

Abstract
Background
Recently, we and others have reported a causal role for oxidized lipids in the pathogenesis of pulmonary hypertension (PH). However, the role of low‐density lipoprotein receptor (LDL‐R) in PH is not known.
Methods and Results
We examined the role of LDL‐R in the development of PH and determined the efficacy of high‐density lipoprotein mimetic peptide 4F in mitigating PH. Explanted human lungs and plasma from patients with PH and control subjects were analyzed for gene expression, histological characteristics, and lipoprotein oxidation. Male LDL‐R null (LDL‐R knockout) mice (12–15 months old) were fed chow, Western diet (WD), WD with 4F, and WD with scramble peptide for 12 weeks. Serial echocardiography, cardiac catheterization, oxidized LDL assay, real‐time quantitative reverse transcription–polymerase chain reaction, and histological analysis were performed. The effect of LDL‐R knockdown and oxidized LDL on human pulmonary artery smooth muscle cell proliferation was assessed in vitro. LDL‐R and CD36 expression levels were significantly downregulated in the lungs of patients with PH. Patients with PH also had increased lung lipid deposits, oxidized LDL, E06 immunoreactivity, and plasma oxidized LDL/LDL ratio. LDL‐R knockout mice on WD developed PH, right ventricular hypertrophy, right ventricular dysfunction, pulmonary vascular remodeling, fibrosis, and lipid deposition in lungs, aortic atherosclerosis, and left ventricular dysfunction, which were prevented by 4F. Interestingly, PH in WD group preceded left ventricular dysfunction. Oxidized LDL or LDL‐R knockdown significantly increased proliferation of human pulmonary artery smooth muscle cells in vitro.
Conclusions
Human PH is associated with decreased LDL‐R in lungs and increased oxidized LDL in lungs and plasma. WD‐fed LDL‐R knockout mice develop PH and right ventricular dysfunction, implicating a role for LDL‐R and oxidized lipids in PH.
Keywords: low‐density lipoprotein receptor, oxidized lipids, oxidized low‐density lipoprotein, pulmonary hypertension, Western diet
Subject Categories: Vascular Disease, Pulmonary Hypertension
Clinical Perspective
What Is New?
This research demonstrates an important role of low‐density lipoprotein (LDL) receptor and oxidized LDL in the pathogenesis of pulmonary hypertension (PH).
PH is associated with decreased LDL receptor and CD36 in human lungs, along with increased inflammation and oxidized lipids. Western diet–fed LDL receptor knockout mice develop PH that precedes left ventricular dysfunction.
Targeting oxidized lipids with high‐density lipoprotein mimetic peptides is a potential novel therapeutic strategy for treating PH.
What Are the Clinical Implications?
There is a growing body of evidence implicating oxidized lipids in the pathogenesis of PH; however, the role of LDL receptor has never been investigated in PH.
This research may take us one step further in understanding how oxidized lipids promote PH.
High‐density lipoprotein mimetic peptides may serve as novel therapeutic agents for PH and right ventricular dysfunction.
Pulmonary hypertension (PH) is a pulmonary vascular disease characterized by a pathologically increased mean pulmonary arterial pressure (≥25 mm Hg)1 The cause of PH is multifactorial and includes pulmonary endothelial cell dysfunction, smooth muscle cell proliferation, extracellular matrix remodeling, and inflammation2 Pulmonary vasoconstriction and remodeling contribute to increased pulmonary vascular resistance, leading to right ventricular (RV) hypertrophy and failure.
Recently, we and others have reported a critical role for oxidized lipids in the pathogenesis of PH.2, 3, 4, 5, 6 RV lipid accumulation and lipotoxicity have also been reported in humans and animal models of PH.7, 8, 9
Low‐density lipoproteins (LDLs) and high‐density lipoproteins (HDLs) are the major source of lipid transport and are platforms for lipid oxidation in the circulation. Both LDL and HDL were reported to be dysfunctional in patients with PH.4 Zhang et al recently investigated the role of lectin‐like oxidized LDL receptor (LDL‐R)‐1 (OLR1) in PH and showed that OLR1 promotes pulmonary artery (PA) smooth muscle cell dedifferentiation under hypoxic conditions.10 However, the role of LDL‐R, which binds and internalizes LDL into the cell, has never been investigated in PH.
HDL levels are significantly depressed in patients with PH, which is associated with worse clinical outcomes.11 HDL's major protein, apolipoprotein A‐1, is attributable for the beneficial effects of HDL on atherosclerosis. The apolipoprotein A‐1 mimetic peptide 4F restores vascular endothelial function and has been shown to have anti‐inflammatory properties in lungs. 4F also decreases airway hyperresponsiveness and oxidative stress.12, 13, 14 4F has recently been shown to rescue PH in rodents.3 However, the precise mechanism of how 4F rescues development of PH is unclear. Our recent study demonstrated that this effect is mediated through the induction of downstream effector of 4F, microRNA‐193‐3p.3
In this report, we show that the expression levels of LDL‐R and fatty acid transporter CD36 are significantly reduced in lungs of patients with PH. Moreover, oxidized LDL in both lungs and plasma is significantly elevated in patients with PH. LDL‐R knockout (LDL‐R KO) mouse model has been studied extensively for the development of hyperlipidemia, atherosclerosis, and left ventricular (LV) dysfunction,15 but it has not been investigated for the development of PH and RV dysfunction. We now show, for the first time, that Western diet (WD)–fed LDL‐R KO mice develop PH. Furthermore, we also demonstrate a potential therapeutic role of 4F peptide on WD‐induced PH in LDL‐R KO mice. Mechanistically, we show knockdown of LDL‐R resulted in significantly increased proliferation of human PA smooth muscle cells (hPASMCs) in vitro, and oxidized LDL treatment is sufficient to induce hPASMC proliferation in vitro.
Methods
Full details of all experimental protocols are presented in Data S1. The data that support the findings of this study are available from the corresponding author on reasonable request.
Human Subjects
After institutional review board approval, human explanted lung tissue from patients with PH (PH group; n=7) were obtained. Discarded donor lung tissue or lung tissue from patients without evidence of PH undergoing lung transplants at UCLA Medical Center were used as controls for the study (control group; n=8). Clinical characteristics of human subjects are given in Table S1. For human samples, either the human subjects gave informed consent or this requirement was waived.
Human Lung Microarray Data Analysis for LDL‐R and CD36
Gene expression data for LDL‐R and CD36 from 13 control human lungs and 18 lungs with pulmonary arterial hypertension were retrieved from Gene Expression Omnibus using the GSE15197 data set.16 A heat map was generated using normalized expression values in the pheatmap package in R.
Oxidized LDL Assay
Human plasma was collected from patients with PH (PH; n=9) and subjects without PH (control; n=12). Plasma oxidized LDL was quantified using the ELISA assay for oxidized LDL, according to the manufacturer's instructions.
Real‐Time Polymerase Chain Reaction
Total RNA was purified from human lungs using the Trizol method. Quantitative real‐time reverse transcription–polymerase chain reaction was performed. The LDL‐R and CD36 gene expression was assessed using gene‐specific primers. GAPDH was used as a reference control for normalization.
Animals and Treatments
All animal studies were performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Middle‐aged male LDL‐R KO mice (12–15 months old) were fed either chow (n=16) or WD (n=27) for 12 weeks. Mice on WD were randomly divided into 3 groups: WD (n=9), WD together with apolipoprotein A1 mimetic (HDL mimetic) peptide D‐4F (WF+4F group; n=11), or WD together with scramble peptide (WD+SCRM group; n=7) in drinking water for 12 weeks. A group of chow‐fed mice received 4F (chow+4F group; n=8). Serial echocardiography was performed to monitor cardiopulmonary hemodynamics and the development of PH and RV dysfunction. Direct RV and LV catheterization was performed terminally, and RV hypertrophy index was calculated as weight ratio of RV/(LV+interventricular septum). Aorta, RV, LV, and lung tissue were collected. Short‐term effects of WD were also studied in LDL‐R KO mice (n=14) over 2 weeks. LV and RV function were noninvasively assessed by echocardiography at baseline, week 1, and week 2, followed by invasive RV systolic pressure (RVSP) recording before euthanasia. For comparative analysis of lung oxidized lipids (E06 immunostaining), lung sections from male Sprague Dawley rats from control, monocrotaline‐induced PH (60 mg/kg; single SC injection and followed up for ≈30 days), and Sugen‐hypoxia (20 mg/kg; single SC injection, followed by 3 weeks of 10% O2 and 2 weeks of normoxia) groups were also used.
Echocardiography and Cardiopulmonary Hemodynamic Measurements
Transthoracic echocardiography was performed to monitor cardiopulmonary hemodynamics using a Vevo 2100 high‐resolution image system. Ejection fraction (EF; %) was measured using M‐mode images. A 30‐MHz linear transducer was used for pulmonary pulsed‐wave Doppler echocardiography of PA flow. PA acceleration time (PAAT) was determined by calculating time taken from the start of flow to maximal velocity using echocardiogram software.
The RVSP and LV systolic pressure were measured directly by inserting a catheter into the RV or LV just before euthanasia. Heart and lung tissues were removed rapidly under deep anesthesia for preservation of protein and RNA integrity.
Gross Histologic Analysis, Tissue Preparation, and Imaging
The RV wall, the LV wall, and the interventricular septum were dissected. The ratio of the RV to LV plus septal weight [RV/(LV+interventricular septum)] was calculated as the Fulton index of RV hypertrophy. Lungs were frozen, and transversal 4‐ to 6‐μm sections were obtained with a cryostat. Paraffin‐embedded control lung sections, obtained from the UCLA (University of California Los Angeles) pathology laboratory, were sectioned at 5 μm. Images were acquired using a confocal microscope (Nikon). Lung tissue sections were stained with Masson's trichrome, Oil Red O, immunofluorescence (CD68, oxidized LDL, and OLR‐1), and immunohistochemistry (EO6).
Immunofluorescence and Immunohistochemical Stainings
Briefly, lung sections were fixed and incubated with normal goat serum to block the background and incubated with primary antibodies at 4°C overnight. Sections were then incubated with the secondary antibody and mounted for imaging using Fluoromount‐G with 4′,6‐diamidino‐2‐phenylindole.
For immunohistochemistry, endogenous peroxidase activity was inhibited by incubating the lung sections with H2O2. The sections were then incubated with normal goat serum to block the nonspecific binding. The sections were incubated with the appropriate primary antibodies at 4°C overnight and incubated with horseradish peroxidase–conjugated secondary antibody for 1 hour at room temperature. Sections were stained with 3,3′‐diaminobenzidine as a substrate and mounted using Permount. The images were acquired using a confocal microscope.
Masson's Trichrome and Oil Red O Staining
Masson's trichrome and Oil Red O stainings were performed according to the manufacturer's protocol, and images were acquired with a confocal microscope.
Quantification of Pulmonary Vascular Remodeling, Lung Fibrosis, and Lipid Deposition
For assessment of pulmonary arteriolar wall thickness, only distal pulmonary arteries <100 μm were quantified using ImageJ software. Percentage of fully muscularized, partially muscularized, and nonmuscularized arteries was also calculated. Pulmonary fibrosis was quantified using a grid over the images of lungs. Results are expressed as the percentage occupied by fibrosis to the total area examined. Percentage lipid deposition was quantified in lung sections using ImageJ software.
Assessment of hPASMC Proliferation With LDL‐R Knockdown
hPASMCs at passage 6 to 7 were seeded in a 96‐well plate at a density of 8000 cells/well. After overnight incubation at 37°C, cells were transfected with 100 nmol/L LDL‐R siRNA or scramble siRNA, following the protocol of Lipofectamine RNAiMAX Transfection Reagent. After 4 hours of transfection, cells were starved with 0.1% SMGS (Smooth muscle growth supplement) Media 231 for 40 hours. Cell viability was measured by Cell Counting Kit‐8 (DoJindo Molecular Technologies). LDL‐R mRNA levels were measured by reverse transcription–polymerase chain reaction from parallel experiments using cells seeded in a 6‐well plate.
Assessment of hPASMC Proliferation With Oxidized LDL Incubation
hPASMCs at passage 5 to 6 were seeded in a 96‐well plate at a density of 8000 cells/well. After 24 hours of incubation at 37°C, cells were starved with 0.1% SMGS Media 231 overnight, followed by oxidized LDL (20, 50, and 100 μg/mL) treatment for 24 hours. Cell viability was measured by Cell Counting Kit‐8.
Reagents
Primary antibodies used were anti‐CD68, anti–oxidized LDL, anti‐OLR1, and anti‐E06. Secondary antibodies used were goat anti‐rat IgG antibody Alexa Fluor 594 and goat anti‐rabbit Alexa Fluor 594 antibody.
Statistical Analysis
Unpaired t‐tests and 1‐way ANOVA tests were used to compare between groups using GraphPad Prism for Windows. When significant differences were detected, individual mean values were compared by post hoc tests (Tukey or Dunnett) that allowed for multiple comparisons. P<0.05 was considered statistically significant. Values are expressed as mean±SEM.
Results
Human PH Is Associated With Decreased Lung LDL‐R and CD36 Expression and Increased Plasma Oxidized LDL/LDL Ratio
As expected, patients with PH had significantly elevated RVSP compared with controls (85±7 in PH versus 34±3 mm Hg in controls; P<0.001; Figure 1A, Table S1). Next, we assessed LDL‐R and CD36 expression in lungs of control and PH groups. Real‐time qPCR showed that LDL‐R and CD36 transcript expression levels were significantly decreased in lungs of patients with PH (both P<0.05; Figure 1B and 1C).
Figure 1.
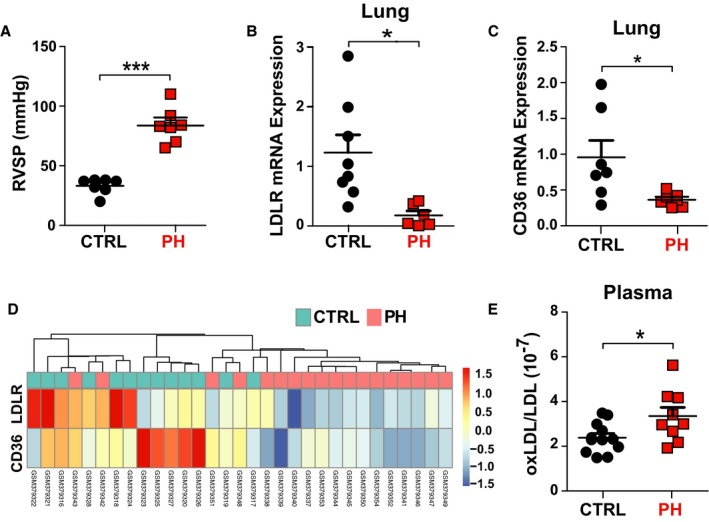
Human pulmonary hypertension (PH) is associated with decreased lung low‐density lipoprotein receptor (LDL‐R) and CD36 expression and increased plasma oxidized LDL/LDL ratio. A, Right ventricular systolic pressure (RVSP; mm Hg; control, n=6; PH, n=6). B, Normalized LDL‐R mRNA expression in human lungs (control, n=8; PH, n=6). C, Normalized CD36 mRNA expression in human lungs (control, n=7; PH, n=7). D, Heat map showing normalized expression of LDL‐R and CD36 in 13 controls and 18 human lungs with PH from the GSE15197 microarray data set. E, Plasma oxidized LDL/LDL ratio in controls and patients with PH (controls, n=12; PH, n=9). *P<0.05, ***P<0.001. CTRL indicates control; ox, oxidized.
Similarly, analysis of the gene expression data for LDL‐R and CD36 from 13 control human lungs and 18 PH lungs, retrieved from Gene Expression Omnibus using the online available GSE15197 data set (Figure 1D), demonstrated significantly decreased LDL‐R (49%; false discovery rate, 0.0022) and CD36 expression (51%; false discovery rate, 0.0023) in PH compared with control lungs.
Next, we measured the plasma levels of circulating LDL and oxidized LDL in controls and patients with PH and calculated the ratio of oxidized LDL/LDL to assess for the oxidized LDL fraction. We found a significant increase in plasma oxidized LDL/LDL ratio in the patients with PH compared with controls (P<0.05; Figure 1E, Table S2).
LDL‐R KO Mice Fed a WD Develop PH and RV Dysfunction
In our human studies, we observed a significant downregulation of LDL‐R expression in lungs of patients with PH, along with an increase in oxidized LDL/LDL ratio. Next, we hypothesized that WD may induce PH in LDL‐R KO mice. We used middle‐aged LDL‐R KO mice (12‐15 months old) to better mimic the clinical scenario. Middle‐aged LDL‐R KO mice fed a WD for 12 weeks developed PH, as demonstrated by a decrease in PAAT (16.1±1.12 ms versus 21.8±1.12 ms; P<0.0001) and an increase in RVSP (40.9±2.04 versus 29.2±0.79 mm Hg; P=0.0003) in the WD and compared with chow (Figure 2A through 2C). PH in LDL‐R KO mice on WD was also associated with an increase in RV hypertrophy index (0.41±0.01 versus 0.23±0.02; P<0.0001) and decrease in RV EF (43.63±1.37% versus 67.7±0.66%; P<0.0001) compared with LDL‐R KO mice on chow diet (Figure 2D and 2E).
Figure 2.
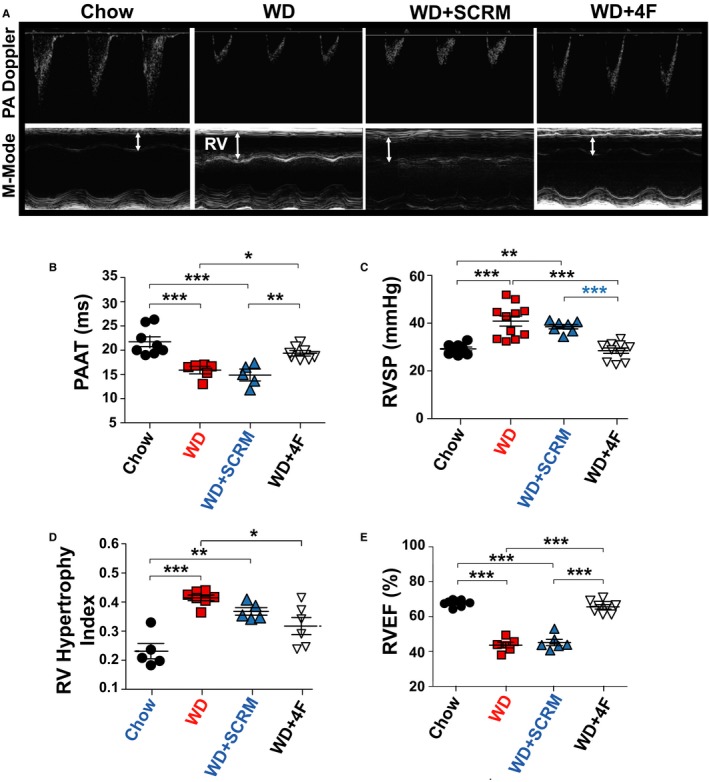
Low‐density lipoprotein receptor knockout (LDL‐R KO) mice fed a Western diet (WD) develop pulmonary hypertension, right ventricular (RV) hypertrophy, and RV dysfunction that is prevented by 4F. A, Pulmonary artery (PA) pulsed‐wave Doppler and M‐mode echocardiographic images of the RV. B, PA acceleration time (PAAT; ms). C, Right ventricular systolic pressure (RVSP; mm Hg). D, Right ventricular hypertrophy Fulton index [RV weight/(left ventricular weight+interventricular septum weight)]. E, Right ventricular ejection fraction (RV EF; %) in LDL‐R KO mice fed with chow (n=5–8), Western diet (WD; n=6–12), WD+scrambled peptide (SCRM) (n=5–7), and WD+4F peptide (n=6–8). *P<0.05, **P<0.01, ***P<0.001.
Our data show, for the first time, that LDL‐R KO mice on WD develop PH, RV hypertrophy, and RV dysfunction.
WD‐Induced PH in LDL‐R KO Mice Is Prevented by 4F
We found 4F treatment prevented development of PH in LDL‐R KO mice fed with WD, whereas scrambled peptide treatment (WD+SCRM group) had no significant effect on the severity of PH (PAAT: 19.4±1.12 ms in WD+4F and 14.9±1.12 ms in WD+SCRM [P=0.0006]; RVSP: 28.5±1.12 in WD+4F and 38.5±0.92 mm Hg in WD+SCRM [P<0.0001]; Figure 2C). Furthermore, 4F therapy resulted in prevention of RV dysfunction (RV EF: WD+SCRM=45.14±1.63%, WD+4F=65.5±1.05% [P<0.0001]) (Figure 2D and 2E). PH in WD‐fed LDL‐R KO mice was also associated with increase in body weight in WD and WD+SCRM groups that was prevented by 4F treatment (Figure S1B).
PH Precedes the Development of LV Dysfunction in WD‐Fed LDL‐R KO Mice
We also examined the effect of WD on LV function of middle‐aged LDL‐R KO mice. We found that WD induced LV dysfunction in LDL‐R KO mice because LV EF was significantly lower compared with mice on chow diet (46.2±2.09% versus 64.6±1.47%; P=0.0001; Figure 3A through 3C). LV dysfunction was prevented by 4F therapy (LV EF: WD+SCRM=46.1±1.86%, WD+4F=63.8±1.72% [P<0.0001]; Figure 3A and 3B). LV pressures were significantly increased in WD and WD+SCRM groups (LV systolic pressure, mm Hg: chow=72.9±2.95, WD=99.3±5.44, WD+SCRM=98.9±3.02, WD+4F=82.7±4.35 [P=0.007 for chow versus WD; P=0.014 for WD+SCRM versus WD+4F) (Figure 3C).
Figure 3.
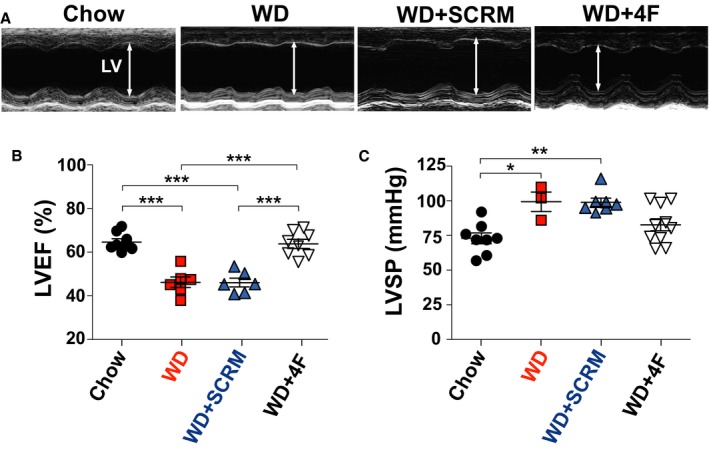
Low‐density lipoprotein receptor knockout (LDL‐R KO) mice fed a Western diet (WD) develop left ventricular (LV) dysfunction that is prevented by 4F. A, M‐mode echocardiographic images of the LV. B, LV ejection fraction (EF; %). C, LV systolic pressure (LVSP; mm Hg) in LDL‐R KO mice fed with chow (n=8), WD (n=3–6), WD+scrambled peptide (SCRM) (n=6–7), and WD+4F peptide (n=8). *P<0.05, **P<0.01, ***P<0.001.
To investigate whether PH in our model is secondary to LV dysfunction, we performed a time course experiment for noninvasive assessment of development of PH through measurement of PAAT and LV function through LV EF over 12 weeks via serial transthoracic echocardiography (Figure 4A and 4B). Interestingly, we observed that the development of PH preceded the development of LV dysfunction in this model. In fact, we observed a significant decrease in PAAT as early as 1 week after initiation of WD, whereas we observed a more gradual decline in LV EF over 12 weeks (Figure 4A and 4B). To further confirm the presence of RV pressure overload as early as 1 and 2 weeks after initiation of WD, we performed invasive right heart catheterization at weeks 1 and 2 after initiation of WD in LDL‐R KO mice. Our results confirmed that RVSP was significantly increased at 1 and 2 weeks after WD compared with baseline (both P<0.001 versus baseline; P<0.001 for 1 versus 2 weeks), whereas LV EF was unchanged (Figure 4C through 4E). Hence, we believe that PH precedes LV dysfunction in this model. This observation supports a direct effect of oxidized lipids in inducing PH in this model, which is in agreement with our previously published work.3
Figure 4.
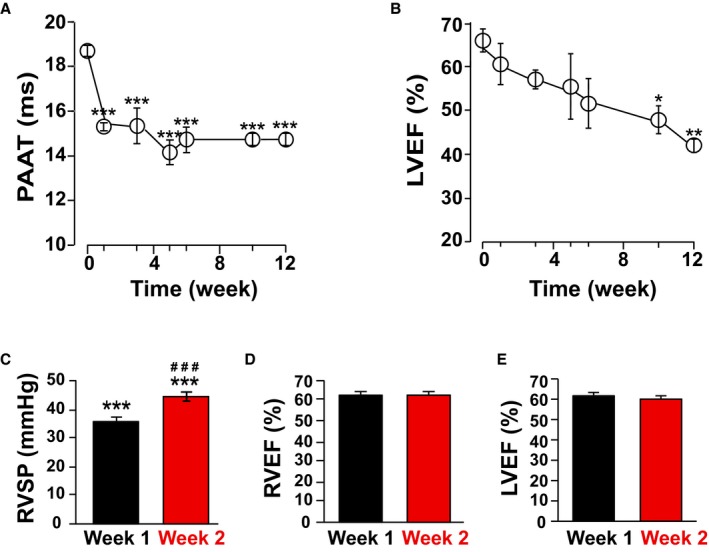
Pulmonary hypertension (PH) precedes the development of left ventricular (LV) dysfunction in Western diet (WD)–fed low‐density lipoprotein receptor knockout (LDL‐R KO) mice. Time course experiment for pulmonary artery acceleration time (PAAT; ms; A) and LV ejection fraction (EF; %; B) measurement over 12‐weeks via serial transthoracic echocardiography in WD‐fed LDL‐R KO mice (n=3) showing that the development of PH precedes the development of LV dysfunction. C–E, Right ventricular systolic pressure (RVSP), RV EF, and LV EF via direct RV catheterization in WD‐fed LDL‐R KO mice (n=14) at weeks 1 and 2. *P<0.05 vs baseline, **P<0.01 vs baseline, ***P<0.001 vs baseline; ### P<0.001 vs week 1.
Pulmonary Vascular Remodeling, Fibrosis, and Lipid Deposition in LDL‐R KO Mice Fed WD Are Prevented by 4F
Pulmonary vascular remodeling is a hallmark of PH. PH in WD‐fed LDL‐R KO mice was associated with pulmonary vascular remodeling, as demonstrated by increased pulmonary arteriolar medial hypertrophy in WD group that was prevented by 4F therapy (% wall thickness: chow=23.3±1.20, WD=31.9±1.21, WD+SCRM=29.6±2.82, WD+4F=24.1±0.79; Figure 5A and 5D). The increase in % wall thickness in WD and WD+SCRM groups was associated with an increase in the percentage of partially and fully muscular arteries (Figure 5E).
Figure 5.
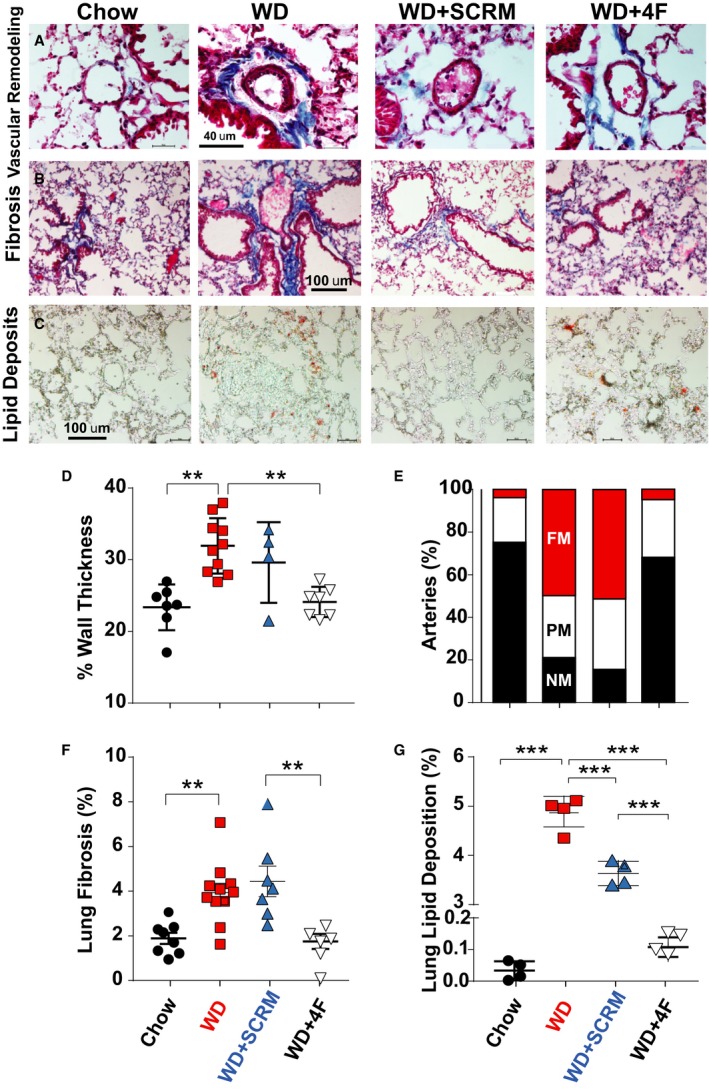
Low‐density lipoprotein receptor knockout mice fed a Western diet (WD) develop pulmonary vascular remodeling, fibrosis, and lipid deposition that are prevented by 4F. Masson's trichrome staining of lung sections showing pulmonary vascular remodeling (A) and pulmonary fibrosis (B), and Oil Red O staining showing lung lipid deposition (C), in chow, WD, WD+scrambled peptide (SCRM), and WD+4F groups. Quantification of percentage wall thickness (chow=7, WD=10, WD+SCRM=4, WD+4F=7; D), percentage of nonmuscular (NM; black), partially muscular (PM; white), and fully muscular (FM; red) arteries (E), percentage fibrosis (chow=7, WD=11, WD+SCRM=7, WD+4F=6; F), and percentage lipid deposition (chow=4, WD=4, WD+SCRM=4, WD+4F=4; G) is also shown. **P<0.01, ***P<0.001.
PH in LDL‐R KO mice on WD was also associated with the development of both interstitial and perivascular pulmonary fibrosis that was prevented by 4F therapy (% fibrosis: chow=23.3±1.20, WD=31.9±1.21, WD+SCRM=29.6±2.82, WD+4F=24.1±0.79; Figure 5B and 5F). We also observed increased lung lipid deposition in WD and WD+SCRM groups that was prevented by 4F treatment (Figure 5C, G). The lung weight was also significantly increased in the WD‐fed LDL‐R KO mice, most likely as a result of vascular remodeling, fibrosis, and lipid deposition (Figure S1C).
LDL‐R KO Mice on WD Demonstrate Increased Lung Inflammation and Oxidized Lipids
Oxidized lipids are known to induce inflammation. We assessed lung inflammation in our mouse model using CD68 as a marker for macrophages. We observed increased CD68 positive alveolar macrophage infiltration in WD‐fed LDL‐R KO mice (chow=118±10 versus WD=278±63 cells/mm2; Figure 6A through 6C). Treatment with 4F prevented the increase in CD68‐positive cells in the lungs, whereas scramble peptide had no effect (WD+4F=143±16; WD+SCRM=248±44 cells/mm2 Figure 6A through 6C).
Figure 6.
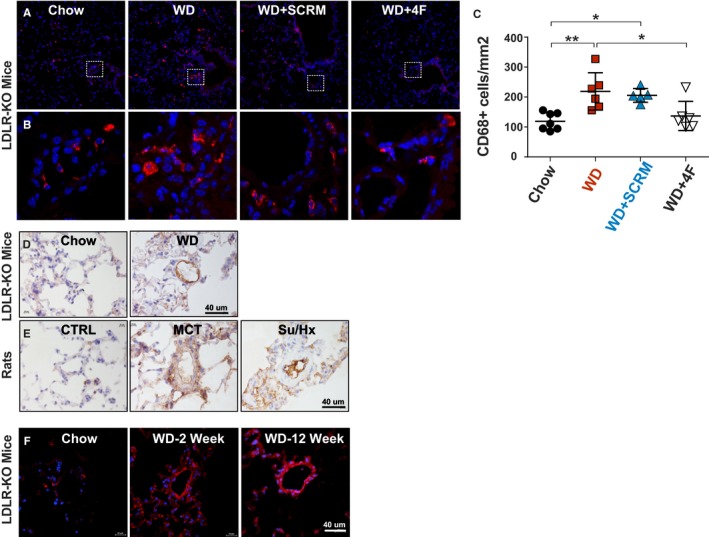
Low‐density lipoprotein receptor knockout (LDL‐R KO) mice on Western diet (WD) demonstrate increased lung inflammation and oxidized lipids. A and B, Immunofluorescence staining for CD68+ macrophages (red) in lung sections from chow, WD, WD+scrambled peptide (SCRM), and WD+4F groups. Nuclei are stained with 4′,6‐diamidino‐2‐phenylindole (DAPI) in blue. C, Quantification of CD68+ macrophages (cells/mm2) is also shown in chow (n=7), WD (n=6), WD+SCRM (n=5), and WD+4F (n=6) groups. D and E, Immunohistochemical analysis of E06 (brown) in chow (n=4) vs WD (n=4) fed LDL‐R KO mouse lungs (D) and rat lungs from control (n=3), monocrotaline‐induced pulmonary hypertension (PH) (n=4), and Sugen‐hypoxia (Su/Hx)–induced PH (n=4) models (E). F, Immunofluorescence staining for lectin‐like oxidized LDL‐R‐1 (red) in lung sections from chow (n=3), WD at 2 weeks (n=5), and WD at 12 weeks (n=4) groups. Nuclei are stained with DAPI in blue. *P<0.05, **P<0.01.
We assessed oxidized phospholipids in the lungs of LDL‐R KO mice on chow versus WD and in 2 well‐established experimental models of PH in rats (monocrotaline‐induced PH and Sugen‐hypoxia–induced PH) compared with controls using EO6 antibody. We found a significant increase in EO6 immunolabeling in the vasculature, perivascular regions, and the macrophages in the PH lungs (Figure 6D and 6E). We also assessed OLR1 in lungs of LDL‐R KO mice and observed an increase in OLR1 staining in pulmonary vascular cells and macrophages in lungs of LDL‐R KO mice on WD for 2 and 12 weeks (Figure 6F).
WD Induces Lipid Deposition in Aorta and Heart of LDL‐R KO Mice
LDL‐R KO mice are known to develop atherosclerosis when challenged with a WD. As expected, we observed increased lipid deposition in the aorta in WD and WD+SCRM groups, which was prevented by 4F therapy (Figure S2). Similarly, we also observed increased lipid deposition in RV and LV in WD and WD+SCRM groups that was prevented by 4F treatment (Figure S2). RV lipid deposition and lipotoxicity have been reported in patients and mice with PH.7, 8 We also stained the heart sections of the mice with trichrome staining but did not observe an increase in either RV or LV fibrosis (Figure S2).
Human PH Is Associated With Increased Lipid Deposition, Inflammation, EO6, and Oxidized LDL Immunolabeling in Lungs
As we observed increased lipid deposition and inflammation in the lungs of LDL‐R KO mice on WD diet, we also assessed human lungs from control and PH groups for lipid deposition and inflammation. We observed lipid deposition in both the lung parenchyma as well as the pulmonary vasculature (Figure 7A). We also assessed oxidized phospholipids in the lungs using EO6 antibody and found a significant increase in immunolabeling in the macrophages and perivascular regions in the PH lungs (Figure 7B). CD68 immunofluorescence staining demonstrated increased CD68‐positive macrophages in the lungs of patients with PH (Figure 7C). As we had observed an increase plasma oxidized LDL/LDL ratio in patients with PH, we assessed the lungs for oxidized LDL immunolabeling and found an overall increase in oxidized LDL in perivascular regions and macrophages in PH lungs (Figure 7D).
Figure 7.
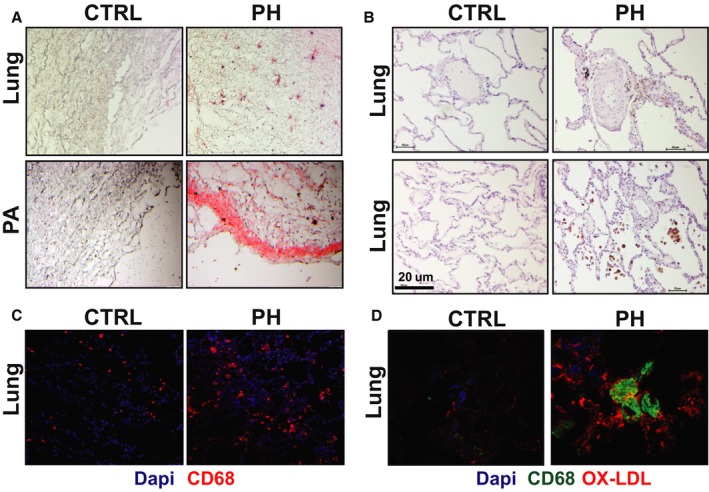
Human pulmonary hypertension (PH) is associated with increased lipid deposition, inflammation, EO6, and oxidized low‐density lipoprotein (LDL) immunolabeling in lungs. A, Oil Red O staining (red) showing lipid deposition in lungs and pulmonary arteries (PAs) of control subjects and patients with PH. B, Immunoperoxidase staining for E06 in lungs of control subjects and patients with PH. C, Immunofluorescence staining for CD68+ macrophages (red) in lung sections from control subjects and patients with PH. Nuclei are stained with 4′,6‐diamidino‐2‐phenylindole (DAPI) in blue. D, Immunofluorescence staining showing coimmunolabeling of CD68 (green) and oxidized LDL (red) in lung sections from control subjects and patients with PH.
Knockdown of LDL‐R or Oxidized LDL Treatment Results in hPASMC Proliferation in Vitro
To assess the mechanistic role of LDL‐R in PA smooth muscle cell proliferation, we performed in vitro cell experiments using hPASMCs. Similar to the human lung tissue findings and the LDL‐R KO mouse model, we performed LDL‐R knockdown in hPASMCs using siRNA technique, which was confirmed by PCR (Figure 8A). Interestingly, LDL‐R knockdown resulted in significantly increased proliferation of hPASMCs in vitro, which is a hallmark of the pulmonary vascular disease of PH (Figure 8B).
Figure 8.
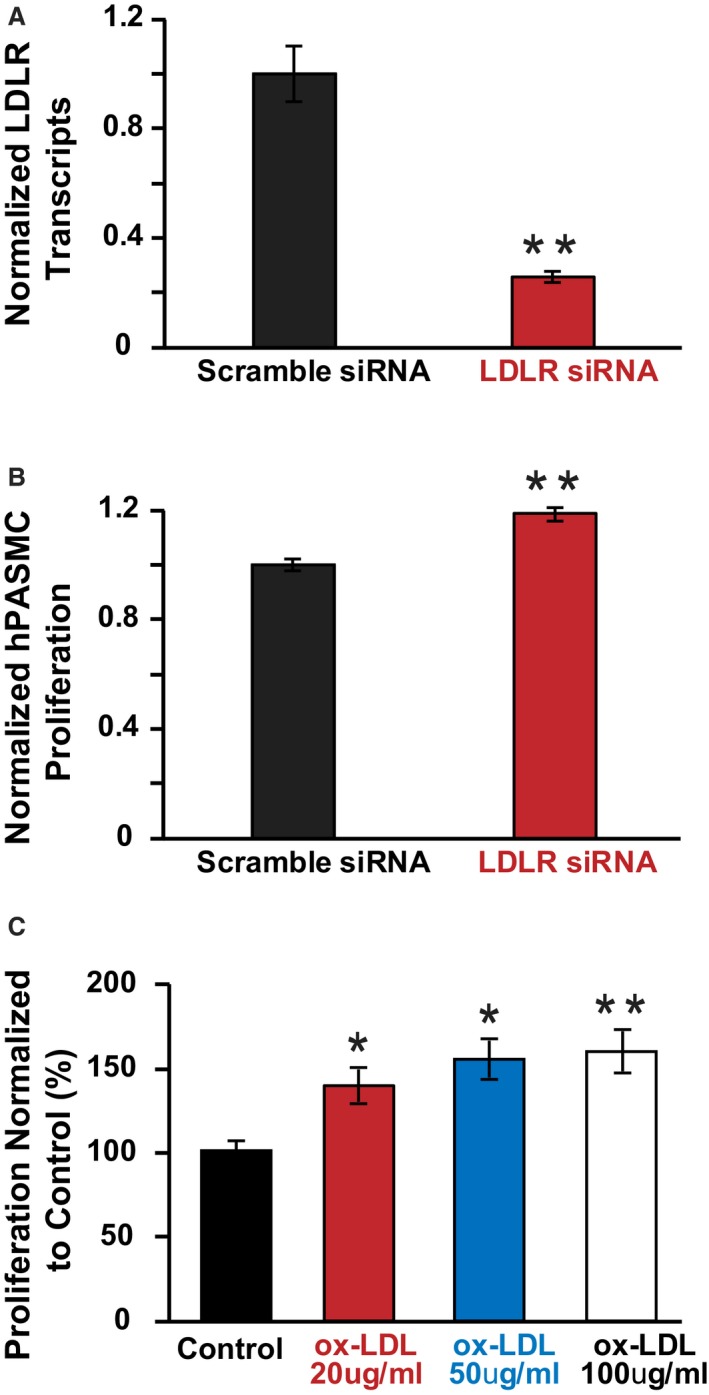
Knockdown of low‐density lipoprotein receptor (LDL‐R) or oxidized LDL treatment results in human pulmonary artery smooth muscle cell (hPASMC) proliferation in vitro. A, Normalized LDL‐R transcript expression in scramble siRNA treated vs LDL‐R siRNA treated hPASMCs (n=average of 6 experiments per group). B, Normalized hPASMC proliferation in scramble siRNA treated vs LDL‐R siRNA treated hPASMCs (n=average of 6 experiments per group). **P<0.01 vs scramble siRNA. C, Normalized hPASMC proliferation in control vs oxidized LDL treated hPASMCs (n=average of 3 experiments per group). *P<0.05 vs control, **P<0.01 vs control.
Next, we investigated the potential of oxidized LDL to induce hPASMC proliferation in vitro. We found that oxidized LDL alone is sufficient to induce hPASMC proliferation in vitro in a dose‐dependent manner (Figure 8C).
Discussion
Herein, we demonstrate, for the first time, that patients with PH have decreased LDL‐R and CD36 expression in their lungs, along with increased plasma oxidized LDL/LDL ratio. There is also evidence of increased inflammation and lipid deposition in the lungs of patients with PH. Furthermore, LDL‐R KO mice fed WD for 12 weeks developed PH that preceded the development of LV dysfunction, both of which were prevented by apolipoprotein A‐1 mimetic (HDL mimetic) peptide 4F therapy. Incubation of hPASMCs with oxidized LDL and LDL‐R knockdown resulted in significantly increased proliferation in vitro.
PH is a multifactorial disease with poor prognosis. The pathogenesis of PH encompasses a complex interplay of molecular and cellular events, leading to pulmonary vascular remodeling, inflammation, and oxidative stress.2 Over the past decade, we and others have demonstrated the critical implication of lipids, particularly oxidized lipids, in the pathogenesis of PH.2, 3, 4, 5, 17, 18 There is a growing body of evidence demonstrating that increased levels of oxidized lipids are found in the plasma and lungs of patients and rodents with PH.3, 4, 5, 17, 18
PH is linked to metabolic syndrome and its associated conditions.19 The high‐fat, high‐cholesterol containing WD has long been implicated in the development of metabolic syndrome, which encompasses a cluster of conditions, including obesity, insulin resistance, hyperlipidemia, and systemic hypertension. All these conditions are risk factors for atherosclerosis.20 Furthermore, WD‐fed LDL‐R KO mice develop obesity and insulin resistance,21, 22 conditions associated with both metabolic syndrome and PH.23, 24 The absence of LDL‐Rs, which regulate the LDL levels in the bloodstream, results in the development of atherosclerosis in LDL‐R KO mice.22 When a critical concentration is reached, LDL phospholipids are oxidized. LDL cholesterol is a well‐known marker of cardiovascular risk, and its levels are decreased in patients with pulmonary arterial hypertension and associated with an increased risk of death.25 We also found a trend toward lower circulating LDL cholesterol levels in patients with PH (Table S1), but an increased circulating oxidized LDL/LDL ratio (Figure 1E).
We found a significant decrease in LDL‐R and fatty acid transporter CD36 expression in the lungs of patients with PH (Figure 1B through 1D). In addition, we found an increase in oxidized LDL in the plasma and lungs of patients with (Figures 1E and 7D). Oxidized phospholipid marker EO6 immunoreactivity was also increased in lungs with PH, specifically in the macrophages (Figure 7B). We found a similar increase in lung EO6 immunolabeling in mice with WD‐induced PH and in 2 established experimental models of PH in rats induced with monocrotaline and Sugen‐hypoxia. We also observed an increase in lung OLR1 immunolabeling in pulmonary vascular cells and macrophages in WD‐fed LDL‐R KO mice, which is consistent with the recent report on OLR1 immunolabeling in human lungs with pulmonary arterial hypertension.26 The decrease in LDL metabolizing machinery in the lungs may have led to decreased reverse cholesterol transport, resulting in elevated oxidized LDL levels. These oxidized phospholipids are known to mediate the development of fatty streaks in atherosclerosis.27 Furthermore, oxidized LDL has been shown to increase vascular smooth muscle cell proliferation and migration in the context of atherosclerosis.28 It is plausible that oxidized LDL may have similar effects on pulmonary vascular smooth muscle cells promoting PH phenotype. In fact, in our cell culture experiments, we observed that incubation of hPASMCs with oxidized LDL resulted in increased proliferation, a hallmark of pulmonary vascular disease in PH. In a separate set of experiments, we demonstrated that LDL‐R knockdown in hPASMCs resulted in significantly increased proliferation in vitro, implicating LDL‐R in pulmonary vascular remodeling. The colocalization of EO6 and oxidized LDL with CD68+ macrophages suggests possibility of a link between inflammation and oxidized lipids (Figure 7). Oxidized LDL induces significant inflammatory response in vascular cells, resulting in the production of monocyte chemoattractant protein‐1. Oxidized LDL also binds monocyte chemoattractant protein‐1 and retains its ability to recruit monocytes.29 In addition, OLR1, known to be an endothelial receptor of oxidized LDL, has recently been linked to the development of PH via increased oxidative stress and phenotypic switching of PA smooth muscle cells.10, 30
Previously, LDL‐R KO mice on WD for prolonged period (18 weeks) have been shown to develop atherosclerosis and LV dysfunction,15 but it has never been tested whether they develop PH and RV dysfunction. In the current study, we used middle‐aged LDL‐R KO mice that are susceptible to oxidative stress. Interestingly, in addition to the development of LV dysfunction, we found these mice also developed PH that was associated with pulmonary vascular remodeling and pulmonary fibrosis. The contribution of LV dysfunction and atherosclerosis to the development of PH and RV dysfunction cannot be ruled out. The presence of pulmonary vascular remodeling and pulmonary fibrosis point toward a possible direct effect of WD‐induced oxidative stress on the lung. To examine whether PH is secondary to LV dysfunction, we serially monitored the mice with echocardiography and demonstrated that WD‐induced decrease in PAAT preceded the decrease in LV EF, highlighting the susceptibility of the pulmonary circulation to the effects of WD. This decrease in PAAT was sustained over the 12‐week period. LV EF, on the other hand, had a more gradual decline over the 12‐week period, only reaching significance at 10 weeks. The abrupt decrease in PAAT in 1 week was confirmed by invasive right heart catheterization, in the absence of any change in LV EF, further confirming that PH precedes the development of LV dysfunction (Figure 4).
Plasma HDL levels are significantly depressed in patients with PH and are associated with increased mortality and clinical deterioration.11 Apolipoprotein A‐I (the major component of HDL) and its mimetic peptides (4F) prevent the formation or inactivate inflammatory LDL‐derived oxidized phospholipids.31 Navab et al demonstrated that 4F inhibited atherosclerosis by reducing inflammation.32 Furthermore, 4F restores vasodilation, reduces inflammation, and prevents heart hypertrophy in LDL‐R KO mice on WD.12, 15, 33 In a previous study, we demonstrated that 4F effectively rescued rats and wild‐type mice with established PH. We discovered that microRNA‐193‐3p was significantly downregulated in lungs of rodents with PH and that 4F restores its expression levels.3 Interestingly, in the current study, we found that the use of 4F peptide prevented the development of PH and RV dysfunction in WD‐fed LDL‐R KO mice (Figure 2).
Hansmann et al conducted experiments with apolipoprotein E–deficient mice, another mouse strain susceptible to the effects of oxidized lipids, and demonstrated that they developed PH when they were fed high‐fat diet.34 However, this had not been tested in LDL‐R KO mice, which share similar associated conditions, such as metabolic syndrome, obesity, and insulin resistance. In the same study, Hansmann et al also demonstrated that PH could be reversed by peroxisome proliferator‐activated receptor‐γ activation.34 The peroxisome proliferator‐activated receptor‐γ inhibits the expression of proinflammatory genes and regulates fat cell development, inhibiting the development of atherosclerosis in LDL‐R KO mice.35
There is accumulating evidence that RV dysfunction in PH may be secondary to maladaptive fatty acid metabolism and lipid accumulation, resulting in RV lipotoxicity.7, 8 Talati et al also found lipid deposition in the BMPR2 (Bone Morphogenetic Protein Receptor) mutant RVs, which was exacerbated in the presence of WD.7 We also observe RV lipid deposition along with RV hypertrophy and dysfunction in our model of PH (Figure 2, Figure S2).
One of the limitations of our study is the use of 4F peptide as a preventive rather than a rescue therapy. We have previously shown that 4F peptide can rescue PH in rodent models.3 On the basis of our previous results, we anticipate that 4F is a potent rescue agent for oxidized lipid–induced PH.
Conclusions
In conclusion, human PH is associated with downregulation of LDL‐R and CD36 in the lungs, along with increased oxidized lipids and inflammatory markers (Figure 9). WD‐fed LDL‐R KO mice develop PH and biventricular dysfunction. Development of PH preceded LV dysfunction, suggesting a susceptibility of the pulmonary circulation to the effects of the WD. HDL mimetic peptide 4F prevented the development of PH and RV dysfunction. Future studies are warranted to understand the precise role of LDL‐Rs and oxidized lipids in PH pathogenesis.
Figure 9.
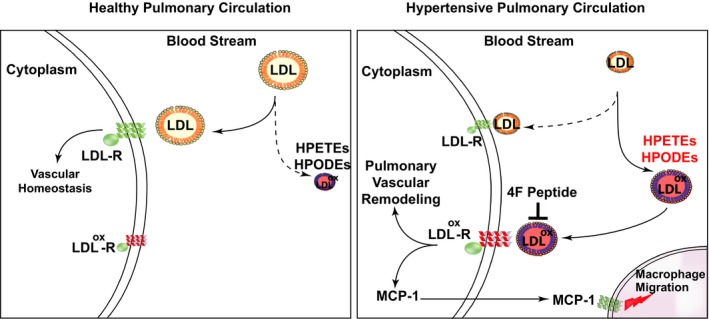
Hypothetical scheme for the role of low‐density lipoprotein (LDL), LDL receptor (LDL‐R), and oxidized LDL in pulmonary hypertension (PH). Schematic showing the involvement of LDL and its receptors in PH. Healthy pulmonary circulation (left) is compared with pulmonary circulation in PH (right). HPETE indicates hydroperoxyeicosatetraenoic acid; HPODE, hydroxyoctadecadienoic acid; MCP‐1, monocyte chemoattractant protein‐1.
Perspectives
Despite considerable advances, the survival of patients with PH remains poor. Significant evidence exists implicating oxidized lipids, metabolic syndrome, and insulin resistance in PH. However, the role of LDL‐R and oxidized LDL has not been investigated in PH. The present study demonstrates that PH is associated with decreased LDL‐R and CD36 in human lungs, along with increased inflammation and oxidized lipids. WD‐fed LDL‐R KO mice develop PH that precedes LV dysfunction. Targeting oxidized lipids with HDL mimetic peptides is a potential novel therapeutic strategy for treating PH.
Sources of Funding
This work was supported by National Institutes of Health (NIH) K08 Grant 1K08, HL141995 01A1 (Dr Umar), and NIH R01 Grant 5R01 HL129051 03 (Drs Reddy and Eghbali).
Disclosures
Dr Reddy is a principal in BruinPharma. The remaining authors have no disclosures to report.
Supporting information
Data S1. Supplemental Methods.
Table S1. Comparison Between Clinical Characteristics of Controls (CTRL) and Patients With PH
Table S2. Statistics
Figure S1. A, Experimental protocol. LDL‐R KO mice were fed either regular chow diet, Western diet (WD) alone or WD in the presence of scrambled peptide, or HDL mimetic 4F peptide for 12 weeks. One group of LDL‐R KO mice was fed with chow diet in the presence of 4F peptide. B, Body weight (g) and (C) Lung weight (g) in LDL‐receptor knockout mice fed with chow (n=8), Western diet (WD, n=12), WD+Scrambled peptide (n=7) and WD+4F peptide (n=8). **P<0.01; ***p<0.001
Figure S2. Oil Red O staining showing lipid deposition (red) in aorta (A), right ventricle (RV, B), and left ventricle (LV, C), and Masson's trichrome staining showing heart fibrosis (blue) in RV (D) and LV (E) in Chow, WD, WD+Scramble and WD+4F groups
(J Am Heart Assoc. 2020;9:e012063 DOI: 10.1161/JAHA.119.012063.)
References
- 1. Hoeper MM, Bogaard HJ, Condliffe R, Frantz R, Khanna D, Kurzyna M, Langleben D, Manes A, Satoh T, Torres F, Wilkins MR, Badesch DB. Definitions and diagnosis of pulmonary hypertension. J Am Coll Cardiol. 2013;62:D42–D50. [DOI] [PubMed] [Google Scholar]
- 2. Sharma S, Ruffenach G, Umar S, Motayagheni N, Reddy ST, Eghbali M. Role of oxidized lipids in pulmonary arterial hypertension. Pulm Circ. 2016;6:261–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sharma S, Umar S, Potus F, Iorga A, Wong G, Meriwether D, Breuils‐Bonnet S, Mai D, Navab K, Ross D, Navab M, Provencher S, Fogelman AM, Bonnet S, Reddy ST, Eghbali M. Apolipoprotein A‐I mimetic peptide 4F rescues pulmonary hypertension by inducing microRNA‐193‐3p. Circulation. 2014;130:776–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ross DJ, Hough G, Hama S, Aboulhosn J, Belperio JA, Saggar R, Van Lenten BJ, Ardehali A, Eghbali M, Reddy S, Fogelman AM, Navab M. Proinflammatory high‐density lipoprotein results from oxidized lipid mediators in the pathogenesis of both idiopathic and associated types of pulmonary arterial hypertension. Pulm Circ. 2015;5:640–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Al‐Husseini A, Wijesinghe DS, Farkas L, Kraskauskas D, Drake JI, Tassel BV, Abbate A, Chalfant CE, Voelkel NF. Increased eicosanoid levels in the sugen/chronic hypoxia model of severe pulmonary hypertension. PLoS One. 2015;10:e0120157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Li Q, Mao M, Qiu Y, Liu G, Sheng T, Yu X, Wang S, Zhu D. Key role of ROS in the process of 15‐lipoxygenase/15‐hydroxyeicosatetraenoiccid‐induced pulmonary vascular remodeling in hypoxia pulmonary hypertension. PLoS One. 2016;11:e0149164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Talati MH, Brittain EL, Fessel JP, Penner N, Atkinson J, Funke M, Grueter C, Jerome WG, Freeman M, Newman JH, West J, Hemnes AR. Mechanisms of lipid accumulation in the bone morphogenetic protein receptor type 2 mutant right ventricle. Am J Respir Crit Care Med. 2016;194:719–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Brittain EL, Talati M, Fessel JP, Zhu H, Penner N, Calcutt MW, West JD, Funke M, Lewis GD, Gerszten RE, Hamid R, Pugh ME, Austin ED, Newman JH, Hemnes AR. Fatty acid metabolic defects and right ventricular lipotoxicity in human pulmonary arterial hypertension. Circulation. 2016;133:1936–1944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hemnes AR, Brittain EL, Trammell AW, Fessel JP, Austin ED, Penner N, Maynard KB, Gleaves L, Talati M, Absi T, Disalvo T, West J. Evidence for right ventricular lipotoxicity in heritable pulmonary arterial hypertension. Am J Respir Crit Care Med. 2014;189:325–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zhang W, Zhu T, Wu W, Ge X, Xiong X, Zhang Z, Hu C. LOX‐1 mediated phenotypic switching of pulmonary arterial smooth muscle cells contributes to hypoxic pulmonary hypertension. Eur J Pharmacol. 2018;818:84–95. [DOI] [PubMed] [Google Scholar]
- 11. Heresi GA, Aytekin M, Newman J, DiDonato J, Dweik RA. Plasma levels of high‐density lipoprotein cholesterol and outcomes in pulmonary arterial hypertension. Am J Respir Crit Care Med. 2010;182:661–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Van Lenten BJ, Navab M, Anantharamaiah GM, Buga GM, Reddy ST, Fogelman AM. Multiple indications for anti‐inflammatory apolipoprotein mimetic peptides. Curr Opin Investig Drugs Lond Engl. 2008;9:1157–1162. [PMC free article] [PubMed] [Google Scholar]
- 13. He D, Zhao M, Wu C, Zhang W, Niu C, Yu B, Jin J, Ji L, Willard B, Mathew AV, Chen YE, Pennathur S, Yin H, He Y, Pan B, Zheng L. Apolipoprotein A‐1 mimetic peptide 4F promotes endothelial repairing and compromises reendothelialization impaired by oxidized HDL through SR‐B1. Redox Biol. 2017;15:228–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Nandedkar SD, Weihrauch D, Xu H, Shi Y, Feroah T, Hutchins W, Rickaby DA, Duzgunes N, Hillery CA, Konduri KS, Pritchard KA. D‐4F, an apoA‐1 mimetic, decreases airway hyperresponsiveness, inflammation, and oxidative stress in a murine model of asthma. J Lipid Res. 2011;52:499–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Han J, Zhang S, Ye P, Liu Y‐X, Qin Y‐W, Miao D‐M. Apolipoprotein A‐I mimetic peptide D‐4F reduces cardiac hypertrophy and improves apolipoprotein A‐I‐mediated reverse cholesterol transport from cardiac tissue in LDL receptor‐null mice fed a western diet. J Cardiovasc Pharmacol. 2016;67:412–417. [DOI] [PubMed] [Google Scholar]
- 16. Rajkumar R, Konishi K, Richards TJ, Ishizawar DC, Wiechert AC, Kaminski N, Ahmad F. Genomewide RNA expression profiling in lung identifies distinct signatures in idiopathic pulmonary arterial hypertension and secondary pulmonary hypertension. Am J Physiol Heart Circ Physiol. 2010;298:H1235–H1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Al‐Naamani N, Sagliani KD, Dolnikowski GG, Warburton RR, Toksoz D, Kayyali U, Hill NS, Fanburg BL, Roberts KE, Preston IR. Plasma 12‐ and 15‐hydroxyeicosanoids are predictors of survival in pulmonary arterial hypertension. Pulm Circ. 2016;6:224–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bowers R, Cool C, Murphy RC, Tuder RM, Hopken MW, Flores SC, Voelkel NF. Oxidative stress in severe pulmonary hypertension. Am J Respir Crit Care Med. 2004;169:764–769. [DOI] [PubMed] [Google Scholar]
- 19. Ussavarungsi K, Thomas CS, Burger CD. Prevalence of metabolic syndrome in patients with pulmonary hypertension. Clin Respir J. 2017;11:721–726. [DOI] [PubMed] [Google Scholar]
- 20. Dandona P, Aljada A, Chaudhuri A, Mohanty P, Garg R. Metabolic syndrome: a comprehensive perspective based on interactions between obesity, diabetes, and inflammation. Circulation. 2005;111:1448–1454. [DOI] [PubMed] [Google Scholar]
- 21. Merat S, Casanada F, Sutphin M, Palinski W, Reaven PD. Western‐type diets induce insulin resistance and hyperinsulinemia in LDL receptor‐deficient mice but do not increase aortic atherosclerosis compared with normoinsulinemic mice in which similar plasma cholesterol levels are achieved by a fructose‐rich diet. Arterioscler Thromb Vasc Biol. 1999;19:1223–1230. [DOI] [PubMed] [Google Scholar]
- 22. Ma Y, Wang W, Zhang J, Lu Y, Wu W, Yan H, Wang Y. Hyperlipidemia and atherosclerotic lesion development in ldlr‐deficient mice on a long‐term high‐fat diet. PLoS One. 2012;7:e35835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Friedman SE, Andrus BW. Obesity and pulmonary hypertension: a review of pathophysiologic mechanisms. J Obes. 2012;2012:505274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zamanian RT, Hansmann G, Snook S, Lilienfeld D, Rappaport KM, Reaven GM, Rabinovitch M, Doyle RL. Insulin resistance in pulmonary arterial hypertension. Eur Respir J Off J Eur Soc Clin Respir Physiol. 2009;33:318–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kopeć G, Waligóra M, Tyrka A, Jonas K, Pencina MJ, Zdrojewski T, Moertl D, Stokwiszewski J, Zagożdżon P, Podolec P. Low‐density lipoprotein cholesterol and survival in pulmonary arterial hypertension. Sci Rep. 2017;7:41650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hemnes AR, Luther JM, Rhodes CJ, Burgess JP, Carlson J, Fan R, Fessel JP, Fortune N, Gerszten RE, Halliday SJ, Hekmat R, Howard L, Newman JH, Niswender KD, Pugh ME, Robbins IM, Sheng Q, Shibao CA, Shyr Y, Sumner S, Talati M, Wharton J, Wilkins MR, Ye F, Yu C, West J, Brittain EL. Human PAH is characterized by a pattern of lipid‐related insulin resistance. JCI Insight. 2019;4.;pii:123611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Navab M, Berliner JA, Subbanagounder G, Hama S, Lusis AJ, Castellani LW, Reddy S, Shih D, Shi W, Watson AD, Van Lenten BJ, Vora D, Fogelman AM. HDL and the inflammatory response induced by LDL‐derived oxidized phospholipids. Arterioscler Thromb Vasc Biol. 2001;21:481–488. [DOI] [PubMed] [Google Scholar]
- 28. Liu J, Ren Y, Kang L, Zhang L. Oxidized low‐density lipoprotein increases the proliferation and migration of human coronary artery smooth muscle cells through the upregulation of osteopontin. Int J Mol Med. 2014;33:1341–1347. [DOI] [PubMed] [Google Scholar]
- 29. Wiesner P, Tafelmeier M, Chittka D, Choi S‐H, Zhang L, Byun YS, Almazan F, Yang X, Iqbal N, Chowdhury P, Maisel A, Witztum JL, Handel TM, Tsimikas S, Miller YI. MCP‐1 binds to oxidized LDL and is carried by lipoprotein(a) in human plasma. J Lipid Res. 2013;54:1877–1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ogura S, Shimosawa T, Mu S, Sonobe T, Kawakami‐Mori F, Wang H, Uetake Y, Yoshida K, Yatomi Y, Shirai M, Fujita T. Oxidative stress augments pulmonary hypertension in chronically hypoxic mice overexpressing the oxidized LDL receptor. Am J Physiol Heart Circ Physiol. 2013;305:H155–H162. [DOI] [PubMed] [Google Scholar]
- 31. Navab M, Ananthramaiah GM, Reddy ST, Van Lenten BJ, Ansell BJ, Fonarow GC, Vahabzadeh K, Hama S, Hough G, Kamranpour N, Berliner JA, Lusis AJ, Fogelman AM. The oxidation hypothesis of atherogenesis: the role of oxidized phospholipids and HDL. J Lipid Res. 2004;45:993–1007. [DOI] [PubMed] [Google Scholar]
- 32. Navab M, Reddy ST, Van Lenten BJ, Buga GM, Hough G, Wagner AC, Fogelman AM. High‐density lipoprotein and 4F peptide reduce systemic inflammation by modulating intestinal oxidized lipid metabolism. Arterioscler Thromb Vasc Biol. 2012;32:2553–2560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ou J, Wang J, Xu H, Ou Z, Sorci‐Thomas MG, Jones DW, Signorino P, Densmore JC, Kaul S, Oldham KT, Pritchard KA. Effects of D‐4F on vasodilation and vessel wall thickness in hypercholesterolemic LDL receptor‐null and LDL receptor/apolipoprotein A‐I double‐knockout mice on Western diet. Circ Res. 2005;97:1190–1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hansmann G, Wagner RA, Schellong S, Perez VA, de J, Urashima T, Wang L, Sheikh AY, Suen RS, Stewart DJ, Rabinovitch M. Pulmonary arterial hypertension is linked to insulin resistance and reversed by peroxisome proliferator‐activated receptor‐gamma activation. Circulation. 2007;115:1275–1284. [DOI] [PubMed] [Google Scholar]
- 35. Li AC, Brown KK, Silvestre MJ, Willson TM, Palinski W, Glass CK. Peroxisome proliferator–activated receptor γ ligands inhibit development of atherosclerosis in LDL receptor–deficient mice. J Clin Invest. 2000;106:523–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Supplemental Methods.
Table S1. Comparison Between Clinical Characteristics of Controls (CTRL) and Patients With PH
Table S2. Statistics
Figure S1. A, Experimental protocol. LDL‐R KO mice were fed either regular chow diet, Western diet (WD) alone or WD in the presence of scrambled peptide, or HDL mimetic 4F peptide for 12 weeks. One group of LDL‐R KO mice was fed with chow diet in the presence of 4F peptide. B, Body weight (g) and (C) Lung weight (g) in LDL‐receptor knockout mice fed with chow (n=8), Western diet (WD, n=12), WD+Scrambled peptide (n=7) and WD+4F peptide (n=8). **P<0.01; ***p<0.001
Figure S2. Oil Red O staining showing lipid deposition (red) in aorta (A), right ventricle (RV, B), and left ventricle (LV, C), and Masson's trichrome staining showing heart fibrosis (blue) in RV (D) and LV (E) in Chow, WD, WD+Scramble and WD+4F groups