

Abstract
Hypertension is an important contributor to cognitive decline but the underlying mechanisms are unknown. Although much focus has been placed on the effect of hypertension on vascular function, less is understood of its effects on non-vascular cells. Because astrocytes and parenchymal arterioles (PA) form a functional unit (neurovascular unit), we tested the hypothesis that hypertension-induced changes in PA tone concomitantly increases astrocyte Ca2+. We used cortical brain slices from eight week old mice to measure myogenic responses from pressurized and perfused PA. Chronic hypertension was induced in mice by 28-day angiotensin II (Ang II) infusion; PA resting tone and myogenic responses increased significantly. In addition, chronic hypertension significantly increased spontaneous Ca2+ events within astrocyte microdomains (MD). Likewise, a significant increase in astrocyte Ca2+ was observed during PA myogenic responses supporting enhanced vessel-to-astrocyte signaling. The transient potential receptor vanilloid 4 (TRPV4) channel, expressed in astrocyte processes in contact with blood vessels, namely endfeet, respond to hemodynamic stimuli such as increased pressure/flow. Supporting a role for TRPV4 channels in aberrant astrocyte Ca2+ dynamics in hypertension, cortical astrocytes from hypertensive mice showed augmented TRPV4 channel expression, currents and Ca2+ responses to the selective channel agonist GSK1016790A. In addition, pharmacological TRPV4 channel blockade or genetic deletion abrogated enhanced hypertension-induced increases in PA tone. Together, these data suggest chronic hypertension increases PA tone and Ca2+ events within astrocytes MD. We conclude that aberrant Ca2+ events in astrocyte constitute an early event towards the progression of cognitive decline.
Keywords: astrocytes, neurovascular unit, hypertension, parenchymal arteriole, myogenic tone
Graphical Abstract
INTRODUCTION
Hypertension-evoked functional and structural changes to the neurovascular unit impair dynamic flow of information between neurons and blood vessels increasing the risks for dysfunction. If left untreated, hypertension plays an important role in the progression of dementia and cognitive decline (Debette et al. 2011; Gąsecki et al. 2013; Iadecola et al. 2016; Iulita and Girouard 2017; Saavedra 2016; Waldstein et al. 2007). Although evidence supports the premise that hypertension and its complications significantly disrupts brain function, the cellular mechanisms are unknown.
Recently, we reported a bi-directional form of communication between blood vessels, astrocytes and neurons referred to as vasculo-neuronal coupling (VNC)(Kim et al. 2016). Different from neurovascular coupling (NVC), where an increase in neuronal activity increases cerebral blood flow (CBF), we proposed VNC constitutively balanced baseline cerebral perfusion with resting neuronal activity levels, thereby safeguarding the brain from a mismatch in metabolic demand and supply (Kim et al. 2015; Kim et al. 2016). In response to a rise in intravascular pressure, cerebral arterioles constrict to maintain CBF constant. This intrinsic property, referred to as cerebral autoregulation (CA), protects the brain from hypoperfusion at low pressures and from hyperpefusion when pressure rises (Cipolla 2009; Faraci et al. 1989; Paulson et al. 1990). Importantly, we demonstrated that during myogenic (pressure-evoked) constrictions, astrocytes respond with an increase in Ca2+ resulting in the release of vasoactive signals which aid in the maintenance of parenchymal arteriole (PA) tone (Kim et al. 2016). Chronic hypertension causes arteriole remodeling (Baumbach and Heistad 1989; Hart et al. 1980; Intengan and Schiffrin 2001) and increases in vascular tone (Chan et al. 2013; De Silva et al. 2016; Diaz-Otero et al. 2017; Iddings et al. 2015; Pires et al. 2015; Sweet et al. 2015). In addition, chronic hypertension evokes a rightward shift of the CA curve (Paulson et al. 1989; Saavedra et al. 2006) protecting the brain from high pressures. This adaptive response however, increases the brain vulnerability to ischemia when pressure drops (Girouard and Iadecola 2006). Astrocyte processes are in physical contact with blood vessels (Kacem et al. 1998; Mathiisen et al. 2010) placing them in an ideal position to sense vascular changes. Because astrocytes respond to increases in intravascular pressure (Kim et al. 2015; Kim et al. 2016), we hypothesized that chronic hypertension-evoked PA adaptive responses are accompanied with constitutive increases in astrocyte Ca2+ events. These sustained astrocyte Ca2+ elevations impair perfusion and contribute to astrocyte dysfunction.
Astrocytes play a central role in brain energy metabolism and homeostatic processes (Belanger et al. 2011; Gordon et al. 2007; McKenna 2013; Pellerin and Magistretti 1994; Wallraff et al. 2006). The early brain slice work of Zonta et al. 2003, showing astrocyte regulate vessel diameter, was then followed by a first in vivo study demonstrating sensory-evoked increases in astrocyte Ca2+ during functional hyperemia (Takano et al. 2006). Today, while still somewhat controversial, both in vivo (Otsu et al. 2015; Takata et al. 2018; Zonta et al. 2002) and brain slice (Attwell et al. 2010; Mulligan and MacVicar 2004; Takano et al. 2006; Zonta et al. 2003) evidence supports a role for astrocytes in the regulation of CBF following increases in neuronal activity, NVC. Moreover, alterations in basal astrocyte Ca2+ levels, independent of those evoked by increases in neuronal activity, modulate vascular tone (Kim et al. 2015; Rosenegger et al. 2015). Together, these findings support constitutive bi-directional flow of information between blood vessels and astrocytes. How this communication modality, and consequent changes in astrocyte Ca2+, is affected by disease conditions such as hypertension is not known. We demonstrated that intravascular pressure-evoked increases in astrocyte Ca2+ are partly mediated via TRPV4 channel activation (Kim et al. 2015). TRPV4 are Ca2+ permeable non-selective cation channels (Plant and Strotmann 2007) involved in numerous functions including, systemic tonicity, mechanosensation, pain, inflammation and vascular tone (Earley et al. 2009; Hill-Eubanks et al. 2014; Kanju and Liedtke 2016; Plant and Strotmann 2007; Ryskamp et al. 2015; Vriens et al. 2004). TRPV4 channels expressed in the vasculature serve as molecular detectors and transducers of arteriole myogenic responses (e.g. mechanical stress)(Kim et al. 2015). Numerous studies support an important role for endothelial TRPV4 channel-mediated arteriole dilation (Bagher et al. 2012; Marrelli et al. 2007; Seki et al. 2017; Sonkusare et al. 2012; Zhang and Gutterman 2011; Zhang et al. 2013; Zou et al. 2015). However, in some vascular beds, TRPV4 channel activation can evoke constriction (Saifeddine et al. 2015; Soni et al. 2017; Zhang et al. 2018). In the absence of NO-mediated signaling, TRPV4 channel coupling with G-protein-couple receptor (GPCR) can amplify intracellular Ca2+ events and generate vasoconstrictor prostanoids (Saifeddine et al. 2015; Zhang et al. 2018). In the cerebral cortex, TRPV4 channels localized to astrocytes (Benfenati et al. 2007; Dunn et al. 2013; Kim et al. 2015) and contribute to the regulation of vascular tone (Dunn et al. 2013; Kim et al. 2015). In addition to pharmacological agonists, astrocyte TRPV4 channels respond to epoxyeicosatrienoic acid (Baylie and Brayden 2011), hemodynamic and mechanical stimuli (e.g. pressure, stretch, swelling) (Jo et al. 2015; Kim et al. 2015). While evidence support enhanced TRPV4 channel function in ischemia (Butenko et al. 2012; Rakers et al. 2017; Yi et al. 2017), little is known on their function in hypertension.
To investigate the effect of hypertension on astrocyte Ca2+ and PA tone, mice were infused saline or Ang II (14 or 28 days). PA vascular tone and astrocyte Ca2+ activity was assessed using an advanced in vitro brain slice preparation (Kim and Filosa 2012; Kim et al. 2015), where a PA is pressurized and perfused (to allow the development of myogenic tone) and astrocyte Ca2+ activity monitored via confocal imaging. Following 28 days Ang II infusion, PA from hypertensive mice had significantly higher tone. In addition, significant increases in spontaneous astrocyte Ca2+ events and in pressure/flow-evoked astrocyte Ca2+ increases were also noted. Consistent with the idea that astrocyte TRPV4 channels contribute to aberrant astrocyte Ca2+ dynamics, augmented TRPV4 channel expression and activity was observed in hypertensive mice. Furthermore, TRPV4 channel pharmacological blockade or genetic deletion abrogated hypertension-evoked increases in PA tone. Together, these data suggest chronic hypertension affects not only PA tone but concomitantly alters intracellular astrocyte Ca2+ dynamics.
MATERIALS & METHODS
Animals
All experiments were conducted in 8 weeks old male mice including C57BL6 (Jackson Laboratories), TRPV4−/− bred on a C57BL6 background (kindly provided by Dr. Wolfgang Liedtke) and GLAST-CreERT2; R26-lsl-GCaMP3 mice (Otsu et al. 2015) following protocols approved by the animal care and use committee of Augusta University. Litter mate controls were used for TRPV4−/− and GLAST-CreERT2; R26-lsl-GCaMP3 mice experiments. Prior to experimentation, animals were housed in a room maintained at 20–22°C with a 12hr:12hr light-dark cycle and given ad libitum access to food and water.
Blood pressure assessment and hypertension induction
Mice were anesthetized using isoflurane and the catheter of a biotelemetry transmitter device (PA-C10, Data Sciences, St. Paul, MN) was implanted in the left carotid artery (Sturgis et al. 2009). After recovery from surgery, mice were housed individually in standard mouse cages in the animal facility under the conditions described above. Blood pressure signals were sampled at 500 Hz for 5 seconds every 2 minutes, 18 hrs per day. After 7 days, baseline mean arterial blood pressure (MAP) measurements were recorded over four consecutive days, followed by the induction of hypertension by angiotensin II (Ang II) administered via subcutaneous osmotic pump (Alzet, models 1002 and 1004). Pumps were loaded with Ang II (Sigma Aldrich, A9525) calculated to be delivered at 600 ng/Kg/min (Zimmerman et al. 2004) for 14 or 28 days.
Brain slice preparation
Following anesthesia with sodium pentobarbital, the brain was removed and cut into 250–300 μm thick coronal slices using a vibratome (Leica VT 1200S, Leica Microsystems, Wetzlar, Germany) in cold artificial cerebrospinal fluid (aCSF) containing (in mM): KCl 3, NaCl 120, MgCl2 1, NaHCO3 26, NaH2PO4 1.25, glucose 10, CaCl2 2 and 400 μM L-ascorbic acid, with osmolarity at 300–305 mOsm, equilibrated with 95% O2-5% CO2. Slices were kept at room temperature (RT) in aCSF until transferred to the microscope chamber. All experiments were conducted at a chamber temperature of 36±1˚C using a single line solution heater (SH-27G, Warner Instruments, Hamden, CT) connected to a DC power supply (1735A, BK Precision, Yorba Linda, CA) and continuously perfused with aCSF using a peristaltic pump (Miniplus 3, Gilson, Middleton, WI) at a rate of 2~3 ml/min.
Vessel cannulation
Arteriole cannulas (ID 1.17 mm and OD 1.50 mm, G150TF-3, Warner Instruments, Hamden, CT) were pulled with a micropipette puller (P-97 puller Sutter Instruments, Novato, CA), beveled (KT brown type micropipette beveller BV-10. Sutter instrument) and then mounted onto a micromanipulator. Prior to cannulation, the cannula resistance was determined from a flow-pressure curve, as previously reported (Kim and Filosa 2012). Parenchymal arterioles (PA) were visualized using a 60X Nikon (NIR Apo, 60X/1.0w, DIC N2, ∞/0 WD 2.8) objective equipped with Infrared Differential Interference Contrast (IR-DIC) optics. Luminal flow was controlled with a syringe pump (PHD 2000, Harvard Apparatus, Holliston, MA). A pressure transducer was placed just prior to the cannula for constant pressure monitoring [Servo Pump (PS/200, Living System Instrumentation, Burlington, VT)]. The internal cannula solution consisted of (in mM): KCl 3, NaCl 135, MgCl2 1, glucose 10, HEPES 10, CaCl2 2, and 1% albumin (Duling et al. 1981) with osmolarity at 300–305 mOsm and pH 7.4 adjusted with NaOH. The tip of the cannula was maneuvered towards the entrance of the PA and slowly introduced into the vessel lumen (Kim and Filosa 2012). Following cannula insertion, a second flow-pressure curve was performed and the total resistance (cannula + vascular bed) determined. The arteriole resistance was determined by subtracting the cannula resistance from the total resistance. Using Ohm’s law (Q = ΔP/R) where Q=flow rate, ΔP=delta pressures and R=resistance, the calculated arteriole resistance was used to determine the Q required to reach an intravascular pressure of 15, 30, 60 or 80 mmHg.
Vascular reactivity measurements
Studies of flow/pressure-induced myogenic tone in C57BL6 mice were performed in saline- or Ang II-treated (14 or 28 days) animals. Experiments in GLAST-CreERT2 (Figure 2B) and TRPV4−/− (Figure 6F) mice were performed in 28 day-treated mice only. Vascular diameter and lumen pressure values were recorded throughout the experiment at a frequency of 1 image/sec using PCO camware and Clampex 9.2 software, respectively. Upon PA cannulation, the arteriole flow rate was set to that needed to establish the desired starting lumen pressure of 30 mmHg (estimated physiological pressure for PA) and sustained until a plateau was established. Immediately after, flow rate was decreased to reach an arteriole lumen pressure of 15 mmHg (low pressure) and then stepwise increased to reach 30, 60 and 80 mmHg (high pressures). Experiments shown in Figure 6A–D were performed in saline and Ang II treated (28 days) C57BL6 mice. Following PA cannulation, lumen pressure was established at 30 mmHg and the arteriole myogenic response to 60 mmHg assessed. Slices were then perfused with the TRPV4 channel blocker (HC067047 10 μM, Tocris Bioscience) for at least 25 minutes before a second pressure-evoked response was performed in the presence of the blocker. For Figure 6E, PA tone was measured before and after the introduction of L-NAME 10 μM (Tocris) and indomethacin 10 μM (Sigma) into the arteriole lumen followed by bath application of the TRPV4 channel blocker. At the end of all cannulation protocols, intravascular pressure was brought back to 30 mmHg and slices perfused with zero Ca2+ aCSF plus 100 μM papaverine to obtain maximum diameter (100%). Diameter values are expressed as % tone from an averaged maximum diameter. To assess structural changes, in a subset of experiments, PA diameter measurements were obtained at 15, 30, 60 and 80 mmHg while slices were superfused with zero Ca2+ aCSF plus 100 μM papaverine.
Figure 2. Hypertension in GLAST-CreERT2; R26-lsl-GCaMP3 mice increases parenchymal arteriole tone and enhances spontaneous Ca2+ oscillations in cortical astrocytes.

(A) Representative images of cortical astrocytes from normotensive and hypertensive GLAST-CreERT2; R26-lsl-GCaMP3 mice labeled with anti-GFP (green) (left). Averaged GFP intensity per astrocyte for both groups (right) (n=12 images from 3 mice), scale bar = 50 μm, unpaired t-test. (B) % parenchymal arteriole (PA) tone across the pressure range 15–80 mmHg, (n=8 vessels, 7 (normotensive (white bar) and 6 hypertensive (black bars) mice). (C) Left, Representative astrocyte image from a GLAST-CreERT2; R26-lsl-GCaMP3 mice showing various regions of interest delineating the cell soma (dashed circle, blue), processes (square, red) and microdomains (MD) (circle, light green), scale bar = 10 μm. Right, Representative traces of spontaneous astrocyte Ca2+ events from soma, processes and MD. (D) Summary data of area under the curve AUC (F/F0*s) for soma, processes and MD from mice treated with saline or Ang II for 14 and 28 days. For saline-treated (white bars) n=10 cells for 14 days and 8 cells for 28 days from 3 and 4 mice, respectively. For Ang II-treated (black bars) n=11 cells for 14 days and 8 cells for 28 days from 3 and 4 mice, respectively. (E) % PA tone from normotensive (open circles) and hypertensive (closed squares) PA before and after introduction of BAPTA into the astrocyte syncytium. (F) Summary of data shown in (E) before (solid bars) and after (dashed bars) Ca2+ chelation. (G) ΔPA arteriole tone from normotensive (white) and hypertensive (black) mice following introduction of BAPTA into astrocytes (n=5 vessels from 4 normotensive and 3 hypertensive mice). Mean ± SEM, Two way ANOVA with Newman-Keuls post-hoc test (for B and D); paired t-test (for F) and unpaired t-test (for G). *P<0.05, **P<0.01 and ***P<0.001.
Figure 6. TRPV4 channel contribution to parenchymal arteriole tone in hypertension.

(A, B) Pressure protocol induced to measure myogenic responses in cortical parenchymal arterioles (PA) in the absence and then presence of the TRPV4 channel blocker HC67047 (10 μM). At the end of the experiment, maximum diameter was determined from values reached in zero Ca2+/papavarine at 30 mmHg. Representative PA diameter traces from normotensive (A) and hypertensive (B) C57BL6 mice (bottom panels). (C, D) Summary data from protocols shown in (A) corresponding to normotensive (C) (n=7 arterioles from 7 animals) and hypertensive (n=7 arterioles from 7 animals) (D) mice subjected to an increase in lumen pressure from 30–60 mmHg in the absence (solid bars) and then presence (hatch bars) of TRPV4 channel blocker. (E) % PA tone before and after introduction of L-NAME (10 μM) and indomethacin (10 μM) to the arteriole lumen followed by bath application (25 min) of HC67047 (10 μM). (F) % PA tone across the pressure range 15–80 mmHg from TRPV4−/− normotensive (white bars) (n=8 vessels, 7 mice) and hypertensive (black bars)(n=8 vessels, 6 mice) mice. Mean ± SEM. For C, D, Paired T-test; for E, One way Anova and Tukey’s multiple comparisons test; for F, Two way ANOVA and Newman-Keuls post-hoc test. *,ψP<0.05, **P<0.01, ****P<0.0001.
Calcium imaging
Experiments were conducted using the Andor Revolution system (Andor Technology Belfast, UK). A Nikon microscope (Eclipse FN 1, Nikon, Tokyo, Japan) was connected to a laser confocal spinning unit (CSU-X1, Yokogawa, Tokyo, Japan) attached to a Sutter filter wheel and an ultrasensitive EMCCD camera (iXonEM, Andor Technology, Belfast, UK) (Kim and Filosa 2012). Calcium imaging was conducted in saline or Ang II treated (14 or 28 days) GLAST-CreERT2; R26-lsl-GCaMP3 mice and visualized using a 40X Nikon objective (NIR Apo, 40X/0.8w, DIC N2, ∞/0 WD 3.5). Expression of the Ca2+ indicator GCaMP3 was induced following 5 consecutive days of tamoxifen injections (~75 mg/kg) to 6–10 week old mice. Immediately after tamoxifen administration of saline or Ang II treatment was started; brains were used 13–15 or 27–29 days post tamoxifen injections. Fluorescence images were obtained using a krypton/argon laser (488 nm excitation and >495 nm emission). Images were acquired at 2 frames/sec for ~10 minutes. For pressure-evoked Ca2+ changes Figure 3, intravascular pressure was stepwise increased from 15 to 30 mmHg, 30 to 60 mmHg, and 60 to 80 mmHg. Pressure protocols were conducted in the same region but in separate acquisition file durations of ~6 minutes to minimize laser-induced cell damage.
Figure 3. Pressure/flow-evoked astrocyte Ca2+ events in normotensive and hypertensive mice.

(A) Intravascular pressure protocol [15 to 30 mmHg (a), 30 to 60 mmHg (b) and 60 to 80 mmHg (c)] applied to parenchymal arterioles while measuring Ca2+ responses in cortical asrtocytes from GLAST-CreERT2; R26-lsl-GCaMP3 mice. (B-D) Summary data for astrocyte Ca2+ area under the curve (AUC) (F/F0*s) in soma, processes and MD from normotensive (white bars) and hypertensive (black bars) astrocytes in response to intravascular pressure changes (a-c). For protocol a, b: n=6 and 7 cells from 4 mice; for protocol c: n=5 and 5 cells from 3 mice, respectively. Mean ± SEM. Two way ANOVA with Newman-Keuls post-hoc test. *P<0.05.
Electrophysiology
Whole-cell currents were obtained using a Multiclamp 700B amplifier (Axon instruments, Foster City, CA). Patch pipettes were made from thin-walled borosilicate glass (outer diameter 1.5 mm, internal diameter 0.86 mm; Sutter instrument BF150–86-7.5) and pulled (P-97 puller Sutter Instruments, Novato CA) to resistances of 6–8 MΩ. For Figure 2, the internal solution consisted of (in mM): K-gluconate 130, HEPES 10, BAPTA 10, KCl 10, MgCl2 0.9, Mg2ATP 4, Na2GTP 0.3, phosphocreatine 20, added 100 μM Alexa-555, with osmolarity 291–295 mOsm and pH adjusted to 7.2 with KOH. For Figure 4, the external solution consisted of (in mM): CsCl 122, MgCl2 2, CaCl2 10, glucose 5, HEPES 10, with osmolarity 315±5 mOsm and 7.4 pH, adjusted with CsOH. The internal solution consisted of (in mM): Cs-gluconate 100, CsCl 26, MgCl2 2, EGTA 1, HEPES 10, with osmolarity 300±5 mOsm and 7.2 pH, adjusted with CsOH. Astrocyte whole-cell currents were evoked from a holding potential of 0 mV and stimulated using a voltage ramp protocol from −100 mV to +100mV for 500 ms after a potential step to −100 mV (500 ms) (Butenko et al. 2012). Current signals were filtered at 1 kHz low pass filter and digitized with a Digidata 1320 board (Axon instrument). pClamp 10.2 (Axon instrument) was used for data acquisition and storage.
Figure 4. Hypertension enhances TRPV4-mediated Ca2+ activity and whole cell currents in cortical astrocytes.

(A) Representative traces of astrocyte Ca2+ events from microdomains (MD) in response to the TRPV4 channel agonist GSK. (B) Summary data for MD astrocyte Ca2+ parameters (maximum Δpeak (F/F0), average Δpeak (F/F0), frequency (Hz) and area under the curve (AUC) (F/F0*s) from normotensive (white bars, n=9 cells from 3 mice) and hypertensive (black bars, n=11 cells, 3 mice) mice before and after bath applied GSK. (C) Representative whole-cell current traces induced with a voltage ramp protocol (inset) under control (black line) and during GSK bath-application (0.1 μM, light gray line), the subtracted current is shown (dashed line) in normotensive (left) and hypertensive (right) mice. (D) Time course of GSK induced currents obtained at −100 mV and +100mV in normotensive (close triangles) and hypertensive (open circles) mice. (E) Summary data showing Δ currents obtained at −100 mV and +100 mV in normotensive (n=7 cells, 3 mice) (white bars) and hypertensive (n=7 cells, 3 mice) (black bars) mice. Mean ± SEM. Two way ANOVA with Newman-Keuls post-hoc test for figure B and Unpaired T-test for figure D. *P<0.05, ** P<0.01 and *** P<0.0001.
Immunohistochemistry
Brains from saline or Ang II-treated mice (28 days) (C57BL6 or GLAST-CreERT2; R26-lsl-GCaMP3 mice) were fixed in 4% paraformaldehyde for 48 hours at 4°C and then transferred to 0.01 M PBS containing 30% sucrose for 72 hours. Brains were stored at −80°C until sectioning. 50 μm coronal sections were obtained using a Leica CM3050 S cryostat (Leica Microsystems, Wetzlar, Germany) and stored in cryoprotectant solution (50 mmol/L PBS, 30% ethylene glycol, 20% glycerol). Before staining, the cryoprotectant solution was removed by washing the tissue with 0.01M PBS. Fixed slices were then blocked for one hour in 0.01 M PBS containing 0.3% Triton X-100, 0.04% NaN3 and 10% horse serum (Vector Labs, Burlingame, CA). Slices were then incubated at room temperature for 48 hours in rabbit anti-TRPV4 (TRPV4; 5 μg/ml; Abcam) and goat anti-glial fibrillary acidic protein (GFAP; 1:750; Abcam) or in rabbit anti-ionized calcium binding adaptor molecule 1 (Iba1; 1:1000; Wako) or in rabbit anti-GFP (1:1500, Life technologies) suspended in 0.3% Triton X-100 0.01M PBS. Following corresponding washes in 0.01 M PBS, slices were incubated for four hours in donkey anti-rabbit Alexa 594 (1:250, Jackson Immuno Research) and anti-goat Alexa Fluor 647 (1:50, Jackson Immuno Research) or anti-rabbit Alexa 488 (1:250, Jackson Immuno Research) also in 0.3% Triton X-100 0.01M PBS. Slices were then washed three times with 0.01 M PBS and subsequently mounted using Vectashield (Vector Labs Burlingame, CA). Image acquisition was conducted using a confocal microscope (Zeiss LSM 510) equipped with a 40x oil immersion objective and Zen acquisition software. 1 μm interval Z-stacks were acquired from cortical layers III-V in both hemispheres. For microglia imaging (Iba1 staining) Z-stacks of 20 focal planes with 2 μm interval were acquired in the cortical layers III-V.
Data analysis
Electrophysiology data were analyzed using Clampfit 10.6, pClamp version 10.6 (Axon instrument). For Ca2+ imaging, fractional fluorescence (F/Fo) was determined by dividing the fluorescence intensity (F) within a region of interest (ROI) by a baseline fluorescence value (Fo) determined from ~100 images showing no activity. To detect Ca2+ oscillations in brain slices from GLAST-CreERT2; R26-lsl-GCaMP3 mice, we used GECIquant software developed by Dr. Baljit Khakh’s laboratory (http://www.physiology.ucla.edu/Labs/khakh/astrocytetools.htm). To address compartmentalized astrocyte Ca2+ events, the analysis was broken down into three main compartments: soma, larger astrocyte processes (area range 5 to 2000 μm2) and astrocyte microdomains (MD) (area range from 0.5 to 4 μm2). GECIquant automatically detects ROIs. After detecting ROIs intensities, values were converted to F/F0. We used Clampfit software to determine peak, frequency and area under the curve (AUC) for the traces generated for astrocyte soma, processes and microdomains. For Figure 3, interrupted acquisition files were generated for each Δpressure and thus, comparisons between pressures were not possible. For vascular tone experiments, arteriole lumen diameter was determined every ten frames using SparkAn developed by Dr. Adrian Bonev (University of Vermont).
For fixed tissue confocal images, a maximum intensity projection image was generated from the z-stack and the number of astrocytes expressing TRPV4 and GFAP determined. A ROI including all TRPV4 and GFAP-positive astrocytes was drawn on each image and the average intensities of both channels quantified. The resulting data from both hemispheres in the same animal was averaged and comparisons between normotensive and hypertensive animals performed. Microglia number and structural analysis was conducted using ImageJ software (National Institute of Health, https://imagej.nih.gov/ij/) as previously described in (Morrison and Filosa 2013; Morrison and Filosa 2016).
GraphPad Prism 7 software (Graphpad software, La Jolla, CA) was used for all statistical analyses. Values are expressed as mean ± S.E.M. A minimum of three mice was used for each experimental data set. Differences between two means within groups were determined using paired Student’s t-test. Differences between groups were determined using Student’s unpaired t-test, one or two way ANOVA with corresponding multiple comparison post hoc test specified in figure legends. Statistical significance was tested at 95% (P < 0.05) confidence level denoted with corresponding symbol in figures.
RESULTS
Hypertension driven increases in parenchymal arteriole tone
Using an advanced in vitro brain slice preparation, which includes flow and pressure within PA, we measured myogenic-evoked changes in PA tone from C57BL6 mice following 14 or 28 days saline or Ang II infusions. As described in Methods, the flow rate of the arteriole cannula was adjusted to attain an intravascular pressure of 15, 30, 60 or (in some cases), 80 mmHg. Representative traces showing myogenic-evoked PA diameter changes from normotensive and hypertensive mice (28 day treatment) are shown in Figure 1A and B, respectively. As previously described (Kim et al. 2015), raising intravascular pressure increased PA tone in both saline- and Ang II-treated groups, a significant increase in pressure-induced tone was observed in the hypertensive vs. normotensive group, Figure 1D. Consistent with the observation that 600 ng/Kg/min Ang II infusion did not significantly increase MAP by day 14 (Suppl. Figure 1), no differences in myogenic constrictions between groups were observed in PA from 14-day Ang II-treated mice, Figure 1C. In addition, no changes in PA passive diameters were noted between groups (Suppl. Figure 2) after 28 days of treatment. Together, these results show that 28 day Ang II treatment significantly increased arterial blood pressure and concomitantly, vascular tone without changing the passive properties of PA.
Figure 1. Hypertension in mice increases parenchymal arteriole tone.

A, B (Upper panels) Pressure protocol applied to induce myogenic responses in cortical parenchymal arterioles (PA). At the end of the experiment, maximum diameter was determined at 30 mmHg upon zero Ca2+/papavarine superfusion. (Bottom panels) Representative PA diameter traces from 28 day saline (A) and Ang II (600 ng/Kg/min) treated (B) C57BL6 mice. (C, D) % PA tone across the pressure range 15–80 mmHg in 14 days (C) (n=8 vessels, 7 saline mice and n=9 vessels, 8 Ang II-treated mice) and 28 days (D) (n=9 vessels, 8 saline mice and n=10 vessels, 9 Ang II treated mice) saline/Ang II-treated mice. Mean ± SEM. Two way ANOVA and Newman-Keuls post-hoc test. *P<0.05, **P<0.01.
Spontaneous and pressure/flow-evoked astrocyte Ca2+ dynamics
We previously reported that flow/pressure-evoked PA constrictions increased intracellular Ca2+ in astrocytes (Kim et al. 2015; Kim et al. 2016). Using cortical brain slices from normotensive and hypertensive GLAST-CreERT2; R26-lsl-GCaMP3 mice, we determined if Ang II treatment influenced astrocyte Ca2+ dynamics in soma, processes and microdomains (MD). We measured both GFP expression and the myogenic response of PA from normotensive and hypertensive (28 days saline or Ang II) GLAST-CreERT2; R26-lsl-GCaMP3 mice. No changes in GFP expression were observed between groups, Figure 2A. In addition, as with arterioles from C57BL6 mice (Figure 1), hypertension significantly increased PA tone at 60 and 80 mmHg compared to corresponding normotensive controls, Figure 2B.
We then measured astrocyte Ca2+ events from saline and Ang II treated GLAST-CreERT2; R26-lsl-GCaMP3 mice. No differences in spontaneous astrocyte Ca2+ dynamics were noted in mice receiving Ang II infusion for 14 days. However, a significant increase in the AUC was observed for spontaneous astrocyte Ca2+ activity in MD from hypertensive (vs. normotensive) mice, Figure 2D. To assess whether higher astrocyte Ca2+ levels, as seen in hypertension (28 day-Ang II), contribute to enhanced PA tone, we next tested whether the introduction (via a patch pipette) of the fast Ca2+ chelator BAPTA (10 mM) to the astrocytic syncytium altered PA tone, Figure 2E. A PA was first cannulated and allowed to develop myogenic tone at 30 mmHg. Following an equilibration period, an astrocyte in the vicinity of the arteriole was patched and BAPTA introduced into the syncytium (~25 minutes). Lowering basal astrocyte Ca2+ significantly reduced resting PA tone in both normotensive and hypertensive groups, Figure 2E,F. A larger astrocyte-to-vessel contribution was noted in the hypertensive group (9.01±1.68% vs. 20.04±3.85% in Δ PA tone), Figure 2G. These data suggest augmented basal astrocyte Ca2+ events contribute to increased PA resting tone.
To determine whether hypertension treatment affects astrocyte sensitivity to intravascular pressure changes in normotensive vs. hypertensive GLAST-CreERT2; R26-lsl-GCaMP3 mice, we determined the astrocyte Ca2+ response to three stepwise intravascular pressure increases, from 15 to 30 mmHg, 30 to 60 mmHg and 60 to 80 mmHg. Figure 3 shows the astrocyte Ca2+ response (area under the curve, AUC) for soma, processes and MD. No differences between normotensive and hypertensive astrocytes Ca2+ events were noted in response to intravascular pressure increases for astrocyte soma and process. On the other hand, a significant increase in the AUC was observed in the hypertensive group when stepping from 30 to 60 mmHg. In addition, the AUC was also significantly greater in the hypertensive group (vs. normotensive) at 60 mmHg. The latter was due to an increase in peak amplitude and not frequency, Suppl. Table 1. Together, these data support enhanced pressure-evoked astrocyte responses in hypertension.
TRPV4 channel contribution to astrocyte Ca2+ and PA tone
Endothelial cell TRPV4 channel activation on cerebral arterioles contribute to arteriole vasodilation (Dunn et al. 2013; Zhang et al. 2013). However, evidence suggests that changes in the intracellular astrocyte Ca2+ concentrations (Girouard et al. 2010) can affect the direction (dilation vs. constriction) by which stimulus-evoked increases in astrocyte Ca2+ modulate PA tone. Thus, we measured PA diameter responses following TRPV4 channel activation or blockade, putatively in astrocytes. Spontaneous astrocyte Ca2+ activity was measured in brain slices from saline/Ang II-treated GLAST-CreERT2; R26-lsl-GCaMP3 mice in the absence and then presence of the specific TRPV4 channel agonist GSK (0.1 μM). GSK significantly increased the AUC and the Max Δ Peak in the MD of hypertensive astrocytes, Figure 4B. Consistent with the notion of increased TRPV4 channel activity, bath application of the specific TRPV4 channel blocker HC067047 (10 μM) reverted hypertension-evoked enhancement of spontaneous Ca2+ events (shown in Figure 2D) in MD, Suppl. Figure 3. To further evaluate TRPV4 channel function, we recorded astrocyte whole cell currents in response to bath applied GSK. A significant increase in the GSK-sensitive current was observed in astrocytes from hypertensive (vs. normotensive) mice. GSK-evoked currents were reduced in the presence of the TRPV4 specific inhibitor HC 067047, Figure 4 C–E.
To assess TRPV4 channel expression, we conducted TRPV4 immunohistochemistry in GLAST-CreERT2; R26-lsl-GCaMP3 and saline/Ang II-treated C57BL6 mice. Immunolabeling was determined in cortical layers III-V as these were the areas assessed for Ca2+ imaging and electrophysiological recordings. As previously reported (Benfenati et al. 2007), astrocytes expressed TRPV4 channels. In GLAST-CreERT2; R26-lsl-GCaMP3 mice, TRPV4 channel expression was observed in both processes and MD (as defined by GFP-TRPV4 channel colocalization), Figure 5A. To determine if astrocyte TRPV4 channel expression is altered in hypertension we measured channel expression along with the glial fibrillary acidic protein (GFAP), a marker of reactive astrocytes, from C57BL6 normotensive and hypertensive mice, Figure 5B. Interestingly, while all TRPV4-positive astrocytes expressed GFAP, not all GFAP-positive astrocytes expressed TRPV4. The number and intensity of TRPV4- and GFAP-positive astrocytes was significantly increased in hypertensive vs. normotensive mice, Figure 5C. As a proxy to inflammation, we measured microglia cell number and structural morphology following immunostaining against Iba1 (Morrison and Filosa 2013; Morrison and Filosa 2016). While no significant differences between groups (normotensive vs. hypertensive) was observed in cortical (layers III-V) microglia, a significant increase in cell number and morphological differences was observed in the CA1 region of the hippocampus, Figure 5D. Together, these data support increased brain inflammation in hypertensive mice.
Figure 5. Hypertension increases TRPV4 channel expression in astrocytes.

(A) Representative image showing TRPV4 channel (red) expression from a GCaMP3-expressing astrocyte (GFP reporter in green), merged and colocalized images also shown, scale bar = 20 μm. (B) Representative images of brain slices from normotensive and hypertensive mice stained against GFAP (blue) and TRPV4 channel (red), colocalization points (magenta) shown. (C) Summary data for TRPV4 and GFAP mean fluorescence intensity and number of astrocytes in normotensive (white bars) and hypertensive (black bars) mice (n= 9 and 10 images from 6 normotensive and 8 hypertensive mice, respectively). (D) Microglia cell count and structural analysis in cortex and CA1 hippocampus regions (n=3 normotensive and n=3 hypertensive mice). Unpaired T-test, *P<0.05.
To determine the functional contribution of TRPV4 channel activity to PA tone, we measured myogenic responses in normotensive and hypertensive C57BL6 mice in the presence of the TRPV4 channel blocker HC067047. As a control, PA were first subjected to an increase in lumen pressure from 30 to 60 mmHg in the absence of the blocker and then, in the presence of HC067047. Albeit a ~25% decrease in tone, no significant changes in resting PA tone were observed in the normotensive group upon exposure to HC067047 at 30 mmHg. Likewise, no differences were observed in the myogenic response when intravascular pressure was increased from 30 to 60 mmHg in the absence vs. presence of the blocker, Figure 6A and C. On the other hand, in hypertensive mice, HC067047 significantly decreased resting tone at 30 mmHg. Moreover, a significant decrease in PA myogenic responses was observed when intravascular pressure was increased from 30 to 60 mmHg in the absence vs. presence of the blocker, Figure 6B and D.
In a previous study we reported that in normotensive rats, bath applied HC067047 (following the development of myogenic tone) significantly decreased PA resting tone which is not observed in normotensive PA in this study. One possible explanation could be the lower resting tone of PA in this study. Differently to our previous approach, here the flow rate needed to achieve the various pressure ranges used was varied between arterioles. This methodological modification lead to slightly higher flow rates (0.35±0.07 μl/min for 15 mmHg, 0.69±0.14 μl/min for 30 mmHg, 1.39±0.27 μl/min for 60 mmHg and 1.76±0.34 μl/min for 80 mmHg) than before (0.1–0.3 μl/min). Thus, differences in resting tone between studies could have resulted from a larger endothelium-dependent contribution. Using PA from normotensive mice we measured the myogenic response in the presence and then absence of endothelium-mediated vasodilatory mechanisms (namely, NOS and COX) followed by TRPV4 channel blockade. As shown in Figure 6E, a significant increase in PA tone (at 30 mmHg) was observed when the cannula (now containing 10 μM LNAME and 10 μM indomethacin) was re-introduced into the arteriole. Under these conditions, bath applied HC067047 significantly decreased PA tone, Figure 6E. Together, these data support TRPV4 channel contribution is tone dependent and channel activity enhanced in hypertension.
Consistent with the idea that TRPV4 channels contribute to the maintenance of PA tone and/or myogenic responses in hypertension, PA from hypertensive TRPV4−/− mice failed to develop substantial increases in tone upon increases in lumen pressure. Contrary to enhanced myogenic responses from hypertensive C57BL6 mice (as shown in Figure 1), the PA myogenic response from hypertensive TRPV4−/− (vs. normotensive) mice was not different, Figure 6F. Taken together, these data support a role for TRPV4 channels in enhanced PA tone in hypertension.
DISCUSSION
The goal of this study was to determine the effects of Ang II-induced hypertension on PA tone and vessel-to-astrocyte signaling, important determinants of neurovascular unit function. Several significant findings were revealed from hypertensive mice. First, consistent with previous studies in various models of hypertension (Chan et al. 2013; De Silva et al. 2016; Diaz-Otero et al. 2017; Pires et al. 2015; Sweet et al. 2015), PA from hypertensive mice show enhanced myogenic tone. Second, both an increase in spontaneous and myogenic-evoked astrocyte MD Ca2+ events were observed. In addition, elevated basal astrocyte Ca2+ activity was associated with a greater contribution to resting PA tone. Third, augmented astrocyte Ca2+ events were linked to enhanced astrocyte TRPV4 channel activity and expression. Fourth, pharmacological blockade or genetic TRPV4 channel deletion (TRPV4−/− mice) resulted in blunted hypertension-dependent increases in PA myogenic tone. Fifth, hypertensive mice showed increased GFAP expression and microglia activation (albeit in hippocampus and not cortex). Together, these data support Ang II-induced hypertension increases cortical PA tone and leads to aberrant bi-directional signaling at the gliovascular interphase.
Little is known about the effect hypertension and circulating Ang II have on cortical astrocyte function. Astrocyte processes are in intimate contact with blood vessels and synapses thereby establishing a functional bridge between the state of brain perfusion and neuronal activity levels. In the healthy brain, dynamic bi-directional signaling between vascular-glial-neuronal cells establishes a homeostatic equilibrium that ensures a balanced metabolic demand with supply. A shift in the activity of any of these cells puts in motion a cascade of events that ultimately, restore equilibrium. Such is the case of the NVC response where a rise in neuronal activity causes the release of glutamate and K+ ions, for example, triggering the activation of pathways that evoke increased perfusion to the brain. Evidence also support the notion that a rise in astrocyte Ca2+ modulate vascular tone during neuronal activity-mediated signaling [e.g. NVC (Iadecola 2017; Nuriya and Hirase 2016; Otsu et al. 2015; Takano et al. 2006)] as well as during resting (independent of neuronal activity) conditions (Kim et al. 2015; Rosenegger et al. 2015). Thus, both during NVC (Dunn et al. 2013; Girouard et al. 2010; Takano et al. 2006) and at steady-state tone, astrocyte Ca2+ levels influence PA diameter. The molecular players (e.g., K+, arachidonic acid-derived metabolites) driving these responses are complex and poorly understood. Understanding the processes associated with a shift in astrocyte Ca2+ homeostasis may provide important insight into their contribution to vascular regulation and consequently, cerebral perfusion. Data from this study support the notion that Ang II-induced hypertension increases myogenic tone in cortical PA and furthers our understanding of how this adaptive vascular response changes the bi-directional flow of information at the gliovascular interphase.
Astrocyte function is compartmentalized, and little is known about the effect disease conditions have on the homeostasis and function of distinct astrocyte compartments (Agarwal et al. 2017; Dunn et al. 2013; Lind et al. 2018; Oheim et al. 2017; Rungta et al. 2016; Shigetomi et al. 2013). Using saline and Ang II-infused GLAST-CreERT2; R26-lsl-GCaMP3 mice, where compartmentalized Ca2+ activity can be readily assessed, we show increased spontaneous astrocyte Ca2+ events in MD of hypertensive mice. Importantly, chronic hypertension altered bi-directional signaling at the gliovascular interphase. In response to PA myogenic responses (i.e., 30 to 60 mmHg), astrocyte Ca2+ events robustly increased (227.35% in hypertensive vs. 48.48% in normotensive astrocytes), Figure 3. Functionally, higher resting astrocyte Ca2+ levels were associated with augmented levels of PA tone as lowering Ca2+ within the astrocytic syncytium decreased tone with a greater effect observed in hypertension, Figure 2G. A number of mechanisms could increase astrocyte Ca2+ during Ang II-induced hypertension including: direct Ang II actions on astrocytes, mechanotransduction via increased stretching of astrocyte processes (as those evoked during adjustments in PA diameter (Kim et al. 2015)) and/or via the action of endothelium-mediated signals [e.g. nitric oxide (Kohler et al. 2006; Marziano et al. 2017; Mendoza et al. 2010) and EETs (Dunn et al. 2013; Earley et al. 2009; Vriens et al. 2005)]. Based on poor evidence for AT1R expression in cortical astrocytes and established Ang II-induced endothelial dysfunction (Chrissobolis et al. 2012; Chrissobolis et al. 2011; De Silva et al. 2016; Girouard et al. 2007), we propose hypertension increases the sensitivity of mechanosensitive TRPV4 channels expressed on astrocytes. The combined effect of hypertension on vascular cells and astrocytes favors their contribution to PA constrictions.
In a previous study, we reported that increased PA intravascular pressure augmented astrocyte Ca2+, partly via a TRPV4-mediated mechanism. TRPV4 channels are expressed in the plasma membrane of astrocytes (Benfenati et al. 2007; Benfenati et al. 2011; Lipski et al. 2006). While channel expression may be specific to a subpopulation of astrocytes, their activation was shown to increase Ca2+ in TRPV4-negative astrocytes via ATP and gap junction-mediated signaling (Shibasaki et al. 2014). Upon activation, TRPV4-mediated Ca2+ increases the release of glutamate, increasing neuronal excitation (Shibasaki et al. 2014). Pathological conditions can both increase or decrease TRPV4 expression/function. During the development of astrogliosis, following hypoxic/ischemic injury, rat hippocampal CA1 region astrocytes increased TRPV4 expression and activity (Butenko et al. 2012; Shirakawa et al. 2010). In hypothalamic 4B cells, Ang II mediate TRPV4 channel trafficking to the membrane and increased cell Ca2+ (Saxena et al. 2014). In myocytes, Ang II modulate TRPV4 channel activity via downstream activation of PKCα bound to the anchoring protein AKAP150 (Tajada et al. 2017). In human umbilical vein, endothelial cells AT1 receptor activation potentiate constriction via TRPV4-mediated Ca2+ entrance (Saifeddine et al. 2015). In cerebral capillary endothelial cells, TRPV4 channels activation are tonically inhibited by the plasma membrane phosphatidylinositol 4,5-biphosphate (PIP2). Channel inhibition is released following Gq-protein-coupled receptors (GqPCR) activation and downstream PIP2 depletion. Importantly, PIP2 was also shown to regulate inwardly rectifying potassium (Kir) channels (Harraz et al. 2018a; Harraz et al. 2018b). Together, these studies support the notion that aberrant TRPV4-mediated signaling may result in both astrocyte and vascular dysfunction.
We determined if in hypertension sustained astrocyte Ca2+ events are driven by increased TRPV4 channel activity and/or expression. Chronic Ang II infusion increased TRPV4-mediated astrocyte Ca2+ in MD, Figure 4. Likewise, GSK-sensitive currents were augmented in astrocytes from hypertensive mice. Increased expression of the inflammatory TRPV4 channel was observed along with increased GFAP-positive astrocytes and microglia activation (albeit in the hippocampus). However, whether TRPV4-GFAP-positive astrocytes constitute a subpopulation of reactive astrocytes that are pro- vs. anti-inflammatory deserves further investigation as GFAP expression does not differentiate between the different types of reactive astrocytes (Clarke et al. 2018). Also possible is that for astrocytes to transition into a pro-inflammatory state, prolonged chronic sustained Ca2+ levels may be needed. This is supported by lack of vascular and astrocyte functional changes in 14 day Ang II-infused mice were basal astrocyte Ca2+ elevation (vs. 28 days) was not evident. Križaj et al. proposed that a Ca2+ overload in retinal glial cells could result from the combined effect of enhanced TRPV4 channel activation with P2X7R and pannexin channels (Krizaj et al. 2014). TRPV4-mediated Ca2+ entry causes the release of ATP (Kim et al. 2015; Shibasaki et al. 2014) which can in turn trigger further Ca2+ increases via purinergic receptors and, ultimately, the release of pro-inflammatory signals (e.g., TNF-α, IL-1β) (Krizaj et al. 2014). Astrocyte-derived pro-inflammatory signals could activate microglia cells (or vice-versa) exacerbating pro-inflammatory processes in the brain. Future studies addressing the cross-talk between astrocytes and microglia in the context of hypertension and neurovascular unit homeostasis are needed. Thus, while TRPV4 channels play an important role in physiological processes, their contribution to pro-inflammatory pathways is also significant and further studies warranted.
Methodological considerations
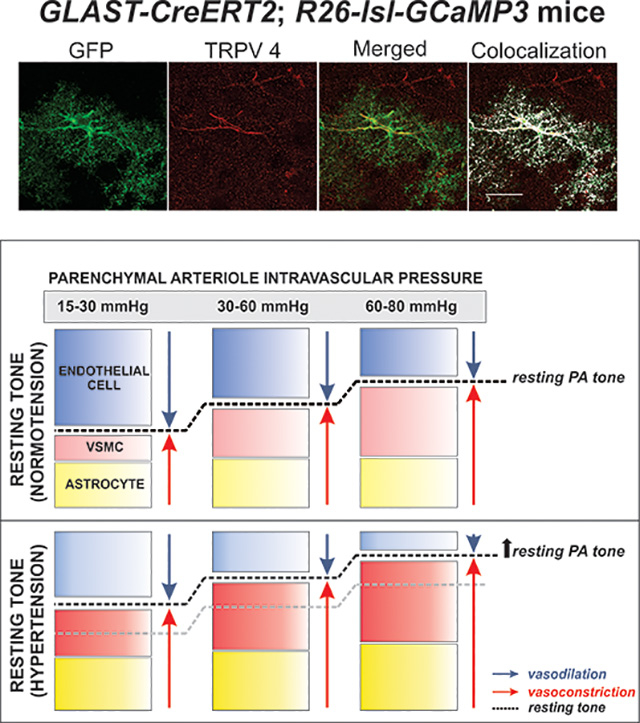
In a previous study, we showed significant reductions in PA tone following TRPV4 channel blockade (Kim et al. 2015). In this study, however, the blocker significantly reduced PA tone in hypertensive but not in normotensive vessels. We reasoned that in this study, lack of significance in normotensive arterioles could have arisen from differences in their resting tone established by the relative contribution from the endothelium in turn determined by the flow rates used. Consistent with this notion, when we functionally blocked the endothelium, TRPV4 blockade significantly reduced PA tone in normotensive mice, Figure 6. These data suggest endothelium-mediated dilations override astrocyte TRPV4-mediated constrictions and that TRPV4 channel contributions are tone dependent. In our brain slice approach, both flow and pressure engage mechanisms associated with flow-mediated endothelial dilation, myogenic constriction and astrocyte-mediated modulation of PA tone, via Ca2+-dependent signaling. Thus, shifting the relative contribution of these various mechanisms may result in one pathway overriding the other and/or masking underlying signaling modalities. We propose a working model (Figure 7) for both normotensive and hypertensive PA were at low intravascular pressures the major contributors to resting tone are the opposing (dilation vs. constriction) effects of endothelium- and astrocyte-mediating signaling, at physiological pressures, myogenic mechanisms along with astrocyte-mediated constriction compete against endothelium-mediated dilation and at high intravascular pressures myogenic- and astrocyte-mediated constricting pathways largely override endothelium-dependent dilations. Under these conditions, hypertension-evoked endothelial dysfunction impairs cerebral perfusion. This effect, along with the well-established rightward shift in the CA curve will increase the brain vulnerability to ischemia particularly at low pressures.
Figure 7. Proposed model depicting the relative contribution of vascular and non-vascular cells defining resting tone in normotensive and hypertensive parenchymal arterioles.

We propose that at low intravascular pressures (15–30 mmHg) resting parenchymal arteriole (PA) tone is determined by signals from endothelial cells and, to a lesser extent, that of vascular smooth muscle cells (VSMC) and astrocytes. As the intravascular pressure increases to physiological ranges for PA (30–60 mmHg), myogenic constriction and increased astrocyte Ca2+ levels augment resting PA tone. At significantly higher intravascular pressure ranges (60–80 mmHg) myogenic constriction and augmented astrocytes Ca2+ signaling events override endothelial-mediated dilations further increasing PA resting tone. In hypertension, the combined effect of endothelial dysfunction (shown by a lighter blue color) with augmented VSMC constriction and resting astrocyte Ca2+ (shown with darker red and yellow colors, respectively) shifts the relative contribution of these three players (endothelium, VSMC and astrocytes) favoring constriction and resulting in higher levels of resting PA tone.
Conclusions and functional considerations
Hypertension-evoked changes at the neurovascular unit can significantly impair the flow of information at the gliovascular interphase compromising perfusion and negatively impacting neuronal function. We show that, at resting conditions (absence of NVC-mediated signaling), Ang II-infusion resulted in enhanced astrocyte TRPV4 channel activity, elevated basal astrocyte MD Ca2+ events and augmented PA tone. While not specifically addressed in this study, elevated basal astrocyte Ca2+ in hypertensive mice may interfere with the molecular mechanism that drive the NVC response and/or are involved in the clearance of metabolic by-products (e.g. glymphatics)(Iliff et al. 2013a; Iliff et al. 2013b). The recent evidence in capillary endothelial cells demonstrating a dynamic interaction between TRPV4 and Kir2.1 channels, whereby GqPCR activation induces PIP2 depletion, a master regulator of both channels (Harraz et al. 2018a; Harraz et al. 2018b; Longden et al. 2017) raises an important question. Could augmented Ang II-mediated signaling impair TRPV4-Kir2.1 interactions? For example, increased TRPV4 channel activity (Harraz et al. 2018a; Harraz et al. 2018b) and reduced Kir2.1-mediated hyperpolarizations (Filosa et al. 2006; Harraz et al. 2018a; Longden et al. 2017) could drive the membrane potential of vascular cells towards more depolarized states, favoring constrictions and consequently, impairing perfusion to the brain. In addition, hypertension-induced changes in PA tone may impair dynamic pulsatile mechanisms involved in waste clearance or glymphatics. While evidence for these mechanism in the context of hypertension is lacking, the concept warrants further consideration as impairments in these important processes can contribute to cognitive decline.
Supplementary Material
Supplementary Table 1. Summary data showing astrocyte Ca2+ parameters (maximum Δpeak (F/F0), average Δpeak (F/F0), frequency (Hz) and area under the curve (AUC) (F/F0*s)) in response to intravascular pressure changes for soma, process and MD in normotensive and hypertensive mice. Two way ANOVA with Newman-Keuls post-hoc test. *P<0.05, **P<0.01 for comparisons within groups (W/G) and ^P<0.05, ^^P<0.01 for comparisons between groups (B/G).
Supplementary figure 1. Progressive hypertension development induced by Ang II infusion. Averaged time course recording of the changes in mean arterial pressure (MAP) from C57BL6 mice treated with 600 ng/Kg/min Ang II (filled circles)(n=5) or saline (open circles) (n=4). Two way ANOVA and Sidak post-hoc test. ** P<0.05, *** P<0.001.
Supplementary figure 2. Hypertension does not change PA passive properties. Average % passive diameter at 15, 30, 60 and 80 mmHg in PA from normotensive (open circles) and hypertensive mice (filled squares) (n=6). Two way ANOVA and Newman-Keuls post-hoc test.
Supplementary figure 3. TRPV4 channel contribution to astrocyte spontaneous Ca2+ events in hypertension. (A) Representative traces of astrocyte Ca2+ events for soma, processes and microdomains from normotensive and hypertensive mice following pre-incubation (25 min) in the TRPV4 channel blocker HC067047 (10 μM). (B) Summary data for soma, processes and MD astrocyte Ca2+ parameters (maximum Δpeak (F/F0), average Δpeak (F/F0), frequency (Hz) and area under the curve (F/F0*s)) in normotensive (white bars, n=7 cells from 3 mice) and hypertensive (black bars, n=6 cells from 3 mice) mice. Mean ± SEM.
Main Points.
Chronic hypertension (via 28-day angiotensin II infusion) increased parenchymal arteriole tone and myogenic responses.
Chronic hypertension significantly increased spontaneous and pressure-evoked Ca2+ events within astrocyte microdomains.
Ca2+ events were associated to increased transient potential receptor vanilloid 4 (TRPV4) channel, expression and activity.
Acknowledgements
We would like to thank Rabei Alaisami for technical support in generation of hypertensive mice and telemetry studies, Khadijah Alexander for her help with immunohistochemistry and Drs. Clinton Webb and Laura Gonzalez Bosc for comments on the manuscript.
Sources of Funding: This work was supported by funding from the National Heart, Lung, and Blood Institute (NHLBI) (R01 HL089067–02) and the National Institute of Neurological Disorders and Stroke (NINDS) (1R01NS082521–01) of the NIH to JAF.
Footnotes
Disclosure: None
REFERENCES
- Agarwal A, Wu PH, Hughes EG, Fukaya M, Tischfield MA, Langseth AJ, Wirtz D, Bergles DE. 2017. Transient Opening of the Mitochondrial Permeability Transition Pore Induces Microdomain Calcium Transients in Astrocyte Processes. Neuron 93:587–605 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attwell D, Buchan AM, Charpak S, Lauritzen M, Macvicar BA, Newman EA. 2010. Glial and neuronal control of brain blood flow. Nature 468:232–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagher P, Beleznai T, Kansui Y, Mitchell R, Garland CJ, Dora KA. 2012. Low intravascular pressure activates endothelial cell TRPV4 channels, local Ca2+ events, and IKCa channels, reducing arteriolar tone. Proc Natl Acad Sci U S A 109:18174–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumbach GL, Heistad DD. 1989. Remodeling of cerebral arterioles in chronic hypertension. Hypertension 13:968–72. [DOI] [PubMed] [Google Scholar]
- Baylie RL, Brayden JE. 2011. TRPV channels and vascular function. Acta Physiol (Oxf) 203:99–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belanger M, Allaman I, Magistretti PJ. 2011. Brain energy metabolism: focus on astrocyte-neuron metabolic cooperation. Cell Metab 14:724–38. [DOI] [PubMed] [Google Scholar]
- Benfenati V, Amiry-Moghaddam M, Caprini M, Mylonakou MN, Rapisarda C, Ottersen OP, Ferroni S. 2007. Expression and functional characterization of transient receptor potential vanilloid-related channel 4 (TRPV4) in rat cortical astrocytes. Neuroscience 148:876–92. [DOI] [PubMed] [Google Scholar]
- Benfenati V, Caprini M, Dovizio M, Mylonakou MN, Ferroni S, Ottersen OP, Amiry-Moghaddam M. 2011. An aquaporin-4/transient receptor potential vanilloid 4 (AQP4/TRPV4) complex is essential for cell-volume control in astrocytes. Proc Natl Acad Sci U S A 108:2563–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butenko O, Dzamba D, Benesova J, Honsa P, Benfenati V, Rusnakova V, Ferroni S, Anderova M. 2012. The increased activity of TRPV4 channel in the astrocytes of the adult rat hippocampus after cerebral hypoxia/ischemia. PLoS One 7:e39959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan SL, Sweet JG, Cipolla MJ. 2013. Treatment for cerebral small vessel disease: effect of relaxin on the function and structure of cerebral parenchymal arterioles during hypertension. FASEB J 27:3917–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chrissobolis S, Banfi B, Sobey CG, Faraci FM. 2012. Role of Nox isoforms in angiotensin II-induced oxidative stress and endothelial dysfunction in brain. J Appl Physiol (1985) 113:184–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chrissobolis S, Miller AA, Drummond GR, Kemp-Harper BK, Sobey CG. 2011. Oxidative stress and endothelial dysfunction in cerebrovascular disease. Front Biosci (Landmark Ed) 16:1733–45. [DOI] [PubMed] [Google Scholar]
- Cipolla MJ. 2009. The Cerebral Circulation. San Rafael CA: 2010 by Morgan & Claypool Life Sciences. [PubMed] [Google Scholar]
- Clarke LE, Liddelow SA, Chakraborty C, Munch AE, Heiman M, Barres BA. 2018. Normal aging induces A1-like astrocyte reactivity. Proc Natl Acad Sci U S A 115:E1896–E1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Silva TM, Kinzenbaw DA, Modrick ML, Reinhardt LD, Faraci FM. 2016. Heterogeneous Impact of ROCK2 on Carotid and Cerebrovascular Function. Hypertension 68:809–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debette S, Seshadri S, Beiser A, Au R, Himali JJ, Palumbo C, Wolf PA, DeCarli C. 2011. Midlife vascular risk factor exposure accelerates structural brain aging and cognitive decline. Neurology 77:461–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz-Otero JM, Fisher C, Downs K, Moss ME, Jaffe IZ, Jackson WF, Dorrance AM. 2017. Endothelial Mineralocorticoid Receptor Mediates Parenchymal Arteriole and Posterior Cerebral Artery Remodeling During Angiotensin II-Induced Hypertension. Hypertension 70:1113–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duling BR, Gore RW, Dacey RG Jr., Damon DN. 1981. Methods for isolation, cannulation, and in vitro study of single microvessels. Am J Physiol 241:H108–116. [DOI] [PubMed] [Google Scholar]
- Dunn KM, Hill-Eubanks DC, Liedtke WB, Nelson MT. 2013. TRPV4 channels stimulate Ca2+-induced Ca2+ release in astrocytic endfeet and amplify neurovascular coupling responses. Proc Natl Acad Sci U S A 110:6157–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Earley S, Pauyo T, Drapp R, Tavares MJ, Liedtke W, Brayden JE. 2009. TRPV4-dependent dilation of peripheral resistance arteries influences arterial pressure. Am J Physiol Heart Circ Physiol 297:H1096–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faraci FM, Baumbach GL, Heistad DD. 1989. Myogenic mechanisms in the cerebral circulation. J Hypertens Suppl 7:S61–4; discussion S65. [PubMed] [Google Scholar]
- Filosa JA, Bonev AD, Straub SV, Meredith AL, Wilkerson MK, Aldrich RW, Nelson MT. 2006. Local potassium signaling couples neuronal activity to vasodilation in the brain. Nat Neurosci. United States. p 1397–1403. [DOI] [PubMed] [Google Scholar]
- Gąsecki D, Kwarciany M, Nyka W, Narkiewicz K. 2013. Hypertension, Brain Damage and Cognitive Decline. Current Hypertension Reports 15:547–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girouard H, Bonev AD, Hannah RM, Meredith A, Aldrich RW, Nelson MT. 2010. Astrocytic endfoot Ca2+ and BK channels determine both arteriolar dilation and constriction. Proc Natl Acad Sci U S A 107:3811–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girouard H, Iadecola C. 2006. Neurovascular coupling in the normal brain and in hypertension, stroke, and Alzheimer disease. J Appl Physiol (1985) 100:328–35. [DOI] [PubMed] [Google Scholar]
- Girouard H, Park L, Anrather J, Zhou P, Iadecola C. 2007. Cerebrovascular nitrosative stress mediates neurovascular and endothelial dysfunction induced by angiotensin II. Arterioscler Thromb Vasc Biol 27:303–9. [DOI] [PubMed] [Google Scholar]
- Gordon GR, Mulligan SJ, MacVicar BA. 2007. Astrocyte control of the cerebrovasculature. Glia 55:1214–21. [DOI] [PubMed] [Google Scholar]
- Harraz OF, Longden TA, Dabertrand F, Hill-Eubanks D, Nelson MT. 2018a. Endothelial GqPCR activity controls capillary electrical signaling and brain blood flow through PIP2 depletion. Proc Natl Acad Sci U S A 115:E3569–E3577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harraz OF, Longden TA, Hill-Eubanks D, Nelson MT. 2018b. PIP2 depletion promotes TRPV4 channel activity in mouse brain capillary endothelial cells. Elife 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart MN, Heistad DD, Brody MJ. 1980. Effect of chronic hypertension and sympathetic denervation on wall/lumen ratio of cerebral vessels. Hypertension 2:419–23. [DOI] [PubMed] [Google Scholar]
- Hill-Eubanks DC, Gonzales AL, Sonkusare SK, Nelson MT. 2014. Vascular TRP channels: performing under pressure and going with the flow. Physiology (Bethesda) 29:343–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iadecola C 2017. The Neurovascular Unit Coming of Age: A Journey through Neurovascular Coupling in Health and Disease. Neuron 96:17–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iadecola C, Yaffe K, Biller J, Bratzke LC, Faraci FM, Gorelick PB, Gulati M, Kamel H, Knopman DS, Launer LJ and others. 2016. Impact of Hypertension on Cognitive Function: A Scientific Statement From the American Heart Association. Hypertension 68:e67–e94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iddings JA, Kim KJ, Zhou Y, Higashimori H, Filosa JA. 2015. Enhanced parenchymal arteriole tone and astrocyte signaling protect neurovascular coupling mediated parenchymal arteriole vasodilation in the spontaneously hypertensive rat. J Cereb Blood Flow Metab. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iliff JJ, Lee H, Yu M, Feng T, Logan J, Nedergaard M, Benveniste H. 2013a. Brain-wide pathway for waste clearance captured by contrast-enhanced MRI. J Clin Invest 123:1299–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iliff JJ, Wang M, Zeppenfeld DM, Venkataraman A, Plog BA, Liao Y, Deane R, Nedergaard M. 2013b. Cerebral arterial pulsation drives paravascular CSF-interstitial fluid exchange in the murine brain. J Neurosci 33:18190–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Intengan HD, Schiffrin EL. 2001. Vascular Remodeling in Hypertension. Hypertension 38:581. [DOI] [PubMed] [Google Scholar]
- Iulita MF, Girouard H. 2017. Treating Hypertension to Prevent Cognitive Decline and Dementia: Re-Opening the Debate. Adv Exp Med Biol 956:447–473. [DOI] [PubMed] [Google Scholar]
- Jo AO, Ryskamp DA, Phuong TT, Verkman AS, Yarishkin O, MacAulay N, Krizaj D. 2015. TRPV4 and AQP4 Channels Synergistically Regulate Cell Volume and Calcium Homeostasis in Retinal Muller Glia. J Neurosci 35:13525–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kacem K, Lacombe P, Seylaz J, Bonvento G. 1998. Structural organization of the perivascular astrocyte endfeet and their relationship with the endothelial glucose transporter: a confocal microscopy study. Glia 23:1–10. [PubMed] [Google Scholar]
- Kanju P, Liedtke W. 2016. Pleiotropic function of TRPV4 ion channels in the central nervous system. Exp Physiol 101:1472–1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KJ, Filosa JA. 2012. Advanced in vitro approach to study neurovascular coupling mechanisms in the brain microcirculation. J Physiol 590:1757–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KJ, Iddings JA, Stern JE, Blanco VM, Croom D, Kirov SA, Filosa JA. 2015. Astrocyte contributions to flow/pressure-evoked parenchymal arteriole vasoconstriction. J Neurosci 35:8245–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KJ, Ramiro Diaz J, Iddings JA, Filosa JA. 2016. Vasculo-Neuronal Coupling: Retrograde Vascular Communication to Brain Neurons. J Neurosci 36:12624–12639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohler R, Heyken WT, Heinau P, Schubert R, Si H, Kacik M, Busch C, Grgic I, Maier T, Hoyer J. 2006. Evidence for a functional role of endothelial transient receptor potential V4 in shear stress-induced vasodilatation. Arterioscler Thromb Vasc Biol 26:1495–502. [DOI] [PubMed] [Google Scholar]
- Krizaj D, Ryskamp DA, Tian N, Tezel G, Mitchell CH, Slepak VZ, Shestopalov VI. 2014. From mechanosensitivity to inflammatory responses: new players in the pathology of glaucoma. Curr Eye Res 39:105–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lind BL, Jessen SB, Lonstrup M, Josephine C, Bonvento G, Lauritzen M. 2018. Fast Ca2+ responses in astrocyte end-feet and neurovascular coupling in mice. Glia 66:348–358. [DOI] [PubMed] [Google Scholar]
- Lipski J, Park TI, Li D, Lee SC, Trevarton AJ, Chung KK, Freestone PS, Bai JZ. 2006. Involvement of TRP-like channels in the acute ischemic response of hippocampal CA1 neurons in brain slices. Brain Res 1077:187–99. [DOI] [PubMed] [Google Scholar]
- Longden TA, Dabertrand F, Koide M, Gonzales AL, Tykocki NR, Brayden JE, Hill-Eubanks D, Nelson MT. 2017. Capillary K+-sensing initiates retrograde hyperpolarization to increase local cerebral blood flow. Nat Neurosci 20:717–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marrelli SP, O’Neil R G, Brown RC, Bryan RM Jr. 2007. PLA2 and TRPV4 channels regulate endothelial calcium in cerebral arteries. Am J Physiol Heart Circ Physiol 292:H1390–7. [DOI] [PubMed] [Google Scholar]
- Marziano C, Hong K, Cope EL, Kotlikoff MI, Isakson BE, Sonkusare SK. 2017. Nitric Oxide-Dependent Feedback Loop Regulates Transient Receptor Potential Vanilloid 4 (TRPV4) Channel Cooperativity and Endothelial Function in Small Pulmonary Arteries. J Am Heart Assoc 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathiisen TM, Lehre KP, Danbolt NC, Ottersen OP. 2010. The perivascular astroglial sheath provides a complete covering of the brain microvessels: an electron microscopic 3D reconstruction. Glia 58:1094–103. [DOI] [PubMed] [Google Scholar]
- McKenna MC. 2013. Glutamate pays its own way in astrocytes. Front Endocrinol (Lausanne) 4:191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendoza SA, Fang J, Gutterman DD, Wilcox DA, Bubolz AH, Li R, Suzuki M, Zhang DX. 2010. TRPV4-mediated endothelial Ca2+ influx and vasodilation in response to shear stress. Am J Physiol Heart Circ Physiol 298:H466–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison HW, Filosa JA. 2013. A quantitative spatiotemporal analysis of microglia morphology during ischemic stroke and reperfusion. J Neuroinflammation 10:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison HW, Filosa JA. 2016. Sex differences in astrocyte and microglia responses immediately following middle cerebral artery occlusion in adult mice. Neuroscience 339:85–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulligan SJ, MacVicar BA. 2004. Calcium transients in astrocyte endfeet cause cerebrovascular constrictions. Nature. England. p 195–9. [DOI] [PubMed] [Google Scholar]
- Nuriya M, Hirase H. 2016. Involvement of astrocytes in neurovascular communication. Prog Brain Res 225:41–62. [DOI] [PubMed] [Google Scholar]
- Oheim M, Schmidt E, Hirrlinger J. 2017. Local energy on demand: Are ‘spontaneous’ astrocytic Ca2+-microdomains the regulatory unit for astrocyte-neuron metabolic cooperation? Brain Res Bull. [DOI] [PubMed] [Google Scholar]
- Otsu Y, Couchman K, Lyons DG, Collot M, Agarwal A, Mallet JM, Pfrieger FW, Bergles DE, Charpak S. 2015. Calcium dynamics in astrocyte processes during neurovascular coupling. Nat Neurosci 18:210–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulson OB, Strandgaard S, Edvinsson L. 1990. Cerebral autoregulation. Cerebrovasc Brain Metab Rev 2:161–92. [PubMed] [Google Scholar]
- Paulson OB, Waldemar G, Schmidt JF, Strandgaard S. 1989. Cerebral circulation under normal and pathologic conditions. Am J Cardiol 63:2C–5C. [DOI] [PubMed] [Google Scholar]
- Pellerin L, Magistretti PJ. 1994. Glutamate uptake into astrocytes stimulates aerobic glycolysis: a mechanism coupling neuronal activity to glucose utilization. Proceedings of the National Academy of Sciences 91:10625–10629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pires PW, Jackson WF, Dorrance AM. 2015. Regulation of myogenic tone and structure of parenchymal arterioles by hypertension and the mineralocorticoid receptor. Am J Physiol Heart Circ Physiol 309:H127–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plant TD, Strotmann R. 2007. Trpv4. Handb Exp Pharmacol:189–205. [DOI] [PubMed] [Google Scholar]
- Rakers C, Schmid M, Petzold GC. 2017. TRPV4 channels contribute to calcium transients in astrocytes and neurons during peri-infarct depolarizations in a stroke model. Glia 65:1550–1561. [DOI] [PubMed] [Google Scholar]
- Rosenegger DG, Tran CH, Wamsteeker Cusulin JI, Gordon GR. 2015. Tonic Local Brain Blood Flow Control by Astrocytes Independent of Phasic Neurovascular Coupling. J Neurosci 35:13463–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rungta RL, Bernier LP, Dissing-Olesen L, Groten CJ, LeDue JM, Ko R, Drissler S, MacVicar BA. 2016. Ca2+ transients in astrocyte fine processes occur via Ca2+ influx in the adult mouse hippocampus. Glia 64:2093–2103. [DOI] [PubMed] [Google Scholar]
- Ryskamp DA, Iuso A, Krizaj D. 2015. TRPV4 links inflammatory signaling and neuroglial swelling. Channels (Austin) 9:70–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saavedra JM. 2016. Evidence to Consider Angiotensin II Receptor Blockers for the Treatment of Early Alzheimer’s Disease. Cell Mol Neurobiol 36:259–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saavedra JM, Benicky J, Zhou J. 2006. Mechanisms of the Anti-Ischemic Effect of Angiotensin II AT( 1 ) Receptor Antagonists in the Brain. Cell Mol Neurobiol 26:1099–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saifeddine M, El-Daly M, Mihara K, Bunnett NW, McIntyre P, Altier C, Hollenberg MD, Ramachandran R. 2015. GPCR-mediated EGF receptor transactivation regulates TRPV4 action in the vasculature. Br J Pharmacol 172:2493–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxena A, Bachelor M, Park YH, Carreno FR, Nedungadi TP, Cunningham JT. 2014. Angiotensin II induces membrane trafficking of natively expressed transient receptor potential vanilloid type 4 channels in hypothalamic 4B cells. Am J Physiol Regul Integr Comp Physiol 307:R945–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seki T, Goto K, Kiyohara K, Kansui Y, Murakami N, Haga Y, Ohtsubo T, Matsumura K, Kitazono T. 2017. Downregulation of Endothelial Transient Receptor Potential Vanilloid Type 4 Channel and Small-Conductance of Ca2+-Activated K+ Channels Underpins Impaired Endothelium-Dependent Hyperpolarization in Hypertension. Hypertension 69:143–153. [DOI] [PubMed] [Google Scholar]
- Shibasaki K, Ikenaka K, Tamalu F, Tominaga M, Ishizaki Y. 2014. A novel subtype of astrocytes expressing TRPV4 (transient receptor potential vanilloid 4) regulates neuronal excitability via release of gliotransmitters. J Biol Chem 289:14470–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shigetomi E, Bushong EA, Haustein MD, Tong X, Jackson-Weaver O, Kracun S, Xu J, Sofroniew MV, Ellisman MH, Khakh BS. 2013. Imaging calcium microdomains within entire astrocyte territories and endfeet with GCaMPs expressed using adeno-associated viruses. J Gen Physiol 141:633–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirakawa H, Nakagawa T, Kaneko S. 2010. [Pathophysiological roles of transient receptor potential channels in glial cells]. Yakugaku Zasshi 130:281–7. [DOI] [PubMed] [Google Scholar]
- Soni H, Peixoto-Neves D, Matthews AT, Adebiyi A. 2017. TRPV4 channels contribute to renal myogenic autoregulation in neonatal pigs. Am J Physiol Renal Physiol 313:F1136–F1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonkusare SK, Bonev AD, Ledoux J, Liedtke W, Kotlikoff MI, Heppner TJ, Hill-Eubanks DC, Nelson MT. 2012. Elementary Ca2+ signals through endothelial TRPV4 channels regulate vascular function. Science 336:597–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturgis LC, Cannon JG, Schreihofer DA, Brands MW. 2009. The role of aldosterone in mediating the dependence of angiotensin hypertension on IL-6. Am J Physiol Regul Integr Comp Physiol 297:R1742–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweet JG, Chan SL, Cipolla MJ. 2015. Effect of hypertension and carotid occlusion on brain parenchymal arteriole structure and reactivity. J Appl Physiol (1985) 119:817–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tajada S, Moreno CM, O’Dwyer S, Woods S, Sato D, Navedo MF, Santana LF. 2017. Distance constraints on activation of TRPV4 channels by AKAP150-bound PKCalpha in arterial myocytes. J Gen Physiol 149:639–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takano T, Tian GF, Peng W, Lou N, Libionka W, Han X, Nedergaard M. 2006. Astrocyte-mediated control of cerebral blood flow. Nat Neurosci 9:260–7. [DOI] [PubMed] [Google Scholar]
- Takata N, Sugiura Y, Yoshida K, Koizumi M, Hiroshi N, Honda K, Yano R, Komaki Y, Matsui K, Suematsu M and others. 2018. Optogenetic astrocyte activation evokes BOLD fMRI response with oxygen consumption without neuronal activity modulation. Glia 66:2013–2023. [DOI] [PubMed] [Google Scholar]
- Vriens J, Janssens A, Prenen J, Nilius B, Wondergem R. 2004. TRPV channels and modulation by hepatocyte growth factor/scatter factor in human hepatoblastoma (HepG2) cells. Cell Calcium 36:19–28. [DOI] [PubMed] [Google Scholar]
- Vriens J, Owsianik G, Fisslthaler B, Suzuki M, Janssens A, Voets T, Morisseau C, Hammock BD, Fleming I, Busse R and others. 2005. Modulation of the Ca2 permeable cation channel TRPV4 by cytochrome P450 epoxygenases in vascular endothelium. Circ Res 97:908–15. [DOI] [PubMed] [Google Scholar]
- Waldstein SR, Rice SC, Thayer JF, Najjar SS, Scuteri A, Zonderman AB. 2007. Pulse Pressure and Pulse Wave Velocity Are Related to Cognitive Decline in the Baltimore Longitudinal Study of Aging. Hypertension 51:99. [DOI] [PubMed] [Google Scholar]
- Wallraff A, Kohling R, Heinemann U, Theis M, Willecke K, Steinhauser C. 2006. The impact of astrocytic gap junctional coupling on potassium buffering in the hippocampus. J Neurosci 26:5438–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi M, Wei T, Wang Y, Lu Q, Chen G, Gao X, Geller HM, Chen H, Yu Z. 2017. The potassium channel KCa3.1 constitutes a pharmacological target for astrogliosis associated with ischemia stroke. J Neuroinflammation 14:203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang DX, Gutterman DD. 2011. Transient receptor potential channel activation and endothelium-dependent dilation in the systemic circulation. J Cardiovasc Pharmacol 57:133–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Papadopoulos P, Hamel E. 2013. Endothelial TRPV4 channels mediate dilation of cerebral arteries: impairment and recovery in cerebrovascular pathologies related to Alzheimer disease. Br J Pharmacol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P, Sun C, Li H, Tang C, Kan H, Yang Z, Mao A, Ma X. 2018. TRPV4 (Transient Receptor Potential Vanilloid 4) Mediates Endothelium-Dependent Contractions in the Aortas of Hypertensive Mice. Hypertension 71:134–142. [DOI] [PubMed] [Google Scholar]
- Zimmerman MC, Lazartigues E, Sharma RV, Davisson RL. 2004. Hypertension caused by angiotensin II infusion involves increased superoxide production in the central nervous system. Circ Res 95:210–6. [DOI] [PubMed] [Google Scholar]
- Zonta M, Angulo MC, Gobbo S, Rosengarten B, Hossmann KA, Pozzan T, Carmignoto G. 2002. Neuron-to-astrocyte signaling is central to the dynamic control of brain microcirculation. Nat Neurosci 6:43–50. [DOI] [PubMed] [Google Scholar]
- Zonta M, Sebelin A, Gobbo S, Fellin T, Pozzan T, Carmignoto G. 2003. Glutamate-mediated cytosolic calcium oscillations regulate a pulsatile prostaglandin release from cultured rat astrocytes. J Physiol 553:407–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou Q, Leung SW, Vanhoutte PM. 2015. Transient Receptor Potential Channel Opening Releases Endogenous Acetylcholine, which Contributes to Endothelium-Dependent Relaxation Induced by Mild Hypothermia in Spontaneously Hypertensive Rat but Not Wistar-Kyoto Rat Arteries. J Pharmacol Exp Ther 354:121–30. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table 1. Summary data showing astrocyte Ca2+ parameters (maximum Δpeak (F/F0), average Δpeak (F/F0), frequency (Hz) and area under the curve (AUC) (F/F0*s)) in response to intravascular pressure changes for soma, process and MD in normotensive and hypertensive mice. Two way ANOVA with Newman-Keuls post-hoc test. *P<0.05, **P<0.01 for comparisons within groups (W/G) and ^P<0.05, ^^P<0.01 for comparisons between groups (B/G).
Supplementary figure 1. Progressive hypertension development induced by Ang II infusion. Averaged time course recording of the changes in mean arterial pressure (MAP) from C57BL6 mice treated with 600 ng/Kg/min Ang II (filled circles)(n=5) or saline (open circles) (n=4). Two way ANOVA and Sidak post-hoc test. ** P<0.05, *** P<0.001.
Supplementary figure 2. Hypertension does not change PA passive properties. Average % passive diameter at 15, 30, 60 and 80 mmHg in PA from normotensive (open circles) and hypertensive mice (filled squares) (n=6). Two way ANOVA and Newman-Keuls post-hoc test.
Supplementary figure 3. TRPV4 channel contribution to astrocyte spontaneous Ca2+ events in hypertension. (A) Representative traces of astrocyte Ca2+ events for soma, processes and microdomains from normotensive and hypertensive mice following pre-incubation (25 min) in the TRPV4 channel blocker HC067047 (10 μM). (B) Summary data for soma, processes and MD astrocyte Ca2+ parameters (maximum Δpeak (F/F0), average Δpeak (F/F0), frequency (Hz) and area under the curve (F/F0*s)) in normotensive (white bars, n=7 cells from 3 mice) and hypertensive (black bars, n=6 cells from 3 mice) mice. Mean ± SEM.