

Introduction
Non-alcoholic steatohepatitis (NASH) is within the spectrum of non-alcoholic fatty liver disease (NAFLD) and can progress to fibrosis, cirrhosis, and even hepatocellular carcinoma (HCC). The prevalence of NASH is rising and has become a large burden to the medical system worldwide. Unfortunately, despite its high prevalence and severe health consequences, there is currently no therapeutic agent approved to treat NASH. Therefore, the development of efficacious therapies is of utmost urgency and importance. Many molecular targets are currently under investigation for their ability to halt NASH progression. One of the most promising and well-studied targets is the bile acid (BA)-activated nuclear receptor, farnesoid X receptor (FXR). In this chapter, the characteristics, etiology, and prevalence of NASH will be discussed. A brief introduction to FXR regulation of BA homeostasis will be described, however, for more detail regarding FXR in BA homeostasis please refer to previous chapters. In this chapter, the mechanisms by which tissue and cell type specific FXR regulates NASH development will be discussed in detail. Several FXR agonists have reached later phase clinical trials for treatment of NASH. The progress of these compounds and summary of released data will be provided. Lastly, this chapter will address safety liabilities specific to the development of FXR agonists.
Background - NASH
Disease characteristics, etiology, and risk factors:
NASH is the inflammatory form of NAFLD characterized by steatosis, hepatocyte ballooning, inflammation, and fibrosis.1, 2 NAFLD is a progressive disease beginning as simple steatosis but can develop into NASH that is characterized by inflammation and other cellular degenerations. NASH can further progress to fibrosis, cirrhosis, and even HCC.1–3 Metabolic syndrome often accompanies the development of NASH. Metabolic syndrome is defined as having 3 of 5 clinical presentations: 1) serum triglycerides greater than 150 mg/dL; 2) serum high density lipoprotein (HDL) less than 40 or 50 mg/dL in men and women, respectively; 3) increase in waist circumference; 4) serum glucose levels greater than 100 mg/dL; and 5) systolic or diastolic blood pressures greater than 130 and 85 mmHg, respectively.4
The mechanisms regulating NAFLD to NASH progression remains unclear. A “two-hit” model was proposed in 1998.5 This model speculates that NASH develops as the result of two sequential liver injuries. The “first hit” in the model being the accumulation of lipids in the liver leading to the development of simple steatosis. The “second hit” is a subsequent insult that induces inflammation. Though this model has been well cited for two decades, it has come under recent scrutiny as it is likely a drastic oversimplification of the processes that lead to NASH. For instance, progression to fibrosis can occur in NAFLD without the development of NASH.6 Patients can also present with cryptogenic fibrosis and have numerous risk factors for NAFLD and NASH but have minimal histological features of NASH.7 Additionally, NASH patients can progress to HCC without the development of cirrhosis.8 These findings indicate that more than just the “two-hit” model underlies disease pathogenesis.
Although the etiologies of NASH are not well understood, many risk factors have been identified. The most common health condition associated with NASH is obesity, followed by type 2 diabetes mellitus, dyslipidemia, metabolic syndrome, polycystic ovary syndrome, while less common conditions include hypothyroidism, hypopituitarism, hypogonadism, pancreatoduodenal resection, psoriasis, and sleep apnea.9 Age, sex, female reproductive status, and ethnicity are also associated with NASH development.1 Lastly, genetic polymorphisms have been identified which correlate to NASH; the most notable being variation in the patatin-like phospholipase domain-containing protein (PNPLA3) gene.10, 11 The prevalence of PNPLA3 polymorphisms amongst different ethnic groups may explain ethnic differences in NAFLD and NASH prevalence.11
Disease prevalence, diagnosis, and current treatment:
With the rise of the obesity epidemic, the prevalence of NASH has greatly increased over the past two decades. Current estimates place the North American prevalence of NAFLD at 24% and of those patients with NAFLD 21% may have NASH.12, 13 Further, of NASH patients worldwide, 40% will likely progress to fibrosis. The U.S. census data in 2017 placed the population at 325 million people.14 Based on the census and the estimates of NASH and fibrosis prevalence, we estimate that roughly 7 million individuals in the United States alone have or will develop NASH with fibrosis. The high prevalence of NAFLD and NASH is not limited to North America with the global prevalence of NAFLD estimated at 25.24%.12 Due to the increasing prevalence of NASH and recent breakthroughs in treatment of HCV, NASH will surpass HCV as the primary indication for which patients are added to the liver transplant waiting list. From 2008 to 2014, the number of patients added to the United States transplant waiting list for treatment of HCV was stable at roughly 3000 patients per year.15 In 2017, this number was decreased to 1705. Conversely, the number of patients who were added to the liver waiting list for the treatment of NASH increased from 643 in 2008 to 2100 in 2017.15 Based on these numbers, it appears that NASH has already surpassed HCV to become the number one indication for patients to receive liver transplant or will do so in the very near future.
The gold standard for diagnosing NASH is histolopathologic evaluation of liver biopsy. The diagnosis of definitive NASH requires the presence of all histologic criteria including steatosis, hepatocellular ballooning, and lobular inflammation. The diagnosis of borderline NASH is given when a patient presents with steatosis and most but not all histologic features of NASH.9 Several scoring systems have been developed to assess NASH histologic severity, including the NASH Clinic Research Network’s NAFLD activity score (NAS), the steatosis, activity, and fibrosis (SAF), and Brunt staging.16–18 Less invasive methods to assess NASH severity are currently under investigation with some being incorporated into clinical trials. Examples include magnetic resonance imaging (spectroscopy and proton density fat fraction), transient elastography, and serum fibrosis biomarkers (procollagen type III N-terminal protein, tissue inhibitor of metalloproteases 1, hyaluronic acid, cytokeratin-18 fragments).19
Despite the rising prevalence and burden NASH places on society and the medical system, there is currently no approved therapeutic agent to treat NASH. The current guideline for the management of NASH recommends changes in lifestyles: weight loss, diet and exercise.9 Vitamin E and thiazolidinediones may provide benefit to NASH patients but risks have to be weighed against the potential benefits. Guidelines recommended against using ursodeoxycholic acid (UDCA), metformin, and omega-3 fatty acids for the treatment of NASH, however, can be used to manage concomitant disease states. The guidelines also recommend against the off-label use of obeticholic acid (OCA) until clinical trial data regarding its use for the treatment of NASH become available. The only treatment for patients with advanced fibrotic NASH is liver transplant.9 With the limited number of organs available for transplant, it is a paramount medical necessity to identify the molecular mechanisms underlying NASH pathogenesis and to develop novel therapies to prevent, mitigate, or reverse NASH development.
Background – The BA-FXR-FGF19 pathway:
BAs are amphipathic detergents produced in the liver via the hydroxylation of cholesterol.20, 21 The two predominant pathways responsible for the conversion of cholesterol to BA are the classical (neutral) and alternative (acidic) pathways. In the classical pathway, cholesterol is sequentially oxidized by cytochrome p450 7A1 (CYP7A1) and CYP8B1 to produce cholic acid (CA). The classical pathway accounts for the synthesis of roughly 75% of the total BA pool and the 7-alpha hydroxylation of cholesterol by CYP7A1 is the rate limiting step in BA synthesis. The alternative or acidic pathway produces chenodeoxycholic acid (CDCA) by the metabolism of cholesterol by CYP27A1 and CYP7B1. CA and CDCA are conjugated to glycine or taurine by the enzyme bile acid-CoA amino acid N-acyltransferase. CA, CDCA, and their conjugates are considered primary BAs. In intestine, certain microbial species express the enzyme bile salt hydrolase (BSH) which mediates the deconjugation of BAs. Gut microbes can further metabolize CA and CDCA to the secondary BAs, deoxycholic acid (DCA) and lithocholic acid (LCA) or UDCA, respectively. In mice, UDCA is also a primary BA.22 Upon reabsorption and re-entry to the liver, secondary BAs can be conjugated.20, 21 The total BA pool therefore consists of numerous species of BAs with unconjugated and conjugated primary and secondary BAs. This will be of later importance as different BA species have different activities (agonist vs. antagonist) and potencies for BA receptors.
BAs undergo significant enterohepatic recirculation with roughly 95% of BAs reabsorbed from the intestine. The majority of BAs are reabsorbed in the ileum into enterocytes by the uptake transporter, apical sodium-dependent BA transporter (ASBT).20 Once inside enterocytes, BAs can activate the nuclear receptor FXR, and within the nucleus, FXR dimerizes with retinoid X receptor (RXR) to interact with DNA at the FXR response elements (FXRRE) to alter gene transcription.23–25 Activation of FXR in enterocytes leads to the up-regulation of fibroblast growth factor 19 (FGF19) in humans and orthologous FGF15 in mice.26 Though orthologs, FGF15 and FGF19 share only 50% sequence homology.27, 28 Both FGF15 and FGF19 are considered endocrine FGFs as they do not bind heparin sulfate and thus can escape extracellular matrix, unlike other families of FGF proteins.29 The structural uniqueness of endocrine FGFs that allow for their systemic circulation also reduces their affinity for fibroblast growth factor receptors (FGFR). Therefore, binding of FGF15 and FGF19 to their predominant receptors FGFR4, and to a lesser extent, FGFR1, requires the obligate co-receptor β-KLOTHO (βKL).29 Upon induction in the intestine, FGF15/19 travels through portal circulation and activates FGFR4-βKL on hepatocytes.26, 30, 31 This leads to activation of the extracellular signal-regulated kinase (ERK) and c-Jun N-terminal kinase (JNK) signal pathways and subsequently reduces BA synthesis by down-regulating the expression of CYP7A1/Cyp7a1 and CYP8B1/Cyp8b1 that encode enzymes, CYP7A1 and CYP8B1.30, 32, 33 FGF15 and FGF19 thereby function as a negative feedback loop shutting down BA synthesis when BA levels are high in the intestinal mucosa. BAs reabsorbed in the intestine activate FXR, transiently increase FGFR4/βKL levels, and prime the liver for subsequent FGF15/19 signaling.34 In humans, FGF19 is also expressed at low levels in the liver and is upregulated during cholestasis.35, 36 FXR activation in hepatocytes also suppresses CYP7A1/Cyp7a1 and CYP8B1/Cyp8b1 expression by inducing small heterodimer partner (SHP).24, 37, 38 In hepatocytes, activation of FXR is primarily responsible for promoting BA biliary excretion, and does not suppress BA synthesis as strongly as FGF15/19 signaling.39
As described above, there are numerous species of BAs in the body which are produced via enzymatic reactions performed by the liver and gut microbiome. Each BA species has different affinity, efficacy, and potency for each BA receptor. The strongest endogenous ligand of FXR is CDCA with an EC50 of roughly 5 μM.40 The most common BA in humans and mice, CA, activates FXR with less efficacy (EC50 = ~200 μM).25, 40, 41 The taurine conjugates of α and β muricholic acid (TαMCA,TβMCA) have been shown to be FXR antagonists.42 MCA is synthesized from precursor CDCA by Cyp2c70, an enzyme expressed in mouse liver but not human liver.43 MCA is thus a murine specific BA species. The BA pool composition in mice is predominantly comprised of weak FXR agonist CA (60%) and FXR antagonist MCA (40%). This contrasts to the BA pool in humans comprised of strong FXR agonist CDCA (40%), CA (40%), and DCA (20%). It is currently unclear if the human bile acid pool contains any endogenous FXR antagonists, however, there is growing evidence which supports that UDCA may function as an FXR antagonist.44
Role of FXR in NASH development in animal models
Systemic FXR:
FXR is expressed in many tissues and cell types in the body. Manipulation of body-wide FXR activity either through pharmacologic or genetic means affects the development of each characteristic of NASH; steatosis, inflammation, fibrosis, and metabolic syndrome. This section will broadly describe the effects of systemic FXR activation or deficiency on NASH development. The roles of tissue specific FXR and the mechanisms by which they influence NASH development will be described in depth in following sections. FXR agonists used in the animal studies described below include WAY-362450, GW4064, OCA, and fexaramine. For details regarding their structure and properties please see previous chapters.
Systemic activation of FXR is protective against the development of hepatic steatosis, inflammation, and fibrosis. In mice fed a high-fat diet (HFD), treatment with OCA and GW4064 reduced the accumulation of hepatic triglycerides and free fatty acids and subsequently reduced steatosis severity.45, 46 Similarly, in low density lipoprotein receptor (LDLR) knockout mice fed a Western Diet, WAY-362450 reduced hepatic triglyceride and cholesterol levels and attenuated steatosis.47 Hepatic inflammation is also reduced by treatment with FXR agonists. In both HFD and methionine & choline deficient diet (MCD) models, GW4064 and WAY-362450 reduced hepatic inflammation.46, 48 Correspondingly, FXR deficient mice had worsened inflammation induced by MCD.49 Activation of whole body FXR ameliorates hepatic fibrogenesis. OCA, WAY-362450, and BAR704 decreased the severity of fibrosis in mouse HFD, MCD, and carbon tetrachloride (CCl4) models, respectively.45, 46, 48 Deficiency of FXR worsened fibrosis induced by MCD or knockout of LDLR.49, 50
Body wide activation of FXR has many beneficial effects on metabolic endpoints. In mice fed a HFD, GW4064 reduced body weights and fat mass. GW4064 also lowered fasting glucose concentrations and improved glucose tolerance. Hepatic gluconeogenesis was also reduced.46 Serum lipids are also altered by modulation of whole body activity of FXR. Activation of FXR by GW4064 and WAY-362450 reduced triglyceride and cholesterol levels in HFD and LDLR knockout, Western diet murine models, respectively.46, 47 However, in addition to lowering very low density lipoprotein (VLDL) and low density lipoproteins (LDL), WAY-362450 also decreased HDL.47 In agreement with the gain-of-function studies, FXR knockout mice had increased serum triglyceride, cholesterol, and free fatty acid levels.51
Hepatic FXR:
Multiple cell types in the liver express FXR, including hepatocytes, hepatic stellate cells, endothelial cells, kupffer cells, and cholangiocytes.52–54 The role of FXR in inflammatory cells and in regulating inflammatory signaling pathways will be discussed in this section. The activation of FXR locally in the liver affects the development of each characteristic of NASH; steatosis, inflammation, fibrosis, and metabolic syndrome. The effects on each of these characteristics will be described below. For a summary of the effects of FXR activation in specific cell types, please see Figure 1.
Figure 1–
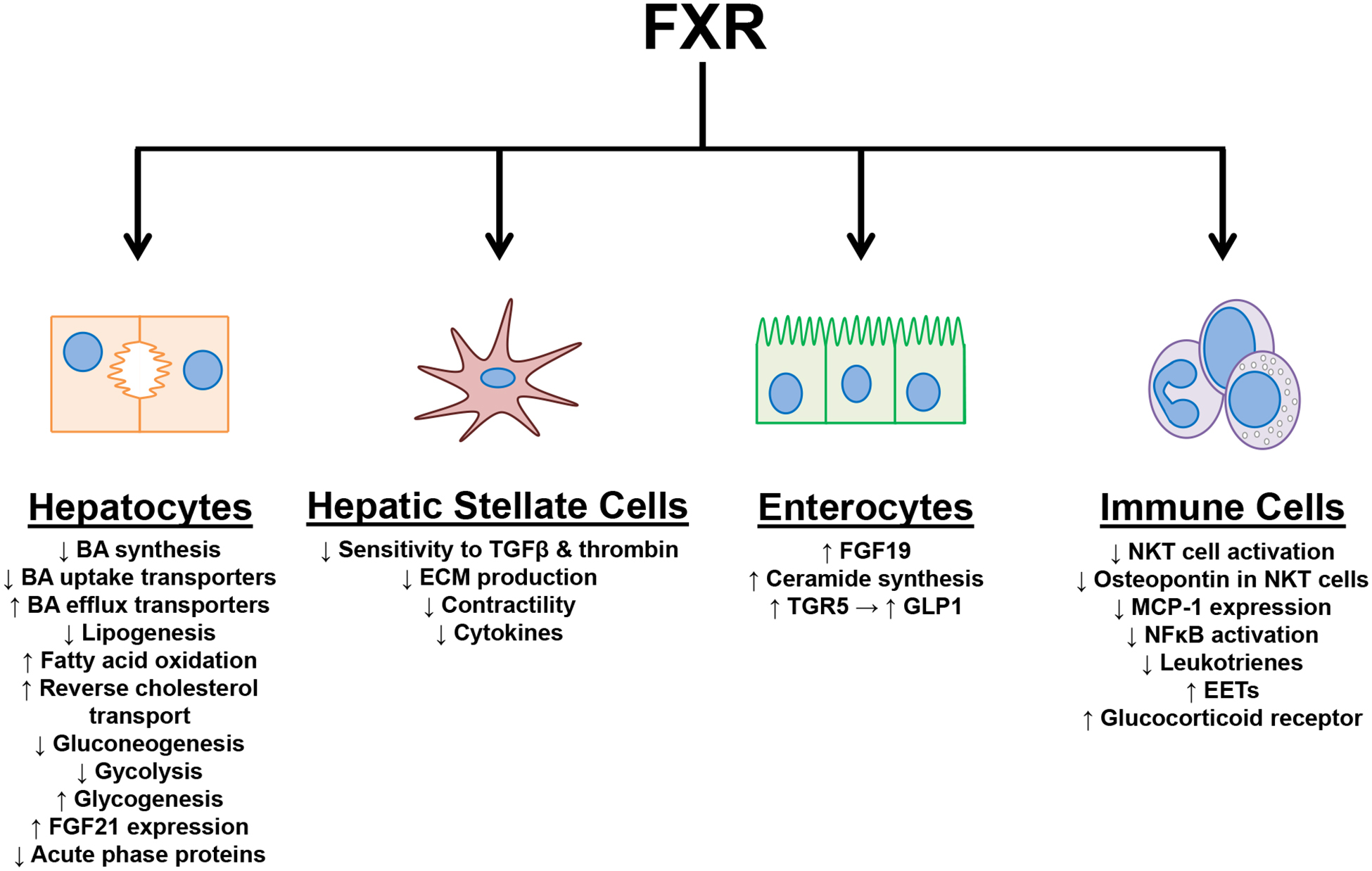
Summary of the effects of FXR activation in specific cell types.
Hepatic FXR activation has been shown to be protective against the development of hepatic steatosis. In a high cholesterol diet model, hepatic FXR deficiency, but not intestinal FXR deficiency, exacerbated hepatic steatosis.55 Hepatic FXR activation mitigates hepatic lipid content by decreasing lipogenesis and increasing fatty acid oxidation.56, 57 By inducing SHP, FXR activation decreased sterol regulatory element-binding protein 1c (SREBP1c) expression and consequently decreased the expression of genes involved in lipogenesis.57 In human hepatocytes, FXR up-regulated peroxisome proliferator-activated receptor alpha (PPARα), which subsequently increased fatty acid oxidation.56 It is important to note that the murine PPARα promoter does not have a functional FXRRE, therefore, in mice PPARα is not an FXR response gene.56 Hepatic FXR also affects lipid homeostasis in the body by enhancing reverse cholesterol transport.58 FXR deficient mice had reduced expression of scavenger receptor class B type 1 (SRB1), hepatic lipase, cholesterol ester hydrolase, sterol carrier protein, and lecithin-cholesterol acyltransferase and increased expression of apolipoproteins (ApoA-IV, ApoE, and ApoC-III).58 Hepatic FXR deficient mice, but not intestinal deficient FXR mice, had increased serum cholesterol compared to wild type mice when fed a high cholesterol diet.55
In addition to regulating hepatic lipid levels, FXR affects hepatic glucose metabolism. FXR activation decreased gluconeogenesis and glycolysis while increasing glycogenesis. CA induced FXR activation in mice reduced the hepatic protein levels of enzymes responsible for gluconeogenesis, peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC1α), phosphoenolpyruvate carboxykinase (PEPCK), and glucose 6-phosphatase (G6Pase). The down-regulation of these proteins by CA did not occur in FXR or SHP deficient mice indicating that FXR regulates gluconeogenesis in the liver via a SHP-dependent pathway.51 Glycogen levels in the liver are increased by FXR activation. In db/db mice, treatment with GW4064 for 5 days increased hepatic glycogen levels by increasing glycogenesis.59 Nonphosphorylated glycogen kinase 3 reduced glycogen synthase activity, however this effect was reduced by phosphorylation.60 Levels of phosphorylated glycogen kinase 3 were increased in the GW4064 treated mice. GW4064 treatment also increased the phosphorylation of insulin receptors 1 and 2. Therefore, FXR may also increase hepatic glycogen levels by enhancing insulin sensitivity.59 In agreement with the previous gain-of-function study, FXR deficient mice had reduced levels of hepatic glycogen.61 FXR activity can also increase hepatic glycogen levels by suppressing glycolysis. Pyruvate dehydrogenase complex (PDC) is an important metabolic switch that regulates the oxidation of glucose for fatty acid synthesis. Pyruvate dehydrogenase kinase 4 (PDK4) inhibits PDC and reduces glycolysis.62 In vitro treatment of human hepatocytes and in vivo treatment of mice with FXR agonist GW4064 increased expression of PDK4 thus decreased glycolysis.63
The metabolic effects of FXR in the liver may also be mediated by fibroblast growth factor 21 (FGF21). The promotor of FGF21 has a functional FXRRE and the expression of FGF21 in the liver has been shown to be regulated by FXR. However, FGF21 is predominantly regulated by PPARα.64, 65 In vivo treatment of mice and in vitro treatment of human hepatocytes with CDCA increases FGF21 expression and secretion.65 Numerous studies have demonstrated the effects of FGF21 on NASH and metabolic endpoints, which has been the subject of many review articles.66–71 In brief, FGF21 increases browning of adipose tissue, energy expenditure, insulin production, glucose uptake by white adipose tissue, gluconeogenesis, ketogenesis, and lipolysis. In NASH models, FGF21 is protective against hepatic steatosis, inflammation, fibrosis and metabolic syndrome.72–74 To our knowledge, in studies using FXR agonists, the extent to which the FXR-FGF21 axis affects NASH or metabolic disease development has not been shown.
FXR activation is anti-inflammatory and affects both innate and adaptive immune responses. Innate immune responses shown to be affected by FXR include the acute phase response and natural killer T-cell (NKT) activation. The acute phase response is a systemic reaction to local or systemic acute infection, illness, or injury.75 During the acute phase response, the expression of acute phase proteins, which are predominantly produced in hepatocytes, is markedly altered; normally increased. In humans, the major acute phase protein is C-reactive protein (CRP) whereas in mice the major acute phase proteins are serum amyloid P component (SAP) and serum amyloid A3 (SAA3).75 FXR activation has been shown to reduce the expression of CRP, SAP, and SAA3. In Hep3B cells, FXR agonism with GW4064 and WAY-362450 mitigated the induction of CRP by interleukin-6.76 Treatment of mice with WAY-362450 reduced LPS stimulated induction of SAP and SAA3 whereas knockout of FXR increased the induction of SAP and SAA3.76 In contrast, FXR activation, at least in mice, has been shown to induce the expression of a cohort of genes involved in acute phase response.77, 78 The exact role of FXR in regulating acute phase response needs further investigation. FXR may also affect the innate immune system by regulating the activation of liver NKT cells. NKT cells have been shown to express both FXR and SHP. In NKT cells, activation of FXR induces SHP, which prevents the binding of c-Jun to the osteopontin promoter.79 Osteopontin has many effects on immune cells including chemotaxis, cellular adhesion, and cell survival.80 In the Con A model of acute hepatitis, OCA treatment reduced the number of FasL positive NKT cells indicating the FXR may mediate NKT cell activation.79
The adaptive immune system is regulated by FXR by several mechanisms; directly altering inflammatory mediator expression, antagonism of the nuclear factor kappa-light-chain-enhancer of activated B cells (NFκB) pathway, and enhancing glucocorticoid signaling. Monocyte chemoattractant protein 1 (MCP-1) is a chemokine that regulates monocyte and macrophage migration and infiltration.81 An FXRRE is present in the promoter of MCP-1. Activation of FXR by CDCA in macrophage cell lines, ANA-1 and RAW264.7, reduced both mRNA and protein levels of MCP-1.82 In primary isolated kupffer cells, OCA mitigated the up-regulation of MCP-1 by both lipopolysaccharide (LPS) and tumor necrosis factor alpha (TNFα).52 In the MCD model of NASH, treatment of mice with FXR agonist WAY-362450 decreased MCP-1 expression in the liver and reduced inflammatory infiltrate.48
Another mechanism by which FXR is anti-inflammatory is through the inhibition of the NFκB signaling pathway. Post-translational modification of FXR can occur at residue K277. This lysine can either be acetylated or SUMOylated. When SUMOylated, FXR can tether to NFκB subunit p65 and prevent the recruitment of p65 to the promoter of its inflammatory response genes. FXR activation increased the amount of SUMOylated FXR and consequently reduced NFκB signaling.83 Treatment of mice with FXR agonists reduced the induction of inflammatory mediators by LPS challenge.84 Similarly, preventing FXR activity or SUMOylation increases inflammatory mediator expression. When challenged with LPS, FXR deficient mice have higher induction of NFκB response genes.84 FXR may also reduce NFκB activation by increasing levels of nuclear factor of kappa light polypeptide gene enhancer in B-cell inhibitor alpha (IκBα), the chaperone protein which prevents the translocation of p65 to the nucleus. In the thioacetamide model of cirrhosis, mice treated with OCA had increased hepatic protein levels of IκBα.52 Lastly, FXR has recently been shown to decrease NFκB pathway activation by increasing the production of anti-inflammatory arachidonic acid derived epoxyeicosatrienoic acids (EETs) and reducing production of inflammatory leukotrienes. During NASH development in humans, the cytochrome p450s which produce EETs are reduced and expression levels inversely correlated to NAS score.85 EETs have been previously shown to reduce NFκB activation.86 In mice fed free fatty acids, OCA increased the expression of cytochrome p450s that synthesize EETs and reduced hepatic inflammation.85
In addition to modulating the activity of the NFκB pathway, FXR regulates glucocorticoid signaling. An FXRRE was identified in the distal portion of the murine and human glucocorticoid receptor promoter.87, 88 As evidenced by chromatin immunoprecipitation and luciferase assay, FXR was recruited to this FXRRE but did not directly alter gene transcription. Instead, the FXRRE functions as an enhancer element and FXR recruitment to this FXRRE mediates chromatin head-to-tail looping, thereby increasing transcriptional efficiency.87 Primary monocytes from wild type and FXR deficient mice were treated with LPS and dexamethasone. Monocytes from FXR deficient mice were less responsive to the anti-inflammatory effects of dexamethasone and had elevated inductions of Il-1β, Tnfα, and interferon-γ.87
Hepatic stellate cells (HSCs) also express FXR in the liver albeit primary isolated rat HSCs express low levels of FXR compared to liver tissue homogenate.89 The rat HSC cell line HSC-T6 and human HSC cell line LX-2 also express FXR.90 Activation of FXR in HSCs affects numerous signaling pathways, which together, function to reduce hepatic fibrosis. The expression of SHP is induced in HSCs by activation of FXR.54, 91 In HSCs, SHP binds to SMAD3 and JunD.54, 91 By binding to SMAD3, SHP prevents SMAD3 from interacting with the transforming growth factor beta (TGFβ) promoter and reduces HSC responsiveness to TGFβ.91 Induction of collagen 1α1 (Col1α1) by TGFβ in HSC-T6 cells was reduced by CDCA.54 In LX-2 cells, OCA treatment reduced TGFβ inductions of COL1α1, alpha smooth muscle actin (αSMA), matrix metalloprotease 2 (MMP2), transforming growth factor beta receptor 2 (TGFβR2), TGFβ, and endothelin-1 (ET-1).91 Through binding to JunD, SHP reduced the binding of activator protein-1 (AP-1) to DNA, thereby preventing HSC activation induced by thrombin.54 OCA treatment of primary rat HSCs and HSC-T6 cells attenuated the induction of tissue inhibitor of metalloproteases 1 (Timp1) by thrombin and increased MMP2 activity in a SHP dependent manner.90
FXR activation also induces the expression of peroxisome proliferator-activated receptor gamma (PPARγ) in HSCs.92 The promoter of PPARγ has been shown to contain a functional FXRRE by a luciferase assay.93 By inducing PPARγ, FXR activation in HSCs reduced the expression of inflammatory cytokines.93 PPARγ is also a negative regulator of collagen expression. During HSC transdifferentiation to an activated phenotype, PPARγ expression is markedly reduced and expression of collagen increases. Treatment of primary rat HSCs with OCA mitigated the down-regulation of PPARγ by random transdifferentiation in culture and reduced collagen expression.92 Primary HSCs were isolated from OCA treated rats that underwent either the porcine serum, bile duct ligation, or CCl4 liver fibrosis models. HSCs from the OCA treated animals had higher expression of PPARγ.92 FXR also decreases extracellular matrix production by increasing the expression of miRNA-29a in HSCs.94 A FXRRE was identified in the miRNA-29a promoter. The expression of extracellular matrix proteins, collagen, elastin, fibrillin, was reduced by miRNA-29a.94
HSC contractility is regulated by FXR. The expression of dimethylarginine dimethylaminohydrolase 2 (DDAH2) is upregulated in HSCs by FXR activation.95 This leads to increased activity of endothelial nitric oxide synthase (eNOS) as DDAH2 degrades asymmetric dimethylarginine and monomethyl-L-arginine, inhibitors of NOS.95, 96 FXR also decreases HSC contractility by decreasing the expression of ET-1.97, 98 Reductions in ET-1 reduces Rho-associated protein kinase pathway activation and reduces the phosphorylation of myosin light chain. FXR activation also reduces phosphorylation of myosin light chain by reducing myosin light chain kinase levels.97 In summary, FXR activation in HSCs reduces extracellular matrix production while increasing extracellular matrix degradation, reduces HSC responsiveness to pro-fibrotic mediators, reduces inflammatory mediator expression, and reduces HSC contractility.
Intestinal FXR:
The role of intestinal FXR during NASH and metabolic disease development is currently unclear. Both inhibition and activation of FXR in the intestine has been shown to have beneficial effects in animal models. In this section we will review the data from studies using both intestinal specific FXR antagonists and agonists.
The beneficial effects of intestinal FXR antagonism on NASH and metabolic diseases are mediated through a microbiome-intestine-liver ceramide axis.99–101 In the intestine, FXR has been shown to upregulate the genes involved in ceramide synthesis.99, 100 Ceramide synthesized in the intestine entered circulation, increased SREBP1c activity in the liver, and subsequently increased lipogenic gene expression.99 Mice fed a HFD were treated with the BA-based FXR antagonist, glycine conjugated MCA (Gly-MCA).100 Gly-MCA reduced hepatic triglyceride accumulation. Gly-MCA also reduced total body weight and fasting insulin levels, improved insulin sensitivity, and led to the browning of adipose tissue. The beneficial effects of Gly-MCA were prevented by co-treatment with ceramide and the FXR agonist GW4064.100 Additionally, treatment of mice with tempol or antibiotics modified the microbiome and increased levels of TβMCA, a FXR antagonist. By increasing TβMCA and inhibiting intestinal FXR, tempol and antibiotic treatment reduced HFD induced hepatic steatosis.99 In a similar study, mice fed a HFD were treated with caffeic acid phenethyl ester (CAPE), a BSH inhibitor.101 CAPE treatment increased ileal levels of TβMCA thereby reducing intestinal FXR activity and ceramide synthesis. CAPE treated mice had reduced body weights, reduced fasting glucose and insulin levels, and improved glucose tolerance. By reducing ceramide levels, CAPE treatment also reduced hepatic endoplasmic reticulum stress and hepatic gluconeogenesis.101 Intestine specific knockout of FXR reduced HFD induced hepatic triglyceride accumulation and steatosis development.102
Reports have also been published demonstrating the benefits of intestinal FXR agonism in animal models. Due to poor systemic bioavailability, fexaramine is an intestinal specific FXR agonist when administered orally.103 In HFD models, mice treated with fexaramine had reduced body weight and body fat mass, increased browning of adipose tissue, and increased energy expenditure.103, 104 Fexaramine treatment reduced expression of genes involved in lipogenesis, triglyceride levels, and steatosis in the liver.103, 104 Glycemic endpoints were also improved by fexaramine; reduced fasting serum insulin and leptin levels, increased serum glucagon-like peptide-1 (GLP1) levels, improved insulin sensitivity, and reduced hepatic gluconeogensis.104 Fexararmine increased intestinal barrier function and decreased circulating levels of inflammatory mediators.103
The effects of intestinal FXR activation described above are mediated through multiple pathways; FXR-Takeda G-protein receptor 5 (TGR5) crosstalk and induction of FGF15/19. TGR5 has been shown to be an FXR response gene. The promoter of TGR5 has a functional FXRRE and FXR activation increases TGR5 mRNA transcript and protein levels.104, 105 Not only does intestinal FXR activation increase TGR5 levels but also increases TGR5 ligands. Fexaramine shifts the BA pool composition to contain markedly higher levels of TLCA and LCA, both strong agonists of TGR5.106, 107 TGR5 activation in the intestine increases serum GLP1 levels. Therefore, the increases in GLP1 levels in fexaramine treated mice are the consequence of enhance TGR5 signaling. Knockout of either Fxr or Tgr5 prevented fexaramine from inducing serum GLP1 concentration and browning of adipose tissue.104 The effects of fexaramine can also be resultant of induction of FGF15. In the intestine, Fgf15 is an FXR target gene. Fexaramine treatment increased intestinal Fgf15 expression and circulating FGF15 protein levels.103, 104 FGF15 and FGF19 have many beneficial effects on NASH and metabolic diseases, which will be described in depth in the following section.
FGF19:
Many of the effects stimulated by activation of intestinal FXR are mediated through the regulation of FGF19. As previously described, FXR activation in the intestine leads to the up-regulation of FGF19.26 Unlike most FGFs, FGF19 does not bind heparin sulfate and therefore can circulate systemically.29 The tissue specific activities of FGF19 are determined by the distribution of FGFR1, FGFR4, and co-receptor βKL throughout the body.31 FGF19 has been shown to regulate the functions of numerous organs paramount to the development of NASH and metabolic diseases including liver, adipose, muscle, and brain. The effects of FGF19 on each of these organs and subsequent effects on NASH and metabolic diseases will be discussed below. For a summary of the effects of FGF19 signaling in specific cell types, please see Figure 2.
Figure 2–
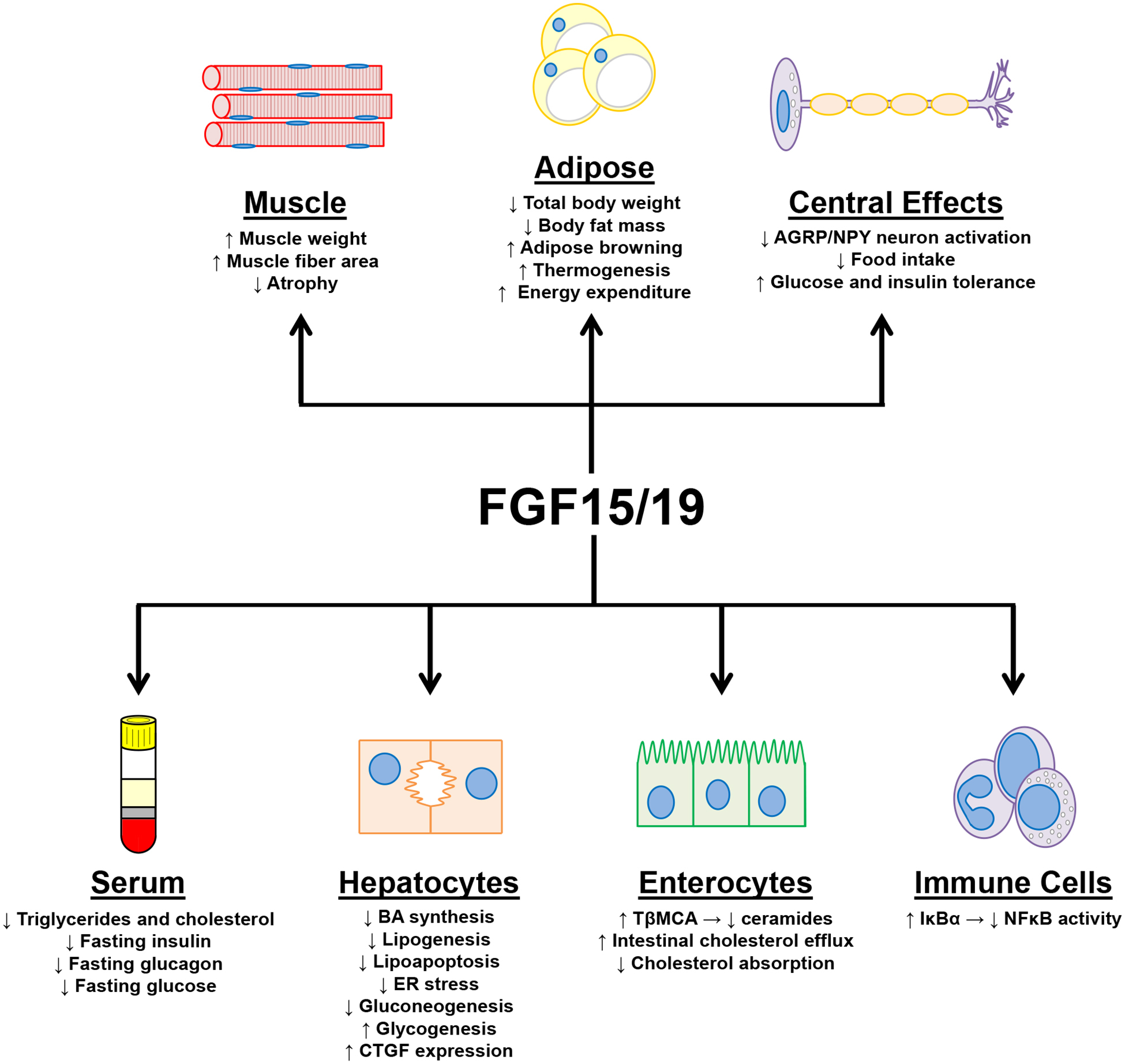
Summary of the effects of FGF19 signaling in specific cell types.
In the liver, FGF15 and FGF19 prevent the development of the major characteristics of NASH; steatosis, inflammation, fibrosis, and metabolic syndrome. FGF19 gain-of-function studies, either from transgenic overexpression or treatment with recombinant or modified FGF19 protein, have shown that FGF19 is protective against triglyceride and cholesterol accumulation in the liver and thereby decreases steatosis.108–110 In agreement, a loss-of-function study found that Fgf15 knockout mice fed a HFD have worsened steatosis.110 A second study which fed a HFD diet to Fgf15 knockout mice did not find worsened steatosis severity but did find altered expression of lipid homeostatic genes.111 FGF15 and FGF19 reduce steatosis by negatively regulating genes involved in lipid synthesis (Fas, Acly, Fatp4, Elovl6, Scd1, Mogat1, Dgat2, Scd1) and lipid uptake (Cd36).108–111 FGF19 has also been shown to reduce steatosis development through altering the composition of the BA pool to contain increased TβMCA. The increased TβMCA levels antagonize intestinal FXR activity and decreased intestinal ceramide synthesis. As previously described (See Section - Intestinal FXR), reduced intestinal ceramide production decreases SREBP1c activation in the liver and subsequently mitigates steatosis.109 In addition to reducing lipid accumulation, FGF19 protects hepatocytes against lipoapoptosis and reduces endoplasmic reticulum stress.109, 110 By altering the BA pool, FGF19 also reduces enterocyte cholesterol absorption, increases transintestinal cholesterol efflux, and increases fecal sterol content.112
FGF15 and FGF19 reduce the development of hepatic inflammation. In a high fat, high fructose, high cholesterol diet mouse model, overexpression of FGF19 or modified FGF19 protein (M70,NGM282) reduced hepatic inflammation severity observed histologically and reduced expression of inflammatory mediators.109 Though not significant, FGF15 deficient mice fed a HFD had trends for worsened inflammation. One mechanism by which FGF19 may mitigate hepatic inflammation is via altering NFκB activity. FGFR4 activation by FGF19 has been shown to reduce NFκB signaling. Activated FGFR4 interacted with inhibitor of nuclear factor kappa-B kinase subunit beta (IKKβ) and decreased IKKβ mediated phosphorylation of IκBα.113
The effect of FGF15 and FGF19 on the development of hepatic fibrosis is currently unclear. In the aforementioned high fat, high fructose, high cholesterol mouse model, FGF19 and M70 overexpression markedly reduced the development of hepatic fibrosis.109 However, in both HFD induced NASH model and CCl4 hepatic fibrosis model, FGF15 deficiency was protective against hepatic fibrosis.111, 114 In a study using the CCl4 model, connective tissue growth factor (CTGF) was shown to be a FGF15 and FGF19 target gene in hepatocytes. Knockout of Fgf15 reduced hepatocyte derived CTGF and ameliorated CCl4 induced fibrosis. Fgf15 knockout also increases total BA pool size and therefore may increase FXR activity in HSCs, which as described previously reduces HSC activation, responsiveness to TGFβ, extracellular matrix production, and contractility. In the FGF19 gain-of-function study, it is possible the reduced fibrosis was resultant of mitigated hepatic steatosis and inflammation, thus reducing the intensity of HSC activating signals.
FGF19 has beneficial effects on the metabolic syndrome: mitigating dyslipidemia, improving glucose homeostasis, reducing total body weights, and reducing body fat mass. Overexpression of FGF19 reduces serum triglyceride and total cholesterol levels.108 In mice fed a diet high in fat, fructose, and cholesterol, FGF19 overexpression reduced triglyceride, total cholesterol, and LDL levels.109 Conversely, FGF15 deficiency in mice increases serum triglyceride levels induced by HFD.111 Fasting serum glucose and insulin levels are decreased by FGF15 and FGF19. Mice overexpressing FGF19 had reduced fasting serum insulin, glucagon, and glucose levels.108, 109 These mice also had improved responses during insulin and glucose tolerance tests.108, 109 Homeostatic model assessment of β cell function and insulin resistance (HOMA-IR), an indicator of insulin resistance, was reduced in transgenic mice.109 The reverse was observed in FGF15 deficient mice which had increased fasting glucose levels and worsened glucose tolerance.111, 115 FGF19 also affects glucose homeostasis in the body by regulating hepatic gluconeogenesis and glycogenesis. In the liver, FGF19 activation of FGFR4-βKL causes the dephosphorylation and inactivation of the cAMP response element binding protein (CREB) and consequently leads to the down-regulation of Pgc1α expression. The lower levels of PGC1α decreases the expression of Pepck and G6Pase, genes involved in gluconeogenesis. Knockout of Fgf15 increased Pgc1α, Pepck, and G6Pase expression.116 Liver glycogenesis is also regulated by FGF15 and FGF19. Liver homogenates from FGF19 treated mice had increased glycogen synthase activity and increased levels of glycogen, whereas FGF15 deficient mice had reduced glycogenesis post glucose challenge.115 The mechanism by which FGF19 increases hepatic glycogenesis is shown to be dependent upon ERK signaling and independent of insulin signaling.115
FGF19 also affects NASH and metabolic disease development by its effects peripherally on adipose and muscle tissue. Adipose tissue does not express FGFR4 and regulation of adipose tissue by FGF19 is mediated by FGFR1-βKL.31, 108 Treatment of mice with FGF19 and transgenic overexpression of FGF19 reduces body fat mass and total body weight.108, 109, 117 When fed a HFD, FGF19 transgenic mice resisted body weight gain and expansion of retroperitoneal and epididymal white adipose tissue.108 Correspondingly, knockout of Fgf15 increased fat mass and total body weight during high fat feeding.110 FGF19 transgenic mice have shown to have increased brown adipose tissue, thermogenesis, and energy expenditure.108, 118
In addition to its effects on adipose tissue, FGF19 also regulates muscle tissue. Treatment of mice with FGF19 increased soleus, tibialis anterior, and gastrocnemius muscle weights in a βKL dependent manner; the number of muscle fibers were not altered by FGF19 but instead fiber area was increased.117 Concordantly, human myotubes treated in vitro with FGF19 have increased area. FGF19 also protects against dexamethasone, obesity, and age induced muscle atrophy. Reductions in atrophy by FGF19 further manifested as improvements in grip strength, an indicator of muscle strength.117
FGF19 not only regulates body weight and glucose homeostasis peripherally but also acts centrally in the brain. A study using radiolabeled iodinated FGF19 examined its pharmacokinetic properties after intravenous injection. After 10 minutes, radiolabeled intact 125I-FGF19 was present in the brain though at low levels. Brain perfusion indicates that FGF19 does cross the blood brain barrier (BBB), but to a limited extent.119 It is important to note that FGF15 and FGF19 are expressed in the developing fetal brain, however, is not expressed in the adult brain.27, 120, 121 It is therefore likely that FGF15 and FGF19 exert their central effects not by crossing the BBB but instead by interacting with neurons that have projections that traverse the BBB. One such neuron type being the agouti-related peptide (AGRP)/ neuropeptide Y (NPY) neurons.122 In the arcuate nucleus of the hypothalamus, AGRP/NPY neurons express FGFR1, FGFR2, and FGFR3 but not FGFR4.123 As shown by immunofluoresence, intraperitoneal injection of FGF19 in mice increased phosphorylation of the FGFR secondary messenger ERK in NPY neurons in the hypothalamic arcuate nucleus.124 FGF19 signaling decreases the activation of AGRP/NPY neurons.124 Expression of c-Fos is a marker of neuron activation.125 HFD fed mice and ob/ob mice have increased NPY/c-Fos co-positive cells in the hypothalamus. FGF19 given by intracerebral ventricular injection (i.c.v.) decreased the number of NPY/c-Fos positive cells in HFD mice.124 The effects of i.c.v. FGF19 on metabolic disease development has been studied in both ob/ob and HFD mouse models.118, 124, 126 In these studies, i.c.v. FGF19 reduced food intake and body weight gain, improved glucose and insulin tolerance, and decreased fasting insulin levels. Inhibition of FGFR in the brain via i.c.v. injection of FGFR inhibitor PD173074 had the opposite effects: increased food intake, total body weight, and worsened insulin tolerance.126 Taurocholic acid (TCA) feeding was shown to increase FGF15 levels and increased glucose tolerance. Tissue-specific knockout of Fgfr1 in AGPR neurons prevented the improvement of glucose tolerance by TCA. These findings indicate the beneficial central effects of FGF15 and FGF19 on glucose homeostasis are likely mediated by FGF19 activation of FGFR1 centrally.123
A bi-specific activating antibody (bFKB1) targeting FGFR1-βKL has been designed and tested in mice and cynomolgus monkeys.127, 128 As the effects of FGF19 on adipose tissue and brain are mediated by FGFR1-βKL, the effects of bFKB1 should mirror the extrahepatic effects of FGF19 but not the hepatic FGFR4 mediated effects. As expected, bFKB1 decreased body weight while increasing browning of adipose tissue, thermogenesis, and energy expenditure. Treatment with bFKB1 also reduces blood glucose and insulin levels, improved glucose tolerance, reduced hepatic triglycerides, and reduced serum lipids. Interestingly, the effects of bFKB1 on brown fat thermogenesis were still present in adipocyte-specific FGFR1 deficient mice and in uncoupling protein 1 (UCP1) deficient mice indicating the effects on thermogenesis may be mediated indirectly.127 In both mice and cynomolgus monkeys, bFKB1 treatment led to inductions of high molecular weight adiponectin. Changes in body weight and energy expenditure are also independent of effects on adiponectin; body weight and energy expenditure changes were also present in adiponectin deficient mice.128 It is possible that the effects of bFKB1 are mediated centrally. Of importance, treatment with bFKB1 did not induce phosphorylation of ERK in the liver.128 This is promising as one of the potential liabilities of FXR agonists and FGF19 analog therapeutics is FGF19-FGFR4 driven hepatocellular carcinoma (See later section - Safety concerns of FXR agonist therapy).
Progress of FXR agonists in human clinical trials:
FXR agonists:
The development of FXR agonists for the treatment of NASH is currently a hotbed of research. Several compounds currently in human clinical trials and one compound, obeticholic acid (OCA), has already been approved for the treatment of another liver disease. These agonists have both steroidal and nonsteroidal pharmacophores and activate FXR systemically. Current progress of these compounds in clinical trials is described below and summarized in Table 1.
Table 1-.
List of completed and on-going clinical trials investigating the use of FXR agonists and FGF19 analogs for the treatment of NASH.
| Mechanism | Compound | Phase | Study Title | Start Date | End Date | NCT ID# |
|---|---|---|---|---|---|---|
| FXR agonist | OCA | 3 | Randomized Global Phase 3 Study to Evaluate the Impact on NASH With Fibrosis of Obeticholic Acid Treatment (REGENERATE) | 9/2015 | 10/2022 | |
| 3 | Study Evaluating the Efficacy and Safety of Obeticholic Acid in Subjects With Compensated Cirrhosis Due to Nonalcoholic Steatohepatitis (REVERSE) | 8/2017 | 7/2021 | |||
| 2 | The Farnesoid X Receptor (FXR) Ligand Obeticholic Acid in NASH Treatment Trial (FLINT) | 3/2011 | 9/2014 | |||
| 1 | Obeticholic Acid in Morbidly Obese Patients and Healthy Volunteers (OCAPUSH) | 8/2015 | 10/2019 | |||
| 1 | Hepatic Impairment Trial of Obeticholic Acid | 6/2013 | 10/2013 | |||
| 1 | Effect of Food on Pharmacokinetics of Obeticholic Acid (OCA) | 8/2013 | 11/2013 | |||
| 1 | Single Dose and Multiple Dose Trial to Assess Pharmacokinetics of Obeticholic Acid (OCA) | 10/2013 | 11/2013 | |||
| Tropifexor (LJN452) | 2 | Safety, Tolerability, and Efficacy of a Combination Treatment of Tropifexor (LJN452) and Cenicriviroc (CVC) in Adult Patients With Nonalcoholic Steatohepatitis (NASH) and Liver Fibrosis (TANDEM) | 8/2018 | 6/2020 | ||
| 2 | Study of Safety and Efficacy of Tropifexor (LJN452) in Patients With Non-alcoholic Steatohepatitis (NASH) (FLIGHT-FXR) | 8/2016 | 9/2019 | |||
| EDP305 | 2 | A Study to Assess the Safety, Tolerability, Pharmacokinetics and Efficacy of EDP-305 in Subjects With Non-Alcoholic Steatohepatitis | 4/2018 | 4/2019 | ||
| 1 | Drug-drug Interaction Study Between EDP-305, Intraconazole and Rifampin in Healthy Volunteers | 7/2017 | 9/2017 | |||
| 1 | A Study of EDP-305 in Subjects With Mild and Moderate Hepatic Impairment Compared With Normal Healthy Volunteers | 6/2017 | 9/2017 | |||
| 1 | Drug-drug Interaction Study Between EDP-305, Midazolam, Caffeine and Rosuvastatin in Healthy Volunteers | 5/2017 | 6/2017 | |||
| 1 | A Study of EDP 305 in Healthy Subjects and Subjects With Presumptive NAFLD | 9/2016 | 6/2017 | |||
| GS-9674 | 2 | Safety and Efficacy of Selonsertib, GS-0976, GS-9674, and Combinations in Participants With Bridging Fibrosis or Compensated Cirrhosis Due to Nonalcoholic Steatohepatitis (NASH) (ATLAS) | 3/2018 | 4/2020 | ||
| 2 | Safety, Tolerability, and Efficacy of Selonsertib, GS-0976, and GS-9674 in Adults With Nonalcoholic Steatohepatitis (NASH) | 7/2016 | 7/2019 | |||
| 2 | Evaluating the Safety, Tolerability, and Efficacy of GS-9674 in Participants With Nonalcoholic Steatohepatitis (NASH) | 10/2016 | 1/2018 | |||
| 1 | Pharmacokinetics and Pharmacodynamics of GS-9674 in Adults With Normal and Impaired Hepatic Function | 7/2016 | 12/2018 | |||
| 1 | Study in Healthy Volunteers to Evaluate the Safety, Tolerability, Pharmacokinetics and Pharmacodynamics of GS-9674, and the Effect of Food on GS-9674 Pharmacokinetics and Pharmacodynamics | 1/2016 | 7/2016 | |||
| Nidufexor (LMB763) | 2 | Safety, Tolerability, Pharmacokinetics and Efficacy of LMB763 in Patients With NASH | 10/2016 | 3/2019 | ||
| Turofexorate (FXR450) | 1 | Study Evaluating the Safety of FXR-450 in Healthy Subjects | 10/2007 | 2/2008 | ||
| EYP001 | 1 | Study Evaluating Safety, Tolerability and Pharmacokinetics of EYP001a in Healthy Male Subjects | 8/2016 | 3/2017 | ||
| FGF19 analog | NGM282 | 2 | Study of NGM282 in Patients With Nonalcoholic Steatohepatitis (NASH) | 5/2015 | 9/2019 | |
| 1/2 | Study of NGM282 in Subjects With Functional Constipation and Healthy Individuals | 12/2015 | 1/2017 | |||
| 1 | SAD and MAD Study of NGM282 in Healthy Adult Participants | 1/2013 | 7/2013 | |||
| FGFR1-βKL activating antibody | NGM313 | 1 | Study of NGM313 in Obese Participants | 9/2017 | 12/2018 | |
| 1 | Study of NGM313 in Healthy Adult Participants | 2/2016 | 4/2017 |
OCA received accelerated approval for the treatment of primary biliary cirrhosis (PBC) in 2016. The accelerated approval was based upon reductions in alkaline phosphatase (ALP) in PBC patients and was given with the condition that improvements in survival or disease outcomes be established.129 To ascertain this information, the FDA required three additional studies; 1) a pharmacokinetic, safety and efficacy study in PBC patients with Child-Pugh classes B and C, 2) a safety and efficacy study of OCA for the monotherapy of PBC in patients intolerant or unresponsive to UDCA, and 3) a study in PBC patients demonstrating that observed decreases in ALP are associated with changes in clinical progression to cirrhosis, transplant, decompensation, or death.130 These trials are to be completed by the end of 2022.130 For the treatment of NASH, OCA has completed both Phase II and Phase III (FLINT) trials with additional Phase III trials underway (REGENERATE and REVERSE).131–134
In the Phase II study, the safety and efficacy of OCA was investigated in patients with NAFLD and type 2 diabetes mellitus.131 64 patients were randomized to placebo (n = 23), 25 mg of OCA (n = 20), and 50 mg of OCA (n = 21). The primary endpoint was changes in insulin sensitivity determined by glucose infusion rate during 2-step euglycemic clamp procedure. Insulin sensitivity was improved in patients in the low dose group and trended for improved in the high dose group. Many additional secondary endpoints were also measured including changes in body weight, serum biomarkers of liver injury, serum biomarkers of BA homeostasis, and fibrosis biomarkers. As expected, OCA increased serum FGF19 levels, suppressed BA synthesis indicated by decreased serum C4 levels (intermediate of BA synthesis used as biomarker of BA synthesis), and reduced serum BA concentrations. OCA had many beneficial effects in patients including reduced body weights, serum triglyceride levels, serum alanine aminotransferase (ALT) and γ-glutamyl-transferase (GGT) activities, and reduction in fibrosis biomarkers. However, of potential concern, OCA increased levels of serum LDL while lowering HDL. Serum ALP levels were also increased in OCA treated patients.131
The “FXR ligand obeticholic acid for non-cirrhotic, non-alcoholic steatohepatitis trial” (FLINT) was a multi-center, randomized, placebo controlled Phase III study.132 142 and 141 patients were randomized to placebo or 25 mg of OCA, respectively, and treated for 72 weeks. The primary outcome of the study was improvement in liver histology defined as a decrease in NAS score by at least 2 points. A greater percentage of patients in the OCA arm compared to the control arm had improved NAS scores and histology scores regarding steatosis, hepatic inflammation, fibrosis, and hepatocyte ballooning. In concordance with the Phase II study, OCA reduced body weights, and serum activities of ALT and GGT.131 There was also a modest decrease in systolic blood pressure in OCA treated patients. Also corresponding to the Phase II trial, OCA increased serum activities of ALP and levels of LDL while decreasing levels of HDL. Contrary to the Phase II study findings, fasting insulin and HOMA-IR were increased in OCA treated patients. The most common side effect was pruritis (23.4% vs 6.3% in placebo), which led to some patients receiving antipruritic mediation or temporary discontinuation of OCA.
Two additional Phase III trials are currently underway investigating the effects of OCA for the treatment of NASH. The REGENERATE trial is a multi-centered, randomized, double blinded, placebo controlled trial that began in September 2015 and is currently recruiting patients. This study aims to follow 2370 participants treated with either placebo, 10 mg of OCA, or 25 mg of OCA for 18 months. Participants will be non-cirrhotic NASH patients with fibrosis scores of 2 or 3. The primary endpoints under investigation are improvements in liver histology and progression to disease related events including common liver complications, HCC, liver transplantation, and death. As the FLINT and REGENERATE trials investigated and will investigate OCA in non-cirrhotic NASH, the REVERSE trial will study the effects of OCA in compensated cirrhotic NASH patients. This trial is a multicenter, randomized, double blinded, placebo controlled study that began in August 2017 and has a targeted estimated enrollment of 540 participants. Patients will be randomized to placebo, 10 mg of OCA, or 25 mg of OCA. The primary endpoint is the percentage of patients with histologic improvement of fibrosis by a score of 1 or more using the NASH Clinical Research Network scoring system. The expected completion dates of the REVERSE and REGENERATE trials are in 2020 and 2022 respectively.
Several non-steroidal FXR agonists have reached clinical trials. Compounds in this class are named using the drug suffix –fexor (i.e. tropifexor, nidufexor, turofexorate). The compound WAY-362450 described in the animal studies above was developed under the name FXR450 or turofexorate. A Phase I study using turofexorate was completed but development was discontinued thereafter.135 The compounds tropifexor (LJN452), nidufexor (LMB763), and EDP305 have completed Phase I trials and are currently in Phase II trials.136–139 A Phase II study was recently completed on GS-9674 (previously known as Px-104 and Px-102), and is currently under investigation in two additional Phase II studies.140–142 GS-9674 is a close analogue of GW4064.143 See Table 1 for a summary of completed and on-going trials with FXR agonists.
FGF19 modified protein:
An analog of FGF19, NGM282, is currently in human clinical trials. A Phase I safety and tolerability study of NGM282 in adults has been completed as well as a 12 week-long Phase II safety, tolerability and efficacy study in NASH patients.144, 145 Findings from the Phase II study mirrored results from preclinical animal studies. NGM282 decreased body weight and BMI. While no changes in hemoglobin A1c were observed, NGM282 reduced serum insulin levels and improved insulin sensitivity as evident by decreased HOMA-IR. NGM282 reduced absolute lipid content in the liver and reduced serum liver injury biomarkers ALT and AST. The levels of serum fibrosis biomarkers (pro-C3, PIIINP, and TIMP1) were reduced by NGM282. Fibrosis severity measured by multiparametric MRI was also decreased by NGM282. Histologic assessment of liver biopsies found that 84% of patients had improved NAS scores and 42% of patients had improved fibrosis stage. The primary difference of the findings from the Phase II study from preclinical animal studies pertains to serum lipid levels. NGM282 increased serum LDL levels in patients, however, concurrent treatment with a statin brought LDL levels back to baseline.146 Common adverse reactions were diarrhea (41% and 36%; 3 mg and 6 mg doses respectively), abdominal pain (30% and 18%; 3 mg and 6 mg doses respectively), and nausea (33% and 14%, 3 mg and 6 mg doses respectively). Due to adverse effects, 32% of patients treated with 6 mg of NGM282 had to interrupt or discontinue therapy.144
As described previously, a FGFR1-βKL bi-specific activating antibody would be expected to have effects comparable to FGF21 and the extrahepatic effects of FGF19 mediated through FGFR1-βKL. NGM313 is an FGFR1-βKL bi-specific activating antibody currently in Phase I trials. NGM313 has already completed a Phase I trial in healthy adults and is now being studied in a Phase I trial in obese individuals.147, 148
Safety concerns of FXR agonist therapy
Experiences with OCA
OCA is currently the only approved FXR agonist on the market and is approved for the treatment of PBC. In September 2017, just under a year and a half after its accelerated approval, an FDA safety communication was released regarding OCA.149 This report described 11 cases of severe liver injury and 19 cases of death associated with OCA therapy. The communication described how these adverse outcomes appear to be due to excessive dosing, in particular frequency of dosing. In the OCA package insert, it is stated that in patients with moderate and severe liver injury, Child-Pugh Class B and C, the serum levels of OCA increase 4 and 17 fold respectively.149 Hence, dose adjustment is required for these patients; the medication is to be dosed weekly instead of daily.129 In the 19 cases of death associated with OCA, 8 cases reported the cause of death. Of these 8 cases, 7 cases involved the daily dosing of OCA in patients with moderate and severe liver injury instead of the recommended weekly dosing. Of the 11 reports of severe liver injury induced by OCA, 6 were cases of patients with moderate or severe liver injury receiving daily dosing of OCA. The safety communication reminded health care providers to assess liver function in all patients before treating with OCA and to follow recommended dose adjustments. In February 2018, a follow-up safety communication was released stating that a black box warning was added to the OCA prescribing information.150 This black box warning highlights the importance of screening liver function, properly selecting dose, and performing monitoring after initiation of therapy.129 This communication urged prescribers to follow dosing on labeling, perform routine biochemical monitoring, re-calculate Child-Pugh class, and adjust dosage accordingly when warranted.150
A second safety concern regarding OCA was identified during Phase II and Phase III NASH clinical trials.131, 132 In both trials, OCA treatment increased serum LDL levels while lowering HDL levels. As most NASH patients have underlying metabolic syndrome and higher rates of cardiovascular disease morbidity and mortality, these changes in serum lipid levels may lead to detrimental consequences. The on-going Phase III REGENERATE trial will study the effects of 18 month long OCA therapy in a targeted 2370 patients with NASH.133 OCA had beneficial effects on liver histology in NASH patients during the FLINT trial and therefore the benefit of OCA may outweigh potential cardiovascular risks. It will be of interest to see how OCA effects the development of NASH and cardiovascular outcomes in the large REGENERATE trial and the risk-benefit of OCA treatment.
Carcinogenicity of FGF19 and relevance of animal carcinogenicity studies
In multiple mouse models, it has been demonstrated that activation of FGFR4/βKL by FGF19, but not FGF15 or NGM282, is carcinogenic.151–154 While FGF15 and FGF19 are orthologs they only share 50% amino acid sequence homology.27, 28 Additionally, FGF15 has an unpaired cysteine not present in the sequence of FGF19. It has been proposed that the unpaired cysteine in FGF15 forms an intermolecular disulfide bond leading to the formation of FGF15 homodimers. In non-reducing gels, it was shown that anti-FGF15 antibodies detect only FGF15 dimers, whereas in reducing gels anti-FGF15 antibodies detect only FGF15 monomers. In both non-reducing and reducing gels, FGF19 is detected as a monomer. This study proposed that FGF15 circulates as a homodimer and therefore may lead to different signaling outcomes than those induced by FGF19. The authors further speculated that the altered configuration of FGF15 is responsible for its lack of carcinogenicity.151 The stark differences in carcinogenicity of FGF15 and FGF19 raise the concern that there is a lack of animal model able to adequately assess the carcinogenicity of FXR agonists. If a FXR agonist is found to be non-carcinogenic in rodent models, one must consider if this is indeed due lack of carcinogenic risk or due to the fact that rodents express non-carcinogenic FGF15 and not carcinogenic FGF19.
Summary
NASH is within the spectrum of NAFLD and is characterized by hepatic steatosis, hepatocyte ballooning, inflammation, fibrosis and is associated with metabolic syndrome. Despite its high prevalence and severe health detriments, there is currently no approved therapy to treat NASH. Great effort has therefore been made to identify mechanisms underlying NASH pathogenesis and to develop efficacious therapies. One target identified to affect NASH development in animal models is the nuclear receptor FXR. Activation of FXR in multiple tissues and cell types attenuates the severity of the major characteristics of NASH. It is for this reason the development of FXR agonists has been an active area of research in the pharmaceutical industry and in academic research. There is currently one FXR agonist, OCA, already on the market for the treatment of PBC. OCA has completed one Phase III clinical trial for the treatment of NASH with two additional Phase III trials on-going. There are several other FXR agonists at various phases of clinical trials. As with most drug targets, FXR agonists have potential safety liabilities. In particular, safety concerns include the worsened serum lipid profile in patients treated with OCA, the carcinogenic risk of FGF19, and the potential lack of relevant animal model for preclinical carcinogenicity studies. Despite these challenges, the development of FXR agonists provides hope for patients with NASH whose only treatment option currently is lifestyle modification and liver transplant. It will be of great interest in the upcoming years to see how FXR agonists perform in on-going clinical trials and whether an FXR agonist becomes the first approved drug for the treatment of NASH.
Abbreviations
- AGRP
Agouti-related peptide
- ALP
Alkaline phosphatase
- ALT
Alanine aminotransferase
- AP-1
Activator protein 1
- ApoA-IV
Apolipoprotein A-IV
- ApoC-III
Apolipoprotein C-III
- ApoE
Apolipoprotein E
- ASBT
Apical sodium dependent bile acid transporter
- BA
Bile acid
- BBB
Blood brain barrier
- bFKB1
Bi-specific activating antibody of FGFR1 and β-Klotho
- BSH
Bile salt hydrolase
- C4
7α-Hydroxy-4-cholesten-3-one
- CA
Cholic acid
- CAPE
Caffeic acid phenethyl ester
- CCl4
Carbon tetrachloride
- CDCA
Chenodeoxycholic acid
- COL1α1
Collagen type 1, α1
- CREB
cAMP response element binding protein
- CRP
C-reactive protein
- CTGF
Connective tissue growth factor
- CYP27A1
Cytochrome P450 27A1
- CYP7A1
Cytochrome P450 7A1
- CYP8B1
Cytochrome P450 8B1
- DCA
Deoxycholic acid
- DDAH2
Dimethylarginine dimethylaminohydrolase 2
- EETs
Epoxyeicosatrienoic acids
- eNOS
Endothelial nitric oxide synthase
- ERK
Extracellular signal-regulated kinases
- Endothelin
1 – ET-1
- FGF15
Fibroblast growth factor 15
- FGF19
Fibroblast growth factor 19
- FGF21
Fibroblast growth factor 21
- FGFR1
Fibroblast growth factor receptor 1
- FGFR4
Fibroblast growth factor receptor 4
- FXR
Farnesoid X receptor
- FXRRE
Farnesoid X receptor response element
- G6Pase
Glucose 6-phosphatase
- GGT
γ-Glutamyl-Transferase
- GLP1
Glucagon-like peptide-1
- Gly-MCA
Glycine conjugated muricholic acid
- HCC
Hepatocellular carcinoma
- HCV
Hepatitis C Virus
- HDL
High density lipoprotein
- HFD
High fat diet
- HOMA-IR
Homeostatic model assessment of β cell function and insulin resistance
- I.C.V.
Intracerebral ventricular injection
- IKKβ
Inhibitor of nuclear factor kappa-B kinase subunit beta
- IκBα
Nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor, alpha
- JNK
c-Jun N-terminal kinase
- βKL
Beta KLOTHO
- LCA
Lithocholic acid
- LDL
Low density lipoprotein
- LDLR
Low density lipoprotein receptor
- LPS
Lipopolysaccharide
- MCA
Muricholic acid
- MCD
Methacholine deficient diet
- MCP-1
Macrophage chemoattractant protein 1
- MMP2
Matrix metalloprotease 2
- NAFLD
Non-alcoholic fatty liver disease
- NAS
NAFLD activity score
- NASH
Non-alcoholic steatohepatitis
- NFκB
Nuclear factor kappa-light-chain-enhancer of activated B cells
- NKT
Natural killer T-cell
- NPY
Neuropeptide Y
- OCA
Obeticholic acid
- PBC
Primary biliary cirrhosis
- PDC
Pyruvate dehydrogenase complex
- PDK4
Pyruvate dehydrogenase kinase 4
- PEPCK
Phosphoenolpyruvate carboxykinase
- PGC1α
Peroxisome proliferator-activated receptor gamma coactivator 1-alpha
- PIIINP
N-terminal propeptide of type III collagen
- PNPLA3
Patatin-like phospholipase domain-containing protein 3
- PPARα
Peroxisome proliferator-activated receptor alpha
- PPARγ
Peroxisome proliferator-activated receptor gamma
- Pro-C3
N-terminal type III collagen propeptide
- RXR
Retinoid X receptor
- SAA3
Serum amyloid A3
- SAF
Steatosis, activity, fibrosis scoring system
- SAP
Serum amyloid P
- SHP
Small heterodimer partner
- αSMA
Alpha smooth muscle actin
- SRB1
Scavenger receptor class B type 1
- SREBP1c
Sterol regulatory element-binding protein 1c
- TCA
Taurocholic acid
- TGFβ
Transforming growth factor beta
- TGFβR2
Transforming growth factor beta receptor 2
- TGR5
Takeda G-protein receptor 5
- TIMP1
Tissue inhibitor of metalloproteases 1
- TNFα
Tumor necrosis factor alpha
- TβMCA
Taurine-conjugated beta-muricholic acid
- UCP1
Uncoupling protein 1
- UDCA
Ursodeoxycholic acid
- VLDL
Very low density lipoprotein
References
- 1.Benedict M & Zhang X Non-alcoholic fatty liver disease: An expanded review. World J Hepatol 9, 715–732 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hashimoto E, Taniai M & Tokushige K Characteristics and diagnosis of NAFLD/NASH. J Gastroenterol Hepatol 28 Suppl 4, 64–70 (2013). [DOI] [PubMed] [Google Scholar]
- 3.Michelotti GA, Machado MV & Diehl AM NAFLD, NASH and liver cancer. Nat Rev Gastroenterol Hepatol 10, 656–65 (2013). [DOI] [PubMed] [Google Scholar]
- 4.Grundy SM et al. Diagnosis and management of the metabolic syndrome: an American Heart Association/National Heart, Lung, and Blood Institute Scientific Statement. Circulation 112, 2735–52 (2005). [DOI] [PubMed] [Google Scholar]
- 5.Day CP & James OF Steatohepatitis: a tale of two “hits”? Gastroenterology 114, 842–5 (1998). [DOI] [PubMed] [Google Scholar]
- 6.Singh S et al. Fibrosis progression in nonalcoholic fatty liver vs nonalcoholic steatohepatitis: a systematic review and meta-analysis of paired-biopsy studies. Clin Gastroenterol Hepatol 13, 643–54.e1–9; quiz e39–40 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Caldwell SH et al. NASH and cryptogenic cirrhosis: a histological analysis. Ann Hepatol 8, 346–52 (2009). [PMC free article] [PubMed] [Google Scholar]
- 8.Mittal S et al. Hepatocellular Carcinoma in the Absence of Cirrhosis in United States Veterans is Associated With Nonalcoholic Fatty Liver Disease. Clin Gastroenterol Hepatol 14, 124–31.e1 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chalasani N et al. The diagnosis and management of nonalcoholic fatty liver disease: Practice guidance from the American Association for the Study of Liver Diseases. Hepatology 67, 328–357 (2018). [DOI] [PubMed] [Google Scholar]
- 10.Dongiovanni P, Anstee QM & Valenti L Genetic predisposition in NAFLD and NASH: impact on severity of liver disease and response to treatment. Curr Pharm Des 19, 5219–38 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Romeo S et al. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat Genet 40, 1461–5 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Younossi ZM et al. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 64, 73–84 (2016). [DOI] [PubMed] [Google Scholar]
- 13.Younossi Z et al. Global burden of NAFLD and NASH: trends, predictions, risk factors and prevention. Nat Rev Gastroenterol Hepatol 15, 11–20 (2018). [DOI] [PubMed] [Google Scholar]
- 14.U.S. Census Bureau. QuickFacts. (2017).
- 15.Organ Procurement and Transplantation Network. National data - Waiting List Additions Listing Year by Diagnosis; January, 1995. - May 31, 2018. [Google Scholar]
- 16.Bedossa P Utility and appropriateness of the fatty liver inhibition of progression (FLIP) algorithm and steatosis, activity, and fibrosis (SAF) score in the evaluation of biopsies of nonalcoholic fatty liver disease. Hepatology 60, 565–75 (2014). [DOI] [PubMed] [Google Scholar]
- 17.Kleiner DE et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 41, 1313–21 (2005). [DOI] [PubMed] [Google Scholar]
- 18.Brunt EM, Janney CG, Di Bisceglie AM, Neuschwander-Tetri BA & Bacon BR Nonalcoholic steatohepatitis: a proposal for grading and staging the histological lesions. Am J Gastroenterol 94, 2467–74 (1999). [DOI] [PubMed] [Google Scholar]
- 19.Vilar-Gomez E & Chalasani N Non-invasive assessment of non-alcoholic fatty liver disease: Clinical prediction rules and blood-based biomarkers. J Hepatol 68, 305–315 (2018). [DOI] [PubMed] [Google Scholar]
- 20.Chiang JY Bile acids: regulation of synthesis. J Lipid Res 50, 1955–66 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chiang JY Recent advances in understanding bile acid homeostasis. F1000Res 6, 2029 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Selwyn FP, Csanaky IL, Zhang Y & Klaassen CD Importance of Large Intestine in Regulating Bile Acids and Glucagon-Like Peptide-1 in Germ-Free Mice. Drug Metab Dispos 43, 1544–56 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Makishima M et al. Identification of a nuclear receptor for bile acids. Science 284, 1362–5 (1999). [DOI] [PubMed] [Google Scholar]
- 24.Wang H, Chen J, Hollister K, Sowers LC & Forman BM Endogenous bile acids are ligands for the nuclear receptor FXR/BAR. Mol Cell 3, 543–53 (1999). [DOI] [PubMed] [Google Scholar]
- 25.Parks DJ et al. Bile acids: natural ligands for an orphan nuclear receptor. Science 284, 1365–8 (1999). [DOI] [PubMed] [Google Scholar]
- 26.Inagaki T et al. Fibroblast growth factor 15 functions as an enterohepatic signal to regulate bile acid homeostasis. Cell Metab 2, 217–25 (2005). [DOI] [PubMed] [Google Scholar]
- 27.Nishimura T, Utsunomiya Y, Hoshikawa M, Ohuchi H & Itoh N Structure and expression of a novel human FGF, FGF-19, expressed in the fetal brain. Biochim Biophys Acta 1444, 148–51 (1999). [DOI] [PubMed] [Google Scholar]
- 28.Xie MH et al. FGF-19, a novel fibroblast growth factor with unique specificity for FGFR4. Cytokine 11, 729–35 (1999). [DOI] [PubMed] [Google Scholar]
- 29.Goetz R et al. Molecular insights into the klotho-dependent, endocrine mode of action of fibroblast growth factor 19 subfamily members. Mol Cell Biol 27, 3417–28 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Song KH, Li T, Owsley E, Strom S & Chiang JY Bile acids activate fibroblast growth factor 19 signaling in human hepatocytes to inhibit cholesterol 7alpha-hydroxylase gene expression. Hepatology 49, 297–305 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kurosu H et al. Tissue-specific expression of betaKlotho and fibroblast growth factor (FGF) receptor isoforms determines metabolic activity of FGF19 and FGF21. J Biol Chem 282, 26687–95 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yu C, Wang F, Jin C, Huang X & McKeehan WL Independent repression of bile acid synthesis and activation of c-Jun N-terminal kinase (JNK) by activated hepatocyte fibroblast growth factor receptor 4 (FGFR4) and bile acids. J Biol Chem 280, 17707–14 (2005). [DOI] [PubMed] [Google Scholar]
- 33.Kong B et al. Mechanism of tissue-specific farnesoid X receptor in suppressing the expression of genes in bile-acid synthesis in mice. Hepatology 56, 1034–43 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fu T et al. FXR Primes the Liver for Intestinal FGF15 Signaling by Transient Induction of betaKlotho. Mol Endocrinol 30, 92–103 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wunsch E et al. Expression of hepatic Fibroblast Growth Factor 19 is enhanced in Primary Biliary Cirrhosis and correlates with severity of the disease. Sci Rep 5, 13462 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schaap FG, van der Gaag NA, Gouma DJ & Jansen PL High expression of the bile salt-homeostatic hormone fibroblast growth factor 19 in the liver of patients with extrahepatic cholestasis. Hepatology 49, 1228–35 (2009). [DOI] [PubMed] [Google Scholar]
- 37.Goodwin B et al. A regulatory cascade of the nuclear receptors FXR, SHP-1, and LRH-1 represses bile acid biosynthesis. Mol Cell 6, 517–26 (2000). [DOI] [PubMed] [Google Scholar]
- 38.Lu TT et al. Molecular basis for feedback regulation of bile acid synthesis by nuclear receptors. Mol Cell 6, 507–15 (2000). [DOI] [PubMed] [Google Scholar]
- 39.Kim I et al. Differential regulation of bile acid homeostasis by the farnesoid X receptor in liver and intestine. J Lipid Res 48, 2664–72 (2007). [DOI] [PubMed] [Google Scholar]
- 40.Lew JL et al. The farnesoid X receptor controls gene expression in a ligand- and promoter-selective fashion. J Biol Chem 279, 8856–61 (2004). [DOI] [PubMed] [Google Scholar]
- 41.Rizzo G et al. Functional characterization of the semisynthetic bile acid derivative INT-767, a dual farnesoid X receptor and TGR5 agonist. Mol Pharmacol 78, 617–30 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sayin SI et al. Gut microbiota regulates bile acid metabolism by reducing the levels of tauro-beta-muricholic acid, a naturally occurring FXR antagonist. Cell Metab 17, 225–35 (2013). [DOI] [PubMed] [Google Scholar]
- 43.Takahashi S et al. Cyp2c70 is responsible for the species difference in bile acid metabolism between mice and humans. J Lipid Res 57, 2130–2137 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mueller M et al. Ursodeoxycholic acid exerts farnesoid X receptor-antagonistic effects on bile acid and lipid metabolism in morbid obesity. J Hepatol 62, 1398–404 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gai Z et al. The effects of farnesoid X receptor activation on arachidonic acid metabolism, NF-kB signaling and hepatic inflammation. Molecular Pharmacology (2018). [DOI] [PubMed] [Google Scholar]
- 46.Ma Y, Huang Y, Yan L, Gao M & Liu D Synthetic FXR agonist GW4064 prevents diet-induced hepatic steatosis and insulin resistance. Pharm Res 30, 1447–57 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Evans MJ et al. A synthetic farnesoid X receptor (FXR) agonist promotes cholesterol lowering in models of dyslipidemia. Am J Physiol Gastrointest Liver Physiol 296, G543–52 (2009). [DOI] [PubMed] [Google Scholar]
- 48.Zhang S, Wang J, Liu Q & Harnish DC Farnesoid X receptor agonist WAY-362450 attenuates liver inflammation and fibrosis in murine model of non-alcoholic steatohepatitis. J Hepatol 51, 380–8 (2009). [DOI] [PubMed] [Google Scholar]
- 49.Wu W et al. Bile acids override steatosis in farnesoid X receptor deficient mice in a model of non-alcoholic steatohepatitis. Biochem Biophys Res Commun 448, 50–5 (2014). [DOI] [PubMed] [Google Scholar]
- 50.Kong B, Luyendyk JP, Tawfik O & Guo GL Farnesoid X receptor deficiency induces nonalcoholic steatohepatitis in low-density lipoprotein receptor-knockout mice fed a high-fat diet. J Pharmacol Exp Ther 328, 116–22 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ma K, Saha PK, Chan L & Moore DD Farnesoid X receptor is essential for normal glucose homeostasis. J Clin Invest 116, 1102–9 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Verbeke L et al. FXR agonist obeticholic acid reduces hepatic inflammation and fibrosis in a rat model of toxic cirrhosis. Sci Rep 6, 33453 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jung D et al. FXR-induced secretion of FGF15/19 inhibits CYP27 expression in cholangiocytes through p38 kinase pathway. Pflugers Arch 466, 1011–9 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fiorucci S et al. The nuclear receptor SHP mediates inhibition of hepatic stellate cells by FXR and protects against liver fibrosis. Gastroenterology 127, 1497–512 (2004). [DOI] [PubMed] [Google Scholar]
- 55.Schmitt J et al. Protective effects of farnesoid X receptor (FXR) on hepatic lipid accumulation are mediated by hepatic FXR and independent of intestinal FGF15 signal. Liver Int 35, 1133–44 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pineda Torra I et al. Bile acids induce the expression of the human peroxisome proliferator-activated receptor alpha gene via activation of the farnesoid X receptor. Mol Endocrinol 17, 259–72 (2003). [DOI] [PubMed] [Google Scholar]
- 57.Watanabe M et al. Bile acids lower triglyceride levels via a pathway involving FXR, SHP, and SREBP-1c. J Clin Invest 113, 1408–18 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lambert G et al. The farnesoid X-receptor is an essential regulator of cholesterol homeostasis. J Biol Chem 278, 2563–70 (2003). [DOI] [PubMed] [Google Scholar]
- 59.Zhang Y et al. Activation of the nuclear receptor FXR improves hyperglycemia and hyperlipidemia in diabetic mice. Proc Natl Acad Sci U S A 103, 1006–11 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Plyte SE, Hughes K, Nikolakaki E, Pulverer BJ & Woodgett JR Glycogen synthase kinase-3: functions in oncogenesis and development. Biochim Biophys Acta 1114, 147–62 (1992). [DOI] [PubMed] [Google Scholar]
- 61.Cariou B et al. Transient impairment of the adaptive response to fasting in FXR-deficient mice. FEBS Lett 579, 4076–80 (2005). [DOI] [PubMed] [Google Scholar]
- 62.Sun W, Liu Q, Leng J, Zheng Y & Li J The role of Pyruvate Dehydrogenase Complex in cardiovascular diseases. Life Sci 121, 97–103 (2015). [DOI] [PubMed] [Google Scholar]
- 63.Savkur RS, Bramlett KS, Michael LF & Burris TP Regulation of pyruvate dehydrogenase kinase expression by the farnesoid X receptor. Biochem Biophys Res Commun 329, 391–6 (2005). [DOI] [PubMed] [Google Scholar]
- 64.Badman MK et al. Hepatic fibroblast growth factor 21 is regulated by PPARalpha and is a key mediator of hepatic lipid metabolism in ketotic states. Cell Metab 5, 426–37 (2007). [DOI] [PubMed] [Google Scholar]
- 65.Cyphert HA et al. Activation of the farnesoid X receptor induces hepatic expression and secretion of fibroblast growth factor 21. J Biol Chem 287, 25123–38 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhang F et al. Minireview: Roles of Fibroblast Growth Factors 19 and 21 in Metabolic Regulation and Chronic Diseases. Mol Endocrinol 29, 1400–13 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Staiger H, Keuper M, Berti L, Hrabe de Angelis M & Haring HU Fibroblast Growth Factor 21-Metabolic Role in Mice and Men. Endocr Rev 38, 468–488 (2017). [DOI] [PubMed] [Google Scholar]
- 68.Potthoff MJ FGF21 and metabolic disease in 2016: A new frontier in FGF21 biology. Nat Rev Endocrinol 13, 74–76 (2017). [DOI] [PubMed] [Google Scholar]
- 69.Markan KR & Potthoff MJ Metabolic fibroblast growth factors (FGFs): Mediators of energy homeostasis. Semin Cell Dev Biol 53, 85–93 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Guan D, Zhao L, Chen D, Yu B & Yu J Regulation of fibroblast growth factor 15/19 and 21 on metabolism: in the fed or fasted state. J Transl Med 14, 63 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Nies VJ et al. Fibroblast Growth Factor Signaling in Metabolic Regulation. Front Endocrinol (Lausanne) 6, 193 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lee JH et al. An engineered FGF21 variant, LY2405319, can prevent non-alcoholic steatohepatitis by enhancing hepatic mitochondrial function. Am J Transl Res 8, 4750–4763 (2016). [PMC free article] [PubMed] [Google Scholar]
- 73.Liu X et al. Lack of fibroblast growth factor 21 accelerates metabolic liver injury characterized by steatohepatities in mice. Am J Cancer Res 6, 1011–25 (2016). [PMC free article] [PubMed] [Google Scholar]
- 74.Fisher FM et al. Fibroblast growth factor 21 limits lipotoxicity by promoting hepatic fatty acid activation in mice on methionine and choline-deficient diets. Gastroenterology 147, 1073–83.e6 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Cray C, Zaias J & Altman NH Acute phase response in animals: a review. Comp Med 59, 517–26 (2009). [PMC free article] [PubMed] [Google Scholar]
- 76.Zhang S, Liu Q, Wang J & Harnish DC Suppression of interleukin-6-induced C-reactive protein expression by FXR agonists. Biochem Biophys Res Commun 379, 476–9 (2009). [DOI] [PubMed] [Google Scholar]
- 77.Armstrong L et al. Effects of Acute-Phase Proteins in Mediating Hepatic FXR’s Protection of Mice from NASH Development. AASLD 2017 Liver Meeting Abstract 643 (2017). [Google Scholar]
- 78.Porez G et al. The hepatic orosomucoid/alpha1-acid glycoprotein gene cluster is regulated by the nuclear bile acid receptor FXR. Endocrinology 154, 3690–701 (2013). [DOI] [PubMed] [Google Scholar]
- 79.Mencarelli A et al. The bile acid sensor farnesoid X receptor is a modulator of liver immunity in a rodent model of acute hepatitis. J Immunol 183, 6657–66 (2009). [DOI] [PubMed] [Google Scholar]
- 80.Wang KX & Denhardt DT Osteopontin: role in immune regulation and stress responses. Cytokine Growth Factor Rev 19, 333–45 (2008). [DOI] [PubMed] [Google Scholar]
- 81.Deshmane SL, Kremlev S, Amini S & Sawaya BE Monocyte chemoattractant protein-1 (MCP-1): an overview. J Interferon Cytokine Res 29, 313–26 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Li L et al. Activation of farnesoid X receptor downregulates monocyte chemoattractant protein-1 in murine macrophage. Biochem Biophys Res Commun 467, 841–6 (2015). [DOI] [PubMed] [Google Scholar]
- 83.Kim DH et al. A dysregulated acetyl/SUMO switch of FXR promotes hepatic inflammation in obesity. Embo j 34, 184–99 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wang YD et al. Farnesoid X receptor antagonizes nuclear factor kappaB in hepatic inflammatory response. Hepatology 48, 1632–43 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Gai Z et al. The effects of farnesoid X receptor activation on arachidonic acid metabolism, NF-kB signaling and hepatic inflammation. Mol Pharmacol (2018). [DOI] [PubMed] [Google Scholar]
- 86.Dai M et al. Epoxyeicosatrienoic acids regulate macrophage polarization and prevent LPS-induced cardiac dysfunction. J Cell Physiol 230, 2108–19 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Renga B et al. FXR mediates a chromatin looping in the GR promoter thus promoting the resolution of colitis in rodents. Pharmacol Res 77, 1–10 (2013). [DOI] [PubMed] [Google Scholar]
- 88.Renga B et al. Glucocorticoid receptor mediates the gluconeogenic activity of the farnesoid X receptor in the fasting condition. Faseb j 26, 3021–31 (2012). [DOI] [PubMed] [Google Scholar]
- 89.Fickert P et al. Farnesoid X receptor critically determines the fibrotic response in mice but is expressed to a low extent in human hepatic stellate cells and periductal myofibroblasts. Am J Pathol 175, 2392–405 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Fiorucci S et al. A farnesoid x receptor-small heterodimer partner regulatory cascade modulates tissue metalloproteinase inhibitor-1 and matrix metalloprotease expression in hepatic stellate cells and promotes resolution of liver fibrosis. J Pharmacol Exp Ther 314, 584–95 (2005). [DOI] [PubMed] [Google Scholar]
- 91.Carino A et al. Disruption of TFGbeta-SMAD3 pathway by the nuclear receptor SHP mediates the antifibrotic activities of BAR704, a novel highly selective FXR ligand. Pharmacol Res 131, 17–31 (2018). [DOI] [PubMed] [Google Scholar]
- 92.Fiorucci S et al. Cross-talk between farnesoid-X-receptor (FXR) and peroxisome proliferator-activated receptor gamma contributes to the antifibrotic activity of FXR ligands in rodent models of liver cirrhosis. J Pharmacol Exp Ther 315, 58–68 (2005). [DOI] [PubMed] [Google Scholar]
- 93.Renga B et al. SHP-dependent and -independent induction of peroxisome proliferator-activated receptor-gamma by the bile acid sensor farnesoid X receptor counter-regulates the pro-inflammatory phenotype of liver myofibroblasts. Inflamm Res 60, 577–87 (2011). [DOI] [PubMed] [Google Scholar]
- 94.Li J et al. Roles of microRNA-29a in the antifibrotic effect of farnesoid X receptor in hepatic stellate cells. Mol Pharmacol 80, 191–200 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Verbeke L et al. Obeticholic acid, a farnesoid X receptor agonist, improves portal hypertension by two distinct pathways in cirrhotic rats. Hepatology 59, 2286–98 (2014). [DOI] [PubMed] [Google Scholar]
- 96.Vallance P, Leone A, Calver A, Collier J & Moncada S Accumulation of an endogenous inhibitor of nitric oxide synthesis in chronic renal failure. Lancet 339, 572–5 (1992). [DOI] [PubMed] [Google Scholar]
- 97.Xu W, Lu C, Zhang F, Shao J & Zheng S Dihydroartemisinin restricts hepatic stellate cell contraction via an FXR-S1PR2-dependent mechanism. IUBMB Life 68, 376–87 (2016). [DOI] [PubMed] [Google Scholar]
- 98.Li J et al. Inhibition of endothelin-1-mediated contraction of hepatic stellate cells by FXR ligand. PLoS One 5, e13955 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Jiang C et al. Intestinal farnesoid X receptor signaling promotes nonalcoholic fatty liver disease. J Clin Invest 125, 386–402 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Jiang C et al. Intestine-selective farnesoid X receptor inhibition improves obesity-related metabolic dysfunction. Nat Commun 6, 10166 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Xie C et al. An Intestinal Farnesoid X Receptor-Ceramide Signaling Axis Modulates Hepatic Gluconeogenesis in Mice. Diabetes 66, 613–626 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Li F et al. Microbiome remodelling leads to inhibition of intestinal farnesoid X receptor signalling and decreased obesity. Nat Commun 4, 2384 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Fang S et al. Intestinal FXR agonism promotes adipose tissue browning and reduces obesity and insulin resistance. Nat Med 21, 159–65 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Pathak P et al. Intestine farnesoid X receptor agonist and the gut microbiota activate G-protein bile acid receptor-1 signaling to improve metabolism. Hepatology (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Pathak P et al. Farnesoid X receptor induces Takeda G-protein receptor 5 cross-talk to regulate bile acid synthesis and hepatic metabolism. J Biol Chem 292, 11055–11069 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Kawamata Y et al. A G protein-coupled receptor responsive to bile acids. J Biol Chem 278, 9435–40 (2003). [DOI] [PubMed] [Google Scholar]
- 107.Maruyama T et al. Identification of membrane-type receptor for bile acids (M-BAR). Biochem Biophys Res Commun 298, 714–9 (2002). [DOI] [PubMed] [Google Scholar]
- 108.Tomlinson E et al. Transgenic mice expressing human fibroblast growth factor-19 display increased metabolic rate and decreased adiposity. Endocrinology 143, 1741–7 (2002). [DOI] [PubMed] [Google Scholar]
- 109.Zhou M et al. Engineered FGF19 eliminates bile acid toxicity and lipotoxicity leading to resolution of steatohepatitis and fibrosis in mice. Hepatol Commun 1, 1024–1042 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Alvarez-Sola G et al. Fibroblast growth factor 15/19 (FGF15/19) protects from diet-induced hepatic steatosis: development of an FGF19-based chimeric molecule to promote fatty liver regeneration. Gut 66, 1818–1828 (2017). [DOI] [PubMed] [Google Scholar]
- 111.Schumacher JD et al. The effect of fibroblast growth factor 15 deficiency on the development of high fat diet induced non-alcoholic steatohepatitis. Toxicol Appl Pharmacol 330, 1–8 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.de Boer JF et al. Intestinal Farnesoid X Receptor Controls Transintestinal Cholesterol Excretion in Mice. Gastroenterology 152, 1126–1138.e6 (2017). [DOI] [PubMed] [Google Scholar]
- 113.Drafahl KA, McAndrew CW, Meyer AN, Haas M & Donoghue DJ The receptor tyrosine kinase FGFR4 negatively regulates NF-kappaB signaling. PLoS One 5, e14412 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Uriarte I et al. Ileal FGF15 contributes to fibrosis-associated hepatocellular carcinoma development. Int J Cancer 136, 2469–75 (2015). [DOI] [PubMed] [Google Scholar]
- 115.Kir S et al. FGF19 as a postprandial, insulin-independent activator of hepatic protein and glycogen synthesis. Science 331, 1621–4 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Potthoff MJ et al. FGF15/19 regulates hepatic glucose metabolism by inhibiting the CREB-PGC-1alpha pathway. Cell Metab 13, 729–38 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Benoit B et al. Fibroblast growth factor 19 regulates skeletal muscle mass and ameliorates muscle wasting in mice. Nat Med 23, 990–996 (2017). [DOI] [PubMed] [Google Scholar]
- 118.Fu L et al. Fibroblast growth factor 19 increases metabolic rate and reverses dietary and leptin-deficient diabetes. Endocrinology 145, 2594–603 (2004). [DOI] [PubMed] [Google Scholar]
- 119.Hsuchou H, Pan W & Kastin AJ Fibroblast growth factor 19 entry into brain. Fluids Barriers CNS 10, 32 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Gimeno L, Brulet P & Martinez S Study of Fgf15 gene expression in developing mouse brain. Gene Expr Patterns 3, 473–81 (2003). [DOI] [PubMed] [Google Scholar]
- 121.Fon Tacer K et al. Research resource: Comprehensive expression atlas of the fibroblast growth factor system in adult mouse. Mol Endocrinol 24, 2050–64 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Faouzi M et al. Differential accessibility of circulating leptin to individual hypothalamic sites. Endocrinology 148, 5414–23 (2007). [DOI] [PubMed] [Google Scholar]
- 123.Liu S et al. A gut-brain axis regulating glucose metabolism mediated by bile acids and competitive fibroblast growth factor actions at the hypothalamus. Mol Metab 8, 37–50 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Marcelin G et al. Central action of FGF19 reduces hypothalamic AGRP/NPY neuron activity and improves glucose metabolism. Mol Metab 3, 19–28 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Bullitt E Expression of c-fos-like protein as a marker for neuronal activity following noxious stimulation in the rat. J Comp Neurol 296, 517–30 (1990). [DOI] [PubMed] [Google Scholar]
- 126.Ryan KK et al. Fibroblast growth factor-19 action in the brain reduces food intake and body weight and improves glucose tolerance in male rats. Endocrinology 154, 9–15 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Chen MZ et al. FGF21 mimetic antibody stimulates UCP1-independent brown fat thermogenesis via FGFR1/betaKlotho complex in non-adipocytes. Mol Metab 6, 1454–1467 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Kolumam G et al. Sustained Brown Fat Stimulation and Insulin Sensitization by a Humanized Bispecific Antibody Agonist for Fibroblast Growth Factor Receptor 1/betaKlotho Complex. EBioMedicine 2, 730–43 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Ocaliva (obeticholic acid)[package insert]. Intercept Pharmaceuticals, Inc, New York, NY: (2018). [Google Scholar]
- 130.FDA approval letter - Ocaliva; NDA 207999.. (2016).
- 131.Mudaliar S et al. Efficacy and safety of the farnesoid X receptor agonist obeticholic acid in patients with type 2 diabetes and nonalcoholic fatty liver disease. Gastroenterology 145, 574–82.e1 (2013). [DOI] [PubMed] [Google Scholar]
- 132.Neuschwander-Tetri BA et al. Farnesoid X nuclear receptor ligand obeticholic acid for non-cirrhotic, non-alcoholic steatohepatitis (FLINT): a multicentre, randomised, placebo-controlled trial. Lancet 385, 956–65 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Intercept Pharmaceuticals. Randomized Global Phase 3 Study to Evaluate the Impact on NASH With Fibrosis of Obeticholic Acid Treatment (REGENERATE). ClinicalTrials.gov Identifier: NCT02548351 (Revised May 28, 2018).
- 134.Intercept Pharmaceuticals. Study Evaluating the Efficacy and Safety of Obeticholic Acid in Subjects With Compensated Cirrhosis Due to Nonalcoholic Steatohepatitis (REVERSE). ClinicalTrials.gov Identifier: NCT03439254 (Revised May 24, 2018).
- 135.Pharmaceuticals W Study Evaluating the Safety of FXR-450 in Healthy Subjects. ClinicalTrials.gov Identifier: NCT00499629 (Revised March 13, 2008).
- 136.Novartis Pharmaceuticals. Study of Safety and Efficacy of Tropifexor (LJN452) in Patients With Non-alcoholic Steatohepatitis (NASH) (FLIGHT-FXR). ClinicalTrials.gov Identifier: NCT02855164 (Revised April 18, 2018).
- 137.Novartis Pharmaceuticals. Safety, Tolerability, and Efficacy of a Combination Treatment of Tropifexor (LJN452) and Cenicriviroc (CVC) in Adult Patients With Nonalcoholic Steatohepatitis (NASH) and Liver Fibrosis (TANDEM). ClinicalTrials.gov Identifier: NCT03517540 (Revised May 17, 2018).
- 138.Novartis Pharmaceuticals. Safety, Tolerability, Pharmacokinetics and Efficacy of LMB763 in Patients With NASH. ClinicalTrials.gov Identifier: NCT02913105 (Revised May 31, 2018).
- 139.Enanta Pharmaceuticals. A Study to Assess the Safety, Tolerability, Pharmacokinetics and Efficacy of EDP-305 in Subjects With Non-Alcoholic Steatohepatitis. ClinicalTrials.gov Identifier: NCT03421431 (Revised March 29, 2018).
- 140.Gilead Sciences. Evaluating the Safety, Tolerability, and Efficacy of GS-9674 in Participants With Nonalcoholic Steatohepatitis (NASH). ClinicalTrials.gov Identifier: NCT02854605 (Revised January 17, 2018).
- 141.Gilead Sciences. Safety, Tolerability, and Efficacy of Selonsertib, GS-0976, and GS-9674 in Adults With Nonalcoholic Steatohepatitis (NASH). ClinicalTrials.gov Identifier: NCT02781584 (Revised February 8, 2018).
- 142.Gilead Sciences. Safety and Efficacy of Selonsertib, GS-0976, GS-9674, and Combinations in Participants With Bridging Fibrosis or Compensated Cirrhosis Due to Nonalcoholic Steatohepatitis (NASH) (ATLAS). ClinicalTrials.gov Identifier: NCT03449446 (Revised June 11, 2018).
- 143.Hambruch E et al. Synthetic farnesoid X receptor agonists induce high-density lipoprotein-mediated transhepatic cholesterol efflux in mice and monkeys and prevent atherosclerosis in cholesteryl ester transfer protein transgenic low-density lipoprotein receptor (−/−) mice. J Pharmacol Exp Ther 343, 556–67 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Harrison SA et al. NGM282 for treatment of non-alcoholic steatohepatitis: a multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. Lancet 391, 1174–1185 (2018). [DOI] [PubMed] [Google Scholar]
- 145.NGM Biopharmaceuticals. Phase 1 SAD and MAD Study of NGM282 in Healthy Adult Participants. ClinicalTrials.gov Identifier: NCT01776528 (Revised December 31, 2013).
- 146.Harrison S et al. NGM282 improves fibrosis and NASH-related histology in 12 weeks in patients with biopsy-confirmed NASH, which is preceded by significant decreases in hepatic steatosis, liver transaminases and fibrosis markers at 6 weeks. Journal of Hepatology 68, S65–S66 (2018). [Google Scholar]
- 147.NGM Biopharmaceuticals. Phase 1 Study of NGM313 in Healthy Adult Participants. ClinicalTrials.gov Identifier: NCT02708576 (Revised September 14, 2017).
- 148.NGM Biopharmaceuticals. Study of NGM313 in Obese Participants. ClinicalTrials.gov Identifier: NCT03298464 (Revised December 7, 2017).
- 149.U.S. Food and Drug Administration. Drug Safety Communication: FDA Drug Safety Communication: FDA warns about serious liver injury with Ocaliva (obeticholic acid) for rare chronic liver disease. (September/21/2017).
- 150.Administration, U.S.F.a.D. Drug Safety Communication - Ocaliva (obeticholic acid): Drug Safety Communication - Boxed Warning Added To Highlight Correct Dosing. (February/1/2018).
- 151.Zhou M et al. Mouse species-specific control of hepatocarcinogenesis and metabolism by FGF19/FGF15. J Hepatol 66, 1182–1192 (2017). [DOI] [PubMed] [Google Scholar]
- 152.Wu X et al. FGF19-induced hepatocyte proliferation is mediated through FGFR4 activation. J Biol Chem 285, 5165–70 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Zhou M et al. Separating Tumorigenicity from Bile Acid Regulatory Activity for Endocrine Hormone FGF19. Cancer Res 74, 3306–16 (2014). [DOI] [PubMed] [Google Scholar]
- 154.Luo J et al. A nontumorigenic variant of FGF19 treats cholestatic liver diseases. Sci Transl Med 6, 247ra100 (2014). [DOI] [PubMed] [Google Scholar]