

Abstract
Background
Patients with muscular dystrophy (MD) represent a vulnerable patient population with no clearly defined care model in modern‐day clinical practice to manage a high burden of heart disease and comorbidities. We demonstrate the effectiveness of cardiac interventions, namely the initiation and optimization of medical and device therapies, as part of a multidisciplinary care approach to improve clinical outcomes in patients with MD.
Methods and Results
We conducted a prospective cohort study at the Neuromuscular Multidisciplinary clinic following patients with dystrophinopathies, limb‐girdle MD, type 1 myotonic dystrophy, and facioscapulohumeral MD. A negative control group classified as non‐MD myopathies without heart disease, was also tracked. Our cohort of 185 patients (median age: 42 years; 79 [42.7%] women), included 145 patients with MD. Cardiomyopathy was present in 65.6% of the patients with dystrophinopathies (21 of 32) and 27.3% of the patients with limb‐girdle MD (9 of 33). Conduction abnormalities were common in type 1 myotonic dystrophy (33.3% [20/60] patients). Cardiac intervention reversed systolic dysfunction, with left ventricular ejection fraction improving from 43% to 50.0% over a 3‐year period. A sustained reduction in healthcare utilization was also observed. The number of outpatient clinic visits decreased from 3.0 to 1.5 visits per year, the duration of hospitalizations was reduced from 14.2 to 0.9 days per year, and the number of cardiac‐related hospitalizations decreased from 0.4 to 0.1 hospitalizations per year associated with low mortality.
Conclusions
Our study demonstrates that cardiac intervention as part of a comprehensive multidisciplinary care approach to treating patients with MD leads to a sustained improvement in clinical outcomes.
Keywords: heart disease, medical therapy, multidisciplinary care, muscular dystrophy, outcome data
Subject Categories: Quality and Outcomes, Mortality/Survival, Cardiomyopathy, Heart Failure, Genetics
Clinical Perspective
What Is New?
Cardiac interventions as part of a multidisciplinary care approach can markedly improve all‐cause clinical outcomes in patients with muscular dystrophy.
Use of medical and device therapies improved systolic dysfunction in different cohorts of patients with muscular dystrophy.
What Are the Clinical Implications?
Design and implementation of multidisciplinary care that includes cardiology should be undertaken to provide optimal care to patients with muscular dystrophy.
Cardiomyopathy and arrhythmias are frequent comorbidities in patients with muscular dystrophy, which requires expedient diagnosis and management with frequent monitoring.
Introduction
Muscular dystrophies (MDs) are inherited neuromuscular diseases with a wide range of systemic manifestations that are often life‐threatening. The multisystem involvement of MD leads to significant disability ranging from limited ambulation to heart and lung disease, resulting in poor quality of life, and increased morbidity and mortality.1, 2, 3
Heart disease, characterized by cardiomyopathy and arrhythmias, is now recognized as the primary cause of mortality in patients with MD.4, 5, 6, 7, 8 Of the 9 main types of MDs, dystrophinopathies, namely Duchenne MD (DMD) and Becker MD, exhibit dilated cardiomyopathy, characterized by decreased left ventricular (LV) ejection fraction (LVEF), and increased end‐diastolic volumes.2, 9 Many subtypes of limb‐girdle MD (LGMD), specifically subtypes 1B, 2E, and 2I have similar cardiac manifestations.4, 10 Conduction abnormalities are observed in patients with type 1 myotonic dystrophy (DM1) such as atrioventricular block and left bundle branch block (LBBB). Importantly, ventricular arrhythmias are a serious complication in patients with DMD, Becker MD, LGMD, and DM1.4, 11, 12 Facioscapulohumeral MD (FSHD) exhibits cardiac conduction abnormalities to a milder degree.13
Given the inherent complexities of MD, the ideal care model for these patients has not been established.2, 14, 15 Our Neuromuscular Multidisciplinary (NMMD) clinic was established to provide multifaceted care by specialist physicians and allied healthcare professionals to patients with MD in a single visit. Our prospective cohort study was designed to determine the impact of specialist cardiology care as part of a novel multidisciplinary care pathway for patients with MD.
Methods
The data that support the findings of this study are available from the corresponding author upon reasonable request. In coordination with the NMMD care clinic established in 2014 at the Kaye Edmonton Clinic, 185 patients were recruited into our prospective study, after providing written consent, over a 4‐year period from November 5, 2014, to December 1, 2018. Following neurological assessments, muscle biopsy, and genetic testing to confirm diagnosis, patients were categorized into the various cohorts of MD including dystrophinopathies (32 patients), LGMD (33 patients), DM1 (60 patients), and FSHD (20 patients). We included a negative control group classified as non‐MD myopathies, which included patients with myositis, mitochondrial myopathies, spinal muscular atrophy, and undefined congenital myopathies (40 patients). These patients had moderately impaired ambulation. The prevalence of respiratory disease was similar to the patients with MD; however, these patients did not have heart disease. Patients were referred to the NMMD clinic with no bias towards patients with overt cardiac or respiratory symptoms to receive specialist care as well as interventions from allied healthcare professionals. Guideline‐based medical therapy was implemented and appropriate device implantations and follow‐up care was performed. The investigation conforms to the principles outlined by our locally appointed ethics committee, the Health Research Ethics Board, at the University of Alberta. All patients provided informed and written consent before being recruited into our study.
Risk Assessment
In addition to demographic and clinical parameters, biochemical, ECG, transthoracic echocardiogram, and cardiac magnetic resonance imaging (MRI) data were collected to create detailed patient profiles. Heart rate and blood pressure were recorded at each visit, and the presence of cardiomyopathy and comorbidities, such as anemia, diabetes mellitus, ambulatory status, respiratory disease, and sleep‐disordered breathing (SDOB), defined as obstructive sleep apnea or nocturnal hypoventilation, were documented. All patients with suspected SDOB underwent polysomnography for further evaluation. Respiratory disease was defined as chronic obstructive pulmonary disease, asthma, recurrent aspiration pneumonia, respiratory muscle weakness, or restrictive lung disease. Appropriate patients were trained in lung volume recruitment and the use of a mechanical insufflator‐exsufflator and provided with ventilatory assist devices such as bilevel positive airway pressure, when appropriate. Overall ambulatory function was evaluated by physiatrists, and interventions in the form of different mobility aids such as cane or walker and wheelchair use, manual or powered, were instituted where needed.
Serum biochemistry, fasting lipid profile, hemoglobin, plasma BNP (B‐type natriuretic peptide) and creatine kinase (CK) were monitored. Anemia was defined as per the hemoglobin level cutoffs established by the World Health Organization. Dyslipidemia was defined in accordance with the 2016 Canadian Cardiovascular Society guidelines.16 Serial 12‐lead resting ECG and Holter data collected over either 24‐ or 48‐hour periods were evaluated. All patients with DM1 received a 48‐hour Holter monitor following their initial NMMD clinic visit. Subsequent Holter studies were completed after follow‐up visits for patients who required further electrophysiological studies because of risk suggestive of arrhythmias. Serial transthoracic echocardiograms and cardiac MRI were used to detect and follow cardiac structure and function. Cardiomyopathy was defined as LVEF <55% or a LV end‐diastolic volume index >105 mL/m²,17 as determined by cardiac MRI. Incident heart failure and arrhythmias were also documented. Heart failure was diagnosed following a comprehensive cardiac assessment, which considered symptoms and signs such as dyspnea, orthopnea, poor appetite, elevated jugular venous pressure, peripheral edema, and abdominal distention. Medical therapy by maximum tolerated dose,18 use and type of device therapy, and use of bilevel positive airway pressure ventilation were documented at each clinic visit.
Outcome Data Assessment
Cardiac systolic function was tracked through the monitoring of LVEF, obtained by cardiac MRI and/or echocardiogram, from 57 patients who had a cardiomyopathy and had imaging data available for the full 3‐year period, spanning from their initial NMMD clinic visit to their 3‐year follow‐up visit. This includes 23 patients with dystrophinopathies, 11 patients with LGMD, 20 patients with DM1, and 3 patients with FSHD. Outcome data, such as the number of unplanned, all‐cause outpatient clinic visits, the duration of hospitalization, and the number of cardiac‐related hospitalizations were collected by the Data Integration and Management Repository (DIMR) analytics branch of Alberta Health Services using our provincial electronic health records. All 185 patients had DIMR data available over a 3‐year period, beginning from November 1, 2015, until December 1, 2018, to capture clinical outcomes. To account for the ongoing enrollment process and variable lengths of follow‐up, outpatient clinic visits and duration of hospitalization were standardized as rates for the patients in each cohort. Outcome rates of the patients with MD and non‐MD myopathies cohort were plotted in 6‐month intervals as time series graphs to quantify the change of healthcare utilization rates, in days per year, following initial intervention and optimization of medical therapies 6 months after their initial clinic visit.
Data Analysis
Continuous variables analyzed were compared using a Mann–Whitney U test or Kruskal–Wallis test where appropriate, and all categorical data were compared using Pearson chi‐square tests. Patient LVEF at the point of clinic enrollment was compared with data collected at the end of the study using a paired t test. Time series plots were used to illustrate the rates associated with clinical outcome data and accompanied by linear regression analysis. A P<0.05 was considered significant through all statistical analysis. Statistical analyses were conducted using SPSS Statistics 25.0 (IBM).
Results
Dystrophinopathies
Patients were recruited from our multidisciplinary care clinic where they received concurrent care from specialists in cardiology, neurology, respirology, and physiatry (Figure 1A). The dystrophinopathies cohort comprising young male patients (27 patients with DMD and 5 patients with Becker MD) (Table 1). The majority of these patients exhibited severe skeletal muscle wasting and motor difficulties at their initial clinic visit, with 27 (84.4%) patients being wheelchair‐bound (Table 1). Dilated cardiomyopathy was highly prevalent, with 21 (65.6%) patients affected based on echocardiogram and cardiac MRI (Figure 1B), illustrated by biventricular remodeling with a moderately reduced LVEF (Table 2), and 6 (42.9%) patients who received a cardiac MRI showing evidence of myocardial fibrosis (Figure S1). In this cohort, echocardiogram confirmed LV dilation and dysfunction (Table 2) and showed no valvular abnormalities (Table S1). The high prevalence of cardiomyopathy resulted in a high rate of incident heart failure diagnosed following the initial NMMD clinic visit (Figure 1C). For the majority of the patients in this cohort, ECG parameters were within normal limits (Table 2) and, concordant with these findings, there was a low incidence of atrial and ventricular arrhythmias in this cohort (Figure 1D). Respiratory disease, including SDOB, was prevalent in this population, with 27 (84.4%) patients affected. Lung volume recruitment and mechanical insufflation‐exsufflation were implemented in 26 (81.3%) and 13 (40.6%) patients, respectively (Table 1).
Figure 1.
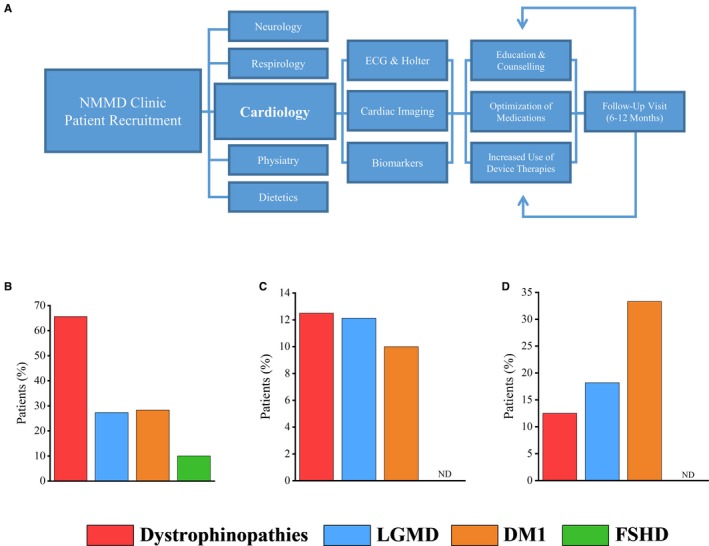
Cardiac assessment of patients with muscular dystrophy (MD) in the Neuromuscular Multidisciplinary (NMMD) clinic. Cardiac assessment and intervention applied through the NMMD clinic care pathway (A). Prevalence of cardiomyopathy in patients with MD (B). Heart failure diagnosed in patients at their initial NMMD clinic visit (C). Arrhythmia burden as captured by ECG, Holter monitoring, and device interrogation (D). DM1 indicates type 1 myotonic dystrophy; FSHD, facioscapulohumeral muscular dystrophy; LGMD, limb‐girdle muscular dystrophy; ND, not detected.
Table 1.
Baseline Characteristics of Patients Referred to the Neuromuscular Multidisciplinary Clinic
| Characteristic | All Patients (N=185) | Dystrophinopathies (n=32) | LGMD (n=33) | DM1 (n=60) | FSHD (n=20) | Non‐MD Myopathies (n=40) | P Valuea | P Valueb |
|---|---|---|---|---|---|---|---|---|
| Follow‐up time, y | 2.2 (1.4–3.5) | 2.2 (1.4–3.4) | 2.2 (1.7–3.6) | 2.3 (1.0–3.2) | 1.8 (0.9–3.4) | 2.3 (1.9–3.5) | 0.43 | 0.17 |
| Men/women, No. | 106 (57.3)/79 (42.7) | 32 (100) | 14 (42.4)/19 (57.6) | 30 (50)/30 (50) | 11 (55)/9 (45) | 19 (47.5)/21 (52.5) | <0.001 | 0.16 |
| Median age, y | 42 (25–54) | 22 (18–28.8) | 44 (27–57) | 42 (34.8–50) | 45 (24.8–52.5) | 54 (33.5–60) | <0.001 | 0.004 |
| Current/former smoker, No. | 22 (11.9) | 0 | 4 (12.1) | 11 (18.3) | 3 (15) | 4 (10) | 0.14 | 0.68 |
| Ambulation, No. | ||||||||
| Cane/walker | 16 (8.6) | 1 (3.1) | 3 (9.1) | 7 (11.7) | 2 (10) | 3 (7.5) | 0.70 | 0.77 |
| mWC/pWC | 49 (26.5) | 27 (84.4) | 12 (36.4) | 4 (6.7) | 3 (15) | 3 (7.5) | <0.001 | 0.002 |
| Comorbidities, No. | ||||||||
| Diabetes mellitus | 18 (9.7) | 0 | 8 (24.2) | 5 (8.3) | 2 (10) | 3 (7.5) | 0.03 | 0.59 |
| Dyslipidemia | 24 (13.0) | 2 (6.3) | 4 (12.1) | 7 (11.7) | 4 (20) | 7 (17.5) | 0.60 | 0.34 |
| Hypertension | 27 (14.6) | 2 (6.3) | 6 (18.2) | 4 (6.7) | 6 (30) | 9 (22.5) | 0.045 | 0.11 |
| Respiratory disease | 100 (54.1) | 27 (84.4) | 8 (24.2) | 32 (53.3) | 6 (30) | 27 (67.5) | <0.001 | 0.05 |
| SDOB | 47 (25.4) | 14 (43.8) | 5 (15.2) | 16 (36.7) | 6 (30) | 6 (15) | 0.07 | 0.09 |
| Anemia | 5 (2.7) | 3 (9.4) | 2 (6.1) | 0 | 0 | 0 | 0.022 | <0.001 |
| Lung volume recruitment, No. | 87 (47.0) | 26 (81.3) | 6 (18.2) | 27 (45) | 5 (25) | 23 (57.5) | <0.001 | 0.13 |
| Mechanical insufflation‐exsufflation, No. | 18 (9.7) | 13 (40.6) | 1 (3.0) | 1 (1.7) | 2 (10) | 1 (2.5) | <0.001 | 0.08 |
| Noninvasive ventilation, No. | 36 (19.5) | 12 (37.5) | 5 (15.2) | 8 (13.3) | 5 (25) | 6 (15) | 0.05 | 0.42 |
| Vitals, median | ||||||||
| HR, beats per min | 76 (67.5–86.5) | 86 (78.8–100) | 76 (70–83.5) | 70 (62.8–80) | 77.5 (71–80) | 73.5 (61–80) | 0.009 | 0.32 |
| SBP, mm Hg | 120 (110–132) | 108 (100–121) | 122 (112.5–131) | 114 (106–126.8) | 130 (120.5–133.8) | 126 (119.5–140) | <0.001 | <0.001 |
| DBP, mm Hg | 75 (70–82) | 71 (64–78) | 80 (70.5–84.5) | 74 (68.5–78) | 83.5 (76.5–87.5) | 76 (72–84) | 0.001 | 0.21 |
| Biomarkers, median | ||||||||
| BNP, pg/mL | 20 (12.5–42.5) | 20 (12–75) | 14.5 (10.5–47.5) | 21 (15–40) | 21 (14.5–35.5) | 20 (13–37) | 0.67 | 0.66 |
| CK, U/L | 274 (126–569.8) | 1277 (377.8–2380.8) | 526 (302.5–1759.5) | 237 (127.5–309) | 174.5 (159–395.3) | 86 (53–211) | <0.001 | <0.001 |
| Creatinine, μmol/L | 50.5 (29.3–67) | 20 (14.8–25) | 37 (25.3–56) | 57.5 (52.5–69.5) | 48 (39–60) | 58 (44.5–68.5) | <0.001 | 0.040 |
| Potassium, mmol/L | 4.3 (3.8–4.5) | 4.3 (4.0–4.5) | 4.1 (3.8–4.4) | 4.3 (3.9–4.5) | 4.3 (3.9–4.7) | 4.2 (4.0–4.4) | 0.86 | 0.78 |
Data are presented as median (interquartile range) or number (percentage). BNP indicates B‐type natriuretic peptide; CK, creatine kinase; DBP, diastolic blood pressure; DM1, type 1 myotonic dystrophy; FSHD, facioscapulohumeral muscular dystrophy; HR, heart rate; LGMD, limb‐girdle muscular dystrophy; mWC, manual wheelchair; pWC, power wheelchair; SBP, systolic blood pressure; SDOB, sleep‐disordered breathing.
Indicates statistical analysis comparing all patients.
Indicates statistical analysis comparing the cohorts of patients with muscular dystrophy (MD) and non‐MD myopathies.
Table 2.
Cardiac Evaluation of MD Patient Cohorts
| Modality | Dystrophinopathies | LGMD | DM1 | FSHD | P Value |
|---|---|---|---|---|---|
| 12‐Lead ECG | (n=32) | (n=33) | (n=60) | (n=20) | |
| Heart rate, beats per min | 83 (71–98) | 75 (60–81) | 68 (63.3–75) | 75 (69.5–85) | 0.005 |
| PR interval, ms | 132 (121.5–144) | 157.5 (140.5–164) | 193 (178.3–223) | 162 (148–170) | <0.001 |
| QRS duration, ms | 97 (87.5–109.3) | 94 (86–102) | 107 (96–118) | 91 (85.5–97.8) | <0.001 |
| QTc interval, ms | 377 (359.5–399) | 394 (374.8–426) | 412 (396–440) | 378 (352–399.5) | <0.001 |
| First‐degree AVB | 0 | 0 | 18 (30) | 0 | <0.001 |
| LAFB | 2 (6.3) | 0 | 6 (10) | 1 (5) | 0.29 |
| LBBB | 3 (9.4) | 1 (3) | 11 (18.3) | 0 | 0.039 |
| RBBB | 2 (6.3) | 2 (6.1) | 2 (3.3) | 2 (10) | 0.71 |
| Echocardiogram | (n=23) | (n=21) | (n=50) | (n=12) | |
| LVIDd, cm | 4.7 (4.2–5.3) | 4.7 (4.4–5.1) | 4.4 (4.0–4.9) | 4.4 (4.3–4.7) | 0.16 |
| LVIDs, cm | 3.5 (3–4.6) | 3 (2.8–3.6) | 2.8 (2.6–3.1) | 2.9 (2.5–3) | 0.003 |
| LVPWd, cm | 0.7 (0.6–0.8) | 0.9 (0.8–0.9) | 0.9 (0.7–1) | 0.8 (0.7–0.9) | 0.003 |
| LVEF, % | 38.1 (26.3–52.3) | 55 (52.8–60) | 55 (50–60) | 60 (56.3–60) | <0.001 |
| LVMI, g/m2 | 63.2 (56–71.1) | 57.1 (52.1–64.5) | 46.7 (38.2–49.9) | 49.1 (41.5–55.7) | 0.003 |
| Cardiac MRI | (n=14) | (n=16) | (n=21) | (n=10) | |
| LA volume index, mL/m2 | 39.8 (35.5–43.5) | 36 (31.3–38.2) | 29 (23.8–31.3) | 29 (27.7–33) | 0.06 |
| LVEDVi, mL/m2 | 91 (76.8–110.8) | 74 (64.5–92.3) | 61 (55–72) | 68 (56.5–79) | 0.001 |
| LVESVi, mL/m2 | 50 (31–61) | 31 (25–44) | 29 (20.5–36.3) | 23.5 (20.5–32.5) | 0.015 |
| LVEF, % | 45 (40–56.5) | 55.5 (52–58.3) | 56 (50–63) | 58.5 (53.5–66) | 0.08 |
| LVMI, g/m2 | 61 (44–67) | 45 (39–54) | 42.5 (39.5–49.8) | 42 (37.5–49.3) | 0.07 |
| RVEDVi, mL/m2 | 76 (68–84) | 67 (60.5–78.5) | 60 (56–75) | 73 (58–94) | 0.09 |
| RVESVi, mL/m2 | 38.5 (35.3–43.3) | 32 (28.5–41.5) | 31 (26.8–35) | 27 (26–41) | 0.13 |
| RVEF, % | 49 (44–53) | 52 (48.5–54) | 50 (47–55) | 53 (49–55) | 0.66 |
Data are presented as median (interquartile range) or number (percentage). AVB indicates atrioventricular block; DM1, type 1 myotonic dystrophy; FSHD, facioscapulohumeral muscular dystrophy; LA, left atrial; LAFB, left anterior fascicular block; LBBB, left bundle branch block; LGMD, limb‐girdle muscular dystrophy; LVEDVi, left ventricular end‐diastolic volume index; LVEF, left ventricular ejection fraction; LVESVi, left ventricular end‐systolic volume index; LVIDd, left ventricular internal dimension at end‐diastole; LVIDs, left ventricular internal dimension at end‐systole; LVMI, left ventricular mass index; LVPWd, left ventricular posterior wall thickness at end‐diastole; MD, muscular dystrophy; MRI, magnetic resonance imaging; PR interval, duration of atrial depolarization; QRS duration, duration of ventricular depolarization; QTc interval, corrected duration between ventricular depolarization and repolarization; RBBB, right bundle branch block; RVEDVi, right ventricular end‐diastolic volume index; RVEF, right ventricular ejection fraction; RVESVi, right ventricular end‐systolic volume index.
Limb‐Girdle MD
Patients with LGMD presented with a moderate burden of muscle weakness as indicated by wheelchair use (Table 1), with muscle wasting more localized to the shoulder and pelvic regions. The LGMD cohort demonstrated a substantial prevalence of dilated cardiomyopathy based on echocardiogram and cardiac MRI (Figure 1B; Table 2), with delayed enhancement present in 3 (18.8%) patients who received a cardiac MRI (Figure S1). The mild cardiomyopathy in patients with LGMD was not detected by routine transthoracic echocardiography (Table 2). Twelve‐lead ECG and Holter monitoring demonstrated 3 patients with atrial fibrillation and 2 patients with ventricular tachycardia (Figure 1D), with a low prevalence of conduction disease (Table 2). Respiratory disease accompanied by SDOB was prevalent in this cohort, with 5 (15.2%) patients requiring pressure ventilation devices (Table 1).
Type 1 Myotonic Dystrophy
Based on echocardiogram and cardiac MRI studies in the DM1 cohort, 17 (28.3%) patients had a cardiomyopathy, as indicated by reduced LVEFs (Figure 1B; Table 2), with 2 patients also displaying myocardial fibrosis (Figure S1). Patients with DM1 exhibited a high prevalence of conduction abnormalities, with prolonged PR intervals and QRS durations (Table 2). Indeed, first‐degree atrioventricular block and LBBB were common abnormalities, occurring in 18 (30%) and 11 (18.3%) patients, respectively, while 6 patients had left anterior fascicular block (Table 2). Atrial fibrillation or flutter and ventricular tachycardia were detected in 14 and 6 patients, respectively. Respiratory disease was accompanied by SDOB in 16 of the patients with DM1, with 8 patients requiring ventilator devices. In addition to DM1, 7 patients with type 2 myotonic dystrophy were evaluated; all patients with type 2 myotonic dystrophy exhibited normal 12‐lead ECGs, echocardiograms, and cardiac MRIs (Table S2).
Fascioscapulohumeral MD
Two patients with FSHD had a cardiomyopathy as indicated through echocardiogram and cardiac MRI studies (Figure 1B), although the degree of cardiomyopathy was milder relative to the other cohorts; both patients had mildly thickened right ventricles (Table 2 and Table S1) and only 1 showed myocardial fibrosis (Figure S1). ECG abnormalities were minimal and associated with a low prevalence of atrial and ventricular arrhythmias (Figure 1D; Table 2). Respiratory disease and SDOB were observed in 6 and 6 patients, respectively, with 5 patients receiving noninvasive pressure ventilation.
Non‐MD Myopathies
The non‐MD myopathies cohort included 40 patients with a significant burden of respiratory disease and moderate degree of limited ambulation (Table 1). These patients were included as a negative control cohort for appropriate comparisons to the broader MD cohort, which had a similar distribution of sex. The patients with non‐MD myopathies had normal cardiac structure and function with no indication of a cardiomyopathy or ECG abnormalities (Table 3).
Table 3.
Cardiac Evaluation of the Non‐MD Myopathy Patient Cohort
| Modality | Non‐MD Myopathies |
|---|---|
| 12‐Lead ECG (n=40) | |
| Heart rate, beats per min | 72 (62–83.5) |
| PR interval, ms | 152 (140–172) |
| QRS duration, ms | 90 (82–98) |
| QTc interval, ms | 396 (382–416) |
| First‐degree AVB | 2 (5) |
| LAFB | 2 (5) |
| LBBB | 1 (2.5) |
| RBBB | 2 (5) |
| Echocardiogram (n=33) | |
| LVIDd, cm | 4.5 (4.1–5) |
| LVIDs, cm | 2.8 (2.6–3.3) |
| LVPWd, cm | 0.8 (0.7–1) |
| LVEF, % | 60 (55–60) |
| LVMI, g/m2 | 51.8 (43.9–54) |
| Cardiac MRI (n=16) | |
| LA volume index, mL/m2 | 36.8 (24–40.9) |
| LVEDVi, mL/m2 | 71 (62.5–78.5) |
| LVESVi, mL/m2 | 27 (21.5–31) |
| LVEF, % | 62 (57–66) |
| LVMI, g/m2 | 43 (32.5–52) |
| RVEDVi, mL/m2 | 74 (65–80) |
| RVESVi, mL/m2 | 31 (26–37) |
| RVEF, % | 57 (53–59.5) |
Values are expressed as median (interquartile range) or number (percentage). AVB indicates atrioventricular block; LA, left atrial; LAFB, left anterior fascicular block; LBBB, left bundle branch block; LVEF, left ventricular ejection fraction; LVEDVi, left ventricular end‐diastolic volume index; LVESVi, left ventricular end‐systolic volume index; LVIDd, left ventricular internal dimension at end‐diastole; LVIDs, left ventricular internal dimension at end‐systole; LVMI, left ventricular mass index; LVPWd, left ventricular posterior wall thickness at end‐diastole; MD, muscular dystrophy; MRI, magnetic resonance imaging; PR interval, duration of atrial depolarization; QRS duration, duration of ventricular depolarization; QTc interval, corrected duration between ventricular depolarization and repolarization; RBBB, right bundle branch block; RVEDVi, right ventricular end‐diastolic volume index; RVEF, right ventricular ejection fraction; RVESVi, right ventricular end‐systolic volume index.
Biomarkers
Plasma CK (normal <250 U/L) was markedly elevated in the dystrophinopathies and LGMD cohorts (Table 1). In contrast, BNP values (normal range: 100–500 pg/mL) were within the normal range in both cohorts (Table 1). There was no substantial elevation of plasma BNP or CK in the DM1 cohort, with CK being increased in only 3 patients. Similarly, patients with FSHD showed normal BNP, while 2 patients exhibited elevated CK values. All patients with non‐MD myopathies had normal BNP levels (Table 1). There was no evidence of hyperkalemia, and serum creatinine remained within the low‐normal range (Table 1).
Optimization of Medical and Device Therapies
Following enrollment in the NMMD clinic, the initiation and optimization of pharmacological therapies were implemented to better manage and improve heart disease (Figure 2). The use of angiotensin‐converting enzyme inhibitors/angiotensin receptor blockers (P=0.039), β‐blockers (P=0.018), and mineralocorticoid receptor antagonists (P=0.001) was increased for eligible patients (Figure 3A) and the dose of angiotensin‐converting enzyme inhibitors/angiotensin receptor blockers was uptitrated (P=0.004) and evaluated relative to the maximum tolerated dose as defined by 2016 American Heart Association guidelines (Figure 3B; Table S3).18 Diuretics and statin therapies were uptitrated in 21 (14.5%) and 25 (17.2%) patients, respectively. Corticosteroid use, with either deflazacort or prednisone, was used in 9 of 27 patients with DMD. Based on clinical guidelines and on a clinical basis of primary and secondary prophylaxis,19 device therapy was used in patients with more severe cardiomyopathies, namely in the DMD and DM1 cohorts (Figure 3C). These included implantable cardioverter‐defibrillators and pacemakers, such as cardiac resynchronization therapy (CRT) devices. Four patients received an implantable cardioverter‐defibrillator and 9 patients received a CRT device.
Figure 2.
Cardiac assessment and management in a multidisciplinary setting improves outcomes in patients with muscular dystrophy (MD). MRI indicates magnetic resonance imaging; NMMD, Neuromuscular Multidisciplinary.
Figure 3.
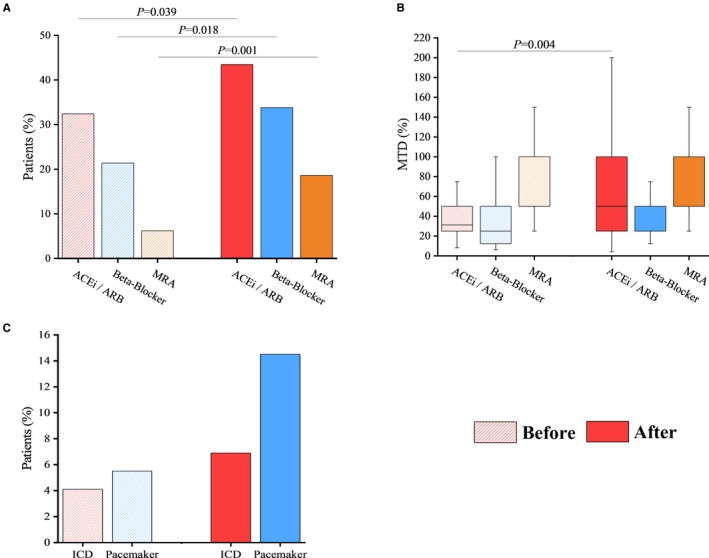
Uptitration of medical therapy and increase in device implantation following the initial clinic visit in patients with muscular dystrophy (MD). Baseline use of angiotensin‐converting enzyme inhibitors (ACEIs) and angiotensin receptor blockers (ARBs), β‐blockers, mineralocorticoid receptor antagonists (MRAs) (before), and their initiation at the initial Neuromuscular Multidisciplinary clinic visit (after) (A); and their uptitration following the initial visit (B). Implantable cardioverter‐defibrillator (ICD) and pacemaker (including cardiac resynchronization therapy) implantation (C). Maximum tolerated dose (MTD) as defined by 2016 American Heart Association guidelines for the diagnosis and treatment of acute and chronic heart failure.
Clinical Outcomes
We assessed the impact of cardiac intervention as part of a multidisciplinary care approach on the clinical outcomes of our MD patient cohort. The median LVEF of patients tracked with a cardiomyopathy in dystrophinopathies, LGMD, DM1, and FSHD cohorts was 39%, 44%, 45%, and 49%, respectively, at the initial NMMD clinic visit, which improved to 50%, 57%, 50%, and 55%, respectively (Figure 4). Overall, the 57 patients with MD tracked with a cardiomyopathy showed a marked improvement in their median LVEF from baseline of 43% to 50% at the end of the 3‐year period (P<0.001) (Figure 4). The rate of unplanned, all‐cause outpatient clinic visits was reduced from 3.0 to 1.5 visits per year among all cohorts of MD. Over the first 18 months, there was minimal difference between the rates of all‐cause outpatient clinic visits between the MD and non‐MD myopathies cohorts (β=0.06; 95% CI, −0.93 to 1.05 [P=0.86]); however, from 18 months onward, there was a marked lowering in all‐cause outpatient clinic visits for the MD cohort (β=−1.09; 95% CI, −2.17 to −0.01 [P=0.048]) (Figure 5A). The dystrophinopathies and DM1 groups showed the most substantial reduction in annual unplanned, all‐cause outpatient clinic visits, from 6.5 to 1.6 visits per year and 3.2 to 0.5 visits per year, respectively. A smaller improvement in the rate of outpatient clinic visits was noted in the LGMD group (2.8 to 1.3 visits per year) and FSHD group (2.5 to 2.2 visits per year). The rate of hospitalization duration decreased from 14.2 to 0.9 days per year among all cohorts of MD. Over the first 18 months, there was no difference between the rates of hospitalization between the MD and non‐MD myopathies cohorts (β=0.65; 95% CI, −6.22 to 7.51 [P=0.78]); however, from 18 months onward, there was a clear divergence in rates illustrating a marked reduction in hospitalizations for the MD cohort (β=−7.72; 95% CI, −13.73 to −1.71 [P=0.021]) (Figure 5B). Both the LGMD and DM1 groups had a large decline in the rates of hospitalization duration from 27.0 to 1.2 days per year and 14.9 to 0.5 days per year, respectively. The dystrophinopathies group showed a moderate decline in the rates of hospitalization duration from 13.4 to 4.8 days per year. The FSHD group maintained a low rate of hospitalization duration at 0.4 to 0 days per year. The rate of cardiac‐related hospitalizations was reduced from 0.4 to 0.1 hospitalizations per year, while there were no cardiac‐related hospitalizations in the non‐MD myopathies cohort (β=0.21; 95% CI, 0.14–0.28 [P<0.001]) (Figure 5C). At the 6‐month period following the optimization of medical therapies, outcome rates of the MD patient cohorts were higher than the non‐MD myopathies cohort (Figure 5). Overall, all MD cohorts showed a marked and sustained reduction in cumulative rates of unplanned, all‐cause outpatient clinic visits, the duration of hospitalization, and the number of cardiac‐related hospitalizations over the 3‐year period (Figure 5). One patient with DMD died of cardiac causes and 1 patient with DMD died of noncardiac causes (2 of 32 patients over 3 years [2.1% per year]). Two patients with LGMD died of noncardiac causes (mortality rate of 2.0% per year), while 3 patients with DM1 died of noncardiac causes, yielding a mortality rate of 1.7% per year.
Figure 4.
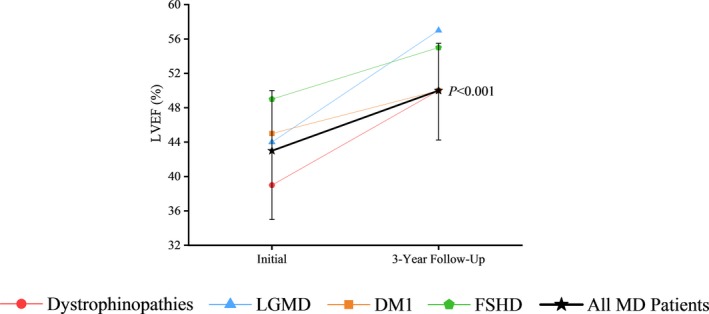
Improvement in left ventricular ejection fraction (LVEF) in the various muscular dystrophy (MD) cohorts in response to multidisciplinary care. LVEF obtained by cardiac magnetic resonance imaging and/or echocardiogram for 57 patients with cardiomyopathy, with 3 years of imaging data, shown as the median LVEF at the time of their initial clinic visit and at their 3‐year follow‐up. DM1 indicates type 1 myotonic dystrophy; FSHD, facioscapulohumeral muscular dystrophy; LGMD, limb‐girdle muscular dystrophy.
Figure 5.
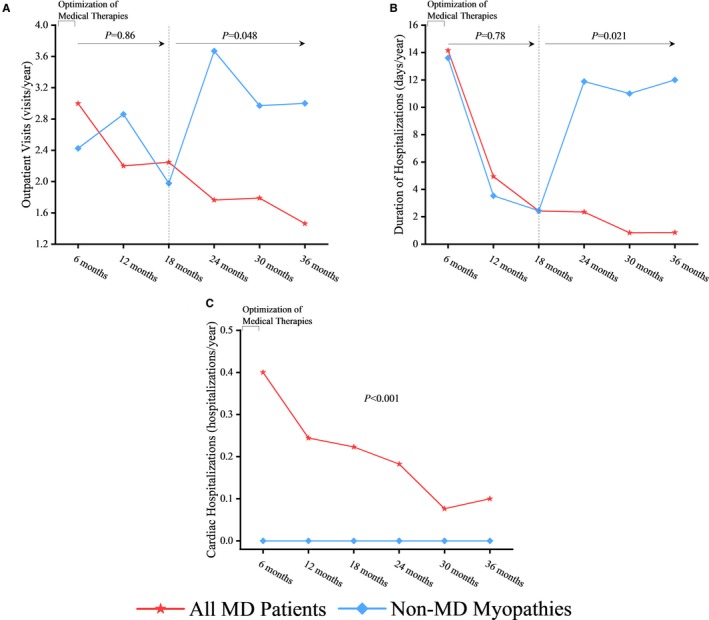
Marked improvement in clinical outcomes in the various muscular dystrophy (MD) cohorts in response to multidisciplinary care. Comparing the rates of unplanned, all‐cause outpatient clinic visits (A), duration of hospitalizations (B), and incidence of cardiac hospitalizations (C) following a 6‐month period of patient assessment and care and within 3 years of the initial Neuromuscular Multidisciplinary clinic visit.
Discussion
Patients with MD represent a vulnerable population because of the complex syndrome of progressive muscle weakness along with cardiac and respiratory comorbidities. Our cohort clearly demonstrates a significant burden of heart disease and our care model facilitates a prompt and careful assessment, followed by the initiation and optimization of medical and device therapy with effective follow‐up care. We included a cohort of patients with non‐MD, without heart disease, but with moderately impaired ambulation and respiratory disease concomitant with relatively low adverse outcomes treated in the same multidisciplinary care environment. Our outcome data demonstrated a high burden of healthcare use in the dystrophinopathies, LGMD, and DM1 cohorts. The incidence of unplanned, all‐cause outpatient clinic visits, the duration of hospitalizations, and the incidence of cardiac‐related hospitalizations are indicative of the severity of patient condition. Cardiac intervention had a direct effect on the rate of cardiac‐related hospitalizations. In addition, all‐cause outcomes were progressively improved in our patients with MD, through optimized medications, illustrated by the divergence of all‐cause outcome rates following 18 months of treatment. Furthermore, the implantation of implantable cardioverter‐defibrillator and pacemaker devices usually occurs within the first 18 months of NMMD clinic enrollment and cardiac assessment. With regards to the non‐MD myopathies cohort, these patients show comparable complex neuromuscular disease phenotypes to the MD cohort and receive the same multidisciplinary care, including a similar degree of respiratory intervention, with the exception of cardiac intervention in the form of medical and device therapy. Unlike the MD cohort, these patients do not have heart disease. We can therefore elucidate that heart disease is an important driver of MD patient outcomes by contrasting them with non‐MD myopathies patient outcomes. This emphasizes the necessity for cardiac intervention and management in the MD patient population, effectively provided through our multidisciplinary care model, in which confounding comorbidities may be treated and stabilized. Importantly, in our dystrophinopathies, LGMD, and DM1 cohorts, mortality rates were markedly lower (1.7–2.1%) compared with historical cohort data collected from patient cohorts in the absence of multidisciplinary care (2.0–3.3%).20, 21, 22, 23
The demographics of all of the MD patient cohorts appear uniform, except for the dystrophinopathies cohort, which comprised younger males as shown by the X‐linked inheritance pattern.2, 24, 25 We used a combination of transthoracic echocardiography and cardiac MRI at the initial clinic visit and during follow‐up to evaluate the degree of cardiomyopathy and the response to therapy. Although cardiac MRI is the preferred imaging modality for patients with MD,24 its feasibility in patients with advanced disease can be challenging because of limited mobility, wheelchair dependence, use of defibrillators and pacemakers, spinal stabilization rods, and mouthpiece ventilation. Therefore, the use of transthoracic echocardiography was an important imaging modality for our patients who were unable to undergo cardiac MRI. We detected a high prevalence of cardiomyopathy in the dystrophinopathies and LGMD cohorts characterized by reduced systolic function as well as LV chamber dilation. Cardiac imaging is particularly important since plasma BNP, a well‐known biomarker of pathological cardiac remodeling,26 was not markedly elevated, illustrating the limitation of BNP in predicting heart disease in our cohort.
Patients with DM1 presented with a high burden of atrial and ventricular arrhythmias. Notably, there was a high prevalence of first‐degree atrioventricular block and LBBB27 The presence of LBBB likely aggravated the cardiomyopathy in patients with DM1 because of the associated electromechanical ventricular dyssynchrony leading to progressive LV dysfunction.28 The demonstrated occurrence of ventricular tachyarrhythmias in our DM1 patient cohort is consistent with the increased risk of sudden cardiac death in this patient population.7, 11, 29 Of our entire MD cohort, there were 10 cardiac defibrillators (6.9% [10/145] patients) and 20 pacemakers (13.8% [20/145] patients) implanted. As such, the use of pacemakers, including CRT, is particularly important while allowing the initiation of β‐blocker therapy. Nine CRT devices were implanted in our patients with DM1 and we have recently shown that CRT improves LVEF in patients with DM1 with LBBB.30
The use of medical therapies in our patients with dystrophinopathies is supported by clinical trial evidence. Angiotensin‐converting enzyme inhibition,31 β‐blockade,32, 33 and mineralocorticoid receptor antagonism34 improves LV systolic function and delays the progression of cardiomyopathy in DMD. Importantly, we extrapolated these findings to other MD cohorts with cardiomyopathies, which was associated with similar improvement in cardiac and clinical outcomes, thereby supporting the widespread use of these therapies in this vulnerable patient population. Our data show that active cardiac care by way of optimized medical and device therapies, prescribed to patients with MD but not patients with non‐MD myopathies, improved clinical outcomes. However, respiratory care, through the assessment and management of respiratory comorbidities, likely also contributed to the improved outcomes in our patients. SDOB and limited airway clearance in our patients can be attributed to muscle weakness affecting the chest wall, diaphragm, and upper airways. The initiation of assisted noninvasive positive pressure ventilation alongside the use of lung volume recruitment strategies and mechanical insufflation‐exsufflation likely contributed to improved clinical outcomes. Although these were prescribed to a similar degree in the MD and non‐MD myopathies cohorts, mechanical insufflation‐exsufflation tended to be prescribed at a greater extent in patients with dystrophinopathies. The involvement of social workers in our clinic allowed patients to receive financial assistance and counseling for psychosocial distress, and enabled adherence to medical therapy. Our dietician provided healthy dietary recommendations and adjusted potassium intake to minimize hyperkalemia, which is particularly important since medications such as angiotensin‐converting enzyme inhibitors may cause electrolyte imbalances and there is no accurate measure of renal function in our patients. Taken together, relieved heart disease burden combined with comorbidity control, facilitated by our novel multidisciplinary care model provided at the NMMD clinic, improved outcomes in patients with MD.
Limitations
Attributable to the systemic manifestations of this disease, patients with MD in our region are generally referred to the NMMD clinic, and it is therefore not feasible to recruit patients with MD exclusively receiving cardiac care. With regards to cardiac imaging for patients with MD, although echocardiography may be the only feasible cardiac imaging modality, there are limitations to the reliability of data collected because of the obstructed acoustic windows as a result of scoliosis, obesity, and lung disease. Another limitation to our study is our modest cohort size. Given that these conditions are relatively uncommon, we believe that our group sizes are reasonable. The rarity of neuromuscular conditions also limits the number of patients referred to the NMMD clinic. This introduces limitations when composing a suitable negative control cohort, such as the non‐MD myopathies cohort we included in this study. Inherent differences between the heterogeneous MD and non‐MD myopathies cohorts include ambulation and age, in which the patients with non‐MD myopathies were older. We have included this patient cohort on the basis that they received the same multidisciplinary care in the same setting as our patients with MD, in the absence of heart disease, allowing for a comparison of the impact of cardiac care. Our outcome data were provided over a 3‐year period, but, given the prospective nature of our study, we will continue to recruit patients and obtain additional outcome data over a longer duration.
Conclusions
Patients with MD experience a considerable burden of heart disease with multiple comorbidities. The treatment of patients using a multidisciplinary care model, such as the one initiated at the NMMD clinic, provides patients with comprehensive care including cardiac assessment and prompt management of their heart disease. Our prospective cohort study demonstrates the effectiveness of cardiac intervention facilitated through a multidisciplinary care pathway to improve health outcomes of patients with MD.
Sources of Funding
This work was supported by the University Hospital Foundation, University of Alberta Hospital.
Disclosures
None.
Supporting information
Table S1. Structural Evaluation of the Mitral, Tricuspid, Aortic, and Pulmonic Valves by Cohort*
Table S2. Comparing 12‐Lead ECG and Systolic Function Data of Patients With DM1 and Patients With DM2*
Table S3. Defined Maximum Tolerated Dose of Medications*
Figure S1. Prevalence of myocardial fibrosis based on cardiac magnetic resonance imaging (MRI).
Acknowledgments
We would like to acknowledge the patients and their families and caregivers for their willing participation in this study.
(J Am Heart Assoc. 2020;9:e014004 DOI: 10.1161/JAHA.119.014004.)
References
- 1. Koeks Z, Bladen CL, Salgado D, van Zwet E, Pogoryelova O, McMacken G, Monges S, Foncuberta ME, Kekou K, Kosma K, Dawkins H, Lamont L, Bellgard MI, Roy AJ, Chamova T, Guergueltcheva V, Chan S, Korngut L, Campbell C, Dai Y, Wang J, Barisic N, Brabec P, Lahdetie J, Walter MC, Schreiber‐Katz O, Karcagi V, Garami M, Herczegfalvi A, Viswanathan V, Bayat F, Buccella F, Ferlini A, Kimura E, van den Bergen JC, Rodrigues M, Roxburgh R, Lusakowska A, Kostera‐Pruszczyk A, Santos R, Neagu E, Artemieva S, Rasic VM, Vojinovic D, Posada M, Bloetzer C, Klein A, Diaz‐Manera J, Gallardo E, Karaduman AA, Oznur T, Topaloglu H, El Sherif R, Stringer A, Shatillo AV, Martin AS, Peay HL, Kirschner J, Flanigan KM, Straub V, Bushby K, Beroud C, Verschuuren JJ, Lochmuller H. Clinical outcomes in Duchenne muscular dystrophy: a study of 5345 patients from the TREAT‐NMD DMD global database. J Neuromuscul Dis. 2017;4:293–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bushby K, Finkel R, Birnkrant DJ, Case LE, Clemens PR, Cripe L, Kaul A, Kinnett K, McDonald C, Pandya S, Poysky J, Shapiro F, Tomezsko J, Constantin C; Group DMDCCW . Diagnosis and management of Duchenne muscular dystrophy, part 2: implementation of multidisciplinary care. Lancet Neurol. 2010;9:177–189. [DOI] [PubMed] [Google Scholar]
- 3. Arbustini E, Di Toro A, Giuliani L, Favalli V, Narula N, Grasso M. Cardiac phenotypes in hereditary muscle disorders. J Am Coll Cardiol. 2018;72:2485–2506. [DOI] [PubMed] [Google Scholar]
- 4. Miskew Nichols B, Nikhanj A, Wang F, Freed DH, Oudit GY. Advanced dilated cardiomyopathy in a patient with Hutterite limb‐girdle muscular dystrophy: use of a left ventricular assist device. Circ Heart Fail. 2018;11:e004960. [DOI] [PubMed] [Google Scholar]
- 5. Silva MC, Meira ZM, Gurgel Giannetti J, da Silva MM, Campos AF, Barbosa Mde M, Starling Filho GM, Ferreira Rde A, Zatz M, Rochitte CE. Myocardial delayed enhancement by magnetic resonance imaging in patients with muscular dystrophy. J Am Coll Cardiol. 2007;49:1874–1879. [DOI] [PubMed] [Google Scholar]
- 6. Mascarenhas DA, Spodick DH, Chad DA, Gilchrist J, Townes PL, DeGirolami U, Mudge GH, Maki DW, Bishop RL. Cardiomyopathy of limb‐girdle muscular dystrophy. J Am Coll Cardiol. 1994;24:1328–1333. [DOI] [PubMed] [Google Scholar]
- 7. Groh WJ, Groh MR, Saha C, Kincaid JC, Simmons Z, Ciafaloni E, Pourmand R, Otten RF, Bhakta D, Nair GV, Marashdeh MM, Zipes DP, Pascuzzi RM. Electrocardiographic abnormalities and sudden death in myotonic dystrophy type 1. N Engl J Med. 2008;358:2688–2697. [DOI] [PubMed] [Google Scholar]
- 8. Groh WJ, Bhakta D. Arrhythmia management in myotonic dystrophy type 1. JAMA. 2012;308:337–338. [DOI] [PubMed] [Google Scholar]
- 9. Backman E, Nylander E. The heart in Duchenne muscular dystrophy: a non‐invasive longitudinal study. Eur Heart J. 1992;13:1239–1244. [DOI] [PubMed] [Google Scholar]
- 10. Nigro V, Savarese M. Genetic basis of limb‐girdle muscular dystrophies: the 2014 update. Acta Myol. 2014;33:1–12. [PMC free article] [PubMed] [Google Scholar]
- 11. Nikhanj A, Sivakumaran S, Miskew‐Nichols B, Siddiqi ZA, Oudit GY. Ventricular tachycardia in patients with type 1 myotonic dystrophy: a case series. Eur Heart J Case Rep. 2019;3:1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Verhaert D, Richards K, Rafael‐Fortney JA, Raman SV. Cardiac involvement in patients with muscular dystrophies: magnetic resonance imaging phenotype and genotypic considerations. Circ Cardiovasc Imaging. 2011;4:67–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Trevisan CP, Pastorello E, Armani M, Angelini C, Nante G, Tomelleri G, Tonin P, Mongini T, Palmucci L, Galluzzi G, Tupler RG, Barchitta A. Facioscapulohumeral muscular dystrophy and occurrence of heart arrhythmia. Eur Neurol. 2006;56:1–5. [DOI] [PubMed] [Google Scholar]
- 14. Smith PE, Calverley PM, Edwards RH, Evans GA, Campbell EJ. Practical problems in the respiratory care of patients with muscular dystrophy. N Engl J Med. 1987;316:1197–1205. [DOI] [PubMed] [Google Scholar]
- 15. Feingold B, Mahle W T, Auerbach S, Clemens P, Domenighetti A A, Jefferies J L, Judge D P, Lal A K, Markham L W, Parks WJ, Tsuda T, Wang Paul J, Yoo SJ. Management of cardiac involvement associated with neuromuscular diseases: a scientific statement from the American Heart Association. Circulation. 2017;136:e200–e231. [DOI] [PubMed] [Google Scholar]
- 16. Anderson TJ, Gregoire J, Pearson GJ, Barry AR, Couture P, Dawes M, Francis GA, Genest J Jr, Grover S, Gupta M, Hegele RA, Lau DC, Leiter LA, Lonn E, Mancini GB, McPherson R, Ngui D, Poirier P, Sievenpiper JL, Stone JA, Thanassoulis G, Ward R. 2016 Canadian Cardiovascular Society guidelines for the management of dyslipidemia for the prevention of cardiovascular disease in the adult. Can J Cardiol. 2016;32:1263–1282. [DOI] [PubMed] [Google Scholar]
- 17. Kawel‐Boehm N, Maceira A, Valsangiacomo‐Buechel ER, Vogel‐Claussen J, Turkbey EB, Williams R, Plein S, Tee M, Eng J, Bluemke DA. Normal values for cardiovascular magnetic resonance in adults and children. J Cardiovasc Magn Reson. 2015;17:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yancy CW, Jessup M, Bozkurt B, Butler J, Casey DE, Colvin MM, Drazner MH, Filippatos G, Fonarow GC, Givertz MM, Hollenberg SM, Lindenfeld J, Masoudi FA, McBride PE, Peterson PN, Stevenson LW, Westlake C. 2016 ACC/AHA/HFSA focused update on new pharmacological therapy for heart failure: an update of the 2013 ACCF/AHA guideline for the management of heart failure. J Am Coll Cardiol. 2016;68:1476–1488. [DOI] [PubMed] [Google Scholar]
- 19. Epstein AE, DiMarco JP, Ellenbogen KA, Estes NA III, Freedman RA, Gettes LS, Gillinov AM, Gregoratos G, Hammill SC, Hayes DL, Hlatky MA, Newby LK, Page RL, Schoenfeld MH, Silka MJ, Stevenson LW, Sweeney MO; American College of Cardiology Foundation, American Heart Association Task Force on Practice Guidelines, Heart Rhythm Society . 2012 ACCF/AHA/HRS focused update incorporated into the ACCF/AHA/HRS 2008 guidelines for device‐based therapy of cardiac rhythm abnormalities: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines and the Heart Rhythm Society. Circulation. 2013;127:e283–e352. [DOI] [PubMed] [Google Scholar]
- 20. Ballard E, Grey N, Jungbluth H, Wraige E, Kapetanakis S, Davidson C, Hart N. Observation cohort study of cause of death in patients with Duchenne muscular dystrophy (DMD). Eur Respir J. 2012;40:P1720. [Google Scholar]
- 21. Fayssoil A, Ogna A, Chaffaut C, Chevret S, Guimarães‐Costa R, Leturcq F, Wahbi K, Prigent H, Lofaso F, Nardi O, Clair B, Behin A, Stojkovic T, Laforet P, Orlikowski D, Annane D. Natural history of cardiac and respiratory involvement, prognosis and predictive factors for long‐term survival in adult patients with limb girdle muscular dystrophies type 2C and 2D. PLoS One. 2016;11:e0153095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mathieu J, Allard P, Potvin L, Prevost C, Begin P. A 10‐year study of mortality in a cohort of patients with myotonic dystrophy. Neurology. 1999;52:1658–1662. [DOI] [PubMed] [Google Scholar]
- 23. Bhakta D, Groh MR, Shen C, Pascuzzi RM, Groh WJ. Increased mortality with left ventricular systolic dysfunction and heart failure in adults with myotonic dystrophy type 1. Am Heart J. 2010;160:1137–1141. [DOI] [PubMed] [Google Scholar]
- 24. Shirokova N, Niggli E. Cardiac phenotype of Duchenne muscular dystrophy: insights from cellular studies. J Mol Cell Cardiol. 2013;58:217–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Schram G, Fournier A, Leduc H, Dahdah N, Therien J, Vanasse M, Khairy P. All‐cause mortality and cardiovascular outcomes with prophylactic steroid therapy in Duchenne muscular dystrophy. J Am Coll Cardiol. 2013;61:948–954. [DOI] [PubMed] [Google Scholar]
- 26. Vanderheyden M, Goethals M, Verstreken S, De Bruyne B, Muller K, Van Schuerbeeck E, Bartunek J. Wall stress modulates brain natriuretic peptide production in pressure overload cardiomyopathy. J Am Coll Cardiol. 2004;44:2349–2354. [DOI] [PubMed] [Google Scholar]
- 27. Nikolaidou T, Ghosh JM, Clark AL. Outcomes related to first‐degree atrioventricular block and therapeutic implications in patients with heart failure. JACC Clin Electrophysiol. 2016;2:181–192. [DOI] [PubMed] [Google Scholar]
- 28. Auffret V, Martins RP, Daubert C, Leclercq C, Le Breton H, Mabo P, Donal E. Idiopathic/iatrogenic left bundle branch block‐induced reversible left ventricle dysfunction: JACC State‐of‐the‐Art Review. J Am Coll Cardiol. 2018;72:3177–3188. [DOI] [PubMed] [Google Scholar]
- 29. Wahbi K, Meune C, Porcher R, Bécane HM, Lazarus A, Laforêt P, Stojkovic T, Béhin A, Radvanyi‐Hoffmann H, Eymard B, Duboc D. Electrophysiological study with prophylactic pacing and survival in adults with myotonic dystrophy and conduction system disease. JAMA. 2012;307:1292–1301. [DOI] [PubMed] [Google Scholar]
- 30. Nikhanj A, Sivakumaran S, Miskew Nichols B, Siddiqi ZA, Oudit GY. Comparison of usefulness of cardiac resynchronization therapy in patients with type 1 myotonic dystrophy with versus without left bundle branch block. Am J Cardiol. 2019;124:1770–1774. [DOI] [PubMed] [Google Scholar]
- 31. Duboc D, Meune C, Lerebours G, Devaux JY, Vaksmann G, Bécane HM. Effect of perindopril on the onset and progression of left ventricular dysfunction in Duchenne muscular dystrophy. J Am Coll Cardiol. 2005;45:855–857. [DOI] [PubMed] [Google Scholar]
- 32. Kajimoto H, Ishigaki K, Okumura K, Tomimatsu H, Nakazawa M, Saito K, Osawa M, Nakanishi T. Beta‐blocker therapy for cardiac dysfunction in patients with muscular dystrophy. Circ J. 2006;70:991–994. [DOI] [PubMed] [Google Scholar]
- 33. Viollet L, Thrush PT, Flanigan KM, Mendell JR, Allen HD. Effects of angiotensin‐converting enzyme inhibitors and/or beta blockers on the cardiomyopathy in Duchenne muscular dystrophy. Am J Cardiol. 2012;110:98–102. [DOI] [PubMed] [Google Scholar]
- 34. Raman SV, Hor KN, Mazur W, Halnon NJ, Kissel JT, He X, Tran T, Smart S, McCarthy B, Taylor MD, Jefferies JL, Rafael‐Fortney JA, Lowe J, Roble SL, Cripe LH. Eplerenone for early cardiomyopathy in Duchenne muscular dystrophy: a randomised, double‐blind, placebo‐controlled trial. Lancet Neurol. 2015;14:153–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Structural Evaluation of the Mitral, Tricuspid, Aortic, and Pulmonic Valves by Cohort*
Table S2. Comparing 12‐Lead ECG and Systolic Function Data of Patients With DM1 and Patients With DM2*
Table S3. Defined Maximum Tolerated Dose of Medications*
Figure S1. Prevalence of myocardial fibrosis based on cardiac magnetic resonance imaging (MRI).