

Abstract
Background
Polygenic risk scores (PRSs) based on risk variants from genome‐wide association studies predict coronary artery disease (CAD) risk. However, it is unknown whether the PRS is associated with specific CAD characteristics.
Methods and Results
We consecutively included 1645 patients with suspected stable CAD undergoing coronary computed tomography angiography. A multilocus PRS was calculated as the weighted sum of CAD risk variants. Plaques were evaluated using an 18‐segment model and characterized by stenosis severity and composition (soft [0%‐19% calcified], mixed‐soft [20%‐49% calcified], mixed‐calcified [50%‐79% calcified], or calcified [≥80% calcified]). Coronary artery calcium score and segment stenosis score were used to characterize plaque burden. For each standard deviation increase in the PRS, coronary artery calcium score increased by 78% (P=4.1e‐26) and segment stenosis score increased by 16% (P=2.4e‐29) in the fully adjusted model. The PRS was associated with a higher prevalence of obstructive plaques (odds ratio [OR ]: 1.78, P=5.6e‐16), calcified (OR: 1.69, P=6.5e‐17), mixed‐calcified (OR: 1.67, P=7.3e‐9), mixed‐soft (OR: 1.45, P=1.6e‐6), and soft plaques (OR: 1.49, P=2.5e‐6), and a higher prevalence of plaque in each coronary vessel (all P<1.0e‐4). However, when analyzing data on a plaque level (3007 segments with plaque in 849 patients) the PRS was not associated with stenosis severity, plaque composition, or localization (all P>0.05).
Conclusions
Our results suggest that polygenic risk based on large genome‐wide association studies increases CAD risk through an increased burden of coronary atherosclerosis rather than promoting specific plaque features.
Clinical Trial Registration
URL: https://www.clinicaltrials.gov. Unique identifier: NCT02264717.
Keywords: atherosclerosis, coronary artery disease, coronary computed tomography angiography, plaque
Subject Categories: Genetics, Coronary Artery Disease, Atherosclerosis, Functional Genomics, Computerized Tomography (CT)
Clinical Perspective
What Is New?
A polygenic risk score, as a measure of genetically determined risk of coronary artery disease, strongly associates with an increased coronary plaque burden.
The association is driven by an increased burden of all types of coronary plaques and a higher prevalence of plaque in all coronary vessels.
The polygenic risk score does not associate with any specific plaque characteristics over others.
What Are the Clinical Implications?
Our findings provide a better understanding of the inheritance of coronary artery disease by suggesting that genetically determined risk increases coronary artery disease risk through the development of atherosclerosis in general rather than promoting specific plaque features.
Introduction
Coronary artery disease (CAD) is characterized by a substantial and complex polygenic contribution to disease risk.1 Studies consistently show that ≈50% of the phenotypic variance in CAD may be explained by genetics (ie, heritability).2, 3 Since 2007, genome‐wide association studies (GWASs) have identified a large number of common genetic variants associated with CAD.4 Independently, the effect of each risk variant is modest. However, assessing the polygenic risk by summing the effect of all risk variants into a polygenic risk score (PRS) predicts incident and recurrent CAD events on top of traditional risk factors.5, 6, 7 and may identify individuals who obtain increased benefit from adherence to a healthy lifestyle or preventive medical treatment.8, 9 Since the PRS is calculated based on common inherited variants, it may allow translation into future clinical practice for estimation of CAD risk and tailoring medical treatment at any time in life and even from birth before any traditional risk factors develop.
Coronary computed tomography angiography (CTA) is a well‐established method to visualize coronary atherosclerosis and the preferred diagnostic modality for patients at suspected low‐to‐intermediate risk of stable CAD.10 Using coronary CTA, coronary plaque burden and composition may directly be quantified, including high‐risk plaque features responsible for myocardial ischemia or subsequent plaque rupture.11, 12 We have previously demonstrated that genetically predisposed individuals carry an elevated coronary plaque burden as measured by coronary CTA, including an increased burden of high‐risk plaque features.13 In prior studies, PRSs based on CAD‐GWASs have been associated with increased coronary artery calcium scores (CACS).8, 14 However, the impact of PRSs on coronary plaque burden or detailed coronary plaque characteristics beyond CACS remains unknown.
Therefore, we aimed to investigate whether a PRS based on CAD‐GWASs is associated with coronary plaque burden and specific plaque characteristics, in patients with suspected CAD referred for coronary CTA.
Methods
Study Design and Patients
The study comprised consecutive patients without known CAD referred for coronary CTA because of symptoms suggestive of stable CAD, who had been included in the Dan‐NICAD trial (Danish Multicenter Study of Non‐Invasive Testing in Coronary Artery Disease). The data that support the findings of the study are available from the corresponding author upon reasonable request. The study protocol including enrollment criteria has previously been described in detail.15 Briefly, the Dan‐NICAD trial was designed to assess the diagnostic accuracy of second‐line tests in a population with suspected obstructive CAD based on coronary CTA. In total, 1675 consecutive patients underwent blood sampling and a clinical interview to assess symptoms and CAD risk factors before coronary CTA.15, 16 A family history of CAD was defined as self‐reported CAD <60 years among first‐degree relatives.
The Committee on Health Research Ethics in the Region of Central Denmark and the Danish Data Protection Agency approved the study. All patients provided written informed consent.
Genotyping, Quality Control, Imputing, and PRS Calculation
Out of 1675 enrolled patients, 1665 patients provided a 4‐mL EDTA whole blood sample for DNA isolation. After DNA extraction, high‐quality DNA was available for 1653 patients for genotyping. Genotyping was performed using the Illumina Global Screening Array (Illumina, Inc., San Diego, CA) at deCODE Genetics. Imputing was carried out using a reference panel consisting of 1KG phase 3 and HapMap to predict nongenotyped single‐nucleotide polymorphisms with minor allele frequency >1%. Principal components were calculated using FlashPCA.17 After quality control, 1645 patients remained.
Calculation of the PRS was performed using LDpred18 in the pipeline at deCODE Genetics as previously described.19 Briefly, the calculation was based on P values and log10 odds ratios from the CARDIoGRAMplusC4D meta‐analysis of CAD.20 The fraction of causal markers was set to 0.001, because this has previously been shown to generate the best discrimination of CAD patients from healthy controls.21
CACS, Coronary CTA, and Image Analysis
Coronary CTA was performed on a 320‐slice‐volume computed tomography scanner (Aquillion One; Toshiba Medical Systems, Japan) as previously described.15, 16 Scans were performed with and without iodine‐containing contrast according to clinical guidelines, and plaque analyses were performed on a Vitrea Advanced Workstation (Vital Images, USA) by an experienced cardiologist blinded to the PRS. CACS was calculated using the Agatston method,22 and coronary CTA analysis was performed on segments with a diameter ≥2 mm using an 18‐segment model.23 Proximal CAD was defined as plaque in the left main artery or any of the proximal segments of the left anterior descending, circumflexus, or right coronary artery (segment 1, 5, 6, or 11). All segments with plaque were visually categorized according to plaque composition as soft (0%‐19% calcified), mixed‐soft (20%‐49% calcified), mixed‐calcified (50%‐79% calcified), or calcified (≥80% calcified). Similarly, segments with plaque were classified based on the visual luminal diameter reduction and categorized as no stenosis (0% diameter reduction; ≈0% area reduction), mild stenosis (1%‐29% diameter reduction; ≈1%‐49% area reduction), moderate stenosis (30‐49% diameter reduction; ≈50‐69% area reduction); and severe stenosis (50%‐100% diameter reduction; ≈70%‐100% area reduction). Obstructive CAD was defined as the presence of any severe stenosis. The segment stenosis score (SSS) was calculated by grading each coronary segment based on plaque stenosis severity (ie, grades 0–3) and summing the grades from all segments.24 CACS and SSS were used as overall measures of coronary plaque burden.
Statistical Analysis
Data are presented as number (proportion) or median (interquartile range [IQR]). Differences in patient characteristics between the PRS groups were compared using the χ2 test or Kruskal‐Wallis test as appropriate. Patients were categorized as having a low‐ (<20th percentile), average‐ (20th–80th percentile), or high PRS (>80th percentile) for reporting of plaque prevalence. Although the PRS is a continuous risk variable, these cutoffs have previously been adopted to define PRS risk categories8; thus, plaque prevalence was also reported for each of these groups for comparability.
Multivariable regression models were used to assess the relationship between the PRS and plaque measures. Since the PRS approximated a normal distribution, the PRS was standardized (ie, mean=0, SD=1) in order to obtain a more meaningful interpretation of the effect size. In model 1 the association of the PRS was adjusted for age, sex, the first 4 principal components of ancestry, as well as an interaction term between age and sex, since these variables had a statistically significant interaction in the analyses on CACS and SSS. Model 2 comprised the same covariables as model 1 but also included adjustment for traditional risk factors including antihypertensive treatment, lipid‐lowering treatment, body mass index, angina symptoms, and active smoking. Linear regression models were used to analyze the relationship between the PRS and CACS and SSS, respectively. Diabetes mellitus and cholesterol levels were not added as covariables because they may act as intermediate steps between genetic predisposition to CAD and atherosclerosis development rather than being confounders.7 Because of the skewed distribution, linear regression was performed with both variables log‐transformed as loge (value + 1). Linear effect of the PRS was confirmed by a spline representation (Figure S1). Beta‐coefficients were back‐transformed for reporting. Hence, the reported effect size corresponds to the percentage change in the geometric mean, after adjustment, per SD increase in the PRS. The models were validated by inspecting QQ‐plots of residuals and by plotting residuals against fitted values and explanatory variables. For dichotomous plaque measures, logistic regression models were used and effect sizes reported as odds ratios.
To evaluate whether the PRS had a larger effect on any specific plaque characteristics over others, we analyzed the data on a plaque‐level (coronary segment‐level) comparing the relationship between the PRS and plaque characteristics among 3007 plaques (from 849 patients with plaque). Logistic and ordinal logistic regression models were used to analyze dichotomous and categorical plaque characteristics taking within‐individual correlation into account using the Huber‐White clustered sandwich variance estimator. The models were adjusted for the same covariables as in the patient‐level analyses. However, since we wanted to evaluate the effect on a plaque level independent of overall plaque burden, a spline representation of the SSS was also added in both models. Effect sizes were reported as odds ratios.
Statistical analyses were performed using Stata version 13.1 (StataCorp, 4905 Lakeway Dr, College Station, TX). Violin plots were generated using R version 3.2.2 (http://www.r-project.org/).
Results
We included 1645 patients in the study. Patient characteristics are displayed in Table 1. Median age was 57 (IQR 50‐64) years and 799 (49%) were males. Patients in higher PRS categories were more likely to have a self‐reported family history of CAD and more frequently received lipid‐lowering treatment (Table 1). Anginal symptoms, age, and triglycerides also differed slightly among PRS categories. No significant difference in body mass index, cholesterol levels, diabetes mellitus status, and smoking was found between the groups.
Table 1.
Patient Characteristics (n=1645)
| Total (n=1645) | Low PRS (n=329) | Average PRS (n=987) | High PRS (n=329) | P Value | |
|---|---|---|---|---|---|
| Age, y | 57 (50–64) | 58 (51–64) | 57 (51–64) | 56 (49–63) | 0.022 |
| Male sex | 799 (49%) | 170 (52%) | 518 (52%) | 158 (48%) | 0.37 |
| Family history of CAD <60 y in first‐degree relative | 601 (37%) | 93 (28%) | 358 (36%) | 150 (46%) | <0.001 |
| Antihypertensive treatment | 411 (25%) | 83 (25%) | 249 (25%) | 79 (24%) | 0.90 |
| Lipid‐lowering treatment | 384 (24%) | 59 (18%) | 236 (24%) | 89 (28%) | 0.015 |
| Diabetes mellitus | 94 (6%) | 15 (5%) | 59 (6%) | 20 (6%) | 0.60 |
| Active smoking | 266 (16%) | 57 (17%) | 156 (16%) | 53 (16%) | 0.82 |
| Symptoms | |||||
| Typical chest pain | 89 (27%) | 72 (22%) | 291 (30%) | 89 (27%) | 0.005 |
| Atypical chest pain | 550 (34%) | 115 (35%) | 306 (31%) | 129 (39%) | |
| Nonanginal chest pain | 293 (18%) | 63 (19%) | 189 (19%) | 41 (13%) | |
| Dyspnea | 340 (21%) | 76 (23%) | 195 (20%) | 69 (21%) | |
| BMI, kg/m2 | 26.3 (23.8–29.4) | 26.9 (24.3–29.6) | 26.2 (23.7–29.3) | 26.5 (23.7–29.5) | 0.21 |
| Total cholesterol, mmol/L | 5.3 (4.7‐6.0) | 5.2 (4.6–5.9) | 5.3 (4.7–6.1) | 5.3 (4.6–6.2) | 0.17 |
| HDL cholesterol, mmol/L | 1.4 (1.1–1.7) | 1.4 (1.2–1.7) | 1.4 (1.1–1.7) | 1.4 (1.1–1.7) | 0.14 |
| LDL cholesterol, mmol/L | 3.3 (2.6–3.9) | 3.1 (2.6–3.8) | 3.3 (2.7–3.9) | 3.4 (2.6–4.0) | 0.055 |
| Triglycerides, mmol/L | 1.3 (1.0–2.0) | 1.3 (0.9–1.9) | 1.3 (1.0–2.0) | 1.4 (1.0–2.2) | 0.049 |
| Creatinine, μmol/L | 74 (65–85) | 75 (65–85) | 74 (65–85) | 75 (66–85) | 0.67 |
Data are presented as number (percentage) or median (interquartile range). Family history was missing in 6 patients, lipid‐lowering treatment was missing in 28 patients, diabetes mellitus was missing in 6 patients, smoking was missing in 7 patients, symptoms was missing in 10 patients, BMI was missing in 9 patients, total cholesterol was missing in 100 patients, HDL cholesterol was missing in 97 patients, LDL cholesterol was missing in 105 patients, triglycerides was missing in 109 patients, and creatinine was missing in 14 patients. BMI indicates body mass index; CAD, coronary artery disease; HDL, high‐density lipoprotein; LDL, low‐density lipoprotein; PRS, polygenic risk score.
Patient‐Level Analyses
The PRS was strongly associated with overall plaque burden as measured by CACS and SSS (Figure 1 and Table 2). Across the PRS deciles, CACS and SSS showed a steady upward increase in distribution (Figure 2). Going from low to average and high genetic risk patients the median CACS increased from 0 (IQR 0‐21) to 0 (IQR 0‐85) and 18 (IQR 0‐178), and SSS increased from 0 (IQR 0‐1) to 1 (IQR 0‐4) and 2 (IQR 0‐7). This association was seen for both males and females and for different age spans (Figure 3). Correspondingly, in the fully adjusted model the geometric mean CACS increased by +78% (95% CI +60% to +98%, P=4.1e‐26) and SSS increased by +16% (95% CI +11% to +21%, P=2.4e‐29) per SD increase in the PRS. The upward shift in PRS distribution with increasing coronary plaque burden can be seen in Figure S2. A higher PRS was associated with a higher prevalence of calcified, mixed‐calcified, mixed‐soft, and soft plaques and an increased prevalence of plaque in proximal‐ and non‐proximal segments as well as in all coronary vessels (Table 3). Moreover, the PRS was associated with the presence of obstructive plaques 1.78 (95% CI 1.55–2.05, P=5.6e‐16) per SD increase in the PRS) with the mean PRS being 0.40 SD higher among patients with obstructive CAD compared with patients without obstructive CAD (P=0.022).
Figure 1.
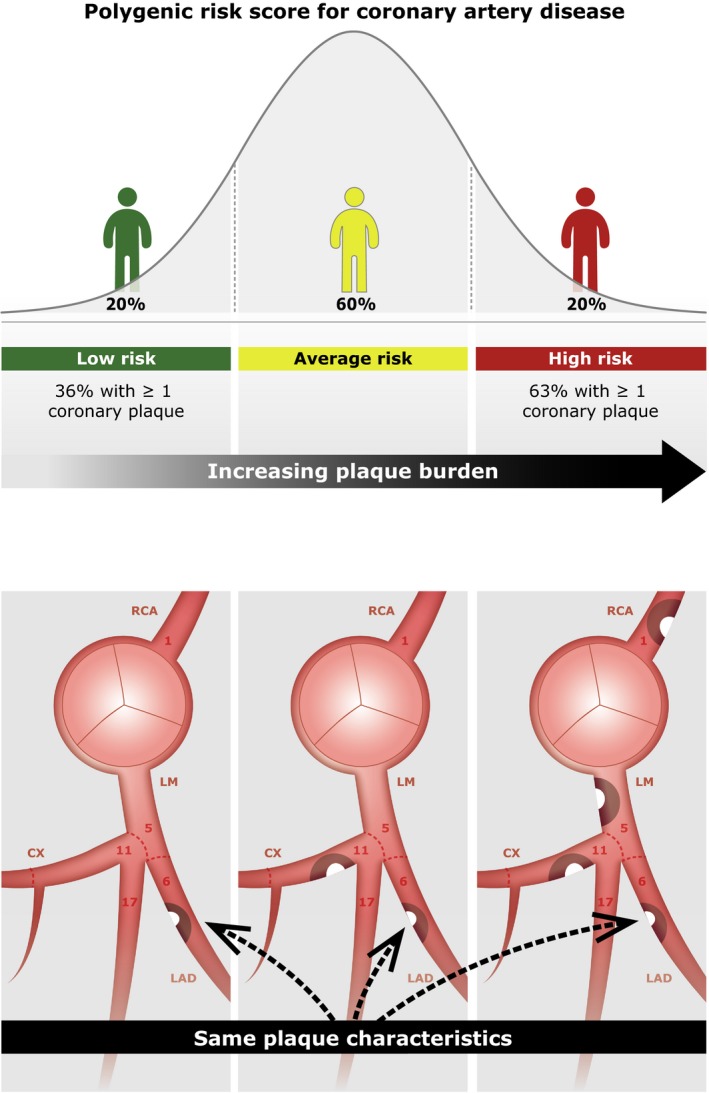
Visual overview. CX indicates circumflex ramus; LAD, left anterior descending; LM, left main; RCA, right coronary artery.
Table 2.
Association of the PRS With Plaque Burden (n=1645 patients)
| Coronary Plaque Burden Median (25–75th Percentile) [1–99th Percentile] | Model 1 Effect Size (95% CI, P Value) | Model 2 Effect Size (95% CI, P Value) | |||
|---|---|---|---|---|---|
| Low PRS (n=329) | Average PRS (n=987) | High PRS (n=329) | |||
| CACS | 0 (0–21) [0–841] | 0 (0–85) [0–2565] | 18 (0–178) [0–2122] | +81% (+63% to +101%, P=2.0e‐27) | +78% (+60% to +98%, P=4.1e‐26) |
| Segment stenosis score | 0 (0 to 1) [0 to 19] | 1 (0 to 4) [0 to 25] | 2 (0–7) [0–26] | +29% (+23% to +34%, P=1.5e‐30) | +16% (+11% to +21%, P=2.4e‐29) |
The effect size corresponds to the change in % in the geometric mean of the variable per SD increase in the PRS. Model 1 was adjusted for age, sex, the first 4 principal components of ancestry, as well as an interaction term between age and sex. Model 2 was further adjusted for antihypertensive treatment, lipid‐lowering treatment, body mass index, symptoms, and active smoking. CACS indicates coronary artery calcium score; PRS, polygenic risk score.
Figure 2.
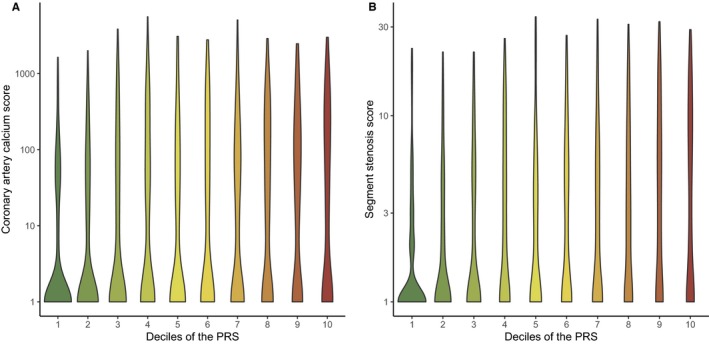
Violin plots displaying the distribution of coronary artery calcium score (A) and segment stenosis score (B) across the deciles of the PRS (n=1645 patients). A log scale was used for the y‐axis for display purposes. All values were transformed as loge (value + 1). PRS indicates polygenic risk score.
Figure 3.
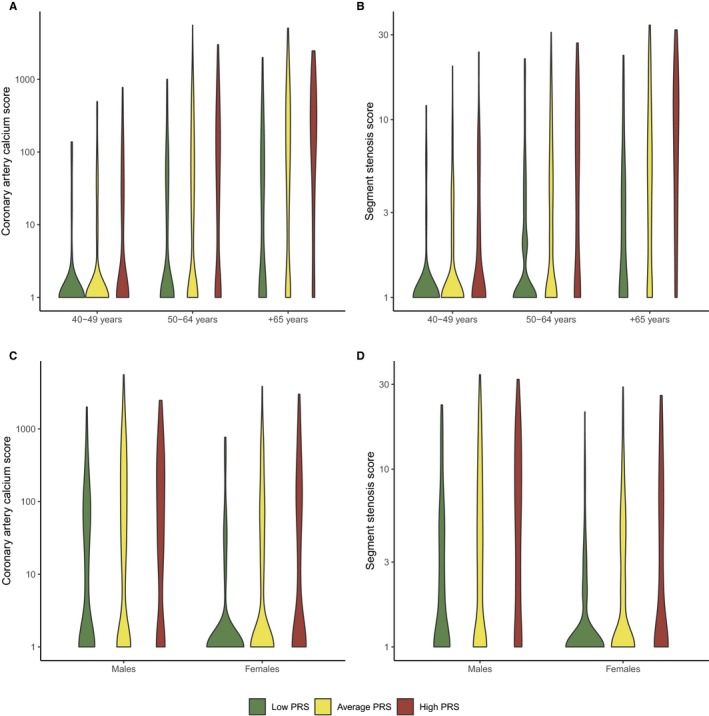
Violin plots displaying the distribution of coronary artery calcium score (A and C) and segment stenosis score (B and D) according to the PRS stratified by age and sex (n=1645 patients). Patients were categorized as low‐ (<20th percentile), average‐ (20th to 80th percentile), or high PRS (>80th percentile). A log scale was used for the y‐axis for display purposes. All values were transformed as loge (value + 1). PRS indicates polygenic risk score.
Table 3.
Association of the PRS With Presence of Plaque (n=1645 Patients)
| Number of Patients (Proportion) | Model 1 OR (95% CI, P Value) | Model 2 OR (95% CI, P Value) | |||
|---|---|---|---|---|---|
| Low PRS (n=329) | Average PRS (n=987) | High PRS (n=329) | |||
| CAD severity | |||||
| Any plaque | 119 (36%) | 522 (53%) | 208 (63%) | 1.76 (1.56–1.98, P=1.3e‐20) | 1.79 (1.58–2.03, P=6.3e‐20) |
| Any obstructive plaque | 40 (12%) | 229 (23%) | 116 (36%) | 1.75 (1.53–2.00, P=3.4e‐16) | 1.78 (1.55–2.05, P=5.6e‐16) |
| Plaque composition | |||||
| Any soft plaque | 37 (11%) | 172 (17%) | 76 (23%) | 1.39 (1.21–1.60, P=2.2e‐6) | 1.40 (1.22–1.62, P=2.5e‐6) |
| Any mixed‐soft plaque | 32 (10%) | 154 (16%) | 68 (21%) | 1.45 (1.25–1.68, P=9.3e‐7) | 1.45 (1.24–1.68, P=1.6e‐6) |
| Any mixed‐calcified plaque | 18 (5%) | 117 (12%) | 57 (17%) | 1.68 (1.42–1.99, P=1.7e‐9) | 1.67 (1.40–1.98, P=7.3e‐9) |
| Any calcified plaque | 91 (28%) | 414 (42%) | 174 (53%) | 1.67 (1.48–1.88, P=1.9e‐17) | 1.69 (1.49–1.91, P=6.5e‐17) |
| Plaque localization | |||||
| Any LM plaque | 24 (7%) | 115 (12%) | 57 (17%) | 1.48 (1.25–1.75, P=4.6e‐6) | 1.46 (1.23–1.73, P=1.5e‐5) |
| Any LAD plaque | 98 (30%) | 476 (48%) | 186 (57%) | 1.75 (1.56–1.97, P=1.6e‐20) | 1.79 (1.58–2.03, P=3.9e‐20) |
| Any CX plaque | 34 (10%) | 202 (20%) | 100 (30%) | 1.74 (1.51–2.00, P=1.4e‐14) | 1.75 (1.51–2.03, P=5.1e‐14) |
| Any RCA plaque | 55 (17%) | 271 (27%) | 133 (40%) | 1.72 (1.51–1.95, P=1.0e‐16) | 1.72 (1.51–1.97, P=7.3e‐16) |
| Any proximal plaque | 103 (31%) | 455 (46%) | 182 (55%) | 1.66 (1.47–1.86, P=5.0e‐17) | 1.67 (1.48–1.89, P=1.9e‐16) |
| Any nonproximal plaque | 79 (24%) | 398 (40%) | 177 (54%) | 1.82 (1.62–2.06, P=1.3e‐22) | 1.85 (1.63–2.10, P=6.4e‐22) |
ORs are reported per SD increase in the PRS. Model 1 was adjusted for age, sex, the first 4 principal components of ancestry, as well as an interaction term between age and sex. Model 2 was further adjusted for antihypertensive treatment, lipid‐lowering treatment, body mass index, symptoms, and active smoking. CAD indicates coronary artery disease; CX, circumflex ramus; LAD, left anterior descending; LM, left main artery; OR, odds ratio; PRS, polygenic risk score; RCA, right coronary artery.
In a sensitivity analysis, we tested whether the association of the PRS with plaque burden differed in patients with and without a family history of CAD. The PRS displayed a positive association with plaque burden both in patients with and without a family history of CAD (Figure S3). Accordingly, adding adjustment for family history of CAD did not alter any of the observed associations between the PRS and plaque burden (Table S1).
Segment‐Level Analyses
Subsequently, we extracted all segments with plaque and analyzed these separately in order to evaluate whether the PRS had a larger effect on any specific plaque characteristics over others. For this analysis we had a total 3007 segments with plaque from 849 patients. Among segments with plaque, the PRS was not associated with plaque stenosis, composition, or location in any specific coronary vessel in any of the adjusted models with SSS included (Table 4). In line with this, a visual representation of the PRS distribution for segments grouped according to plaques characteristics or plaque location showed almost identical curves in all comparisons (Figure S4).
Table 4.
Segment‐Level Analyses of the Association Between the PRS and Plaque Characteristics (n=3007 Segments With Plaque Among 849 Patients)
| Number of Segments With Plaque (proportion) | Model 1 OR (95% CI, P Value) | Model 2 OR (95% CI, P Value) | |||
|---|---|---|---|---|---|
| Low PRS (n=330) | Average PRS (n=1818) | High PRS (n=859) | |||
| Severity | |||||
| No stenosis | 18 (5%) | 67 (4%) | 34 (4%) | ||
| Mild stenosis | 180 (55%) | 948 (52%) | 369 (43%) | ||
| Moderate stenosis | 52 (16%) | 250 (14%) | 140 (16%) | ||
| Severe stenosis | 80 (24%) | 553 (30%) | 316 (37%) | ||
| More severe stenosis categorya | 0.98 (0.90–1.07, P=0.72) | 1.00 (0.92–1.09, P=0.94) | |||
| Plaque composition | |||||
| Calcified | 205 (62%) | 1166 (64%) | 531 (62%) | ||
| Mixed‐calcified | 26 (8%) | 172 (9%) | 79 (9%) | ||
| Mixed‐soft | 47 (14%) | 224 (12%) | 121 (14%) | ||
| Soft | 52 (16%) | 256 (14%) | 128 (15%) | ||
| More noncalcified composition categorya | 0.99 (0.88–1.12, P=0.90) | 1.01 (0.90–1.14, P=0.82) | |||
| Plaque location | |||||
| LM | 24 (7%) | 115 (6%) | 57 (7%) | 0.95 (0.83–1.09, P=0.50) | 0.95 (0.82–1.08, P=0.43) |
| LAD | 164 (50%) | 873 (48%) | 399 (46%) | 1.05 (0.98–1.13, P=0.16) | 1.05 (0.98–1.13, P=0.16) |
| CX | 51 (15%) | 325 (18%) | 167 (19%) | 1.00 (0.91–1.09, P=0.99) | 1.01 (0.92–1.10, P=0.86) |
| RCA | 91 (28%) | 505 (28%) | 236 27%) | 0.96 (0.88–1.03, P=0.26) | 0.95 (0.88–1.03, P=0.22) |
| Proximal | 157 (48%) | 835 (46%) | 375 (44%) | 1.01 (0.95–1.07, P=0.86) | 1.01 (0.95–1.07, P=0.85) |
Each segment with plaque contributes with an observation (ie, the PRS from 1 individual with >1 plaque contributes with the PRS for each plaque). ORs are reported per SD increase in the PRS. Model 1 was adjusted for age, sex, a spline representation of the segment stenosis score, the first 4 principal components of ancestry, as well as an interaction term between age and sex. Model 2 was further adjusted for antihypertensive treatment, lipid‐lowering treatment, body mass index, symptoms, and active smoking. All analyses were performed accounting for within‐individual correlation using the clustered sandwich estimator. CX indicates circumflex ramus; LAD, left anterior descending; LM, left main artery; OR, odds ratio; RCA, right coronary artery; PRS, polygenic risk score.
Analyses were performed using ordered logistic regression.
Discussion
In the present study we investigated the association between polygenic risk of CAD quantified by a PRS and characteristics of coronary atherosclerosis as assessed by coronary CTA. The main finding was a strong association of the PRS with overall coronary plaque burden. The association was driven by an increased burden of all types of plaque and a higher prevalence of plaque in all coronary vessels. However, the PRS did not associate with any specific plaque characteristics over others.
The risk of CAD observed in patients with a high polygenic risk was notable. These patients were twice as likely to have any CAD and 3 times as likely to have obstructive CAD compared with patients at low polygenic risk. The any CAD and obstructive CAD prevalence of 64% and 35% in high PRS patients may be comparable to the risk observed among other patients at increased cardiovascular risk. In the CONFIRM (Coronary Angiography Evaluation For Clinical Outcomes: An International Multicenter) Registry, the prevalence of any CAD and obstructive CAD was 76% and 52% in 5‐year‐older diabetes mellitus patients,25 and 56% and 19% in hypertensive patients at a similar age.26 In a study of asymptomatic statin‐treated patients with familial hypercholesterolemia, the presence of any CAD and obstructive CAD was 85% and 26%, respectively.27 Thus, a high PRS may confer a risk of CAD presence and severity to an extent similar to that obtained from traditional risk factors. This is also in accordance with an observation from the UK biobank, in which high PRS patients had a 2.5‐fold higher CAD risk compared with the remaining 80% of the population, an effect similar to or stronger than any single traditional risk factor.21
A few prior studies have investigated the relationship between a CAD‐PRS and measures of plaque burden. In a coronary angiography study of early‐onset acute coronary syndrome patients, a 30‐variant PRS and a family history of CAD were independently associated with the presence of multivessel disease.28 A study consisting of asymptomatic individuals without evident CAD from the BioImage cohort demonstrated a dose‐responsive relationship between a 50‐variant PRS and CACS, which was independent of lifestyle risk factors.8 Another analysis on the same cohort demonstrated that the same CAD‐based PRS was associated with carotid artery plaque burden determined by ultrasonography with a 9.7% (95% CI 2.2–17.8%) increase in plaque area per SD increase in the PRS.14 Similarly, a Swedish general population study demonstrated a CAD‐based 13‐variant PRS to be associated with carotid bulb intima‐media thickness and carotid plaques,29 which underscores the common polygenic background of atherosclerosis. To our knowledge the present study is the first to dissect the relationship between polygenic risk of CAD and the complex phenotypes of coronary atherosclerosis. Extending prior findings, our study suggests that polygenic risk increases CAD risk through an increased burden of all coronary atherosclerosis components rather than promoting any specific adverse plaque characteristics. This is plausible since coronary plaque burden as quantified by CACS and SSS are well‐established and strong predictors of an adverse outcome.24, 30 However, the hypothesis needs to be supported by longitudinal studies.
Sibling studies among patients undergoing coronary angiography have suggested that heritability of stenoses in the left main artery (h2=47% [95% CI 29–64%]), coronary calcifications (h2=51% [95% CI 37–65%]), and plaques with expansive vessel remodeling (h2=54% [95% CI 38–70%]) is high, whereas extent of CAD and plaques in the nonproximal segments appeared only modestly heritable or nonheritable (h2 estimates <10%, nonsignificant).3, 31 These findings appear to be in contrast to our findings, where the PRS was similarly associated with all plaque characteristics including atherosclerosis in nonproximal segments. Although the explanation for the discrepancy is not obvious, we did not demonstrate any association between the PRS and specific plaque locations or characteristics per se. One possible explanation for this may be that PRSs are built on risk variants identified from CAD‐GWASs, in which cases of CAD patients comprise a heterogeneous group in terms of atherosclerosis subphenotypes. Consequently, the PRSs also represent the sum of a heterogeneous set of variants predisposing to CAD development through a number of different pathways that may each drive the formation of different atherosclerosis subtypes. For example, studies have shown that the lead variant at the ABO locus, identified in CAD‐GWASs, is specifically associated with myocardial infarction in contrast to the majority of other CAD risk variants;20, 32 an effect possibly driven by an influence on platelet aggregation.33 It is possible that future GWASs based on patients with detailed phenotyping by coronary CTA will help identify variants with specific effects on atherosclerosis and thereby further aid explaining the underlying mechanisms behind the development of different CAD phenotypes.
Another possible explanation for the lack of association between the PRS and specific plaque locations and characteristics may be that polygenic CAD risk only acts as a driver of coronary atherosclerosis development, whereas specific locations and characteristics are governed by a number of nongenetic factors including dietary and lifestyle habits. Lastly, different plaque locations and characteristics may in part simply be explained by different disease stages, a possibility that may not be elucidated in the present study because of the cross‐sectional design.
Besides the pathophysiological insights into polygenic CAD risk and its effects on plaque burden and atherosclerosis characteristics, our study may also have some clinical implications and set the direction for future research. The knowledge of a high PRS may alert clinicians of patients at high risk of accelerated atherosclerosis development, who may benefit from recommendations on a healthy lifestyle or preventive medical treatment before atherosclerosis develops or progresses into a clinically significant disease state. In the setting of patients with chest symptoms, the PRS may aid discriminating between patients with and without obstructive CAD requiring coronary CTA and potentially downstream coronary revascularization. Particularly, since the vast majority of patients undergoing coronary CTA do not have obstructive CAD, it may be speculated that a low PRS could be used for risk assessment to down‐classify the estimated pretest probability of obstructive CAD and avoid the need for coronary CTA. However, further studies are required in order to delineate the clinical utilities of the PRS.
Strengths and Limitations
The main strengths of the study include the well‐validated multilocus PRS and the inclusion of consecutive patients referred for coronary CTA in a standardized protocol. However, a number of limitations deserve attention. The patient population comprised patients without prior CAD but with symptoms suggestive of stable CAD. This may have introduced selection bias since patients carrying the most vulnerable plaques are more likely to develop myocardial infarction as a first manifestation of CAD, which would render them ineligible for study inclusion. Consequently, patients with the highest PRSs may have been excluded from the study. Also, compared with a general population cohort, a selection based on symptoms may have caused index event bias (ie, the distortion of risk factor distribution among included patients).34 This could arise because symptomatic patients with obstructive CAD developed from a high polygenic risk do not require the same burden of traditional risk factors as patients with a low polygenic risk for disease to develop. In such case the observed association between the PRS and plaque burden may likely be driven towards the null (ie, no difference), suggesting that the associations between PRS and plaque burden in this study is a conservative estimate. A second limitation is the fact that the PRS is built on CAD‐GWASs in which some control individuals may have subclinical CAD. It may be speculated that control individuals with subclinical CAD carry a certain phenotype of atherosclerosis, which in such case might affect the association between the PRS and CAD characteristics. A third limitation was that CACS and SSS were used as surrogates of coronary plaque burden, although neither CACS nor SSS directly measure plaque volume. In particular, CACS does not quantify noncalcified plaque material and SSS does not capture plaques with extrinsic plaque remodeling nor grade plaque length. Therefore, these measures might deviate from the true volume of coronary atherosclerosis. Finally, a limitation is the sample size. Although the sample is substantial for a study with this amount of phenotypic details, polygenic traits may require hundreds of thousands of samples to detect small effects. Therefore, although we did not detect any association of the PRS with specific plaque characteristics per se, it does not preclude the existence of minor effects, which we cannot detect with the current sample size.
Conclusions
A PRS based on CAD‐GWASs was strongly associated with coronary plaque burden as assessed by coronary CTA. The association was driven by an increased prevalence of all plaque subtypes throughout the coronary artery tree. However, in the segment‐level analyses adjusted for total plaque burden, the PRS was not associated with stenosis severity, plaque composition, or localization. Therefore, our findings suggest that the polygenic risk captured by PRSs increases CAD risk through the overall formation of coronary atherosclerosis rather than promoting specific plaque characteristics.
Sources of Funding
This work was supported by The Danish Heart Foundation (Grant no. 15‐R99‐A5837‐22920), Riisfort Foundation, The Memorial Fund: “Kirsten Anthonius Mindelegat”, The Health Research Fund of the Central Denmark Region, and Acarix A/S. The funding sources did not take part in this study and did not see or approve the manuscript before submission.
Disclosures
Daníel Guðbjartsson, Hilma Holm, and Kári Stefánsson are employees at deCODE Genetics/Amgen Inc. The remaining authors have no disclosures to report.
Supporting information
Table S1. Association of the PRS With Coronary Plaque Burden and Presence of Plaque Characteristics Including Adjustment for Family History (n=1645 Patients)
Figure S1. Spline representation of the effect of the standardized PRS on plaque burden.
Figure S2. Distribution of the smoothed and standardized PRS according to plaque burden.
Figure S3. Distribution of coronary plaque burden according to the PRS stratified by FHx (n=1645 patients). Boxes and whiskers represent the median, interquartile range, and adjacent values. Outliers were omitted from the graph for display purposes. FHx indicates family history of coronary artery disease <60 years among first‐degree relatives; PRS polygenic risk score.
Figure S4. Distribution of the smoothed and standardized PRS according to plaque characteristics (n=3007 segments with plaque among 849 patients). Each segment with plaque contributes to the distribution (ie, the PRS from 1 individual with more than 1 segment with plaque contributes with the PRS for each segment with plaque). CX indicates circumflexus; LAD, left anterior descending; LM, left main; PRS, polygenic risk score; RCA, right coronary artery.
Acknowledgments
We thank Marie Zöga Diederichsen for excellent help with establishing and organizing the biobank. We are grateful to Vibeke Lyngaard, Marianne Engbjerg Andersen, Stine Malling Søndergaard, Louise Jensen, Anne Sofie Mensberg, Helle Thuesen, Hanne Arp, Annette Odgaard (Dan‐NICAD study nurses), and the clinical staff at the enrolling centers for their patience and collaboration. Bjarni Vilhjalmsson is thanked for his helpful inputs on the data analysis and the manuscript. Finally, this study would not have been possible without the willing cooperation of the patients in the Central Region of Denmark.
(J Am Heart Assoc. 2020;9:e014795 DOI: 10.1161/JAHA.119.014795.)
References
- 1. Marenberg ME, Risch N, Berkman LF, Floderus B, de Faire U. Genetic susceptibility to death from coronary heart disease in a study of twins. N Engl J Med. 1994;330:1041–1046. [DOI] [PubMed] [Google Scholar]
- 2. Zdravkovic S, Wienke A, Pedersen NL, Marenberg ME, Yashin AI, de Faire U. Heritability of death from coronary heart disease: a 36‐year follow‐up of 20 966 Swedish twins. J Intern Med. 2002;252:247–254. [DOI] [PubMed] [Google Scholar]
- 3. Fischer M, Broeckel U, Holmer S, Baessler A, Hengstenberg C, Mayer B, Erdmann J, Klein G, Riegger G, Jacob HJ, Schunkert H. Distinct heritable patterns of angiographic coronary artery disease in families with myocardial infarction. Circulation. 2005;111:855–862. [DOI] [PubMed] [Google Scholar]
- 4. van der Harst P, Verweij N. Identification of 64 novel genetic loci provides an expanded view on the genetic architecture of coronary artery disease. Circ Res. 2018;122:433–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Christiansen MK, Nyegaard M, Larsen SB, Grove EL, Würtz M, Neergaard‐Petersen S, Hvas A‐M, Jensen HK, Kristensen SD. A genetic risk score predicts cardiovascular events in patients with stable coronary artery disease. Int J Cardiol. 2017;241:411–416. [DOI] [PubMed] [Google Scholar]
- 6. Abraham G, Havulinna AS, Bhalala OG, Byars SG, De Livera AM, Yetukuri L, Tikkanen E, Perola M, Schunkert H, Sijbrands EJ, Palotie A, Samani NJ, Salomaa V, Ripatti S, Inouye M. Genomic prediction of coronary heart disease. Eur Heart J. 2016;37:3267–3278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Inouye M, Abraham G, Nelson CP, Wood AM, Sweeting MJ, Dudbridge F, Lai FY, Kaptoge S, Brozynska M, Wang T, Ye S, Webb TR, Rutter MK, Tzoulaki I, Patel RS, Loos RJF, Keavney B, Hemingway H, Thompson J, Watkins H, Deloukas P, Di Angelantonio E, Butterworth AS, Danesh J, Samani NJ; UK Biobank CardioMetabolic Consortium CHD Working Group . Genomic risk prediction of coronary artery disease in 480,000 adults: implications for primary prevention. J Am Coll Cardiol. 2018;72:1883–1893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Khera AV, Emdin CA, Drake I, Natarajan P, Bick AG, Cook NR, Chasman DI, Baber U, Mehran R, Rader DJ, Fuster V, Boerwinkle E, Melander O, Orho‐Melander M, Ridker PM, Kathiresan S. Genetic risk, adherence to a healthy lifestyle, and coronary disease. N Engl J Med. 2016;375:2349–2358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mega JL, Stitziel NO, Smith JG, Chasman DI, Caulfield MJ, Devlin JJ, Nordio F, Hyde CL, Cannon CP, Sacks FM, Poulter NR, Sever PS, Ridker PM, Braunwald E, Melander O, Kathiresan S, Sabatine MS. Genetic risk, coronary heart disease events, and the clinical benefit of statin therapy: an analysis of primary and secondary prevention trials. Lancet. 2015;385:2264–2271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Task Force Members , Montalescot G, Sechtem U, Andreotti F, Arden C, Budaj A, Bugiardini R, Crea F, Cuisset T, Di Mario C, Ferreira JR, Gersh BJ, Gitt AK, Hulot J‐S, Marx N, Opie LH, Pfisterer M, Prescott E, Ruschitzka F, Sabaté M, Senior R, Taggart DP, van der Wall EE, Vrints CJM; ESC Committee for Practice Guidelines , Zamorano JL, Achenbach S, Baumgartner H, Bax JJ, Dean V, Deaton C, Fagard R, Ferrari R, Hoes AW, Kirchhof P, Knuuti J, Linhart A, Nihoyannopoulos P, Piepoli MF, Ponikowski P, Sirnes PA, Tamargo JL, Tendera M, Torbicki A, Windecker S; Document Reviewers , Valgimigli M, Bueno H, Claeys MJ, Donner‐Banzhoff N, Erol C, Frank H, Funck‐Brentano C, Gaemperli O, Gonzalez‐Juanatey JR, Hamilos M, Hasdai D, Husted S, James SK, Kervinen K, Kolh P, Kristensen SD, Lancellotti P, Maggioni AP, Pries AR, Romeo F, Ryden L, Simoons ML, Steg PG, Timmis A, Wijns W, Yildirir A. 2013 ESC guidelines on the management of stable coronary artery disease: the Task Force on the management of stable coronary artery disease of the European Society of Cardiology. Eur Heart J. 2013;34:2949–3003. [DOI] [PubMed] [Google Scholar]
- 11. Park H‐B, Heo R, ó Hartaigh B, Cho I, Gransar H, Nakazato R, Leipsic J, Mancini GBJ, Koo B‐K, Otake H, Budoff MJ, Berman DS, Erglis A, Chang H‐J, Min JK. Atherosclerotic plaque characteristics by CT angiography identify coronary lesions that cause ischemia: a direct comparison to fractional flow reserve. JACC Cardiovasc Imaging. 2015;8:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Motoyama S, Ito H, Sarai M, Kondo T, Kawai H, Nagahara Y, Harigaya H, Kan S, Anno H, Takahashi H, Naruse H, Ishii J, Hecht H, Shaw LJ, Ozaki Y, Narula J. Plaque characterization by coronary computed tomography angiography and the likelihood of acute coronary events in mid‐term follow‐up. J Am Coll Cardiol. 2015;66:337–346. [DOI] [PubMed] [Google Scholar]
- 13. Christiansen MK, Jensen JM, Nørgaard BL, Dey D, Bøtker HE, Jensen HK. Coronary plaque burden and adverse plaque characteristics are increased in healthy relatives of patients with early onset coronary artery disease. JACC Cardiovasc Imaging. 2017;10:1128–1135. [DOI] [PubMed] [Google Scholar]
- 14. Natarajan P, Young R, Stitziel NO, Padmanabhan S, Baber U, Mehran R, Sartori S, Fuster V, Reilly DF, Butterworth AS, Rader DJ, Ford I, Sattar N, Kathiresan S. Polygenic risk score identifies subgroup with higher burden of atherosclerosis and greater relative benefit from statin therapy in the primary prevention setting. Circulation. 2017;135:2091–2101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Nissen L, Winther S, Isaksen C, Ejlersen JA, Brix L, Urbonaviciene G, Frost L, Madsen LH, Knudsen LL, Schmidt SE, Holm NR, Maeng M, Nyegaard M, Bøtker HE, Bøttcher M. Danish study of Non‐Invasive testing in Coronary Artery Disease (Dan‐NICAD): study protocol for a randomised controlled trial. Trials. 2016;17:262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Nissen L, Winther S, Westra J, Ejlersen JA, Isaksen C, Rossi A, Holm NR, Urbonaviciene G, Gormsen LC, Madsen LH, Christiansen EH, Maeng M, Knudsen LL, Frost L, Brix L, Bøtker HE, Petersen SE, Bøttcher M. Diagnosing coronary artery disease after a positive coronary computed tomography angiography: the Dan‐NICAD open label, parallel, head to head, randomized controlled diagnostic accuracy trial of cardiovascular magnetic resonance and myocardial perfusion scintigraphy. Eur Heart J Cardiovas Imaging. 2018;19:369–377. [DOI] [PubMed] [Google Scholar]
- 17. Abraham G, Qiu Y, Inouye M. FlashPCA2: principal component analysis of Biobank‐scale genotype datasets. Bioinformatics. 2017;33:2776–2778. [DOI] [PubMed] [Google Scholar]
- 18. Vilhjálmsson BJ, Yang J, Finucane HK, Gusev A, Lindström S, Ripke S, Genovese G, Loh P‐R, Bhatia G, Do R, Hayeck T, Won H‐H; Schizophrenia Working Group of the Psychiatric Genomics Consortium, Discovery, Biology, and Risk of Inherited Variants in Breast Cancer (DRIVE) study , Kathiresan S, Pato M, Pato C, Tamimi R, Stahl E, Zaitlen N, Pasaniuc B, Belbin G, Kenny EE, Schierup MH, De Jager P, Patsopoulos NA, McCarroll S, Daly M, Purcell S, Chasman D, Neale B, Goddard M, Visscher PM, Kraft P, Patterson N, Price AL, Liu J. Modeling linkage disequilibrium increases accuracy of polygenic risk scores. Am J Hum Genet. 2015;97:576–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gudmundsson J, Sigurdsson JK, Stefansdottir L, Agnarsson BA, Isaksson HJ, Stefansson OA, Gudjonsson SA, Gudbjartsson DF, Masson G, Frigge ML, Stacey SN, Sulem P, Halldorsson GH, Tragante V, Hólm H, Eyjolfsson GI, Sigurdardottir O, Olafsson I, Jonsson T, Jonsson E, Barkardottir RB, Hilmarsson R, Asselbergs FW, Geirsson G, Thorsteinsdottir U, Rafnar T, Thorleifsson G, Stefansson K. Genome‐wide associations for benign prostatic hyperplasia reveal a genetic correlation with serum levels of PSA. Nat Commun. 2018;9:4568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. CARDIoGRAMplusC4D Consortium . A comprehensive 1000 Genomes‐based genome‐wide association meta‐analysis of coronary artery disease. Nat Genet. 2015;47:1121–1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Khera AV, Chaffin M, Aragam KG, Haas ME, Roselli C, Choi SH, Natarajan P, Lander ES, Lubitz SA, Ellinor PT, Kathiresan S. Genome‐wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Nat Genet. 2018;50:1219–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Agatston AS, Janowitz WR, Hildner FJ, Zusmer NR, Viamonte M, Detrano R. Quantification of coronary artery calcium using ultrafast computed tomography. J Am Coll Cardiol. 1990;15:827–832. [DOI] [PubMed] [Google Scholar]
- 23. Raff GL, Abidov A, Achenbach S, Berman DS, Boxt LM, Budoff MJ, Cheng V, DeFrance T, Hellinger JC, Karlsberg RP; Society of Cardiovascular Computed Tomography . SCCT guidelines for the interpretation and reporting of coronary computed tomographic angiography. J Cardiovasc Comput Tomogr. 2009;3:122–136. [DOI] [PubMed] [Google Scholar]
- 24. Min JK, Shaw LJ, Devereux RB, Okin PM, Weinsaft JW, Russo DJ, Lippolis NJ, Berman DS, Callister TQ. Prognostic value of multidetector coronary computed tomographic angiography for prediction of all‐cause mortality. J Am Coll Cardiol. 2007;50:1161–1170. [DOI] [PubMed] [Google Scholar]
- 25. Blanke P, Naoum C, Ahmadi A, Cheruvu C, Soon J, Arepalli C, Gransar H, Achenbach S, Berman DS, Budoff MJ, Callister TQ, Al‐Mallah MH, Cademartiri F, Chinnaiyan K, Rubinshtein R, Marquez H, Delago A, Villines TC, Hadamitzky M, Hausleiter J, Shaw LJ, Kaufmann PA, Cury RC, Feuchtner G, Kim Y‐J, Maffei E, Raff G, Pontone G, Andreini D, Chang H‐J, Chow BW, Min J, Leipsic J. Long‐term prognostic utility of coronary CT angiography in stable patients with diabetes mellitus. JACC Cardiovasc Imaging. 2016;9:1280–1288. [DOI] [PubMed] [Google Scholar]
- 26. Nakanishi R, Baskaran L, Gransar H, Budoff MJ, Achenbach S, Al‐Mallah M, Cademartiri F, Callister TQ, Chang H‐J, Chinnaiyan K, Chow BJW, Delago A, Hadamitzky M, Hausleiter J, Cury R, Feuchtner G, Kim Y‐J, Leipsic J, Kaufmann PA, Maffei E, Raff G, Shaw LJ, Villines TC, Dunning A, Marques H, Pontone G, Andreini D, Rubinshtein R, Bax J, Jones E, Hindoyan N, Gomez M, Lin FY, Min JK, Berman DS. Relationship of hypertension to coronary atherosclerosis and cardiac events in patients with coronary computed tomographic angiography. Hypertension. 2017;70:293–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Neefjes LA, Ten Kate G‐JR, Rossi A, Galema‐Boers AJ, Langendonk JG, Weustink AC, Moelker A, Nieman K, Mollet NR, Krestin GP, Sijbrands EJ, de Feyter PJ. CT coronary plaque burden in asymptomatic patients with familial hypercholesterolaemia. Heart. 2011;97:1151–1157. [DOI] [PubMed] [Google Scholar]
- 28. Hindieh W, Pilote L, Cheema A, Al‐Lawati H, Labos C, Dufresne L, Engert JC, Thanassoulis G. Association between family history, a genetic risk score, and severity of coronary artery disease in patients with premature acute coronary syndromes. Arterioscler Thromb Vasc Biol. 2016;36:1286–1292. [DOI] [PubMed] [Google Scholar]
- 29. Hamrefors V, Hedblad B, Engström G, Almgren P, Sjögren M, Melander O. A myocardial infarction genetic risk score is associated with markers of carotid atherosclerosis. J Intern Med. 2012;271:271–281. [DOI] [PubMed] [Google Scholar]
- 30. Budoff MJ, Shaw LJ, Liu ST, Weinstein SR, Mosler TP, Tseng PH, Flores FR, Callister TQ, Raggi P, Berman DS. Long‐term prognosis associated with coronary calcification: observations from a registry of 25,253 patients. J Am Coll Cardiol. 2007;49:1860–1870. [DOI] [PubMed] [Google Scholar]
- 31. Fischer M, Mayer B, Baessler A, Riegger G, Erdmann J, Hengstenberg C, Schunkert H. Familial aggregation of left main coronary artery disease and future risk of coronary events in asymptomatic siblings of affected patients. Eur Heart J. 2007;28:2432–2437. [DOI] [PubMed] [Google Scholar]
- 32. Reilly MP, Li M, He J, Ferguson JF, Stylianou IM, Mehta NN, Burnett M‐S, Devaney JM, Knouff CW, Thompson JR, Horne BD, Stewart AFR, Assimes TL, Wild PS, Allayee H, Nitschke PL, Patel RS; Myocardial Infarction Genetics Consortium, Wellcome Trust Case Control Consortium , Martinelli N, Girelli D, Quyyumi AA, Anderson JL, Erdmann J, Hall AS, Schunkert H, Quertermous T, Blankenberg S, Hazen SL, Roberts R, Kathiresan S, Samani NJ, Epstein SE, Rader DJ. Identification of ADAMTS7 as a novel locus for coronary atherosclerosis and association of ABO with myocardial infarction in the presence of coronary atherosclerosis: two genome‐wide association studies. Lancet. 2011;377:383–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Christiansen MK, Larsen SB, Nyegaard M, Neergaard‐Petersen S, Würtz M, Grove EL, Hvas A‐M, Jensen HK, Kristensen SD. The ABO locus is associated with increased platelet aggregation in patients with stable coronary artery disease. Int J Cardiol. 2019;286:152–158. [DOI] [PubMed] [Google Scholar]
- 34. Dahabreh IJ, Kent DM. Index event bias as an explanation for the paradoxes of recurrence risk research. JAMA. 2011;305:822–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Association of the PRS With Coronary Plaque Burden and Presence of Plaque Characteristics Including Adjustment for Family History (n=1645 Patients)
Figure S1. Spline representation of the effect of the standardized PRS on plaque burden.
Figure S2. Distribution of the smoothed and standardized PRS according to plaque burden.
Figure S3. Distribution of coronary plaque burden according to the PRS stratified by FHx (n=1645 patients). Boxes and whiskers represent the median, interquartile range, and adjacent values. Outliers were omitted from the graph for display purposes. FHx indicates family history of coronary artery disease <60 years among first‐degree relatives; PRS polygenic risk score.
Figure S4. Distribution of the smoothed and standardized PRS according to plaque characteristics (n=3007 segments with plaque among 849 patients). Each segment with plaque contributes to the distribution (ie, the PRS from 1 individual with more than 1 segment with plaque contributes with the PRS for each segment with plaque). CX indicates circumflexus; LAD, left anterior descending; LM, left main; PRS, polygenic risk score; RCA, right coronary artery.