 4 bind photoreceptors in the retina, whereas two nonvisual arrestins (arrestin2 and 3 or ß-arrestin1 and 2, respectively) bind virtually all other GPCRs. Arrestin binding physically prevents the receptor–G protein interaction, leading to desensitization of the receptor-mediated activation of G proteins, and promotes the subsequent receptor endocytosis via clathrin-coated vesicles4−6
In addition to desensitization, arrestin binding to the receptor was proposed to “switch” the receptor from the G protein signaling mode that transmits transient signals from the plasma membrane to an arrestin signaling mode that transmits a distinct set of signals as the receptor internalizes.7,8 Arrestin-mediated signaling downstream of GPCR activation is arguably the most thoroughly studied in the case of mitogen-activated, extracellular signal-regulated kinase 1 and 2 (ERK1/2) cascades. In fact, several research groups have reported the successful “isolation” of G protein-independent ß-arrestin-mediated signaling downstream of GPCR activation using the so-called ß-arrestin-biased agonists, i.e., compounds that do not support receptor coupling to G proteins.9−13 Similarly, receptor mutants incapable of G protein coupling have been used to isolate G protein-independent ß-arrestin effects.14−16
Uncoupling from G protein activation was achieved for several receptors by mutating the highly conserved DRY motif found in all rhodopsin/family A GPCRs.17−19 The DRY motif, located at the cytoplasmic end of the third transmembrane helix (TM3), participates in an ionic lock with Glu in TM6 to stabilize the inactive conformation, as separation of the cytoplasmic parts of TM3 and TM6 is required for GPCR activation.1,20,21 Interestingly, the prevention of this movement selectively abrogated G protein activation by the parathyroid hormone receptor22,23 but not GRK2-mediated receptor phosphorylation or ß-arrestin recruitment. Functional selectivity proposes that the receptor can adopt multiple conformations upon ligand binding, which in turn facilitate a selective activation of either G protein or ß-arrestin-dependent signaling pathways.12,13,24,25 In this view, charge neutralizing mutations within the DRY motif would apparently result in the G protein-uncoupled receptor, which is, however, still capable of ß-arrestin binding and supporting ß-arrestin-mediated signaling.
In our attempt to elucidate the possibility of functional selectivity downstream of the histamine 1 receptor (H1R), we engineered and characterized a H1R DRY mutant with the conserved Asp and Arg residues of the DRY motif simultaneously replaced with Ala residues. As the DRY to AAY mutation that supposedly uncouples GPCR from G protein-mediated signaling was first described for the angiotensin II type 1 receptor (AT1AR), we included the AT1AR DRY mutant in our analyses. We evaluated the subcellular localization and signaling capacity of both DRY mutants using different fluorescence resonance energy-transfer (FRET)-based live cell assays. A detailed comparison of the signaling dynamics downstream of the receptor was carried out. Our study sheds new light on the use of DRY mutants for studying G protein-independent signaling.
Results
Subcellular Localization of WT and DRY Mutants of H1R and AT1AR
First, we evaluated the subcellular localization of H1R DRY and AT1AR DRY mutants in living cells using confocal microscopy. To this end, H1R and AT1AR sequences were fused at their C-terminus with a red fluorescent protein (RFP), mCherry (mCh), whereas H1R DRY and AT1AR DRY sequences were fused similarly to a cyan fluorescent protein, mTurquoise2 (mTQ2). H1R DRY-mTQ2 and AT1AR DRY-mTQ2 were coexpressed with the plasma membrane marker (Lck-mVenus) and, respectively, H1R-mCh or AT1AR-mCh in the human embryonic kidney (HEK) 293TN cells. Both H1R DRY and AT1AR DRY mutants showed largely overlapping subcellular localization with the respective WT receptors (Pearson’s coefficients were 0.62 and 0.80 for H1 receptors and AT1A receptors, respectively): they were located at the plasma membrane as well as in the intracellular (presumably endocytic) compartments. Plasma membrane localization of WT and (to a lesser extent) DRY mutants of H1R (Figure 1A shows a representative cell), as well as AT1AR and AT1AR DRY (Figure 1B shows a representative cell), was confirmed in the colocalization analysis with the plasma membrane marker. The localization of WT and DRY mutants of H1R in the endosomal compartment was examined by the colocalization analysis with the endosomal marker, Rab7.26,27 To this end, H1R-mCh or H1R DRY-mCh were coexpressed with Lck-mVenus and mTQ2-Rab7 in HeLa cells. Colocalization with Rab7, indicating endosomal localization, was more pronounced in the case of H1R DRY receptor (Pearson’s coefficient of 0.67) than that of the WT H1R (Pearson’s coefficient of 0.45; Figure 1C,D shows the representative cells).
Figure 1.
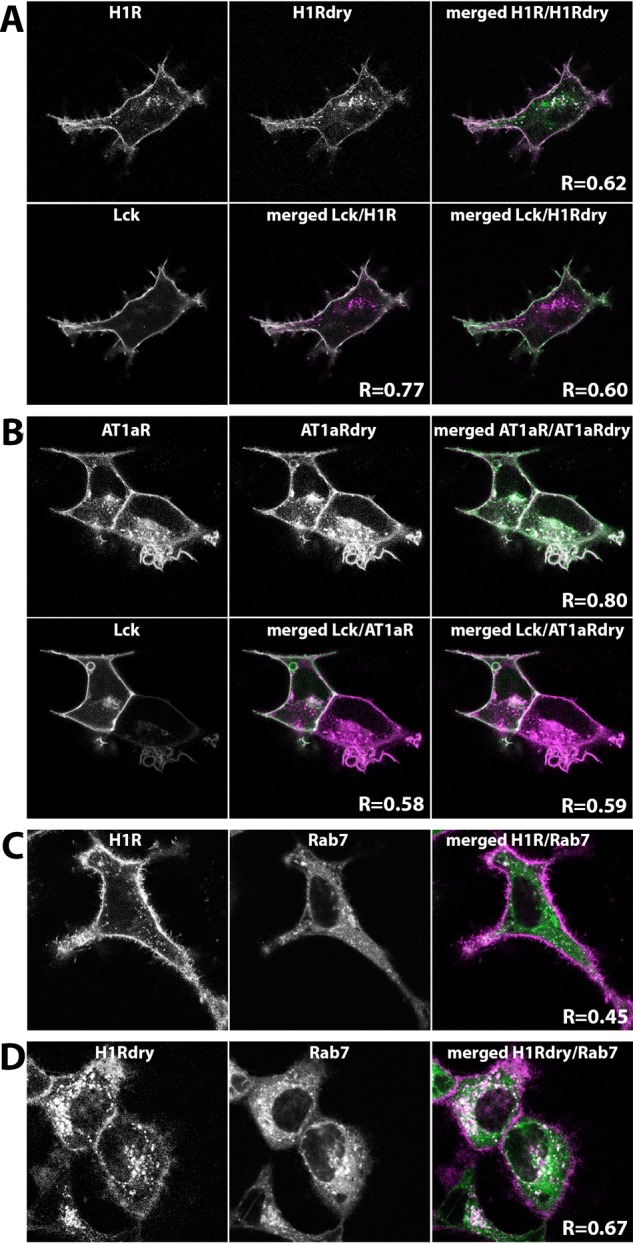
Cellular localization of wild-type (WT) and DRY mutants of H1R and AT1AR in HEK293TN cells. (A) Confocal images depicting the subcellular localization of WT H1R-mCh, H1R DRY-mTQ2, and Lck-mVenus coexpressed in HEK293TN cells. Upper panel (from left to right): H1R-mCh localization; H1R Dry-mTQ2 localization; the merged image of H1R-mCh (set to magenta) and H1R DRY-mTQ2 (set to green) localizations, where the shared localization is depicted in white. Colocalization of WT H1R and H1R DRY is quantified as Pearson’s coefficient (R). Lower panel (from left to right): localization of plasma membrane marker, Lck-mVenus; the merged image of H1R-mCh (set to magenta) and Lck-mVenus (set to green) localizations; the merged image of H1R DRY-mTQ2 (set to magenta) and Lck-mVenus (set to green) localizations. Colocalization of WT H1R or H1R DRY with the plasma membrane marker is quantified as Pearson’s coefficient (R). The size of the images is 60 μm × 60 μm. (B) Confocal images depicting the subcellular localization of WT AT1AR-mCh, AT1AR DRY-mTQ2, and Lck-mVenus coexpressed in HEK293TN cells. Upper panel (from left to right): AT1AR-mCh localization; AT1AR DRY-mTQ2 localization; the merged image of AT1AR-mCh (set to magenta) and AT1AR DRY-mTQ2 (set to green) localizations, where the shared localization is depicted in white. Colocalization of WT AT1AR and AT1AR DRY is quantified as Pearson’s coefficient (R). Lower panel (from left to right): localization of Lck-mVenus; the merged image of AT1AR-mCh (set to magenta) and Lck-mVenus (set to green) localizations; the merged image of AT1AR DRY-mTQ2 (set to magenta) and Lck-mVenus (set to green) localizations. Colocalization of WT AT1AR or AT1AR DRY with the plasma membrane marker is quantified as Pearson’s coefficient (R). The size of the images is 60 μm × 60 μm. (C) Confocal images depicting the subcellular localization of H1R-mCh (left) and endosomal marker, mTQ2-Rab7 (middle), coexpressed in HeLa cells. The right panel shows the merged image of H1R-mCh (set to magenta) and mTQ2-Rab7 (set to green) localizations, where the shared localization is depicted in white. Colocalization of WT H1R with the endosomal marker is quantified as Pearson’s coefficient (R). The size of the images is 60 μm × 60 μm. (D) Confocal images depicting the subcellular localization of H1R DRY-mCh (left) and mTQ2-Rab7 (middle) coexpressed in HeLa cells. The right panel shows the merged image of H1R DRY-mCh (set to magenta) and mTQ2-Rab7 (set to green) localizations, where the shared localization is depicted in white. Colocalization of H1R DRY with the endosomal marker is quantified as Pearson’s coefficient (R). The size of the images is 60 μm × 60 μm.
β-Arrestin Recruitment to WT and DRY Mutants of H1R and AT1AR
To examine whether the DRY mutants are capable of recruiting ß-arrestins, we coexpressed WT or mutant receptors together with Lck-mVenus and ß-arr1 or ß-arr2 fused at their C-terminus with mTQ2 in HEK293TN cells. In agreement with the previous reports,28 we observed a uniform cytosolic localization of both ß-arrestin fusions, with ßarr1-mTQ2 but not ßarr2-mTQ2, showing additional nuclear localization (Figure 2). We observed rapid relocation of both isoforms to the cell periphery upon stimulation of H1R-expressing cells with 100 μM histamine and weak ß-arrestin relocation in H1R DRY-expressing cells (Figure 2A–D). For quantification of the recruitment, see Figure 2E. Recruitment was visible as the histamine-induced localization of ß-arrestin in discrete puncta at and near the plasma membrane (as indicated by their colocalization with Lck-mVenus; Figure 2F,G) and was fully reversed upon addition of a H1R-specific antagonist, pyrilamine (PY) (see Supporting Information Movies S1–S4). In the case of both WT and DRY mutants of AT1AR, stimulation with 1 μM angiotensin II (AngII) resulted in very robust and rapid ß-arr1 and ß-arr2 relocations to the plasma membrane (as indicated by their colocalization with Lck-mVenus; see Pearson’s coefficients in Figure 3) and subsequently to an endosomal compartment (Figure 3A–G; see Supporting Information Movies S5–S8), in agreement with the previous reports.17,18,28,29
Figure 2.

ß-arrestin recruitment upon activation of WT and DRY mutants of H1R in HEK293TN cells. (A–D) Confocal images depicting the subcellular localization of WT H1R-mCh (A, B, upper panels) or H1R DRY-mCh (C, D, upper panels) coexpressed together with ßarr1-mTQ2 (A, C, lower panels) or ßarr2-mTQ2 (B, D, lower panels) in HEK293TN cells. Subcellular localization before (left) and after (right; 125 s after for the WT and at 250 s after for the DRY mutant) stimulation with 100 μM histamine. Arrowheads point to the histamine-induced localization of ß-arrestin at or near the plasma membrane. The size of the images is 60 μm × 60 μm. (E) Dotplot showing histamine-induced relocation of ß-arrestin to the plasma membrane. WT H1R-mCh or H1R DRY-mCh was coexpressed together with ßarr1-mTQ2 or ßarr2-mTQ2 in HEK293TN cells. The cytosolic fraction of ß-arrestin was measured at 250 s after stimulation with 100 μM histamine and normalized to the total ß-arrestin content prior to stimulation; centerlines show the median. (F) Line plot showing colocalization of ß-arrestin1 and H1R at the plasma membrane. Fluorescence intensity of ß-arrestin1 (blue trace), Lck (black trace), and H1R (red trace) along the line shown in the insets. The line was drawn through the ß-arrestin puncta indicated with the arrowhead in panel (A). (G) Line plot showing colocalization of ß-arrestin2 and H1R at the plasma membrane. Fluorescence intensity of ß-arrestin2 (blue trace), Lck (black trace), and H1R (red trace) along the line shown in the insets. The line was drawn through the ß-arrestin puncta indicated with the arrowhead in panel (B).
Figure 3.

ß-arrestin recruitment upon activation of WT and DRY mutants of AT1AR in HEK293TN cells. (A, B) Confocal images depicting subcellular localization of WT AT1AR-mCh (upper panels) coexpressed together with ßarr1-mTQ2 (A, lower panel) or ßarr2-mTQ2 (B, lower panel) in HEK293TN cells. From left to right: subcellular localization before, 125 s after, and 500 s after stimulation with 1 μM AngII. The last (right) panel: the merged image of AT1AR-mCh (set to magenta) and ßarr1-mTQ2 (A) or ßarr2-mTQ2 (B) (set to green) localization 125 and 500 s after AngII stimulation, where the shared localization is depicted in white. Pearson’s coefficient (R) of colocalization between ß-arrestin and Lck was calculated for each of the three time points. Arrowheads point to the AngII-induced localization of ß-arrestin at or near the plasma membrane (A) or in the intracellular compartment (B). The size of the images is 60 μm × 60 μm. (C, D) Confocal images depicting subcellular localization of AT1AR DRY-mCh (upper panels) coexpressed together with ßarr1-mTQ2 (C, lower panel) or ßarr2-mTQ2 (D, lower panel) in HEK293TN cells. From left to right: subcellular localization before, 125 s after, and 500 s after stimulation with 1 μM AngII. The last (right) panel: the merged image of AT1AR DRY-mCh (set to magenta) and ßarr1-mTQ2 (C) or ßarr2-mTQ2 (D) (set to green) localization 125 and 500 s after AngII stimulation, where the shared localization is depicted in white. The size of the images is 60 μm × 60 μm. (E) Dotplot showing AngII-induced relocation of ß-arrestin to the plasma membrane. WT AT1AR-mCh or AT1AR DRY-mCh was coexpressed together with ßarr1-mTQ2 or ßarr2-mTQ2 in HEK293TN cells. The cytosolic fraction of ß-arrestin was measured at 250 s after stimulation with 1 μM AngII and normalized to the total ß-arrestin content prior to stimulation; centerlines show the median. (F) Line plot showing colocalization of ß-arrestin1 and AT1AR at the plasma membrane. Fluorescence intensity of ß-arrestin (blue trace), AT1AR (red trace), and Lck (black trace) was measured along the line shown in the inset. The line was drawn through the ß-arrestin puncta indicated with the arrowhead in panel (A). (G) Line plot showing colocalization of ß-arrestin1 and AT1AR in the endosomal compartment. Fluorescence intensity of ß-arrestin (blue trace) and AT1AR (red trace) was measured along the line shown in the inset. The line was drawn through the ß-arrestin puncta indicated with the arrowhead in panel (B).
Gq Activation by WT and DRY Mutants of H1R and AT1AR
Having established correct localization and the capacity to recruit β-arrestins for both the WT and the DRY mutants, we characterized the functional activity of H1R DRY and AT1AR DRY mutants in a subset of signaling pathways. Both H1R and AT1AR can couple to Gq/11 and Gi proteins.30−34 The coupling of WT and DRY mutants to Gq proteins was evaluated using a FRET reporter for Gq activation.35,36 Stimulation of HEK293TN cells expressing only the reporter with 100 μM histamine or 1 μM AngII did not result in any detectable FRET ratio change (Figure 4A; see Supporting Information Figure S1A for individual measurements of all cells). On the contrary, the robust agonist-induced FRET signal was measured in cells coexpressing H1R-p2A-mCh (see Materials and Methods) or AT1AR-p2A-mCh together with the Gq reporter. Surprisingly, also the stimulation of cells expressing H1R DRY-p2A-mCh and AT1AR DRY-p2A-mCh resulted in reproducible FRET signals, although they were much reduced in comparison to the responses mediated by the respective WT receptors: H1R DRY- and AT1AR DRY-mediated FRET signals reached, respectively, 12 and 17% of the WT response. A limitation of this assay is that it requires overexpression of the Gq heterotrimer, i.e., Gαq-mTQ, Gß1, and YFP-Gγ2. Still, our results demonstrate that the DRY mutants have a guanine exchange factor activity toward a heterotrimeric G protein.
Figure 4.

Signaling activity of WT and DRY mutants of H1R and AT1AR in HEK293TN cells. (A) Gq activation by H1R, H1R DRY, AT1AR, and AT1AR DRY receptors. Time traces show the average ratio change of yellow fluorescent protein (YFP)/cyan fluorescent protein (CFP) fluorescence (and 95% confidence interval as a ribbon) in cells expressing the Gq sensor alone [N = 20 for histamine, N = 31 for AngII] or coexpressing the sensor together with H1R-p2A-mCh [N = 43], H1R DRY-p2A-mCh [N = 66], AT1AR-p2A-mCh [N = 27], or AT1AR DRY-p2A-mCh [N = 51]. Left: 100 μM histamine was added at 38 s and 10 μM pyrilamine (PY) at 238 s; right: 1 μM AngII was added at 38 s. Gray boxes mark the duration of, respectively, histamine or AngII stimulation. (B) Ca2+ changes downstream of H1R, H1R DRY, AT1AR, and AT1AR DRY receptors. Time traces show the average ratio change of YFP/CFP fluorescence (and 95% confidence interval as a ribbon) in cells expressing the YC3.6 sensor alone [N = 57 for histamine, N = 24 for AngII] or coexpressing the sensor together with H1R-p2A-mCh [N = 45], H1R DRY-p2A-mCh [N = 20], AT1AR-p2A-mCh [N = 31], or AT1AR DRY-p2A-mCh [N = 39]. Left: 100 μM histamine was added at 38 s and 10 μM PY at 238 s; right: 1 μM AngII was added at 38 s. Gray boxes mark the duration of, respectively, histamine or AngII stimulation. (C) Protein kinase C (PKC) activation downstream of H1R, H1R DRY, AT1AR, and AT1AR DRY receptors. Time traces show the average ratio change of YFP/CFP fluorescence (and 95% confidence interval as a ribbon) in cells expressing the CKAR sensor alone [N = 26 for histamine, N = 39 for AngII] or coexpressing the sensor together with H1R-p2A-mCh [N = 63], H1R DRY-p2A-mCh [N = 44], AT1AR-p2A-mCh [N = 27], or AT1AR DRY-p2A-mCh [N = 50] and untreated or treated with FR900359 [N = 34 for H1R, N = 32 for H1R DRY, N = 20 for AT1AR, N = 27 for AT1AR DRY]. Histamine (100 μM) (upper panel) or 1 μM AngII (lower panel) was added at 38 s, and 100 nM phorbol myristate acetate (PMA) (a potent PKC activator) was added at 338 s. Gray boxes mark the duration of, respectively, histamine or AngII stimulation. The observed PKC activation was abolished with a specific PKC inhibitor (10 μM Ro31-8425; N = 15 for histamine, N = 20 for AngII). Note: the continuous increase of YFP/CFP fluorescence observed in all time traces results from donor photobleaching. (D) RhoA activation downstream of H1R, H1R DRY, AT1AR, and AT1AR DRY receptors. Time traces show the average ratio change of YFP/CFP fluorescence (± and 95% confidence interval as a ribbon) in cells expressing the DORA-RhoA sensor alone [N = 17 for histamine, N = 16 for AngII] or coexpressing the sensor together with H1R-p2A-mCh [N = 36], H1R DRY-p2A-mCh [N = 16], AT1AR-p2A-mCh [N = 26], or AT1AR-p2A-mCh [N = 27]. Left: 100 μM histamine was added at 38 s and 10 μM PY at 238 s; right: 1 μM AngII was added at 38 s. Gray boxes mark the duration of, respectively, histamine or AngII stimulation. (E) Gi1 activation by H1R, H1R DRY, AT1AR, and AT1AR DRY receptors. Time traces show the average ratio change of YFP/CFP fluorescence (and 95% confidence interval as a ribbon) in cells expressing the Gi sensor alone [N = 14 for histamine, N = 19 for AngII] or coexpressing the sensor together with H1R-p2A-mCh [N = 43], H1R DRY-p2A-mCh [N = 39], AT1AR-p2A-mCh [N = 15], or AT1AR DRY-p2A-mCh [N = 44]. Left: 100 μM histamine was added at 38 s and 10 μM PY at 238 s; right: 1 μM AngII was added at 38 s. Gray boxes mark the duration of, respectively, histamine or AngII stimulation.
Calcium and PKC Signaling Downstream of WT and DRY Mutants of H1R and AT1AR
Gq/11 protein-mediated activation of phospholipase Cß results in the triggering of inositol triphosphate signaling and Ca2+-dependent protein kinase C (PKC) pathway. To evaluate the Gq coupling of the DRY mutants in the absence of the Gq protein overexpression, we subsequently employed FRET reporters for Ca2+- and PKC-dependent phosphorylation. Calcium signaling was monitored with the Yellow Chameleon 3.60 (YC3.6) FRET reporter (Figure 4B; Supporting Information Figure S1B). The stimulation of HEK293TN cells coexpressing the reporter and WT H1R or AT1AR with appropriate agonists resulted in an immediate sharp increase of intracellular Ca2+ levels that subsequently decreased in a biphasic mode: the initial fast drop (to approximately 50–60% of the maximum) within 60 s of stimulation followed by a much slower decrease. Stimulation of cells coexpressing the reporter and AT1AR DRY mutant receptors with 1 μM AngII resulted in a similarly sharp increase of intracellular Ca2+ levels, although the response had lower amplitude and was more transient (returned to baseline within 60 s of the stimulation).
The histamine-induced YC3.6 signal in cells coexpressing the reporter and H1R DRY receptors was also much reduced in comparison to the H1R-mediated response and showed more oscillatory character (see Supporting Information Figure S1B). To quantify the YC3.6 signals, we integrated all values after the addition of the agonist and divided them by the number of cells measured: H1R DRY- and AT1AR DRY-mediated YC3.6 signals were, respectively, 21 and 17% of the WT response. We occasionally measured Ca2+ increases in the absence of overexpressed receptors in cells. Of 57 cells, only 9 responded to 100 μM histamine, and of 24 cells, 7 cells responded to 1 μM AngII (Supporting Information Figure S1B). These sporadic YC3.6 signals could result from the activation of endogenous H1R, H2R, or AT1AR (presumably) expressed in HEK293TN cells.32 However, the YC3.6 signals measured in cells coexpressing the YC3.6 sensor and H1R DRY or AT1AR DRY mutant receptors were clearly different from these endogenous responses, both in kinetics and the percentage of responsive cells (>95%).
Next, the PKC activation was studied with the CKAR FRET reporter. We observed robust agonist-induced FRET ratio changes in cells coexpressing H1R or AT1AR together with CKAR but not in cells expressing only the reporter (Figure 4C; Supporting Information Figure S1C). We have confirmed that the observed PKC activity was Gq-protein-mediated, as the CKAR signal could be abolished with a Gαq-specific inhibitor, FR900359.37 The CKAR signal was similarly abolished using a PKC-specific inhibitor, Ro31-8425. Using this reporter, we confirmed the ability of H1R DRY and AT1AR DRY to couple to Gq proteins: the peak of activity reached approximately 50% (for H1R DRY) or 60% (for AT1AR DRY) of the WT-receptor-mediated response and returned faster to the baseline values (Figure 4C).
Together, these results point to the activation of Gq signaling by DRY mutants of the H1R and AT1AR, albeit at reduced levels compared to their WT counterparts.
RhoA Signaling Downstream of WT and DRY Mutants of H1R and AT1AR
As both H1 and AT1A receptors can activate Rho GTPase, RhoA, via the G-protein (Gq/11 for H1R, G12/13 for AT1AR)-dependent pathway,33,38 we characterized the capacity of WT and DRY mutants for RhoA activation using a DORA-RhoA FRET sensor.38 A limitation of this assay is that it requires the overexpression of the RhoA biosensor, resulting in elevated levels of RhoA. The stimulation of cells expressing only the reporter with 100 μM histamine or 1 μM AngII did not result in any detectable FRET ratio change (Figure 4D; Supporting Information Figure S1D). On the contrary, the robust agonist-induced FRET signal was measured in cells coexpressing H1R or AT1AR together with the reporter. The stimulation of cells coexpressing the reporter and H1R DRY or AT1AR DRY receptors also resulted in FRET ratio changes, although these FRET signals were much weaker than those mediated by the respective WT receptors (decreased to 8 and 5% for the H1R DRY and AT1AR DRY receptors, respectively). These results are in line with the aforementioned activation of the classical Gq effectors, calcium and PKC, downstream of H1R DRY or AT1AR DRY.
Gi Activation by WT and DRY Mutants of AT1AR and H1R
Additionally, we evaluated the ability of H1R DRY and AT1AR DRY receptors to activate Gi protein using a FRET sensor for Gi1 activation.31 The stimulation of cells expressing only the Gi reporter with 100 μM histamine or 1 μM AngII did not result in any detectable FRET ratio change, whereas the robust agonist-induced FRET ratio change was measured in cells coexpressing the receptor (H1R or AT1AR) together with the reporter (Figure 4E; Supporting Information Figure S1E). On the contrary, the stimulation of cells expressing H1R DRY and AT1AR DRY mutant receptors did not result in detectable FRET ratio changes. Similar to the analysis using the Gq sensor, the activity of the DRY mutants on the Gi1 protein required overexpression of the Gi heterotrimer.
ERK Activation by WT and DRY Mutants of H1R and AT1AR
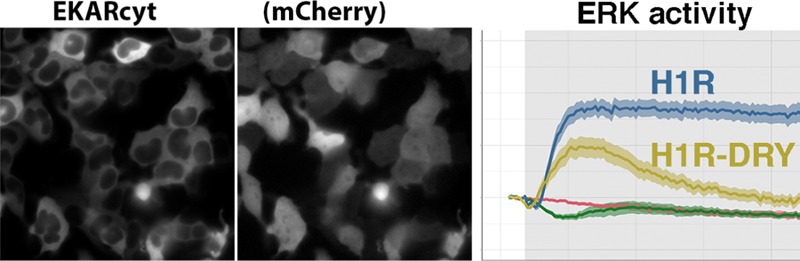
Finally, we characterized the ability of H1R DRY and AT1AR DRY receptors to activate the cytosolic and nuclear pool of ERK1/2 using FRET reporters, respectively, EKARcyt and EKARnuc.39 The localization of the reporters is shown in Figure 5A. To measure both fast and late phases of ERK-mediated phosphorylation, we monitored EKAR signals for more than 20 min. The stimulation of cells expressing only the EKARcyt reporter with 100 μM histamine or 1 μM AngII did not result in any FRET signal above the baseline (Figure 5B,D; Supporting Information Figure S2). In fact, we observed a transient drop (25% of maximal value) of the FRET ratio change in cells expressing only this sensor immediately after histamine stimulation. In cells expressing only the EKARnuc reporter and stimulated with 100 μM histamine or 1 μM AngII, the FRET ratio change stayed constant or showed a slight increase (Figure 5C,E; Supporting Information Figure S2), rather than showing a gradual drop due to mild photobleaching (observed in vehicle-stimulated cells). This could indicate some weak agonist-induced phosphorylation of the EKARnuc reporter.
Figure 5.

ERK1/2 activation downstream of WT and DRY mutants of H1R and AT1AR in HEK293TN cells. (A) Wide-field images of the subcellular localization of EKARcyt and EKARnuc reporters in HEK293TN cells. Localization of the coexpressed free mCherry (mCh) shows the labeling of the complete cell. The size of the images is 105 μm × 105 μm. (B, C) ERK1/2 activation downstream of WT and DRY mutants of H1R. Time traces show the average ratio change of YFP/CFP fluorescence (and 95% confidence interval as a ribbon) in cells expressing the EKARcyt [N = 26] or EKARnuc [N = 20] sensor; coexpressing H1R-p2A-mCh and EKARcyt or EKARnuc and untreated [N = 49 for EKARcyt, N = 25 for EKARnuc] or treated with YM254890 [N = 7]; coexpressing H1R DRY-p2A-mCh and EKARcyt or EKARnuc and untreated [N = 16 for EKARcyt, N = 22 for EKARnuc] or treated with YM254890 [N = 11]. Histamine (100 μM) or vehicle (--) [N = 39 for EKARcyt, N = 5 for EKARnuc] was added at 90 s; gray boxes mark the duration of histamine stimulation. (D, E) ERK1/2 activation downstream of WT and DRY mutants of AT1AR. Time traces show the average ratio change of YFP/CFP fluorescence (and 95% confidence interval as a ribbon) in cells expressing EKARcyt [N = 41] or EKARnuc [N = 17] sensor; coexpressing AT1AR-p2A-mCh and EKARcyt or EKARnuc and untreated [N = 70 for EKARcyt, N = 65 for EKARnuc] or treated with YM254890 [N = 14]; coexpressing AT1AR DRY-p2A-mCh and EKARcyt or EKARnuc and untreated [N = 22 for EKARcyt, N = 26 for EKARnuc] or treated with YM254890 [N = 10]. AngII (1 μM) or vehicle (--) [N = 39 for EKARcyt, N = 5 for EKARnuc] was added at 90 s; gray boxes mark the duration of AngII stimulation.
On the contrary, we measured robust FRET signals with both EKARcyt and EKARnuc reporters in cells coexpressing the sensor together with H1R or AT1AR and stimulated with the respective agonist. In the case of H1R-mediated ERK1/2 activation, EKARcyt and EKARnuc signals showed similar kinetics but differed in amplitude (Figure 5B,C; Supporting Information Figure S2A), with the EKARnuc signal being approximately 50% higher than the EKARcyt signal. Both signals showed a transient drop immediately after histamine addition. Subsequently, both signals rose sharply, reaching maximal levels approx. 5 min after histamine addition and then started to slowly decay. H1R DRY-mediated ERK activation, although reduced, resembled the response downstream of WT H1R (Figure 5B,C; Supporting Information Figure S2A): both EKARcyt and EKARnuc signals showed an initial drop, after which they started increasing, reaching more than 50% of the respective H1R-mediated responses within 5 min of histamine addition. H1R DRY-mediated EKAR signals showed, however, a much faster decline than the H1R-mediated signals. Treatment with a specific Gq protein inhibitor, YM254890,40 reduced H1R-mediated EKARcyt signals (Figure 5B), in agreement with the Gq-dependent activation of the ERK pathway downstream of H1R.41 Interestingly, inhibition with YM254890 completely abolished the EKARcyt signal in cells expressing H1R DRY receptors (Figure 5B), suggesting that the ERK signal induced by the DRY mutant requires the activation of Gq.
The stimulation of cells expressing EKARcyt or EKARnuc reporter and AT1AR with 1 μM AngII (Figure 5D,E; Supporting Information Figure 2B) resulted in EKAR signals resembling histamine-induced responses of H1R-expressing cells. However, AngII-induced responses differed in amplitude from the respective histamine-induced responses (the AngII-induced EKARcyt signal being higher and the EKARnuc signal being lower) and showed slower decay. Both EKARcyt and EKARnuc signals showed similar kinetics and amplitude, although a transient drop (13% of maximal value) immediately upon AngII addition was observed only with the former reporter. Cells expressing AT1AR DRY and stimulated with AngII were able to activate the cytosolic ERK pool to approximately 80% of the respective AT1AR response (Figure 5D; Supporting Information Figure 2B). We did not measure any AT1AR DRY-mediated EKARnuc signal above the response of cells expressing only the EKARnuc reporter and stimulated with AngII. Both AT1AR- and AT1AR DRY-mediated EKARcyt signals were reduced in YM254890-treated cells (Figure 5D).
Discussion
Engineered GPCRs that have a mutation in the DRY motif have been proposed as tools to study G protein-independent signaling. It is important to verify to what extent mutations in the DRY motif inhibit G protein signaling. Here, we report the signaling dynamics of two class A receptors, H1R and AT1AR, in which the DRY motif is changed to AAY. Our results demonstrate that DRY mutants of H1R and AT1AR are capable of activating heterotrimeric G proteins, resulting in downstream signaling, including ERK activation.
The DRY motif in AT1AR is required for receptor activation, although there are contradictory results regarding the level of impairment achieved with identical or similar mutations. Ohyama and colleagues42 using Chinese hamster ovary (CHO-K1) cells reported a severe reduction in the G protein coupling (as indicated by the insensitivity of AngII binding to GTPγS) and inositol phosphate (IP) production for AT1AR mutants with either Asp125 or Arg126 residue replaced by either Ala or Gly. On the contrary, Gaborik and colleagues29 using COS-7 cells observed severe impairment of IP production and ERK1/2-dependent Elk1 promoter activation only with a double DRY/AAY mutant. Importantly, AT1AR DRY/AAY or DRY/GGY double mutants expressed in HEK293 or COS-7 cells were reported to be unable to activate G protein (measured at the level of 35SGTPγS binding, IP production, or Ca2+ accumulation) but capable of ß-arrestin recruitment and ERK1/2 activation.17−19 However, the residual activity (approx. 25% of the WT response measured at the maximal value) of the DRY/AAY mutant in the FLIPR assay that measures increases of intracellular Ca2+ levels was observed.18 Additionally, Bonde and colleagues19 reported increased Sar1–Ile4–Ile8 (SII) AngII-induced IP production downstream of DRY/AAY mutant activation compared to WT AT1AR in COS-7 cells, indicating the ability of this receptor mutant to couple to the Gq/11 protein.
Here, we observed substantial, albeit reduced compared to WT receptor responses, Gq protein coupling of H1R DRY and AT1AR DRY mutants in HEK293TN cells using the Gq FRET reporter. The fact that we did not observe similar coupling of these mutants to Gi1 could suggest signaling bias (preference for Gq coupling over Gi1). However, it could also result from a lower dynamic range of the Gi1 FRET reporter. The observed H1R DRY and AT1AR DRY couplings to the Gq protein were achieved in cells transiently expressing all three subunits of the Gq heterotrimer. Previously, the overexpression of the Gαq protein was shown to increase AngII-induced signaling for all tested AT1AR mutants except the DRY/AAY mutant.19 To confirm the coupling of these receptor mutants to the Gq protein in the absence of its overexpression, we characterized the signaling ability of H1R DRY and AT1AR DRY receptors using FRET reporters for Gq effectors, i.e., PKC and Ca2+. Both CKAR and YC3.6 sensors reported efficient coupling of H1R DRY and AT1AR DRY mutants to the Gq protein. Signaling efficiencies of these mutant receptors measured with CKAR and YC3.6 sensors were increased in comparison to the Gq FRET signal, likely as a result of the amplification of the signaling outcome downstream of the initial (reduced) coupling of these receptor mutants to the Gq proteins.
H1R DRY and AT1AR DRY receptors were also able to weakly activate RhoA. Endogenous H1 receptors in HeLa cells were shown to activate RhoA through the p63RhoGEF- or Trio-mediated, Gq protein-dependent mechanism.38 Similarly, AT1AR was shown to increase the RhoA activity via the G12/13-dependent, RhoGEF-mediated pathway in vascular smooth muscle cells and in cardiac myocytes in culture and in vivo.43 However, Bregeon and colleagues44 have recently demonstrated the G protein-independent activation of RhoA downstream of AT1AR in cultured vascular smooth muscle cells. As we have not investigated the ability of H1R DRY and AT1AR DRY mutants to activate RhoA in the presence of Gq or G12/13 protein inhibitors (FR900359 and RGS domains of p115RhoGEF, respectively), we cannot currently conclude on the mechanism of this activation.
Taken together, our results demonstrate the ability of H1R DRY and AT1AR DRY mutant receptors to signal via Gq proteins, with the strength of the signal strongly depending on the signaling event tested (with more downstream effectors showing higher signals due to the signal amplification). At the same time, we have noted a very transient character of this Gq coupling of the DRY mutants: histamine-induced Gq and CKAR signals, as well as AngII-induced CKAR and YC3.6 signals, decayed within a minute of agonist stimulation. Such transient signals would likely be missed or blunted in the conventional biochemical assays that generally have a poor temporal resolution. Therefore, our results once again demonstrate the strength of single-cell imaging using biosensors.45
With respect to the ß-arrestin recruitment, we observed agonist-induced relocation of both ß-arrestin isoforms to the cell periphery of H1R DRY- and AT1AR DRY-expressing HEK293TN cells. H1R DRY- and AT1AR DRY-mediated relocations were, however, less robust than the ß-arrestin recruitment upon WT receptor activation. Previously, the bioluminescence resonance energy transfer between the ßarr2-green fluorescent protein (GFP) fusion protein and AT1AR DRY fusion to Renilla luciferase, indicating receptor–ßarr2 interaction (or at least close proximity), was reported by Bonde and colleagues.19 The agonist-induced relocation of ßarr1- and/or ßarr2-GFP fusion proteins in AT1AR- and AT1AR DRY-expressing cells was also demonstrated using confocal microscopy.17,18 Both research groups showed the change in ß-arrestin cellular distribution, from uniformly cytosolic to presumably sequestered in the endocytotic compartment, upon agonist stimulation. However, this redistribution was only demonstrated after 30 min of stimulation and in fixed cells. On the contrary, our results showed the agonist-induced relocation of both ß-arrestin isoforms to the cell periphery as early as 2 min of stimulation. Additionally, we reported an overlap of ß-arrestin puncta with the receptor location at the cell periphery and, in the case of AT1AR- and AT1AR DRY-expressing cells, also in the intracellular (presumably endosomal) compartment, indicating colocalization of ß-arrestin and receptor fluorescent fusions.
Finally, we confirmed the ability of WT and DRY mutant receptors to activate ERK. Importantly, AngII-induced EKAR signals (both EKARcyt and EKARnuc), reporting on ERK-mediated phosphorylation, significantly resembled AngII-induced ERK phosphorylation demonstrated in HEK293 cells stably expressing AT1AR using conventional immunoblotting.46 We similarly observed an immediate increase of phosphorylation upon 1 μM AngII addition, reaching a maximum value at 5 min of stimulation, and followed by slow decay. Therefore, our results support the notion that EKAR sensors can faithfully report ERK activation in living cells. An intriguing observation was the transient drop in histamine-induced EKARcyt and EKARnuc signals, as well as the AngII-induced EKARcyt signal, indicating agonist-induced transient inhibition of ERK activity or dephosphorylation of the existing phosphoERK pool.
The G protein-dependent and ßarrestin-dependent ERK activations were postulated to differ with respect to both its kinetics and spatial distribution.15,46−49 In HEK293 cells expressing the AT1AR, beta2 adrenergic receptor, or vasopressin V2 receptor, G protein-dependent activation was reported to be rapid in onset but transient (with most of the signal waning within 10 min of stimulation) and to generate phosphorylated ERK distributed throughout the cytosol and nucleus. On the contrary, the ßarrestin-dependent component was reported to reach a maximum at 10 min, persist for at least 30 min without attenuation, and activate only the cytosolic ERK1/2 pool.
We measured reproducible agonist-induced EKARcyt signals downstream of both H1R DRY and AT1AR DRY. Importantly, the H1R DRY-mediated EKARcyt signal was fully abolished in the absence of Gq protein activation. Treatment with the Gq protein inhibitor, YM254890, reduced the AT1AR DRY-mediated EKARcyt signal, also indicating its partial dependence on G protein activation. It would be interesting to find out the effect of inhibiting both Gq and Gi on the ability of AT1AR DRY receptors to trigger the ERK pathway. With respect to ERK activation in the nucleus, the histamine-induced EKARnuc signal in cells expressing the H1R DRY mutant receptor achieved 60% of the WT response. Since the spatial and temporal aspects of ERK signaling are related to cell fate (differentiation versus proliferation),50−52 it would be interesting to find out whether the differences between native and DRY receptor variants on the ERK response have consequences for cell behavior. Another interesting future direction would be to examine the signaling activity downstream of the wild-type and DRY receptors stimulated with arrestin-biased agonists.
Several research groups postulated the existence of a G protein-independent, ß-arrestin-dependent ERK activation downstream of several GPCRs, including AT1AR.9,11,14,15,17 Some of these studies employed the DRY receptor mutants to claim G protein independence of this ERK activation. However, the residual ability of AT1AR DRY and H1R DRY mutant receptors demonstrated in this study challenges the notion that the DRY/AAY mutation leads to the complete uncoupling of the receptor from G proteins. Importantly, the stimulation of different GPCRs in the absence of G protein activation (including “zero functional G” background) was recently reported not to result in any ERK activation,53,54 challenging the concept of G protein-independent ERK activation. It would be interesting to investigate whether the distinct ERK activation observed downstream of DRY mutant activation could be reproduced by mimicking the suboptimal and transient activation of the Gq protein, such as demonstrated in this study.
Materials and Methods
Constructs
Plasmids encoding HsH1R-mCherry and H1R-p2A-mCherry are reported before32 and are available from addgene (https://www.addgene.org/browse/article/22426/). The AT1AR-p2A-mCherry was reported before55 and is available from addgene (plasmid #112934). The p2A viral sequence ensures that the mCherry is separated from the receptor protein during translation, generating an untagged receptor and free RFP that reports on receptor translation levels. The direct fusion, AT1AR-mCherry, was made by the restriction-enzyme-based cloning and is available from addgene (plasmid #137782). The DRY/AAY mutation was introduced into H1R and AT1AR sequences using site-directed mutagenesis; after the mutations were confirmed by sequencing, H1R DRY and AT1AR DRY sequences were cloned into pN1-mCherry and pN1-p2A-mCherry vectors. Plasmids encoding AT1aR-AAY-mCherry, AT1aR-AAY-p2A-mCherry, H1R-AAY-mCherry, and H1R-AAY-p2A-mCherry are deposited at addgene (plasmids #137783, #137784, #137788, and #137787). Plasmids encoding Rnßarr1-mYFP (plasmid #36916) and Rnßarr2-mYFP (plasmid #36917) were obtained from addgene.org, and plasmids encoding ßarr1-mTQ2 and ßarr2-mTQ2 were generated by cloning ß-arr1 and ß-arr2 sequences into the pN1-mTurquoise2 Clontech vector using PCR amplification and KpnI and AgeI sites. Plasmids encoding ßarr1-mTQ2 and ßarr2-mTQ2 are available from addgene (plasmids #137789 and #137792). Plasmids encoding the Gq sensor (Goedhart et al. 2011) and the DORA-RhoA sensor (Unen et al., 2015) were reported before. Plasmids encoding the endosomal marker mTurquoise2-Rab727 (plasmid #112959), the calcium biosensor YC3.6 (plasmid #67899), and the Gi1 biosensor (plasmid #69623) are available from www.addgene.org. Lck-mVenus (plasmid #84337) was reported as a plasma membrane marker.56 The CKAR biosensor57 was a kind gift from Alexandra Newton (University of California, San Diego). Cerulean-Venus versions of the EKARcyt (#18679) and EKARnuc (#18681) biosensors39 were obtained from addgene.org.
Reagents
Histamine, angiotensin II (AngII), pyrilamine (PY), phorbol myristate acetate (PMA), pertussis toxin (PTX), and Ro31-8425 were purchased from Sigma-Aldrich/Merck. The specific Gαq inhibitor, FR900359 (UBO-QIC), was purchased from the University of Bonn (http://www.pharmbio.uni-bonn.de/signaltransduktion). FR900359 was added to cells (in microscopy medium) at least 10 min before the measurements at a concentration of 1 μM. Ro31-8425 was added to cells (in the microscopy medium) at least 25 min before the measurements at a concentration of 10 μM. Experiments with the PTX inhibitor were carried out using cells cultured in a serum-free Dulbecco’s modified Eagle’s medium (DMEM) and incubated o/n with 100 ng/μL PTX.
Cell Culture and Sample Preparation
HEK293TN cells (System Biosciences, LV900A-1) and HeLA cells (American Tissue Culture Collection: Manassas, VA) were cultured using Dulbecco’s modified Eagle’s medium (DMEM) supplied with glutamax, 10% fetal bovine serum, penicillin (100 U/mL), and streptomycin (100 μg/mL) and incubated at 37 °C and 5% CO2. All cell culture reagents were obtained from Invitrogen (Bleiswijk, NL). Cells were transfected in a 35 mm dish holding a glass coverslip (24 mm ⌀, Menzel-Gläser, Braunschweig, Germany), using polyethylenimine (3 μL of PEI:1 μL of DNA) according to the manufacturer’s protocol. For each transfection, we used 500 ng of the receptor (H1R, H1R DRY, AT1AR, or AT1AR DRY)-carrying plasmid. Other plasmids were transfected at 100 ng (ßarr1 fusion constructs), 150 ng (ßarr2 fusion constructs), 200 ng (Lck-mVenus, CKAR, YC3.6), 250 ng (EKARcyt, EKARnuc), 300 ng (DORA-RhoA), and 750 ng (Gq reporter, Gi1 reporter). The samples were imaged 1 day after transfection: coverslips were mounted in an Attofluor cell chamber (Invitrogen, Breda, NL) and submerged in 1 mL microscopy medium (20 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid, pH = 7.4, 137 mM NaCl, 5.4 mL KCl, 1.8 mM CaCl2, 0.8 mM MgCl2, and 20 mM glucose; Sigma-Aldrich/Merck).
Confocal and Wide-Field Microscopy
Relocation experiments were performed as described.58 Ratiometric FRET measurements were performed on a previously described wide-field microscope.38 The typical exposure time was 100 ms, and the camera binning was set to 4 × 4. Fluorophores were excited with a 420/30 nm light and reflected onto the sample by a 455DCLP dichroic mirror. CFP emission was detected with a BP470/30 filter, and YFP emission was detected with a BP535/30 filter by rotating the filter wheel. RFP was excited with a 570/10 nm light reflected onto the sample by a 585 dichroic mirror, and RFP emission was detected with a BP620/60 nm emission filter. All acquisitions were corrected for the background signal and bleedthrough of CFP emission in the YFP channel. All experiments were performed at 37 °C and in (at least) triplicate.
Image Analysis and Data Visualization
ImageJ (National Institute of Health) was used to analyze the raw microscopy images and to calculate Pearson’s coefficient as a measure of colocalization. Background subtractions, bleedthrough correction, and calculation of the normalized ratio per time point per cell were done in Excel (Microsoft Office). Plots were prepared with the PlotTwist59 and PlotsOfData60 web apps. Time series show the average response as a thicker line and a ribbon for the 95% confidence interval around the mean.
Acknowledgments
The authors thank Alexandra Newton (University of California, San Diego) for providing constructs.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.9b03146.
Confocal recordings of subcellular localization of ßarr1-mTQ2, co-expressed together with H1R-mCh in HEK293TN cells (Movie S1) (AVI)
Confocal recordings of subcellular localization of ßarr2-mTQ2, co-expressed together with H1R-mCh in HEK293TN cells (Movie S2) (AVI)
Confocal recordings of subcellular localization of ßarr1-mTQ2, co-expressed together with H1Rdry-mCh in HEK293TN cells (Movie S3) (AVI)
Confocal recordings of subcellular localization of ßarr2-mTQ2, co-expressed together with H1Rdry-mCh in HEK293TN cells (Movie S4) (AVI)
Confocal recordings of subcellular localization of ßarr1-mTQ2, co-expressed together with AT1AR-mCh in HEK293TN cells (Movie S5) (AVI)
Confocal recordings of subcellular localization of ßarr2-mTQ2, co-expressed together with AT1AR-mCh in HEK293TN cells (Movie S6) (AVI)
Confocal recordings of subcellular localization of ßarr1-mTQ2, co-expressed together with AT1ARdry-mCh in HEK293TN cells (Movie S7) (AVI)
Confocal recordings of subcellular localization of ßarr2-mTQ2, co-expressed together with AT1ARdry-mCh in HEK293TN cells (Movie S8) (AVI)
Signaling activity of WT and DRY mutants of H1R and AT1AR in HEK293TN cells (Figure S1); and ERK activation downstream of WT and DRY mutants (Figure S2) (PDF)
Author Contributions
A.P.-B. designed and performed the experiments, analyzed the data, and wrote the manuscript. L.J. and A.O.C. provided technical assistance with cell culture maintenance and confocal microscopy. J.G. participated in study design, data interpretation, and data visualization. All authors approved the final manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Weis W. I.; Kobilka B. K. The Molecular Basis of G Protein–Coupled Receptor Activation. Annu. Rev. Biochem. 2018, 87, 897–919. 10.1146/annurev-biochem-060614-033910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilger D.; Masureel M.; Kobilka B. K. Structure and Dynamics of GPCR Signaling Complexes. Nat. Struct. Mol. Biol. 2018, 25, 4–12. 10.1038/s41594-017-0011-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komolov K. E.; Benovic J. L. G Protein-Coupled Receptor Kinases: Past, Present and Future. Cell. Signal. 2018, 41, 17–24. 10.1016/j.cellsig.2017.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J.; Barak L. S.; Anborgh P. H.; Laporte S. A.; Caron M. G.; Ferguson S. S. Cellular Trafficking of G Protein-Coupled Receptor/Beta-Arrestin Endocytic Complexes. J. Biol. Chem. 1999, 274, 10999–11006. 10.1074/jbc.274.16.10999. [DOI] [PubMed] [Google Scholar]
- Kang D. S.; Tian X.; Benovic J. L. Role of β-Arrestins and Arrestin Domain-Containing Proteins in G Protein-Coupled Receptor Trafficking. Curr. Opin. Cell Biol. 2014, 27, 63–71. 10.1016/j.ceb.2013.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurevich V. V.; Gurevich E. V. Arrestins: Critical Players in Trafficking of Many GPCRs. Prog. Mol. Biol. Transl. Sci. 2015, 132, 1–14. 10.1016/BS.PMBTS.2015.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiter E.; Ahn S.; Shukla A. K.; Lefkowitz R. J. Molecular Mechanism of β-Arrestin-Biased Agonism at Seven-Transmembrane Receptors. Annu. Rev. Pharmacol. Toxicol. 2012, 52, 179–197. 10.1146/annurev.pharmtox.010909.105800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson Y. K.; Luttrell L. M. The Diverse Roles of Arrestin Scaffolds in G Protein-Coupled Receptor Signaling. Pharmacol. Rev. 2017, 69, 256–297. 10.1124/pr.116.013367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whalen E. J.; Rajagopal S.; Lefkowitz R. J. Therapeutic Potential of β-Arrestin- and G Protein-Biased Agonists. Trends Mol. Med. 2011, 17, 126–139. 10.1016/j.molmed.2010.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saulière A.; Bellot M.; Paris H.; Denis C.; Finana F.; Hansen J. T.; Altié M.-F.; Seguelas M.-H.; Pathak A.; Hansen J. L.; et al. Deciphering Biased-Agonism Complexity Reveals a New Active AT1 Receptor Entity. Nat. Chem. Biol. 2012, 622–630. 10.1038/nchembio.961. [DOI] [PubMed] [Google Scholar]
- Zimmerman B.; Beautrait A.; Aguila B.; Charles R.; Escher E.; Claing A.; Bouvier M.; Laporte S. A. Differential β-Arrestin–Dependent Conformational Signaling and Cellular Responses Revealed by Angiotensin Analogs. Sci. Signal. 2012, 5, 221 10.1126/scisignal.2002522. [DOI] [PubMed] [Google Scholar]
- Littmann T.; Göttle M.; Reinartz M. T.; Kälble S.; Wainer I. W.; Ozawa T.; Seifert R. Recruitment of β-Arrestin 1 and 2 to the B2-Adrenoceptor: Analysis of 65 Ligands. J. Pharmacol. Exp. Ther. 2015, 355, 183–190. 10.1124/jpet.115.227959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J.; Hanada K.; Staus D. P.; Makara M. A.; Dahal G. R.; Chen Q.; Ahles A.; Engelhardt S.; Rockman H. A. Gα(i) Is Required for Carvedilol-Induced β(1) Adrenergic Receptor β-Arrestin Biased Signaling. Nat. Commun. 2017, 8, 1706 10.1038/s41467-017-01855-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aplin M.; Bonde M. M.; Hansen J. L. Molecular Determinants of Angiotensin II Type 1 Receptor Functional Selectivity. J. Mol. Cell. Cardiol. 2009, 46, 15–24. 10.1016/j.yjmcc.2008.09.123. [DOI] [PubMed] [Google Scholar]
- Shenoy S. K.; Drake M. T.; Nelson C. D.; Houtz D. A.; Xiao K.; Madabushi S.; Reiter E.; Premont R. T.; Lichtarge O.; Lefkowitz R. J. Beta-Arrestin-Dependent, G Protein-Independent ERK1/2 Activation by the Beta2 Adrenergic Receptor. J. Biol. Chem. 2006, 281, 1261–1273. 10.1074/jbc.M506576200. [DOI] [PubMed] [Google Scholar]
- Siuda E. R.; McCall J. G.; Al-Hasani R.; Shin G.; Il Park S.; Schmidt M. J.; Anderson S. L.; Planer W. J.; Rogers J. A.; Bruchas M. R. Optodynamic Simulation of β-Adrenergic Receptor Signalling. Nat. Commun. 2015, 6, 8480 10.1038/ncomms9480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei H.; Ahn S.; Shenoy S. K.; Karnik S. S.; Hunyady L.; Luttrell L. M.; Lefkowitz R. J. Independent β-Arrestin 2 and G Protein-Mediated Pathways for Angiotensin II Activation of Extracellular Signal-Regulated Kinases 1 and 2. Proc. Natl. Acad. Sci. U.S.A. 2003, 100, 10782–10787. 10.1073/pnas.1834556100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee M.-H.; El-Shewy H. M.; Luttrell D. K.; Luttrell L. M. Role of Beta-Arrestin-Mediated Desensitization and Signaling in the Control of Angiotensin AT1a Receptor-Stimulated Transcription. J. Biol. Chem. 2008, 283, 2088–2097. 10.1074/jbc.M706892200. [DOI] [PubMed] [Google Scholar]
- Bonde M. M.; Hansen J. T.; Sanni S. J.; Haunsø S.; Gammeltoft S.; Lyngsø C.; Hansen J. L. Biased Signaling of the Angiotensin II Type 1 Receptor Can Be Mediated through Distinct Mechanisms. PLoS One 2010, 5, e14135 10.1371/journal.pone.0014135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrens D. L.; Altenbach C.; Yang K.; Hubbell W. L.; Khorana H. G. Requirement of Rigid-Body Motion of Transmembrane Helices for Light Activation of Rhodopsin. Science 1996, 274, 768–770. 10.1126/science.274.5288.768. [DOI] [PubMed] [Google Scholar]
- Ballesteros J. A.; Shi L.; Javitch J. A. Structural Mimicry in G Protein-Coupled Receptors: Implications of the High-Resolution Structure of Rhodopsin for Structure-Function Analysis of Rhodopsin-like Receptors. Mol. Pharmacol. 2001, 60, 1–19. 10.1124/mol.60.1.1. [DOI] [PubMed] [Google Scholar]
- Sheikh S. P.; Vilardarga J. P.; Baranski T. J.; Lichtarge O.; Iiri T.; Meng E. C.; Nissenson R. A.; Bourne H. R. Similar Structures and Shared Switch Mechanisms of the Beta2-Adrenoceptor and the Parathyroid Hormone Receptor. Zn(II) Bridges between Helices III and VI Block Activation. J. Biol. Chem. 1999, 274, 17033–17041. 10.1074/jbc.274.24.17033. [DOI] [PubMed] [Google Scholar]
- Vilardaga J. P.; Frank M.; Krasel C.; Dees C.; Nissenson R. A.; Lohse M. J. Differential Conformational Requirements for Activation of G Proteins and the Regulatory Proteins Arrestin and G Protein-Coupled Receptor Kinase in the G Protein-Coupled Receptor for Parathyroid Hormone (PTH)/PTH-Related Protein. J. Biol. Chem. 2001, 276, 33435–33443. 10.1074/jbc.M011495200. [DOI] [PubMed] [Google Scholar]
- Smith J. S.; Lefkowitz R. J.; Rajagopal S. Biased Signalling: From Simple Switches to Allosteric Microprocessors. Nat. Rev. Drug Discovery 2018, 17, 243–260. 10.1038/nrd.2017.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurevich V. V.; Gurevich E. V. Arrestin-Mediated Signaling: Is There a Controversy?. World J. Biol. Chem. 2018, 9, 25–35. 10.4331/wjbc.v9.i3.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanlandingham P. A.; Ceresa B. P. Rab7 Regulates Late Endocytic Trafficking Downstream of Multivesicular Body Biogenesis and Cargo Sequestration. J. Biol. Chem. 2009, 284, 12110–12124. 10.1074/jbc.M809277200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chertkova A. O.; Mastop M.; Postma M.; van Bommel N.; van der Niet S.; Batenburg K. L.; Joosen L.; Gadella T. W. J.; Okada Y.; Goedhart J. Robust and Bright Genetically Encoded Fluorescent Markers for Highlighting Structures and Compartments in Mammalian Cells. bioRxiv 2017, 160374 10.1101/160374. [DOI] [Google Scholar]
- Oakley R. H.; Laporte S. A.; Holt J. A.; Caron M. G.; Barak L. S. Differential Affinities of Visual Arrestin, Beta Arrestin1, and Beta Arrestin2 for G Protein-Coupled Receptors Delineate Two Major Classes of Receptors. J. Biol. Chem. 2000, 275, 17201–17210. 10.1074/jbc.M910348199. [DOI] [PubMed] [Google Scholar]
- Ga′borik Z.; Jagadeesh G.; Zhang M.; Spät A.; Catt K. J.; Hunyady L. The Role of a Conserved Region of the Second Intracellular Loop in AT1 Angiotensin Receptor Activation and Signaling. Endocrinology 2003, 144, 2220–2228. 10.1210/en.2002-0135. [DOI] [PubMed] [Google Scholar]
- Seifert R.; Strasser A.; Schneider E. H.; Neumann D.; Dove S.; Buschauer A. Molecular and Cellular Analysis of Human Histamine Receptor Subtypes. Trends Pharmacol. Sci. 2013, 33–58. 10.1016/j.tips.2012.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Unen J.; Stumpf A. D.; Schmid B.; Reinhard N. R.; Hordijk P. L.; Hoffmann C.; Gadella T. W. J.; Goedhart J. A New Generation of FRET Sensors for Robust Measurement of Gαi1, Gαi2 and Gαi3 Activation Kinetics in Single Cells. PLoS One 2016, 11, e0146789 10.1371/journal.pone.0146789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Unen J.; Rashidfarrokhi A.; Hoogendoorn E.; Postma M.; Gadella T. W. J.; Goedhart J. Quantitative Single-Cell Analysis of Signaling Pathways Activated Immediately Downstream of Histamine Receptor Subtypes. Mol. Pharmacol. 2016, 90, 162–176. 10.1124/mol.116.104505. [DOI] [PubMed] [Google Scholar]
- Hunyady L.; Catt K. J. Pleiotropic AT1 Receptor Signaling Pathways Mediating Physiological and Pathogenic Actions of Angiotensin II. Mol. Endocrinol. 2006, 20, 953–970. 10.1210/me.2004-0536. [DOI] [PubMed] [Google Scholar]
- Kawai T.; Forrester S. J.; O’Brien S.; Baggett A.; Rizzo V.; Eguchi S. AT1 Receptor Signaling Pathways in the Cardiovascular System. Pharmacol. Res. 2017, 125, 4–13. 10.1016/j.phrs.2017.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adjobo-Hermans M. J. W.; Goedhart J.; van Weeren L.; Nijmeijer S.; Manders E. M. M.; Offermanns S.; Gadella T. W. J. Real-Time Visualization of Heterotrimeric G Protein Gq Activation in Living Cells. BMC Biol. 2011, 9, 32 10.1186/1741-7007-9-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goedhart J.; van Weeren L.; Adjobo-Hermans M. J. W.; Elzenaar I.; Hink M.; Gadella T. W. J. Quantitative Co-Expression of Proteins at the Single Cell Level--Application to a Multimeric FRET Sensor. PLoS One 2011, 6, e27321 10.1371/journal.pone.0027321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrage R.; Schmitz A.-L.; Gaffal E.; Annala S.; Kehraus S.; Wenzel D.; Bullesbach K. M.; Bald T.; Inoue A.; Shinjo Y.; et al. The Experimental Power of FR900359 to Study Gq-Regulated Biological Processes. Nat. Commun. 2015, 6, 639 10.1038/ncomms10156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Unen J.; Reinhard N. R.; Yin T.; Wu Y. I.; Postma M.; Gadella T. W. J.; Goedhart J. Plasma Membrane Restricted RhoGEF Activity Is Sufficient for RhoA-Mediated Actin Polymerization. Sci. Rep. 2015, 5, 14693 10.1038/srep14693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey C. D.; Ehrhardt A. G.; Cellurale C.; Zhong H.; Yasuda R.; Davis R. J.; Svoboda K. A Genetically Encoded Fluorescent Sensor of ERK Activity. Proc. Natl. Acad. Sci. U.S.A. 2008, 105, 19264–19269. 10.1073/pnas.0804598105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura A.; Kitano K.; Takasaki J.; Taniguchi M.; Mizuno N.; Tago K.; Hakoshima T.; Itoh H. Structural Basis for the Specific Inhibition of Heterotrimeric Gq Protein by a Small Molecule. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 13666–13671. 10.1073/pnas.1003553107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietraszewska-Bogiel A.; Goedhart J. Seeing β-Arrestin in Action: The Role of β-Arrestins in Histamine 1 Receptor Signaling. bioRxiv 2019, 775049 10.1101/775049. [DOI] [Google Scholar]
- Ohyama K.; Yamano Y.; Sano T.; Nakagomi Y.; Wada M.; Inagami T. Role of the Conserved DRY Motif on G Protein Activation of Rat Angiotensin II Receptor Type 1A. Biochem. Biophys. Res. Commun. 2002, 292, 362–367. 10.1006/bbrc.2002.6670. [DOI] [PubMed] [Google Scholar]
- Kimura K.; Eguchi S. Angiotensin II Type-1 Receptor Regulates RhoA and Rho-Kinase/ROCK Activation via Multiple Mechanisms. Focus on “Angiotensin II Induces RhoA Activation through SHP2-Dependent Dephosphorylation of the RhoGAP P190A in Vascular Smooth Muscle Cells. Am. J. Physiol. Cell Physiol. 2009, 297, C1059–C1061. 10.1152/ajpcell.00399.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bregeon J.; Loirand G.; Pacaud P.; Rolli-Derkinderen M. Angiotensin II Induces RhoA Activation through SHP2-Dependent Dephosphorylation of the RhoGAP P190A in Vascular Smooth Muscle Cells. Am. J. Physiol. Cell Physiol. 2009, 297, C1062–C1070. 10.1152/ajpcell.00174.2009. [DOI] [PubMed] [Google Scholar]
- Greenwald E. C.; Mehta S.; Zhang J. Genetically Encoded Fluorescent Biosensors Illuminate the Spatiotemporal Regulation of Signaling Networks. Chem. Rev. 2018, 118, 11707–11794. 10.1021/acs.chemrev.8b00333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn S.; Shenoy S. K.; Wei H.; Lefkowitz R. J. Differential Kinetic and Spatial Patterns of Beta-Arrestin and G Protein-Mediated ERK Activation by the Angiotensin II Receptor. J. Biol. Chem. 2004, 279, 35518–35525. 10.1074/jbc.M405878200. [DOI] [PubMed] [Google Scholar]
- Luttrell L. M.; Roudabush F. L.; Choy E. W.; Miller W. E.; Field M. E.; Pierce K. L.; Lefkowitz R. J. Activation and Targeting of Extracellular Signal-Regulated Kinases by β-Arrestin Scaffolds. Proc. Natl. Acad. Sci. U.S.A. 2001, 98, 2449–2454. 10.1073/pnas.041604898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tohgo A.; Pierce K. L.; Choy E. W.; Lefkowitz R. J.; Luttrell L. M. Beta-Arrestin Scaffolding of the ERK Cascade Enhances Cytosolic ERK Activity but Inhibits ERK-Mediated Transcription Following Angiotensin AT1a Receptor Stimulation. J. Biol. Chem. 2002, 277, 9429–9436. 10.1074/jbc.M106457200. [DOI] [PubMed] [Google Scholar]
- Ren X.-R.; Reiter E.; Ahn S.; Kim J.; Chen W.; Lefkowitz R. J. Different G Protein-Coupled Receptor Kinases Govern G Protein and β-Arrestin-Mediated Signaling of V2 Vasopressin Receptor. Proc. Natl. Acad. Sci. U.S.A. 2005, 102, 1448–1453. 10.1073/pnas.0409534102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu H.; Chung M.; Dobrzyński M.; Fey D.; Blum Y.; Lee S. S.; Peter M.; Kholodenko B. N.; Jeon N. L.; Pertz O. Frequency Modulation of ERK Activation Dynamics Rewires Cell Fate. Mol. Syst. Biol. 2015, 11, 838 10.15252/msb.20156458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebisuya M.; Kondoh K.; Nishida E. The Duration, Magnitude and Compartmentalization of ERK MAP Kinase Activity: Mechanisms for Providing Signaling Specificity. J. Cell Sci. 2005, 118, 2997–3002. 10.1242/jcs.02505. [DOI] [PubMed] [Google Scholar]
- Santos S. D. M.; Verveer P. J.; Bastiaens P. I. H. Growth Factor-Induced MAPK Network Topology Shapes Erk Response Determining PC-12 Cell Fate. Nat. Cell Biol. 2007, 9, 324–330. 10.1038/ncb1543. [DOI] [PubMed] [Google Scholar]
- Alvarez-Curto E.; Inoue A.; Jenkins L.; Raihan S. Z.; Prihandoko R.; Tobin A. B.; Milligan G. Targeted Elimination of G Proteins and Arrestins Defines Their Specific Contributions to Both Intensity and Duration of G Protein-Coupled Receptor Signaling. J. Biol. Chem. 2016, 291, 27147–27159. 10.1074/jbc.M116.754887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grundmann M.; Merten N.; Malfacini D.; Inoue A.; Preis P.; Simon K.; Rüttiger N.; Ziegler N.; Benkel T.; Schmitt N. K.; et al. Lack of Beta-Arrestin Signaling in the Absence of Active G Proteins. Nat. Commun. 2018, 9, 341 10.1038/s41467-017-02661-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mastop M.; Reinhard N. R.; Zuconelli C. R.; Terwey F.; Gadella T. W. J.; Van Unen J.; Adjobo-Hermans M. J. W.; Goedhart J. A FRET-Based Biosensor for Measuring Gα13 Activation in Single Cells. PLoS One 2018, 13, 0193705 10.1371/journal.pone.0193705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Unen J.; Botman D.; Yin T.; Wu Y. I.; Hink M. A.; Gadella T. W. J. Jr; Postma M.; Goedhart J. The C-Terminus of the Oncoprotein TGAT Is Necessary for Plasma Membrane Association and Efficient RhoA-Mediated Signaling. BMC Cell Biol. 2018, 19, 6 10.1186/s12860-018-0155-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Violin J. D.; Zhang J.; Tsien R. Y.; Newton A. C. A Genetically Encoded Fluorescent Reporter Reveals Oscillatory Phosphorylation by Protein Kinase C. J. Cell Biol. 2003, 161, 899–909. 10.1083/jcb.200302125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Unen J.; Yin T.; Wu Y. I.; Mastop M.; Gadella T. W. J.; Goedhart J. Kinetics of Recruitment and Allosteric Activation of ARHGEF25 Isoforms by the Heterotrimeric G-Protein Gαq. Sci. Rep. 2016, 6, 36825 10.1038/srep36825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goedhart J. PlotTwist—a Web App for Plotting and Annotating Time-Series Data. bioRxiv 2019, 745612 10.1101/745612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Postma M.; Goedhart J. PlotsOfData—A Web App for Visualizing Data Together with Their Summaries. PLoS Biol. 2019, 17, e3000202 10.1371/journal.pbio.3000202. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.