

ABSTRACT
Sonic hedgehog (Shh) signal transduction specifies ventral cell fates in the neural tube and is mediated by the Gli transcription factors that play both activator (GliA) and repressor (GliR) roles. Cilia are essential for Shh signal transduction and the ciliary phosphatidylinositol phosphatase Inpp5e is linked to Shh regulation. In the course of a forward genetic screen for recessive mouse mutants, we identified a functional null allele of inositol polyphosphate-5-phosphatase E (Inpp5e), ridge top (rdg), with expanded ventral neural cell fates at E10.5. By E12.5, Inpp5erdg/rdg embryos displayed normal neural patterning and this correction over time required Gli3, the predominant repressor in neural patterning. Inpp5erdg function largely depended on the presence of cilia and on smoothened, the obligate transducer of Shh signaling, indicating that Inpp5e functions within the cilium to regulate the pathway. These data indicate that Inpp5e plays a more complicated role in Shh signaling than previously appreciated. We propose that Inpp5e attenuates Shh signaling in the neural tube through regulation of the relative timing of GliA and GliR production, which is important in understanding how the duration of Shh signaling regulates neural tube patterning.
KEY WORDS: Inpp5e, Phosphatidylinositol phosphatase, Cilia, Sonic hedgehog, Neural tube patterning
Highlighted Article: Inositol polyphosphate-5-phosphatase E attenuates sonic hedgehog signal transduction through a combination of positive and negative regulatory roles that likely control the relative timing of Gli processing.
INTRODUCTION
Shh signaling plays a major role in determining ventral cell fate identity in the developing neural tube (Echelard et al., 1993; Roelink et al., 1994). The floor plate cells at the ventral midline require high Shh concentration during a critical developmental time window (Ribes et al., 2010). Other ventral cell fates are specified due to both the concentration and duration of Shh signaling. For example, Nkx2.2-positive V3 interneuron precursors can be specified by either high Shh concentration or low Shh concentration over time (Dessaud et al., 2007). Thus, the ability of a cell to monitor the duration of signaling is crucial but we currently lack a detailed understanding of how cells interpret the duration of Shh signal.
The downstream effectors of Shh signaling are the Gli transcription factors Gli1, Gli2 and Gli3, which possess context-dependent activator and repressor functions. In the neural tube, Gli2 is the primary activator and Gli3 is the major repressor (Ding et al., 1998; Matise et al., 1998; Litingtung and Chiang, 2000; Persson et al., 2002; Bai et al., 2004). Full-length Gli protein is processed to a mature activator (GliA) form or cleaved to a repressor (GliR) form based on the presence or absence of Shh ligand. GliR production in the absence of ligand depends on the orphan G-protein receptor Gpr161 increasing cAMP levels and protein kinase A (PKA) activity (Mukhopadhyay et al., 2013). PKA phosphorylates full-length Gli to promote its cleavage to GliR and further phosphorylates full-length Gli to prevent it from being processed to GliA (Mukhopadhyay et al., 2013; Niewiadomski et al., 2014). Thus, Shh mutants produce only GliR and do not specify ventral cell fates (Litingtung and Chiang, 2000; Chiang et al., 1996). In contrast, upon Shh stimulation, decreased ciliary cAMP levels antagonize PKA activity, preventing GliR and enabling GliA production (Moore et al., 2016). Thus, the physiological Shh response at any given position in the neural tube is determined by an effective Gli ratio, which integrates opposing gradients of activating GliA and repressive GliR.
The primary cilium is a microtubule-based projection found on almost all cells and is intimately associated with Shh signaling (Huangfu et al., 2003; Corbit et al., 2005; Rohatgi et al., 2007; Haycraft et al., 2005). Mutations in proteins required for cilia biogenesis and function lead to a breakdown in Shh signal transduction regulation and either an increase or an absence of ventral neural cell fates (reviewed by Bangs and Anderson, 2017). Several components of the Shh signaling pathway traffic to cilia and move dynamically upon pathway stimulation. These include the Shh receptor patched 1 (Ptch1) and the obligate transducer of the pathway, smoothened (Smo) (Corbit et al., 2005; Rohatgi et al., 2007). Smo enriches in cilia upon Shh stimulation and this enhanced localization is necessary but not sufficient for its activation. Concomitantly, Gli proteins are enriched at the ciliary tip (Haycraft et al., 2005). Upon genetic ablation of cilia, few ventral cell fates are specified because Gli proteins are not processed to either GliA or to GliR (Huangfu and Anderson, 2005; Liu et al., 2005).
The ciliary membrane is biochemically distinct from the plasma membrane as its phosphatidyl inositol (PI) composition is enriched in phosphatidyl inositol 4 phosphate [PI(4)P] (Chávez et al., 2015; Garcia-Gonzalo et al., 2015). PI(4)P is important in regulating the ciliary localization of Ptch1, as well as ciliary enrichment and activation of Smo (Jiang et al., 2016). In Drosophila, PI(4)P is converted between PI(4)P and PI by Sac1 phosphatase and Stt4 kinase. Sac1 mutants accumulate PI(4)P, leading to increased Shh signaling response (Yavari et al., 2010). Similarly, ptc mutants accumulate PI(4)P and increase Shh pathway activation (Yavari et al., 2010). This mechanism is conserved in mammalian NIH3T3 cells, where PI(4)P treatment induces pathway activation along with Smo ciliary enrichment (Jiang et al., 2016). PI(4)P binds to Ptch1 as well as to Smo (Jiang et al., 2016). Stimulation with Shh increases binding of PI(4)P to Smo and decreases binding to Ptch1, making this molecule an attractive candidate for explaining how Ptch1 inhibits Smo, a crucial step in Shh signaling (Jiang et al., 2016).
The ciliary inositol polyphosphate-5-phosphatase E (Inpp5e) converts PI(3,4,5)P3 and PI(4,5)P2 (hereafter PIP2) to PI(3,4)P2 and PI(4)P, respectively (Kisseleva et al., 2000). Inpp5e localizes to cilia where it maintains PI(4)P levels (Chávez et al., 2015; Garcia-Gonzalo et al., 2015; Jacoby et al., 2009; Bielas et al., 2009). Previous work investigated the role of Inpp5e in regulating Shh signaling. Mouse embryonic fibroblasts (MEFs) and neural stem cells lacking Inpp5e with either of two distinct null alleles, Inpp5etm1.1Cmit (hereafter Inpp5eΔEx2-6) or Inpp5etm1.2Ssch (hereafter Inpp5eΔEx7-8), display a diminished transcriptional response upon Shh stimulation (Chávez et al., 2015; Dyson et al., 2017; Garcia-Gonzalo et al., 2015). This correlates with increased ciliary PIP2 levels recruiting Tulp3 and the Shh antagonist Gpr161 into cilia (Garcia-Gonzalo et al., 2015; Dyson et al., 2017). Gpr161 resides in the cilium in the absence of Shh and generates cAMP (Mukhopadhyay et al., 2013). Increased levels of cAMP activate PKA, resulting in cleavage to GliR and repression of the Shh response (Tuson et al., 2011; Wang et al., 2000). Consistent with this, the ventral-most neural cell fates requiring the highest level of Shh response are lost in Inpp5eΔEx2-6/ΔEx2-6 embryos (Dyson et al., 2017). The simplest interpretation of these data is that Inpp5e normally plays an activating role in Shh signaling. However, ventral neural cell fates requiring intermediate levels of Shh response are dorsally expanded in the Inpp5eΔEx2-6/ΔEx2-6 mutants, suggesting a more complicated mechanism is at play (Dyson et al., 2017).
Here, we investigate the role of Inpp5e in the Shh signaling pathway using an Inpp5e point mutant we identified in a forward genetic screen. We genetically demonstrate that this mutation generates a functional null allele and that loss of Inpp5e activity leads to an initial expansion of ventral neural tube cell fates, indicating Inpp5e negatively regulates the Shh response. Interestingly, we found that the ventral pattern corrects over time and we demonstrate this mechanism requires Gli3, the predominant repressor in neural patterning. Inpp5e localizes to cilia, which we show are required for Inpp5e function in regulating Shh signaling. We found that when Inpp5e function is absent, Smo is required for the highest Shh response but not for more intermediate Shh patterning responses. From these data, we propose that Inpp5e plays a crucial role in controlling the level of Shh response over time by attenuating the pathway through control of the timing of GliA/GliR gradient production. These data provide a mechanistic framework from which to understand how the duration of Shh signaling regulates ventral neural cell fate.
RESULTS
Inpp5e negatively regulates the Shh signaling response in the E10.5 neural tube
We have previously published the Inpp5eM2 line, and showed it carries an A-to-G transition in the mouse Inpp5e gene as well as a change in the Slc2a6 gene 0.625 Mb away (Sun et al., 2012). To eliminate the Slc2a6 mutation, we backcrossed to FVB and generated the recombinant chromosome carrying only the Inpp5e mutation. To distinguish this recombinant line from the Inpp5eM2 line, we refer to this line as ridge top (rdg), Inpp5erdg, to reflect the exencephaly that resembles a ridge top hat.
We evaluated neural tube patterning in Inpp5erdg/rdg mutant embryos at embryonic day (E) 10.5 by staining hindlimb sections with antibodies for specific cell fates. Shh is initially expressed in the notochord and subsequently in the floor plate, which also expresses FoxA2 (Echelard et al., 1993; Roelink et al., 1994; Sasaki and Hogan, 1994; Riddle et al., 1993). In wild type and Inpp5erdg/rdg mutants, the floor plate was visible and expressed both Shh and FoxA2 (Fig. 1A,B,F,G). Additionally, we observed FoxA2-positive cells that did not co-express Shh scattered dorsally within the ventricular zone in Inpp5erdg/rdg mutants (Fig. 1G). We quantified the dorsal boundary of the domain, taking the differences in neural tube shape and size into account, and reported this number as a percentage of the total neural tube distance (detailed in the Materials and Methods). Indeed, we found a significant expansion of the FoxA2 dorsal boundary between control and Inpp5erdg/rdg mutants (Fig. 1K). In wild-type sections, p3 cells adjacent to the floorplate expressed Nkx2.2, whereas in Inpp5erdg/rdg mutant sections, Nkx2.2-positive cells expanded dorsally in a dispersed manner, similar to what we observed with FoxA2 (Fig. 1C,H,K). Additional ventral cell fates expressing Nkx6.1 and Olig2 also expanded dorsally in the Inpp5erdg/rdg mutants (Fig. 1H,I,K). The dorsal boundary for all the expanded cell fates correlated to the ventral boundary of Pax6, which is repressed by Shh signaling (Lek et al., 2010; Ericson et al., 1997). We quantified the ventral boundary analogously to the dorsal boundary (detailed in the Materials and Methods) and found the Pax6 ventral boundary is shifted dorsally in Inpp5erdg/rdg mutants compared with wild-type controls (Fig. 1E,J,L). Taken together, these data demonstrate an expansion of Shh-dependent ventral cell fates in Inpp5erdg/rdg mutant embryos, suggesting an expansion of the Shh activity gradient (Patterson et al., 2009).
Fig. 1.

Inpp5e negatively regulates the Shh signaling response in the E10.5 neural tube. (A-J) Caudal (hindlimb) sections of E10.5 control (A-E, n=7) and Inpp5erdg/rdg (F-J, n=4) neural tubes. (A,F) Inpp5erdg/rdg mutants display normal Shh expression in the floor plate. (B-D,G-I) FoxA2, Olig2, Nkx2.2 and Nkx6.1 are expanded dorsally in Inpp5erdg/rdg mutants. The FoxA2, Olig2 and Nkx2.2 domains are intermixed with a few dorsally scattered FoxA2 cells (G, asterisks). (E,J) The Pax6 domain is shifted dorsally in Inpp5erdg/rdg mutants. (K,L) Quantitation of dorsal and ventral boundary as a percentage of total lumen distance (see Materials and Methods for details). (M,O) Whole-mount and (N,P) neural tube sections of Inpp5e+/+;Ptch1LacZ/+ (n=4) and Inpp5erdg/rdg;Ptch1LacZ/+ embryos (n=3) stained for β-galactosidase activity. (M-P) Inpp5erdg/rdg;Ptch1LacZ/+ mutants display a dorsal expansion of β-galactosidase activity. The extent of the gradient is indicated by a black triangle. (Q) Quantitation of N and P, indicating dorsal boundary. (R,S) Wild-type (R; n=3) and Inpp5erdg/rdg (S; n=3) neural tube sections stained with antibodies against Smo and Arl13b. Bracket depicts region containing Smo-positive cilia. (R′,R″,S′,S″) Enlarged views of cilia from regions indicated by dotted boxes in R and S. (T) Schematic describing quantification of the region of Smo staining and graph reflecting percentage of ventricular lumen displaying ciliary Smo enrichment (see Materials and Methods for details). (K,L,Q,T) Bar indicates mean of biological replicates analyzed by two tailed unpaired t-test with Welch's correction. Individual data points are shown. *P<0.05, **P<0.01, ***P<0.001. Scale bars: 100 µm for A-J,N,P,R,S; 400 µm for M,O; 10 µm for R′,R″,S′,S″.
We also examined neural tube patterning at a more rostral axial level: the forelimb level (Fig. S1). We found no significant shift in the dorsal boundary of FoxA2 and Nkx2.2 or in the ventral boundary of Pax6 in Inpp5erdg/rdg when compared with control embryos (Fig. S1A,B,D-F,H-J). We did observe a shift in the dorsal boundary of Olig2 progenitors similar to the caudal neural tube (Fig. S1C,G,I). This is consistent with observations in mutants of other cilia-related proteins, which also display a more severe neural patterning phenotype in the caudal neural tube than seen in the rostral neural tube (Caspary et al., 2007; Patterson et al., 2009; Eggenschwiler and Anderson, 2000; Xin et al., 2017; Huang et al., 2002; Bulgakov et al., 2004).
In order to monitor the Shh activity gradient, we used a Ptch1LacZ allele as Ptch1 is a transcriptional target of the Shh signaling pathway (Goodrich et al., 1997, 1996). Upon X-gal staining of whole E10.5 wild-type and Inpp5erdg/rdg mutant embryos, we detected blue staining, reflecting the Shh transcriptional response (Fig. 1M,O). In wild-type embryos, we observed staining in known Shh signaling centers, including the notochord and the zone of polarizing activity in the limb buds (Fig. 1M). These regions also stained blue in the Inpp5erdg/rdg; Ptch1LacZ/+ mutant embryos, indicating Shh activity (Fig. 1O). In neural tube sections of wild-type Ptch1LacZ/+ embryos, we saw a steep LacZ expression gradient with the strongest blue staining at the ventral midline of the neural tube (Fig. 1N). In contrast, the staining expanded dorsally in the Inpp5erdg/rdg; Ptch1LacZ/+ mutant embryos (Fig. 1P,Q), consistent with the dorsal expansion of ventral cell fates we observed. We observed analogous dorsal expansion of Ptch1 expression via in situ hybridization of the Inpp5erdg/rdg neural tube, confirming that the LacZ expression expansion reflected Ptch1 expression (Fig. S2). Furthermore, we found neural patterning in Inpp5erdg/rdg; Ptch1LacZ/+ mutant embryos matched that in Inpp5erdg/rdg mutant embryos, indicating that Ptch1 haploinsufficiency does not modify the Inpp5erdg/rdg phenotype (Fig. S3). We also found that Pax7, a dorsal marker inhibited by Shh, was absent in Inpp5erdg/rdg; Ptch1LacZ/+ mutant embryos (Fig. S3R) (Ericson et al., 1996). Taken together, these data indicate dorsally expanded Shh activity in Inpp5erdg/rdg; Ptch1LacZ/+ and in Inpp5erdg/rdg mutants.
At a mechanistic level, Smo ciliary enrichment correlates with Smo activation, so we examined Smo staining in neural tube cilia (Corbit et al., 2005). In wild-type sections, we observed ciliary Smo enrichment near the ventral midline (Fig. 1R,R′). However, in Inpp5erdg/rdg embryos, we found Smo-positive cilia expanded dorsally along the ventricular zone (Fig. 1S,S′). To quantify the Smo expansion, we measured the distance over which we observed ciliary Smo enrichment and report this as a percentage of the total distance of the neural tube lumen (Fig. 1T). We found ciliary Smo enrichment in the ventral 10% of the neural tube in wild-type sections and in the ventral 40% of the neural tube in Inpp5erdg/rdg sections (P<0.001) (Fig. 1T). This dorsal expansion of ciliary Smo enrichment correlates with the expanded Shh activity response, suggesting that the ciliary Smo is activated. Taken together, these data indicate that the normal role of Inpp5e is to negatively regulate the Shh signaling response in the E10.5 neural tube.
Ventral neural patterning in E12.5 Inpp5erdg/rdg mutant embryos indicates recovery of patterning over time
To understand how Inpp5e regulates Shh signaling over time, we evaluated neural patterning at E12.5. In Inpp5erdg/rdg embryos, we found FoxA2 expression was restricted to the floor plate, as in control embryos (Fig. 2A,F). Similarly, we found Nkx2.2-positive cells only in the p3 domain adjacent to the floor plate in control and Inpp5erdg/rdg mutant embryos (Fig. 2B,G). Analysis of the dorsal boundary of these domains show that, although there is some variability in the mutants, no significant difference was found between these two groups (Fig. 2K). Overall, Olig2- and Nkx6.1-positive cells appeared in their normal domains with a slight but significant expansion of the pMN domain of Inpp5erdg/rdg embryos compared with controls (Fig. 2B,C,G,H,K). However, the increase in domain size appears to be due to only a few dorsally scattered cells and there was no apparent intermingling of cells as was seen at E10.5 (Fig. 1C,H). We therefore measured the amount of overlap seen between Nkx2.2 and Olig2 domains in both E10.5 and E12.5 embryos by measuring the distance between the dorsal boundary of Nkx2.2 and the ventral boundary of Olig2. At E10.5, the Nkx2.2 and Olig2 domains significantly overlap in the mutants (Fig. 1C,H: Fig. 2M). However, at E12.5 there is no longer a significant overlap between these domains (Fig. 2B,G,M). To further investigate the differences between the progenitor cells at E10.5 and E12.5, we counted the number of FoxA2-, Nkx2.2- and Olig2-positive progenitor cells. At E10.5 there is a significant increase in the number of FoxA2- and Nkx2.2-positive cells but not Olig2-positive cells in Inpp5erdg/rdg animals when compared with controls (Fig. 2N). However, by E12.5 there is no longer a significant difference between control and Inpp5erdg/rdg animals for any of the markers analyzed. To determine whether there was an increase in differentiated neurons at E12.5, we stained embryos with HB9, which marks differentiated motor neurons (Fig. 2D,I). There was no difference seen in the staining pattern or the number of cells between Inpp5erdg/rdg mutant and control animals (Fig. 2O). Although we see a dorsal shift of the Olig2 and Nkx6.1 boundaries at E12.5, these data indicate that the Shh response in Inpp5erdg/rdg mutant embryos is sufficiently dampened and more comparable with wild type by E12.5.
Fig. 2.

Normal ventral neural patterning in E12.5 Inpp5erdg/rdg embryos reflects a recovery of Shh response. Caudal (hindlimb) sections of E12.5 control (A-E, n=6) and Inpp5erdg/rdg (F-J, n=4) neural tubes. (A,B,E-G,J) FoxA2, Nkx2.2 and Pax6 expression domains look similar in control and Inpp5erdg/rdg neural tube sections. (D,I) HB9 cells are specified in the correct domain in control and Inpp5erdg/rdg neural tube sections. (B,C,G,H) Olig2 and Nkx6.1 domains in Inpp5erdg/rdg show only a few cells scattered dorsally. Scale bar: 100 µm. (K,L) Quantitation of dorsal and ventral boundary. (M) Quantification of distance of overlap between Nkx2.2 and Olig2 domains as a percentage of total lumen distance. Circles represent control; triangles indicate Inpp5erdg/rdg. (N,O) Total number of positive nuclei per section. Bar indicates mean of biological replicates with individual data points shown. (K,L,O) Analysis by two-tailed unpaired t-test with Welch's correction. (M,N) Analysis with ANOVA using Tukey's correction for multiple comparisons. *P<0.05, **P<0.01, ****P<0.0001; ns, not significant.
In order to test whether the dorsal boundary of the Shh response is normal in Inpp5erdg/rdg embryos at E12.5, we stained neural tube sections with antibody against Pax6. We found Pax6 expression in Inpp5erdg/rdg embryos appeared the same as in wild-type controls (Fig. 2E,J,L). Thus, the abnormal ventral patterning we observed in E10.5 Inpp5erdg/rdg embryos recovers by E12.5. Taken together, these data imply that Inpp5e is not simply a negative regulator of the Shh response in the neural tube.
Inpp5erdg is a functional null allele sensitive to strain background
The Inpp5erdg allele changed aspartic acid to glycine at residue 511 within the phosphatase domain of Inpp5e, Inpp5eD511G (Fig. 3A,B). This region of the protein sequence is highly conserved across species (Fig. 3C). At E12.5, most Inpp5erdg/rdg embryos exhibited exencephaly (88%, 61/69) and either microphthalmia or anophthalmia (80%, 33/41). Seventy-seven percent (31/40) of embryos exhibited a rough neural tube with 30% showing spina bifida at the hindlimb level (13/40) (Fig. 3E). The Inpp5erdg/rdg embryos died around E13.5. In order to determine whether the D511G mutation was causative, we crossed Inpp5erdg/+ to mice carrying an Inpp5e-deleted allele (Inpp5eΔEx7-8/+) and examined the compound heterozygote embryos. Inpp5erdg/ΔEx7-8 embryos display exencephaly, anophthalmia, spina bifida and a rough neural tube, indicating the alleles failed to complement and that Inpp5erdg is an allele of Inpp5e (Fig. 3F).
Fig. 3.

Inpp5erdg, which carries a D511G mutation, is a functional null allele of Inpp5e. (A) Schematic showing the protein domain structure of Inpp5e. D511G in Inpp5erdg is the result of an A-to-G transition in the coding strand (NM_033134.3:c2164A>G). (B) Exon structure of Inpp5e, based on NCBI reference sequence NM_033134.3, showing location of deleted exons in two previously characterized alleles. (C) Alignment of protein sequence surrounding position 511 of mouse Inpp5e (highlighted) showing that aspartic acid is conserved across multiple species. (D-G) Whole-mount images of E12.5 embryos. (E) Inpp5erdg/rdg mutants display exencephaly and microphthalmia (n=20). These animals are on a mixed FVB/C3H background so that the retinal pigment epithelium cells are visible. (F,G) Inpp5erdg/ΔEx7-8 (n=4) and FVB-Inpp5eΔEx7-8/ΔEx7-8 (n=9) embryos resemble Inpp5erdg/rdg mutants, indicating the alleles fail to complement. (H) qRT-PCR of Gli1 in Inpp5erdg/+ (n=3) and Inpp5erdg/rdg (n=3) MEFs in the presence (+) and absence (−) of Shh treatment. Bar indicates mean of biological replicates with individual data points shown, analyzed by ANOVA using Tukey's correction for multiple comparisons. **P<0.01; ns, not significant. (I-K) Inpp5erdg/rdg mutants on C57BL/6J background survive to E16.5 and display exencephaly, microphthalmia and spina bifida to varying degrees of severity (n=6). Arrow indicates spina bifida. (I′-K′) Close-up images showing hindlimb preaxial polydactyly. Arrow indicates extra digit. Scale bars: 1 mm for D-K; 500 µm for I′-K′.
Upon stimulation of cultured cells with the Shh agonist SAG, cells lacking Inpp5e show a diminished Shh response (Chávez et al., 2015; Dyson et al., 2017; Garcia-Gonzalo et al., 2015). To determine the Shh response of cells carrying Inpp5erdg, we generated MEFs from Inpp5erdg/rdg and control littermate embryos and tested their ability to respond to Shh stimulation. As expected, we found control MEFs increased expression of Gli1, a direct Shh target gene, upon stimulation with Shh-conditioned media (Fig. 3H) (P<0.01). However, the Inpp5erdg/rdg MEFs displayed no change in Gli1 levels when treated with Shh (Fig. 3H). Similar results have been shown for cell lines lacking Inpp5e function (Chávez et al., 2015; Dyson et al., 2017; Garcia-Gonzalo et al., 2015). Thus, the Inpp5erdg allele phenocopies Inpp5e null alleles in cultured cells.
The fact that Inpp5erdg/rdg embryos died at E13.5 contrasts with the two previously studied Inpp5e null alleles: Inpp5eΔEx7-8 which deletes exons 7-8, and Inpp5eΔEx2-6, which deletes exons 2-6 (Fig. 3B). Both of these deletion mutants die at birth and both were analyzed on a predominantly C57BL/6 background (with some possible contribution from 129/Sv) (Dyson et al., 2017; Jacoby et al., 2009). Our analysis of the Inpp5erdg allele was on an FVB background. To distinguish whether such phenotypic distinctions reflected differences in allelic function or strain background, we backcrossed the Inpp5eΔEx7-8 allele onto FVB for three generations. We found that these FVB-Inpp5eΔEx7-8/ΔEx7-8 embryos died at E13.5-14.5 and displayed exencephaly, anophthalmia, spina bifida and a rough neural tube (Fig. 3G). We also examined neural tube patterning in E10.5 FVB-Inpp5eΔEx7-8/ΔEx7-8 embryos and found expanded ventral neural cell fates comparable with Inpp5erdg/rdg for all markers except FoxA2 and Pax6, which were shifted dorsally but to a lesser extent than in Inpp5erdg/rdg mutants (Fig. S4A-J,U,V). We also observed relatively normal patterning in FVB-Inpp5eΔEx7-8/ΔEx7-8 embryos at E12.5, which was not significantly different from Inpp5erdg/rdg, indicating the recovery of the Shh response also occurs in this allele (Fig. S4K-V). Thus, the Inpp5eΔEx7-8 allele on the FVB background phenocopies the Inpp5erdg allele. We performed the reciprocal experiment and crossed the Inpp5erdg allele onto the C57BL/6 background for four generations. We identified live B6-Inpp5erdg/rdg embryos at E16.5 (6/25, 24%) that displayed exencephaly (5/6), spina bifida (2/6), microphthalmia or anophthalmia (6/6) and hindlimb preaxial polydactyly (5/6) (Fig. 3I-K′). In contrast, we found no Inpp5erdg/rdg embryos at E16.5 on the FVB background (0/51). Taken together, these data indicate that Inpp5e-dependent phenotypes are sensitive to genetic background and that Inpp5erdg is a functional null allele.
Altered ciliary enrichment of Tulp3 and Gpr161 in Inpp5erdg/rdg neural tubes
Inpp5e removes the 5-phosphate from PI(3,4,5)P3 and PI(4,5)P2, and loss of Inpp5e function results in increased ciliary PIP2 (Garcia-Gonzalo et al., 2015; Chávez et al., 2015; Kisseleva et al., 2000). Tulp3 traffics G-protein-coupled receptors (GPCRs) into cilia in a PIP2-dependent manner (Mukhopadhyay et al., 2010). Increased PIP2 levels in Inpp5e-deficient cilia increases the amount of Tulp3 seen (Chávez et al., 2015; Garcia-Gonzalo et al., 2015). To determine whether Inpp5erdg affected Tulp3 localization in a similar manner, we stained E10.5 neural tube sections with antibodies against Tulp3. We detected no Tulp3 in wild-type neural tube cilia (Fig. 4A-C). In contrast, we observed Tulp3 staining in 90% of Arl13b-positive luminal cilia in the ventral neural tube of the Inpp5erdg/rdg mutants (Fig. 4D-G). As Tulp3 is a known PIP2-binding protein, this result is consistent with Inpp5erdg being a loss-of-function allele that increases ciliary PIP2.
Fig. 4.

Altered ciliary Tulp3 and Gpr161 in Inpp5erdg/rdg neural tubes. (A-F,H-M) Images of cilia found in the ventral-most region of E10.5 caudal (hindlimb) sections of wild-type (A-C,H-J, n=3) and Inpp5erdg/rdg (D-F,K-M, n=3) neural tubes. Insets show a single cilium digitally magnified. (A-F) Tulp3 is found in almost all cilia in the neural tube of Inpp5erdg/rdg embryos but is absent from wild-type cilia. (G) Quantification of A-F (see Materials and Methods for details). (H-M) Gpr161 is found in an increased number of cilia in the neural tube of Inpp5erdg/rdg. (N) Quantification of H-M. Bar indicates mean of biological replicates with individual data points shown, analyzed using a two-tailed unpaired t-test with Welch's correction. ***P<0.001. Scale bars: 10 µm.
Increased PIP2 in Inpp5e-deficient cilia recruits the Shh antagonist Gpr161 via a PIP2/Tulp3-dependent mechanism (Chávez et al., 2015; Garcia-Gonzalo et al., 2015). To determine whether an increase in ciliary levels of Gpr161 occurred coincident with the increase in Tulp3, we examined the localization of Gpr161 in ventral neural tube cilia. In wild-type embryos, 3.4% of cilia were Gpr161 positive, whereas 34% of cilia stained for Gpr161 in Inpp5erdg/rdg embryos (P<0.001) (Fig. 4H-N). These data imply that, in the neural tube, the increased PIP2 in Inpp5erdg/rdg mutant cilia efficiently recruits Tulp3 to cilia and inefficiently localizes Gpr161 to cilia.
Ift172-dependent and -independent functions of Inpp5e
Inpp5e is predominately localized to the cilium (Bielas et al., 2009; Jacoby et al., 2009), and Shh signaling is tightly associated with the cilium (Huangfu et al., 2003; Goetz and Anderson, 2010). To determine whether the Inpp5erdg/rdg neural tube patterning phenotype requires cilia, we generated Inpp5erdg/rdg;Ift172wim/wim double mutant embryos. Ift172 is an intraflagellar transport protein required for ciliary assembly and maintenance. Ift172wim/wim single mutants do not produce a cilium, show exencephaly without a groove at the ventral midline and do not specify ventral neural cell fates, with the exception of Nkx6.1-positive cells, which are Shh dependent and cilia independent (Fig. 5E,F) (Huangfu et al., 2003; Norman et al., 2009; Briscoe et al., 2000). In contrast, Inpp5erdg/rdg single mutants displayed pronounced exencephaly with a prominent groove at the midline (Fig. 5C). In the neural tube of Inpp5erdg/rdg mutants at E9.5, we observed FoxA2 expression at the ventral midline, which expanded dorsally albeit diffusely through the majority of the neural tube (Fig. 5D). We identified an additional dorsal expansion of ventral cells expressing Nkx2.2 and Olig2 intermingled with the FoxA2-positive cells (Fig. 5D,D′). We found Inpp5erdg/rdg;Ift172wim/wim double mutant embryos resemble Ift172wim/wim embryos showing exencephaly lacking a ventral midline groove (Fig. 5G). In the neural tube, we found Inpp5erdg/rdg;Ift172wim/wim double mutants specified no FoxA2- or Nkx2.2-positive cells, and had normal numbers of Nkx6.1-positive cells similar to Ift172wim/wim single mutants (Fig. 5H-H″), suggesting that the Inpp5erdg/rdg phenotype is Ift172 dependent and that Inpp5e functions within cilia. However, we observed Olig2-positive cells in Inpp5erdg/rdg;Ift172wim/wim double mutants similar to the pattern in Inpp5erdg/rdg single mutants, suggesting some Ift172-independent Inpp5e function.
Fig. 5.
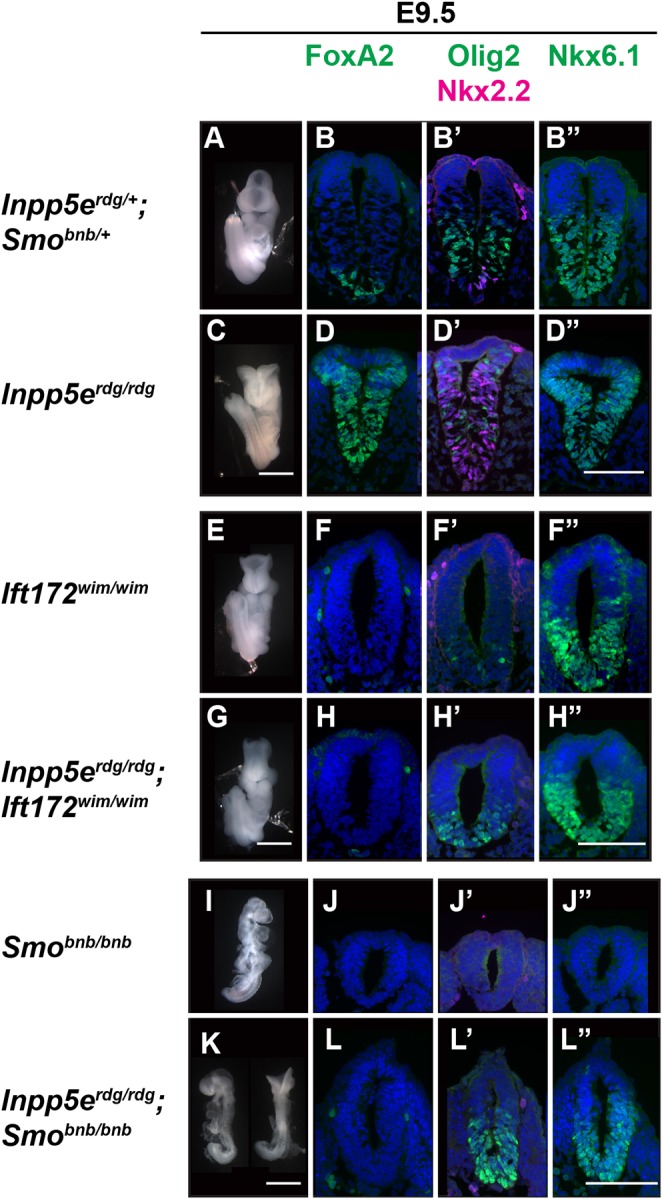
Inpp5e is cilia dependent and depends on smoothened to specify cell fates requiring the highest Shh activity, but not those requiring moderate Shh activity. E9.5 whole-mount images and neural tube staining of caudal (hindlimb) sections. (A,C) Ventral view of control and Inpp5erdg/rdg embryos highlighting exencephaly with prominent midline groove in the mutant (Inpp5erdg/rdg, n=63). (B-B″,D-D″) Neural tube staining shows ventral expansion of FoxA2, Nkx2.2, Olig2 and Nkx6.1 cell fates in Inpp5erdg/rdg embryos (control, n=4; Inpp5erdg/rdg, n=4). (E) Ventral view of an Ift172wim/wim embryo showing exencephaly without midline groove (n=20). (G) Inpp5erdg/rdg;Ift172wim/wim mutants display exencephaly that resembles the Ift172wim/wim mutant (n=6). (F-F″,H-H″) Neural tube staining of both Ift172wim/wim and Inpp5erdg/rdg; Ift172wim/wim show that these mutants fail to properly specify ventral cell fates (Ift172wim/wim, n=1; Inpp5erdg/rdg;Ift172wim/wim, n=3). (I) Side view of Smobnb/bnb mutants reveals a small unturned body with a closed head (n=73). (K) Side and dorsal views of Inpp5erdg/rdg;Smobnb/bnb embryos showing unturned bodies (n=9). The dorsal view highlights the exencephaly seen in some mutants (n=5/9). (J-J″) Smobnb/bnb embryos do not specify ventral cell fates (n=1). (L-L″) Inpp5erdg/rdg;Smobnb/bnb mutants lack FoxA2 and Nkx2.2 specification but specify Olig2 and Nkx6.1 cells (n=5). Scale bars: 1 mm (whole embryo); 100 µm (neural tube sections).
Smo-dependent and -independent functions of Inpp5e
Ciliary Smo enrichment correlates with Smo activation (Corbit et al., 2005). Given that Inpp5e appeared to act within the cilium, we explored the relationship of Inpp5e and Smo by intercrossing Inpp5erdg/+;Smobnb/+ transheterozygous animals, which should generate double mutant embryos at a frequency of 1 out of 16 embryos. We identified fewer Inpp5erdg/rdg;Smobnb/bnb mutants (3%) than expected (6.25%), which is statistically significant (χ2test, P=0.02). Smo bentbody, Smobnb, is a null allele and Smobnb/bnb embryos have a closed, mis-shapen head and fail to complete embryonic turning before dying by E9.5 (Caspary et al., 2002) (Fig. 5I). In neural tube sections, Smobnb/bnb embryos lack ventral cell fate specification so do not express the Shh-dependent cell markers FoxA2, Nkx2.2 and Olig2 (Fig. 5J,J′). At E9.5, Inpp5erdg/rdg;Smobnb/bnb double mutant embryos were small with partially turned or unturned bodies similar to Smobnb/bnb mutants (66%, 6/9). Unlike Smobnb/bnb single mutant embryos, Inpp5erdg/rdg;Smobnb/bnb double mutant embryos displayed exencephaly (55%, 5/9) (Fig. 5K). In the neural tube of Inpp5erdg/rdg;Smobnb/bnb mutants, we detected no expression of FoxA2 or Nkx2.2, cell fates requiring the highest Shh response (Fig. 5L,L′). The lack of ventral cell fates resembled the Smobnb/bnb mutant and indicated that Inpp5e requires Smo function to specify these cell fates. In contrast, unlike Smobnb/bnb mutant neural tube sections, we observed Olig2 and Nkx6.1 expression in Inpp5erdg/rdg;Smobnb/bnb double mutant embryos (Fig. 5J′,J″,L′,L″). Thus, specific ventral cell fates in Inpp5erdg/rdg can be specified without Smo. The most parsimonious interpretation of these data is that the absence of Inpp5e function permits derepression of Shh signaling but only to a level permitting an intermediate Shh response.
Neural tube patterning recovery at E12.5 is dependent on Gli3
Both the Inpp5erdg/rdg;Ift172wim/wim and Inpp5erdg/rdg;Smobnb/bnb double mutant embryos express Olig2-positive pMN cells, suggesting that there may be derepression of these cell fates when Inpp5e is lost. In the neural tube, Gli3 is the major repressor of Shh signaling (Litingtung and Chiang, 2000; Persson et al., 2002), and we previously showed that the GliR gradient plays a crucial role between E10.5 and E12.5 to properly specify cell fate (Su et al., 2012). We performed western blots in order to examine Gli processing in Inpp5erdg/rdg animals. Using antibodies that detect full-length Gli2 and Gli3 (185 kD Gli2 and 190 kD Gli3), and cleaved Gli3 protein (83 kD Gli3R), we evaluated whole-embryo protein extracts at E10.5 and E12.5 (Fig. 6A,B). Full-length Gli protein does not represent activated Gli protein. We observed no change in full-length Gli2 (Gli2FL) or Gli3 (Gli3FL) between wild type and Inpp5erdg/rdg at either E10.5 or E12.5. We saw no change in the 83kDa Gli3 band in Inpp5erdg/rdg compared with wild type at either E10.5 or E12.5. These data suggest that the overall levels of Gli2FL, Gli3FL and Gli3R are unaltered in homogenized Inpp5erdg/rdg embryos.
Fig. 6.

Normal ventral neural patterning in E12.5 Inpp5erdg/rdg embryos reflects a recovery of Shh response and is Gli3 dependent. (A) Western blot analysis of Gli2 and Gli3 in wild-type (n=3) and Inpp5erdg/rdg (n=3) whole-embryo extracts. Asterisk denotes a non-specific band. (B) Quantification of A. Circles represent wild type, triangles indicate Inpp5erdg/rdg. Bar indicates mean of biological replicates with individual data points shown. ns, not significant. (C-J) Caudal (hindlimb) sections of E12.5 Inpp5erdg/+;Gli3Δ/Δ (C-F, n=2) and Inpp5erdg/rdg;Gli3Δ/Δ (G-J, n=3) neural tubes. (C-F) Inpp5erdg/+;Gli3Δ/Δ neural tube sections resemble control (Fig. 2A-E; Fig. S5A-D). (G-J) FoxA2, Olig2 and Nkx6.1 cells are dorsally scattered (expanded), Pax6 ventral boundary is dorsally shifted in E12.5 Inpp5erdg/rdg;Gli3Δ/Δ neural tube, and the lumen shape resembles that of Inpp5erdg/rdg at E10.5 (Fig. 1). Scale bar: 100 µm.
In order to determine whether the Gli3 repressor is functionally altered in Inpp5erdg/rdg embryos, we evaluated Inpp5erdg/rdg;Gli3Δ/Δ double mutants. At E12.5, we found the Gli3Δ/Δ mutants specified FoxA2-, Nkx2.2-, Olig2- and Nkx6.1-positive cells in the same domains as the control and Inpp5erdg/rdg littermates (Fig. 6C-E; Fig. S5A-C), as well as normal domain expression of Pax6 (Fig. 6F; Fig. S5D). This is consistent with previous reports of Gli3Δ/Δ mutants displaying normal specification of these cell fates (Litingtung and Chiang, 2000; Persson et al., 2002). In contrast, we found dorsally scattered FoxA2- and Nkx2.2-positive cells, and an expansion of both Olig2- and Nkx6.1-positive cells in E12.5 Inpp5erdg/rdg;Gli3Δ/Δ neural tubes (Fig. 6G-I), as well as a dorsal shift in Pax6 staining boundary (Fig. 6J). This indicates that the recovery of patterning we observed in Inpp5erdg/rdg mutant embryos by E12.5 does not occur in Inpp5erdg/rdg;Gli3Δ/Δ mutant embryos. These data demonstrate that the Inpp5erdg/rdg recovery phenotype at E12.5 is Gli3 dependent and strongly implicates the GliR gradient in the recovery.
DISCUSSION
Our data point to Inpp5e regulating the Shh response through a more complicated mechanism than previously appreciated. In vitro, we found Inpp5erdg/rdg MEFs did not respond to Shh stimulation, consistent with previous reports that Inpp5e plays a positive role in Shh signal transduction (Garcia-Gonzalo et al., 2015; Dyson et al., 2017; Chávez et al., 2015). However, in vivo we found Inpp5erdg/rdg embryos displayed an expansion of ventral cell fates in the neural tube, indicating loss of negative regulation of the pathway. Importantly, we observed distinct cell fates requiring high, intermediate and low levels of Shh response in the Inpp5erdg/rdg mutant neural tube, signifying graded pathway regulation remained. Furthermore, we found that the mispatterning of the E10.5 Inpp5erdg/rdg neural tube recovered by E12.5 and that the recovery depended on Gli3, which functions predominantly as a repressor in the neural tube. This finding is consistent with our genetic test of the relationship between Inpp5e and Smo, where we found the Inpp5erdg/rdg;Smobnb/bnb neural tube exhibited cell fates requiring intermediate levels of Shh response such as Olig2 and Nkx6.1. As Smo is essential for Shh signal transduction, this result implies a derepression of intermediate cell fates occurs when Inpp5e function is lost in combination with loss of Smo function. Inpp5e localizes to cilia and we showed that the Inpp5erdg/rdg phenotype largely depended on the presence of cilia, consistent with Inpp5e regulating Shh signaling from within the cilium. We showed the Inpp5e mutant alleles are sensitive to strain background, which, along with the complementation test, enabled us to demonstrate that Inpp5erdg is a functional null allele.
The simplest model to explain the expansion of ventral fates we observed in Inpp5erdg/rdg mutants is that Inpp5e normally serves as a negative regulator of Shh signal transduction. However, that interpretation is complicated by the recovery of patterning we observed in E12.5 Inpp5erdg/rdg embryos along with our finding that Inpp5e function is required for the Shh response in cell culture, which aligns with previously published work (Chávez et al., 2015; Dyson et al., 2017; Garcia-Gonzalo et al., 2015). These data indicate that Inpp5e regulates the Shh response over time and can act as both a positive and negative regulator of the pathway. Thus, any model of Inpp5e function must reconcile these distinct observations.
Other negative regulators of Shh signaling fall into two classes: those whose loss leads to complete constitutive activation of the pathway, such as Ptch1, Sufu or Gnas (which encodes Gαs) mutants, and those whose loss is slightly less severe, such as Tulp3, Gpr161 or Rab23 mutants (Mukhopadhyay et al., 2013; Norman et al., 2009; Patterson et al., 2009; Goodrich et al., 1997; Cooper et al., 2005; Svärd et al., 2006). Ptch1, SuFu and Gαs maintain the pathway in an ‘off’ state when ligand is not present, so their loss leads to complete pathway activation: increased GliA production, almost no GliR production and specification of neural fates requiring the highest Shh response. In contrast, Tulp3, Gpr161 and Rab23 adjust the output of the pathway without being essential; they attenuate the pathway. Inpp5e appears to function in this second category: both the Inpp5erdg and Inpp5eΔEx2-6 alleles allow multiple ventral neural cell fates to be specified, indicating that the GliA/GliR ratio is altered consistent with the expanded Shh activity gradient we observed (Dyson et al., 2017). Furthermore, our findings that the recovery of neural patterning in Inpp5erdg/rdg embryos between E10.5 and E12.5 is Gli3 dependent, along with Inpp5erdg/rdg;Smobnb/bnb mutant embryos exhibiting derepression of Olig2 and Nkx6.1 expression, are consistent with lowered GliR production in Inpp5erdg/rdg mutants. Reduced GliR production alters the effective GliA/GliR ratio, consistent with the expansion of ventral cell fates in Inpp5erdg/rdg mutants. The GliA/GliR ratio appears highly variable based on the intermingling of cell fates in the Inpp5erdg/rdg neural tube. Taken together, these observations indicate that Inpp5e is an attenuator of Shh signaling in specifying neural cell fates and hint that it is crucial for the temporal control of the activator to repressor ratio in the neural tube.
Similar to previous findings in cultured cells, we found that Inpp5erdg/rdg mutant MEFs exhibited no Shh transcriptional response, so we were surprised that the mutant neural tube exhibited an expanded Shh transcriptional response. Although on the surface this is contradictory, it adds Inpp5erdg to the list of mutations in Shh signal transduction components that reveal distinctions in their neural and fibroblast phenotypes. (Gigante et al., 2018; Larkins et al., 2011; Pusapati et al., 2018). The variance in cell sensitivity to Shh ligand between fibroblasts (NIH/3T3 cells) and neural progenitors lacking Gpr161 is proposed to be due to cell type-specific differences in PKA activity (Pusapati et al., 2018). Differential PKA activity could also explain the distinct phenotypes in fibroblasts and neural progenitors lacking functional Inpp5e. Loss of Inpp5e function in fibroblasts elevates ciliary PIP2 levels, leading to increased recruitment of Tulp3 and Gpr161, which in turn increases PKA activity resulting in an absence of Shh target gene expression (Garcia-Gonzalo et al., 2015; Mukhopadhyay et al., 2013). Although we observed a higher percentage of Tulp3-positive cilia in the Inpp5erdg/rdg neural tube compared with wild type, the recruitment of Gpr161 to cilia was limited. Concomitantly, we found a significant increase in the region over which Smo enrichment in cilia was visible in the Inpp5erdg/rdg ventral neural tube. The combination of less ciliary Gpr161 and more ciliary Smo would be predicted to result in lower PKA activity in the neural progenitors compared with the MEFs, which could render the cells more sensitive to Shh ligand. This would also be consistent with a low level of GliR production in the E10.5 Inpp5erdg/rdg neural tube.
Taken together, our data support a model in which loss of Inpp5e function results in the alteration of the normal effective Gli ratio formed from the additive result of the GliA and GliR concentration gradients (Fig. 7). A delay in GliR gradient formation would alter the kinetics of the effective Gli ratio such that there is an initial excess of GliA function that normalizes over time as the standard GliA/GliR ratio forms (Fig. 7B). This model reconciles the seemingly discordant aspects of the Inpp5e phenotypes in several ways. First, in Inpp5erdg/rdg mutants at E9.5 and E10.5, low GliR production would derepress known GliR targets: Nkx6.1 and Olig2. This derepression was also evident in the Inpp5erdg/rdg;Ift172wim/wim mutants, as well as the Inpp5erdg/rdg;Smobnb/bnb mutants. At the same time, low GliR production would increase the effective GliA/GliR ratio, which would specify more FoxA2- and Nkx2.2-positive cell fates, as we saw at E9.5 and E10.5 in Inpp5erdg/rdg mutants. Second, the fact that the recovery of Inpp5erdg/rdg neural patterning is Gli3-dependent argues that the GliR gradient does eventually form. Finally, the fact that Inpp5erdg/rdg and Inpp5eΔEx7-8/ΔEx7-8 MEFs do not respond to Shh ligand is consistent with fibroblasts being more efficient than neural progenitors at GliR production.
Fig. 7.

Model for the role of Inpp5e over time in Shh-dependent ventral neural tube patterning. (A) The relative amounts of GliA to GliR specify neural cell fates at any particular position along the ventral-dorsal axis. The combined ratio is the effective Gli ratio (Gli+ or Gli−), which integrates the relative ratio of GliA to GliR production. (B) (Top) Normally, the effective Gli ratio does not change over time, leading to wild-type cell fates. (Bottom) The expanded ventral neural progenitor cell fates in Inpp5erdg/rdg mutants at E9.5 normalize by E12.5 due to changes in the effective Gli ratio. Although multiple mechanisms are possible, the simplest explanation posits that loss of Inpp5e initially alters GliR gradient production and, over time, the GliR gradient is normalized and the ventral cell fates are returned to their correct positions by E12.5.
The intermingling of cell fates in the Inpp5erdg/rdg neural tube suggests that the GliA/GliR ratio is likely quite dynamic and variable from cell to cell. The Gli3-dependent recovery of pattern in E12.5 neural tube argues that Gli3 (the predominant repressor) is biologically significant, even if statistically significant differences cannot be seen on western blots. We note the formal possibility that Inpp5e also changes the kinetics of GliA production; however, changes in the kinetics of GliR production are sufficient to explain the apparent increase in the levels of effective GliA that result in ventral cell fate expansion. Cells can also integrate the level of Shh signaling over time by taking into account the duration of Gli activity in a process known as temporal adaptation (Stamataki et al., 2005; Dessaud et al., 2007). Cells exposed to lower concentrations of Shh lose Gli responsiveness faster than cells exposed to high concentrations of Shh, allowing cells to interpret both concentration and duration of exposure (Dessaud et al., 2007). Thus, the model we propose awaits techniques that monitor GliA and GliR in situ at the cellular level and over time to tease these two possibilities apart.
In conclusion, our data provide genetic evidence that Inpp5e attenuates Shh signaling in the developing mouse neural tube. Our data add to the existing function of Inpp5e and argue that it plays both positive and negative regulatory roles in Shh signal transduction, likely through controlling the timing of Gli processing and thus the sensitivity of cells to respond to Shh ligand, both in the dorsal-ventral axis and over time. These data also highlight the phenotypic variability among Inpp5e mutant cell types and alleles, expanding our knowledge on the role of Shh signaling duration in regulating ventral neural cell fate.
MATERIALS AND METHODS
Mouse lines and maintenance
All mice were cared for in accordance with NIH guidelines and Emory's Institutional Animal Care and Use Committee (IACUC). Alleles used were: Inpp5erdg (MGI: 6295836), Ptch1LacZ (Ptch1tm1Mps) (MGI: 1857447), Inpp5eΔEx7-8 (Inpp5etm1.2Ssch) (MGI: 4360187), Ift172wim (MGI: 2682066), Smobnb (MGI: 2137553), Gli3fl (Gli3tm1Alj) (MGI: 3798847) and CAGGCre-ERTM [Tg(CAG-cre/Esr1*)5Amc] (MGI: 2182767). Inpp5eΔEx7-8/+ animals were generated by crossing Inpp5efl/fl (Inpp5etm1.1Ssch) (MGI: 4360186) animals to CAGGCre-ERTM animals and treating pregnant dams with tamoxifen as previously described (Su et al., 2012). Inpp5efl/fl animals were received at Emory University and rederived on C57BL/6J. They, and the derived Inpp5eΔEx7-8/+ mice, are maintained on FVB/NJ. Inpp5erdg/rdg;Gli3Δ/Δ embryos were generated by crossing Inpp5erdg/+;Gli3fl/+ and Inpp5erdg/+;Gli3fl/+;CAGGCre-ERTM animals, and treating pregnant dams with tamoxifen at E7.5, as previously described (Su et al., 2012). CAGGCre-ERTM, Ptch1LacZ, Ift172wim, Smobnb and Gli3fl were on a C3H/HeJ background when this project began and are currently maintained with Inpp5erdg on FVB/NJ. Genotyping was performed as previously described or by Transnetyx (Goodrich et al., 1997; Blaess et al., 2008; Jacoby et al., 2009; Kasarskis et al., 1998; Hayashi and McMahon, 2002; Sun et al., 2012). Timed mating of heterozygous intercrosses was performed with animals less than 1 year old to generate embryos of the indicated embryonic stage.
Mouse dissection, X-gal staining, in situ hybridization and immunofluorescence
Embryos were dissected in ice-cold phosphate-buffered saline (PBS) and processed for either X-gal staining, in situ hybridization or immunofluorescence. Embryos were stained using X-gal as previously described (Goodrich et al., 1997). After fixing in 4% paraformaldehyde (PFA) overnight at 4°C, embryos were incubated in 30% sucrose in 0.1 M phosphate buffer (pH 7.3) at 4°C overnight prior to being embedded in OCT (Tissue-Tek) and 40 µm sections were obtained using a Leica CM1850 cryostat.
Whole-mount in situ hybridization using digoxigenin-labeled probes was performed on embryos using standard protocols (Mukhopadhyay et al., 2013). Antisense riboprobes were made using the Ptch1 template (from Andrew McMahon's lab, University of Southern California, CA, USA; and from Deanna Grant, Andrew Peterson's lab, Genentech, South San Francisco, CA, USA) and detected using NBT/BCIP reagent. Embryos were fixed in 4% PFA/0.1% glutaraldehyde for 1 h at room temperature and processed through sucrose and OCT as described above. Sections (40 µm) were obtained on a Leica CM1950 cryostat.
For immunofluorescence, embryos were fixed for 1 h in 4% PFA on ice. Embryos were processed through sucrose and OCT as described above, before sectioning at 10 µm. Sections were incubated with primary and secondary antibodies diluted in PBS with 0.1% Triton X and either 1% or 10% heat-inactivated goat serum. The following primary antibodies were used: mouse anti-Shh clone 5E1 (1:10), mouse anti-FoxA2 clone 4C7 (1:10), mouse anti-Nkx2.2 clone 74.5A5, mouse anti-Nkx6.1 clone F65A2, mouse anti-Pax6 (1:100) and mouse anti-Pax7 (1:10) from Developmental Studies Hybridoma Bank; rabbit anti-FoxA2 (Cell Signaling, 3143; 1:500), rabbit anti-Olig2 (Millipore, AB9610; 1:300), rabbit anti-Smo (a kind gift from K. Anderson, Sloan Kettering Institute, NY, USA; 1:1000), mouse anti-Arl13b (Neuromab, N295B/66; 1:2000), rabbit anti-Tulp3 (a kind gift from J. Eggenschwiler, University of Georgia, Atlanta, USA; 1:500) and rabbit anti-Gpr161 (a kind gift from S. Mukhopadhyay, UT Southwestern Medical Center, Dallas, USA; 1:200). The secondary antibodies were conjugated to Alexa Fluor 488, 568 or 594 (ThermoFisher; 1:200). Hoechst 33342 (Sigma; 1:3000) was included in the incubation of slides with secondary antibody.
Quantifications
To quantify the dorsal-most boundary for each domain, the measurement tool in FIJI image processing software package was used to draw a line from the center of the floor plate region to the furthest nuclei, defined by Hoechst staining, expressing the marker of interest (Schindelin et al., 2012). To normalize for different shapes and sizes of neural tubes, a line was drawn around the entire neural tube lumen and the distance determined above was converted to a percentage of half this distance. Cell numbers were determined as the number of Hoechst-positive nuclei also expressing the gene of interest, counted using the cell counting tool in FIJI and reported as absolute number per section. To quantify the Smo expansion, the total distance over which ciliary Smo enrichment was observed was determined and reported as a percentage of the total distance of the neural tube lumen. To quantify Tulp3 and Gpr161, the number of Arl13b-positive luminal cilia that were also positive for either Tulp3 or Gpr161 in the ventral 10% of the neural tube were counted using the counting plugin in FIJI. Three sections from each embryo (wild type, n=3; Inpp5erdg/rdg, n=3), between 400 and 500 cilia, were counted for each condition per genotype. One to five sections from three to seven biological replicates were used for quantifications, with data from the same biological sample averaged. Statistical significance was evaluated on averaged biological replicates in PRISM v8 using either two-tailed unpaired t-test with Welch's correction or ANOVA using Tukey's correction for multiple comparisons.
MEF generation, RNA isolation and qPCR quantitation
Mouse embryonic fibroblasts (MEFs) were isolated from E12.5 embryos and immortalized as previously described (Mariani et al., 2016). Genotypes were verified by PCR and lines were regularly monitored for mycoplasma contamination. For Shh treatment, Inpp5erdg/+ and Inpp5erdg/rdg MEFs were grown at a density of 0.5×106 cells/ml and treated for 24 h with Shh-conditioned medium containing 0.5% fetal bovine serum (Larkins et al., 2011).
For qPCR, whole RNA was extracted from MEFs and qPCR was carried out as previously described (Bay et al., 2018; Gigante et al., 2018). The following primers were used (5′-3′): Gli1 (GCCACACAAGTGCACGTTTG and AAGGTGCGTCTTGAGGTTTTCA); Gapdh (CGTCCCGTAGACAAAATGGT and GAATTTGCCGTGAGTGGAGT) (Bay et al., 2018). Each reaction was performed in technical triplicate. Gli1 values were normalized to Gapdh within each sample. Statistical significance was evaluated in PRISM v8.1.1 by applying a two-way ANOVA with Tukey's correction for multiple analysis on three biological replicates.
Western blotting
Western blotting was performed as previously described (Chang et al., 2016; Mariani et al., 2016; Bay et al., 2018) with the following antibodies: Gli2 (R&D Systems, AF3635, 1:500), Gli3 (R&D Systems, AF3690, 1:1000) and HRP-conjugated donkey anti-goat IgG (Jackson ImmunoResearch, 1:5000). Lysates were made using RIPA buffer with Roche protease inhibitors (Chang et al., 2016). Values displayed are volume intensity, as measured from a chemiluminescent image and normalized, to total protein, as measured on a stain-free gel. Statistical significance was evaluated in PRISM v8.1.1 by applying a two-way ANOVA with Tukey's correction for multiple analysis on three biological replicates.
Supplementary Material
Acknowledgements
We are grateful to K. Anderson for the Smo antibody, to S. Mukhopadhyay for the Gpr161 antibody and to J. Eggenschwiler for the Tulp3 antibody. We are also grateful to S. Hwang for help with the in situ hybridizations, to C. Lin for his initial Inpp5erdg observations and to S. Mukhopadhyay for critical comments on the manuscript along with members of the Caspary lab. We also had additional support from the Emory University Integrated Cellular Imaging Microscopy Core of the Emory Neuroscience NINDS (Core Facilities grant P30NS055077). The content is solely the responsibility of the authors and does not necessarily reflect the official views of the National Institutes of Health.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
Conceptualization: S.C., T.C.; Methodology: S.C., A.B.L., T.C.; Validation: S.C., A.L., K.A.F.; Formal analysis: S.C.; Investigation: S.C., A.B.L., K.A.F.; Resources: S.S.; Writing - original draft: S.C., T.C.; Writing - review & editing: S.C., A.B.L., T.C.; Visualization: S.C., A.L., K.A.F.; Supervision: T.C.; Project administration: T.C.; Funding acquisition: T.C.
Funding
This work was supported by funding from National Institutes of Health (R01NS090029, R01GM110663 and R35GM122549) as well as by a March of Dimes Foundation grant (FY15-343). Deposited in PMC for release after 12 months.
Supplementary information
Supplementary information available online at http://dev.biologists.org/lookup/doi/10.1242/dev.183301.supplemental
References
- Bai C. B., Stephen D. and Joyner A. L. (2004). All mouse ventral spinal cord patterning by hedgehog is gli dependent and involves an activator function of gli3. Dev. Cell 6, 103-115. 10.1016/S1534-5807(03)00394-0 [DOI] [PubMed] [Google Scholar]
- Bangs F. and Anderson K. V. (2017). Primary cilia and mammalian hedgehog signaling. Cold Spring Harb. Perspect. Biol. 9, a028175 10.1101/cshperspect.a028175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bay S. N., Long A. B. and Caspary T. (2018). Disruption of the ciliary gtpase arl13b suppresses sonic hedgehog overactivation and inhibits medulloblastoma formation. Proc. Natl. Acad. Sci. USA 115, 1570-1575. 10.1073/pnas.1706977115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bielas S. L., Silhavy J. L., Brancati F., Kisseleva M. V., Al-Gazali L., Sztriha L., Bayoumi R. A., Zaki M. S., Abdel-Aleem A., Rosti R. O. et al. (2009). Mutations in inpp5e, encoding inositol polyphosphate-5-phosphatase e, link phosphatidyl inositol signaling to the ciliopathies. Nat. Genet. 41, 1032-1036. 10.1038/ng.423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaess S., Stephen D. and Joyner A. L. (2008). Gli3 coordinates three-dimensional patterning and growth of the tectum and cerebellum by integrating shh and fgf8 signaling. Development 135, 2093-2103. 10.1242/dev.015990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briscoe J., Pierani A., Jessell T. M. and Ericson J. (2000). A homeodomain protein code specifies progenitor cell identity and neuronal fate in the ventral neural tube. Cell 101, 435-445. 10.1016/S0092-8674(00)80853-3 [DOI] [PubMed] [Google Scholar]
- Bulgakov O. V., Eggenschwiler J. T., Hong D.-H., Anderson K. V. and Li T. (2004). Fkbp8 is a negative regulator of mouse sonic hedgehog signaling in neural tissues. Development 131, 2149-2159. 10.1242/dev.01122 [DOI] [PubMed] [Google Scholar]
- Caspary T., García-García M. J., Huangfu D., Eggenschwiler J. T., Wyler M. R., Rakeman A. S., Alcorn H. L. and Anderson K. V. (2002). Mouse dispatched homolog1 is required for long-range, but not juxtacrine, hh signaling. Curr. Biol. 12, 1628-1632. 10.1016/S0960-9822(02)01147-8 [DOI] [PubMed] [Google Scholar]
- Caspary T., Larkins C. E. and Anderson K. V. (2007). The graded response to sonic hedgehog depends on cilia architecture. Dev. Cell 12, 767-778. 10.1016/j.devcel.2007.03.004 [DOI] [PubMed] [Google Scholar]
- Chang C.-F., Chang Y.-T., Millington G. and Brugmann S. A. (2016). Craniofacial ciliopathies reveal specific requirements for gli proteins during development of the facial midline. PLoS Genet. 12, e1006351 10.1371/journal.pgen.1006351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chávez M., Ena S., Van Sande J., De Kerchove D'exaerde A., Schurmans S. and Schiffmann S. N. (2015). Modulation of ciliary phosphoinositide content regulates trafficking and sonic hedgehog signaling output. Dev. Cell 34, 338-350. 10.1016/j.devcel.2015.06.016 [DOI] [PubMed] [Google Scholar]
- Chiang C., Litingtung Y., Lee E., Young K. E., Corden J. L., Westphal H. and Beachy P. A. (1996). Cyclopia and defective axial patterning in mice lacking sonic hedgehog gene function. Nature 383, 407-413. 10.1038/383407a0 [DOI] [PubMed] [Google Scholar]
- Cooper A. F., Yu K. P., Brueckner M., Brailey L. L., Johnson L., McGrath J. M. and Bale A. E. (2005). Cardiac and cns defects in a mouse with targeted disruption of suppressor of fused. Development 132, 4407-4417. 10.1242/dev.02021 [DOI] [PubMed] [Google Scholar]
- Corbit K. C., Aanstad P., Singla V., Norman A. R., Stainier D. Y. R. and Reiter J. F. (2005). Vertebrate smoothened functions at the primary cilium. Nature 437, 1018-1021. 10.1038/nature04117 [DOI] [PubMed] [Google Scholar]
- Dessaud E., Yang L. L., Hill K., Cox B., Ulloa F., Ribeiro A., Mynett A., Novitch B. G. and Briscoe J. (2007). Interpretation of the sonic hedgehog morphogen gradient by a temporal adaptation mechanism. Nature 450, 717 10.1038/nature06347 [DOI] [PubMed] [Google Scholar]
- Ding Q., Motoyama J., Gasca S., Mo R., Sasaki H., Rossant J. and Hui C. C. (1998). Diminished sonic hedgehog signaling and lack of floor plate differentiation in gli2 mutant mice. Development 125, 2533-2543. [DOI] [PubMed] [Google Scholar]
- Dyson J. M., Conduit S. E., Feeney S. J., Hakim S., Ditommaso T., Fulcher A. J., Sriratana A., Ramm G., Horan K. A., Gurung R. et al. (2017). Inpp5e regulates phosphoinositide-dependent cilia transition zone function. J. Cell Biol. 216, 247-263. 10.1083/jcb.201511055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Echelard Y., Epstein D. J., St-Jacques B., Shen L., Mohler J., Mcmahon J. A. and Mcmahon A. P. (1993). Sonic hedgehog, a member of a family of putative signaling molecules, is implicated in the regulation of cns polarity. Cell 75, 1417-1430. 10.1016/0092-8674(93)90627-3 [DOI] [PubMed] [Google Scholar]
- Eggenschwiler J. T. and Anderson K. V. (2000). Dorsal and lateral fates in the mouse neural tube require the cell-autonomous activity of the open brain gene. Dev. Biol. 227, 648-660. 10.1006/dbio.2000.9918 [DOI] [PubMed] [Google Scholar]
- Ericson J., Morton S., Kawakami A., Roelink H. and Jessell T. M. (1996). Two critical periods of sonic hedgehog signaling required for the specification of motor neuron identity. Cell 87, 661-673. 10.1016/S0092-8674(00)81386-0 [DOI] [PubMed] [Google Scholar]
- Ericson J., Rashbass P., Schedl A., Brenner-Morton S., Kawakami A., Van Heyningen V., Jessell T. M. and Briscoe J. (1997). Pax6 controls progenitor cell identity and neuronal fate in response to graded shh signaling. Cell 90, 169-180. 10.1016/S0092-8674(00)80323-2 [DOI] [PubMed] [Google Scholar]
- Garcia-Gonzalo F. R., Phua S. C., Roberson E. C., Garcia G. III, Abedin M., Schurmans S., Inoue T. and Reiter J. F. (2015). Phosphoinositides regulate ciliary protein trafficking to modulate hedgehog signaling. Dev. Cell 34, 400-409. 10.1016/j.devcel.2015.08.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gigante E. D., Long A. B., Ben-Ami J. and Caspary T. (2018). Hypomorphic smo mutant with inefficient ciliary enrichment disrupts the highest level of vertebrate hedgehog response. Dev. Biol. 437, 152-162. 10.1016/j.ydbio.2018.03.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goetz S. C. and Anderson K. V. (2010). The primary cilium: a signalling centre during vertebrate development. Nat. Rev. Genet. 11, 331-344. 10.1038/nrg2774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodrich L. V., Johnson R. L., Milenkovic L., Mcmahon J. A. and Scott M. P. (1996). Conservation of the hedgehog/patched signaling pathway from flies to mice: induction of a mouse patched gene by hedgehog. Genes Dev. 10, 301-312. 10.1101/gad.10.3.301 [DOI] [PubMed] [Google Scholar]
- Goodrich L. V., Milenković L., Higgins K. M. and Scott M. P. (1997). Altered neural cell fates and medulloblastoma in mouse patched mutants. Science 277, 1109-1113. 10.1126/science.277.5329.1109 [DOI] [PubMed] [Google Scholar]
- Hayashi S. and Mcmahon A. P. (2002). Efficient recombination in diverse tissues by a tamoxifen-inducible form of cre: a tool for temporally regulated gene activation/inactivation in the mouse. Dev. Biol. 244, 305-318. 10.1006/dbio.2002.0597 [DOI] [PubMed] [Google Scholar]
- Haycraft C. J., Banizs B., Aydin-Son Y., Zhang Q., Michaud E. J. and Yoder B. K. (2005). Gli2 and gli3 localize to cilia and require the intraflagellar transport protein polaris for processing and function. PLoS Genet. 1, e53 10.1371/journal.pgen.0010053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y., Roelink H. and Mcknight G. S. (2002). Protein kinase a deficiency causes axially localized neural tube defects in mice. J. Biol. Chem. 277, 19889-19896. 10.1074/jbc.M111412200 [DOI] [PubMed] [Google Scholar]
- Huangfu D. and Anderson K. V. (2005). Cilia and hedgehog responsiveness in the mouse. Proc. Natl. Acad. Sci. USA 102, 11325-11330. 10.1073/pnas.0505328102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huangfu D., Liu A., Rakeman A. S., Murcia N. S., Niswander L. and Anderson K. V. (2003). Hedgehog signalling in the mouse requires intraflagellar transport proteins. Nature 426, 83-87. 10.1038/nature02061 [DOI] [PubMed] [Google Scholar]
- Jacoby M., Cox J. J., Gayral S., Hampshire D. J., Ayub M., Blockmans M., Pernot E., Kisseleva M. V., Compère P., Schiffmann S. N. et al. (2009). Inpp5e mutations cause primary cilium signaling defects, ciliary instability and ciliopathies in human and mouse. Nat. Genet. 41, 1027-1031. 10.1038/ng.427 [DOI] [PubMed] [Google Scholar]
- Jiang K., Liu Y., Fan J., Zhang J., Li X.-A., Evers B. M., Zhu H. and Jia J. (2016). Pi(4)p promotes phosphorylation and conformational change of smoothened through interaction with its c-terminal tail. PLoS Biol. 14, e1002375 10.1371/journal.pbio.1002375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasarskis A., Manova K. and Anderson K. V. (1998). A phenotype-based screen for embryonic lethal mutations in the mouse. Proc. Natl. Acad. Sci. USA 95, 7485-7490. 10.1073/pnas.95.13.7485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kisseleva M. V., Wilson M. P. and Majerus P. W. (2000). The isolation and characterization of a cdna encoding phospholipid-specific inositol polyphosphate 5-phosphatase. J. Biol. Chem. 275, 20110-20116. 10.1074/jbc.M910119199 [DOI] [PubMed] [Google Scholar]
- Larkins C. E., Aviles G. D. G., East M. P., Kahn R. A. and Caspary T. (2011). Arl13b regulates ciliogenesis and the dynamic localization of shh signaling proteins. Mol. Biol. Cell 22, 4694-4703. 10.1091/mbc.e10-12-0994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lek M., Dias J. M., Marklund U., Uhde C. W., Kurdija S., Lei Q., Sussel L., Rubenstein J. L., Matise M. P., Arnold H.-H. et al. (2010). A homeodomain feedback circuit underlies step-function interpretation of a shh morphogen gradient during ventral neural patterning. Development 137, 4051-4060. 10.1242/dev.054288 [DOI] [PubMed] [Google Scholar]
- Litingtung Y. and Chiang C. (2000). Specification of ventral neuron types is mediated by an antagonistic interaction between shh and gli3. Nat. Neurosci. 3, 979-985. 10.1038/79916 [DOI] [PubMed] [Google Scholar]
- Liu A., Wang B. and Niswander L. A. (2005). Mouse intraflagellar transport proteins regulate both the activator and repressor functions of gli transcription factors. Development 132, 3103-3111. 10.1242/dev.01894 [DOI] [PubMed] [Google Scholar]
- Mariani L. E., Bijlsma M. F., Ivanova A. A., Suciu S. K., Kahn R. A. and Caspary T. (2016). Arl13b regulates shh signaling from both inside and outside the cilium. Mol. Biol. Cell 27, 3687-3790. 10.1091/mbc.e16-03-0189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matise M. P., Epstein D. J., Park H. L., Platt K. A. and Joyner A. L. (1998). Gli2 is required for induction of floor plate and adjacent cells, but not most ventral neurons in the mouse central nervous system. Development 125, 2759-2770. [DOI] [PubMed] [Google Scholar]
- Moore B. S., Stepanchick A. N., Tewson P. H., Hartle C. M., Zhang J., Quinn A. M., Hughes T. E. and Mirshahi T. (2016). Cilia have high camp levels that are inhibited by sonic hedgehog-regulated calcium dynamics. Proc. Natl. Acad. Sci. USA 113, 13069-13074. 10.1073/pnas.1602393113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhopadhyay S., Wen X., Chih B., Nelson C. D., Lane W. S., Scales S. J. and Jackson P. K. (2010). Tulp3 bridges the ift-a complex and membrane phosphoinositides to promote trafficking of g protein-coupled receptors into primary cilia. Genes Dev. 24, 2180-2193. 10.1101/gad.1966210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhopadhyay S., Wen X., Ratti N., Loktev A., Rangell L., Scales S. J. and Jackson P. K. (2013). The ciliary g-protein-coupled receptor gpr161 negatively regulates the sonic hedgehog pathway via camp signaling. Cell 152, 210-223. 10.1016/j.cell.2012.12.026 [DOI] [PubMed] [Google Scholar]
- Niewiadomski P., Kong J. H., Ahrends R., Ma Y., Humke E. W., Khan S., Teruel M. N., Novitch B. G. and Rohatgi R. (2014). Gli protein activity is controlled by multisite phosphorylation in vertebrate hedgehog signaling. Cell Rep 6, 168-181. 10.1016/j.celrep.2013.12.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norman R. X., Ko H. W., Huang V., Eun C. M., Abler L. L., Zhang Z., Sun X. and Eggenschwiler J. T. (2009). Tubby-like protein 3 (tulp3) regulates patterning in the mouse embryo through inhibition of hedgehog signaling. Hum. Mol. Genet. 18, 1740-1754. 10.1093/hmg/ddp113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson V. L., Damrau C., Paudyal A., Reeve B., Grimes D. T., Stewart M. E., Williams D. J., Siggers P., Greenfield A. and Murdoch J. N. (2009). Mouse hitchhiker mutants have spina bifida, dorso-ventral patterning defects and polydactyly: Identification of tulp3 as a novel negative regulator of the sonic hedgehog pathway. Hum. Mol. Genet. 18, 1719-1739. 10.1093/hmg/ddp075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persson M., Stamataki D., Te Welscher P., Andersson E., Bose J., Ruther U., Ericson J. and Briscoe J. (2002). Dorsal-ventral patterning of the spinal cord requires gli3 transcriptional repressor activity. Genes Dev. 16, 2865-2878. 10.1101/gad.243402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pusapati G. V., Kong J. H., Patel B. B., Gouti M., Sagner A., Sircar R., Luchetti G., Ingham P. W., Briscoe J. and Rohatgi R. (2018). G protein-coupled receptors control the sensitivity of cells to the morphogen sonic hedgehog. Sci. Signal. 11, eaao5749 10.1126/scisignal.aao5749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribes V., Balaskas N., Sasai N., Cruz C., Dessaud E., Cayuso J., Tozer S., Yang L. L., Novitch B., Marti E. et al. (2010). Distinct sonic hedgehog signaling dynamics specify floor plate and ventral neuronal progenitors in the vertebrate neural tube. Genes Dev. 24, 1186-1200. 10.1101/gad.559910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riddle R. D., Johnson R. L., Laufer E. and Tabin C. (1993). Sonic hedgehog mediates the polarizing activity of the zpa. Cell 75, 1401-1416. 10.1016/0092-8674(93)90626-2 [DOI] [PubMed] [Google Scholar]
- Roelink H., Augsburger A., Heemskerk J., Korzh V., Norlin S., Ruiz I. Altaba A., Tanabe Y., Placzek M., Edlund T. et al. (1994). Floor plate and motor neuron induction by vhh-1, a vertebrate homolog of hedgehog expressed by the notochord. Cell 76, 761-775. 10.1016/0092-8674(94)90514-2 [DOI] [PubMed] [Google Scholar]
- Rohatgi R., Milenkovic L. and Scott M. P. (2007). Patched1 regulates hedgehog signaling at the primary cilium. Science 317, 372-376. 10.1126/science.1139740 [DOI] [PubMed] [Google Scholar]
- Sasaki H. and Hogan B. L. M. (1994). Hnf-3 beta as a regulator of floor plate development. Cell 76, 103-115. 10.1016/0092-8674(94)90176-7 [DOI] [PubMed] [Google Scholar]
- Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saalfeld S., Schmid B. et al. (2012). Fiji: An open-source platform for biological-image analysis. Nat. Methods 9, 676-682. 10.1038/nmeth.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamataki D., Ulloa F., Tsoni S. V., Mynett A. and Briscoe J. (2005). A gradient of gli activity mediates graded sonic hedgehog signaling in the neural tube. Genes Dev. 19, 626-641. 10.1101/gad.325905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su C.-Y., Bay S. N., Mariani L. E., Hillman M. J. and Caspary T. (2012). Temporal deletion of arl13b reveals that a mispatterned neural tube corrects cell fate over time. Development 139, 4062-4071. 10.1242/dev.082321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun M., Mondal K., Patel V., Horner V. L., Long A. B., Cutler D. J., Caspary T. and Zwick M. E. (2012). Multiplex chromosomal exome sequencing accelerates identification of enu-induced mutations in the mouse. G3 (Bethesda) 2, 143-150. 10.1534/g3.111.001669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svärd J., Heby-Henricson K., Persson-Lek M., Rozell B., Lauth M., Bergström A., Ericson J., Toftgård R. and Teglund S. (2006). Genetic elimination of suppressor of fused reveals an essential repressor function in the mammalian hedgehog signaling pathway. Dev. Cell 10, 187-197. 10.1016/j.devcel.2005.12.013 [DOI] [PubMed] [Google Scholar]
- Tuson M., He M. and Anderson K. V. (2011). Protein kinase a acts at the basal body of the primary cilium to prevent gli2 activation and ventralization of the mouse neural tube. Development 138, 4921-4930. 10.1242/dev.070805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B., Fallon J. F. and Beachy P. A. (2000). Hedgehog-regulated processing of gli3 produces an anterior/posterior repressor gradient in the developing vertebrate limb. Cell 100, 423-434. 10.1016/S0092-8674(00)80678-9 [DOI] [PubMed] [Google Scholar]
- Xin D., Christopher K. J., Zeng L., Kong Y. and Weatherbee S. D. (2017). Ift56 regulates vertebrate developmental patterning by maintaining iftb complex integrity and ciliary microtubule architecture. Development 144, 1544-1553. 10.1242/dev.143255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yavari A., Nagaraj R., Owusu-Ansah E., Folick A., Ngo K., Hillman T., Call G., Rohatgi R., Scott M. P. and Banerjee U. (2010). Role of lipid metabolism in smoothened derepression in hedgehog signaling. Dev. Cell 19, 54-65. 10.1016/j.devcel.2010.06.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.