

Abstract
Background
Antiplatelet therapy with aspirin (acetylsalicylic acid [ASA]) is less efficient in some coronary patients, which increases their risk of developing thrombosis. Elevated blood levels of thromboinflammatory mediators, like soluble CD40L (sCD40L), may explain such variabilities. We hypothesized that in the presence of elevated levels of sCD40L, the efficacy of ASA may vary and aimed to determine the effects of ASA on CD40L signaling and aggregation of platelets.
Methods and Results
The effects of ASA on CD40L‐treated human platelets, in response to suboptimal concentrations of collagen or thrombin, were assessed at levels of aggregation, thromboxane A2 secretion, and phosphorylation of p38 mitogen‐activated protein kinase, nuclear factor kappa B, transforming growth factor‐β–activated kinase 1, and myosin light chain. sCD40L significantly elevated thromboxane A2 secretion in platelets in response to suboptimal doses of collagen and thrombin, which was reversed by ASA. ASA did not inhibit the phosphorylation of p38 mitogen‐activated protein kinase, nuclear factor kappa B, and transforming growth factor‐β–activated kinase 1, with sCD40L stimulation alone or with platelet agonists. sCD40L potentiated platelet aggregation, an effect completely reversed and partially reduced by ASA in response to a suboptimal dose of collagen and thrombin, respectively. The effects of ASA in sCD40L‐treated platelets with collagen were related to inhibition of platelet shape change and myosin light chain phosphorylation.
Conclusions
ASA does not affect platelet sCD40L signaling but prevents its effect on thromboxane A2 secretion and platelet aggregation in response to collagen, via a mechanism implying inhibition of myosin light chain. Targeting the sCD40L axis in platelets may have a therapeutic potential in patients with elevated levels of sCD40L and who are nonresponsive or less responsive to ASA.
Keywords: aspirin, platelet, platelet aggregation, platelet inhibitor, thromboxane
Subject Categories: Cell Signalling/Signal Transduction, Inflammation, Mechanisms, Platelets, Thrombosis
Clinical Perspective
What Is New?
Acetylsalicylic acid (ASA) inhibits thromboxane A2 secretion of soluble CD40L (sCD40L)‐treated platelets stimulated by suboptimal doses of agonists.
ASA has no effect on platelet sCD40L signaling.
ASA inhibits the potentiating action of sCD40L on platelet aggregation, an effect related to inhibition of platelet shape change and myosin light chain phosphorylation.
What Are the Clinical Implications?
Elevated levels of plasma sCD40L in coronary patients may influence ASA efficiency.
Targeting sCD40L signaling might have a therapeutic potential in coronary patients who are nonresponsive or less responsive to ASA.
Introduction
Platelets play a key role in cardiovascular diseases, being at the center of atherothrombotic events and mediating thromboinflammatory responses that amplify atherosclerosis and subsequent clinical complications, including ischemic heart disease, acute coronary syndrome, myocardial infarction, and stroke.1, 2, 3 Therefore, antiplatelet agents have been the main focus of secondary prevention of cardiovascular diseases.4 Aspirin (acetylsalicylic acid [ASA]) is the most widely used antiplatelet drug for prevention of cardiovascular diseases; it reduces the risk of myocardial infarction, stroke, and cardiovascular death in a broad range of patients at high risk for future cardiovascular events.5, 6 Nevertheless, ASA prevents only ≈25% of coronary events and ischemic strokes when used in secondary prevention.7 ASA acts by irreversibly inhibiting cyclooxygenase‐1 enzyme that catalyzes the conversion of arachidonic acid to prostaglandins G1/G2, which are subsequently converted by thromboxane synthase to thromboxane A2.8 A large number of patients continue to experience thromboembolic events despite ASA therapy in a phenomenon known as “aspirin resistance,” which could be attributable to several reasons such as drug interactions; ASA pharmacokinetics9; or polymorphisms affecting several proteins such as cyclooxygenase‐1, thromboxane A2 synthesis machinery, and platelet receptors (αIIbβ3, α2β1, glycoprotein Ib‐IX‐V).10, 11, 12, 13, 14
Patients with inflammatory diseases exhibit higher‐than‐expected rates of ischemic cardiovascular events that are not attributable to traditional risk factors but rather to inflammatory stimuli that trigger pathways partaking in the pathogenesis of atherosclerosis.15, 16 Patients who are resistant to antiplatelet therapy have a hyperactive platelet phenotype,17, 18 which may be related to elevated levels of circulating primers.19, 20, 21 Platelet primers do not stimulate platelet activation, but, in combination with physiologic stimuli, significantly enhance platelet function.22 One of these remarkable primers is the thromboinflammatory mediator CD40L and its classical receptor CD40,23, 24 which have gained significant attention for their involvement in the pathogenesis of inflammation, atherosclerosis, and atherothrombosis.25, 26, 27
CD40L, also known as CD154 or gp39, is a type II transmembrane protein and a member of the tumor necrosis factor gene superfamily. It is expressed on a broad range of cells, mainly T cells and platelets.25 Like other members of the tumor necrosis factor family, CD40L has a soluble form (sCD40L) that is mainly produced by platelets. This molecule was initially thought to be solely implicated in immune and inflammatory responses,28 as it was mostly expressed by immune cells.29 However, CD40L has a much broader role. In particular, we have previously shown that enhanced levels of sCD40L exacerbate platelet activation and aggregation through the CD40/tumor necrosis factor receptor‐associated factor 2/Ras‐related C3 botulinum toxin substrate 1/p38 MAPK (p 38 mitogen‐activated protein kinase) signaling pathways.30 More recently, we have also shown that the priming action of sCD40L on platelets involves nuclear factor kappa B (NF‐κB),31 a central mediator of inflammation linked to thrombotic processes,32 in a CD40‐ and transforming growth factor‐β–activated kinase 1 (TAK1)‐dependent manner.33
Although CD40L has an important role as a platelet primer through its pronounced enhancement of platelet activation and aggregation in response to suboptimal concentrations of agonists, its effect may influence the antiplatelet efficiency of ASA. Henceforth, the objective of the present study was to determine the effect of ASA on CD40L in platelet signaling, secretion, and aggregation.
Methods
The data, analytic methods, and study materials will be made available upon request from the corresponding author to other researchers for the purpose of reproducing the results or replicating the procedure.
Preparation of Human Platelets
Venous blood was drawn from healthy volunteers, free from medication known to interfere with platelet function for at least 3 weeks before the experiment. All participants provided written informed consent, according to a protocol that was approved by the Human Ethical Committee of the Montreal Heart Institute, in accordance with the declaration of Helsinki for experiments involving humans. Washed platelets were prepared as previously described.30, 31 Briefly, platelet‐rich plasma was obtained by centrifugation of acid‐citrate‐dextrose anticoagulated blood. Platelets were then pelleted from platelet‐rich plasma, to which 1 μg/mL of prostaglandin E1 was added, washed with Hank's Balanced Salt Solution–Hank's sodium citrate buffer, and finally resuspended in Hank's Balanced Salt Solution –Hank's buffer containing 2 mmol/L MgCl2 and 2 mmol/L CaCl2. Platelets were rested at 37°C for at least 30 minutes before further manipulation.
Measurement of Thromboxane A2
The stable hydrolysis product of thromboxane A2, thromboxane B2, was used as a readout of thromboxane A2 production. It was measured using a commercial thromboxane B2 ELISA kit (R&D Systems, Minneapolis, MN). Platelet samples from the aggregation reactions were quenched at 5 minutes with 2 μmol/L indomethacin and 5 mmol/L EDTA to inhibit further production of thromboxane A2. Samples were centrifuged (5 minutes, 200g at 4°C) and the supernatant was removed and stored at −80°C for subsequent analysis.
Phosphorylation of p38‐MAPK, NF‐κB, TAK‐1, and MLC
Western blots were performed to assess the phosphorylation levels of p38‐MAPK, NF‐κB, TAK‐1, and MLC. Briefly, platelets (1000×106/mL) were stimulated as indicated and lysed immediately by the addition of 1/4 volume of 4XSDS‐PAGE loading buffer containing 5% β‐mercaptoethanol. All samples were boiled for 5 minutes. Protein lysates were then resolved in 10% SDS‐PAGE gels and transferred to nitrocellulose membranes. The membranes were blocked with 5% nonfat dry milk for 1 hour, washed 3 times with TBS‐T (150 mmol/L NaCl, 20 mmol/L Tris, pH 7.4, 0.1% Tween‐20) and incubated with appropriate primary antibody overnight at 4°C. We used primary antibodies against phospho‐p38‐MAPKthreonine 180/182, phospho‐IκBαserine 32/36, phospho‐TAK1threonine 184/187, phospho‐MLCserine 19 threonine 18, MLC, and β‐actin (Cell Signaling Technology, Danvers, MA). Following washing steps, membranes were labeled with horseradish peroxidase–conjugated secondary antibody for 1 hour, washed, and bound peroxidase activity was detected by enhanced chemiluminescence (PerkinElmer Life Sciences, Hopkinton, MA). All membranes were stripped and reprobed for β‐actin, a commonly chosen loading control. Data were presented as ratios of phosphorylated proteins to respective β‐actin.
Measurement of Platelet Aggregation
We monitored aggregation of washed human platelets on a 4‐channel optical aggregometer (Chrono‐Log Corp., Havertown, PA) under shear (1000 rpm) at 37°C. Platelet suspensions (250×106/mL) were pretreated with ASA (30 μmol/L; Tocris Bioscience, St Louis, MO)8, 34 or ML7 (selective inhibitor of MLC kinase, 50 μmol/L; Tocris Bioscience)35 for 5 minutes followed by treatment with sCD40L (1000 ng/mL, R&D Systems) for 30 minutes at 37°C.33 After that, platelet aggregation was triggered by a suboptimal dose of collagen (0.25±0.1 μg/mL, Chrono‐Log Corp.), or α‐thrombin (0.025±0.01 U/mL, Sigma‐Aldrich, St Louis, MO). The suboptimal dose of agonist that does not induce >30% aggregation was selected before each experiment from a dose‐response curve of platelet aggregation in response to collagen or thrombin (Figure S1). Traces were recorded until stabilization of platelet aggregation.30, 31, 36
Statistical Analysis
Statistical analysis was performed using SPSS Statistics 25 (IBM Corporation, Armonk, NY. Results are presented as median±interquartile range. Statistical comparisons were done using the Kruskal–Wallis test followed by Dunn's post hoc test. The specific statistical tests used, the median of data, the number of experiments, and the P values are specified in the figure legends. A P<0.05 was considered statistically significant.
Results
ASA Inhibits Thromboxane A2 Secretion Induced by sCD40L
We investigated whether sCD40L increases thromboxane A2 formation and whether ASA might reverse this effect. In unstimulated platelets, the production of thromboxane A2 (4.5±3.1 ng/mL) was unaffected by sCD40L in the presence or absence of ASA (Figure 1A). In stimulated platelets, the production of thromboxane A2 increased to 15.7±6.3 ng/mL in response to a suboptimal concentration of collagen (Figure 1C), which was reduced significantly by 5‐fold to 3.5±3.1 in the presence of ASA. Likewise, thromboxane A2 levels remain unchanged (5.9±3.9 ng/mL) with ASA pretreatment in response to a suboptimal concentration of thrombin (Figure 1B). However, sCD40L significantly elevated thromboxane A2 production in platelets in the presence of suboptimal doses of collagen (23.2±8.1 ng/mL; Figure 1C) and thrombin (24.2±7.9 ng/mL, Figure 1B), an effect that was completely reversed by ASA.
Figure 1.
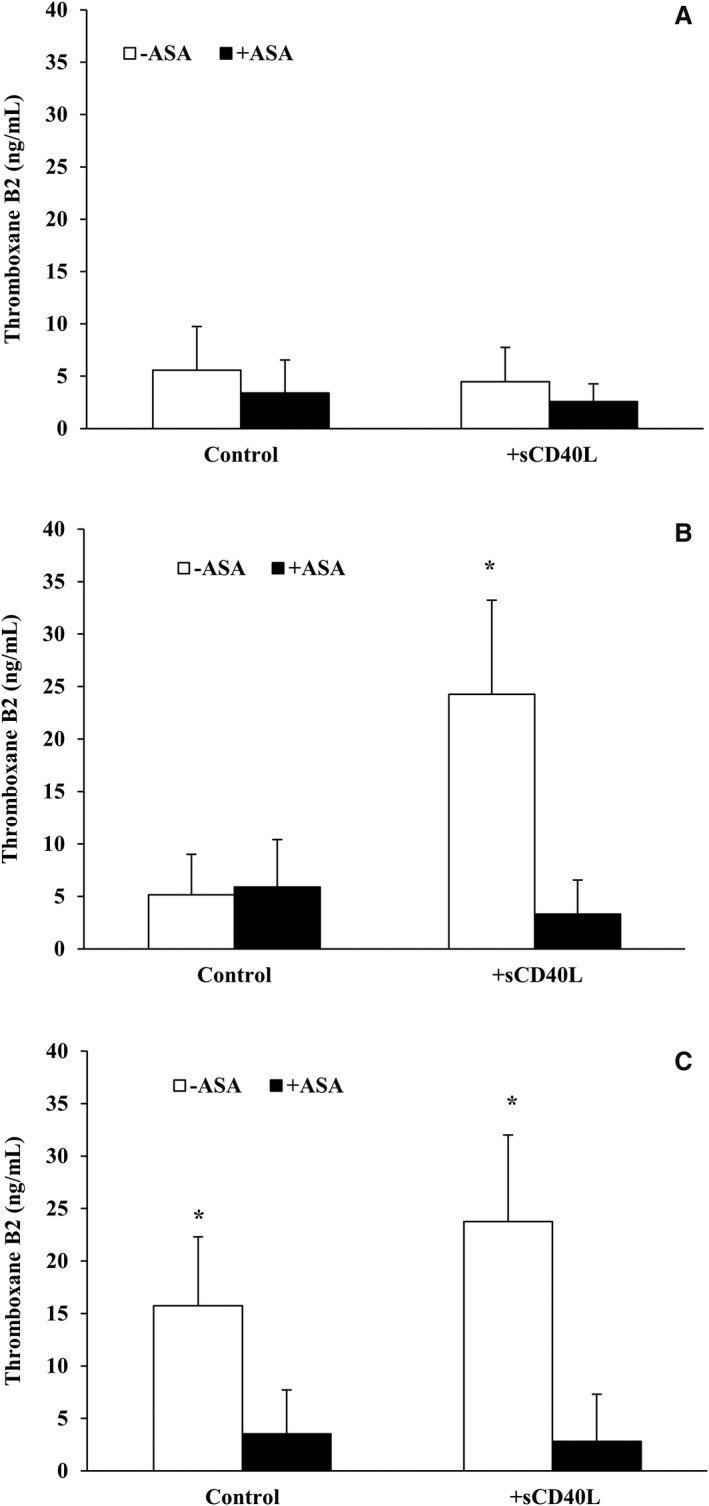
ASA inhibits sCD40L‐induced thromboxane B2 formation in platelets in response to a suboptimal dose of agonists. Washed human platelets (250×106/mL) were left untreated or pretreated with ASA (30 μmol/L) for 5 minutes at 37°C. They were then stimulated with sCD40L (1000 ng/mL) or not for 30 minutes. Platelets were left unstimulated (A) or challenged with a suboptimal dose of thrombin (0.025±0.01 U/mL) (B) or collagen (0.25±0.1 μg/mL) (C). Aggregation was stopped with indomethacin (2 μmol/L) 5 minutes following the adding of suboptimal doses of agonists. Platelets were centrifuged at 14 000g for 5 minutes at 4°C and supernatant was collected. Thromboxane B2 in the supernatant was then measured using a thromboxane B2 ELISA kit. (n=10, median±IQR). *P<0.05 vs other treatements (Kruskal–Wallis followed by Dunn's post hoc test). ASA indicates acetylsalicylic acid; IQR, interquartile range; sCD40L, soluble CD40L.
ASA Does Not Affect CD40L Signaling
We have previously shown that NF‐κB, p38‐MAPK, and TAK1 are required for sCD40L‐induced platelet activation and potentiation of platelet aggregation.30, 31, 33 We aimed to verify whether the impact of ASA passes via modulation of sCD40L signaling in platelets. To this end, we assessed the phosphorylation levels of IκBα (Figure 2), p38‐MAPK (Figure 3), and TAK1 (Figure 4). We have first shown that sCD40L induced robust phosphorylations of IκBα, p38‐MAPK, and TAK1, which were not affected by the addition of suboptimal concentrations of collagen or thrombin. Treatment of platelets with ASA had no impact on the phosphorylation of those proteins, either following sCD40L stimulation alone or with sCD40L and a suboptimal dose of collagen or thrombin.
Figure 2.
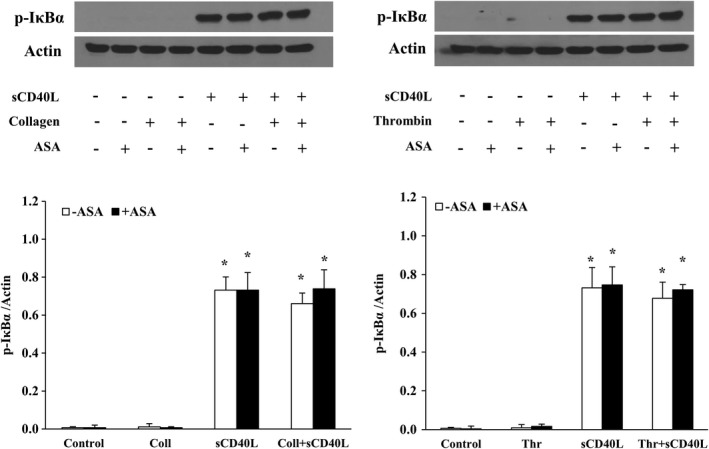
ASA does not affect NF‐κB activation by sCD40L. Washed human platelets (1000×106/mL) were pretreated or not with ASA (30 μmol/L) for 10 minutes, then stimulated with sCD40L (1000 ng/mL) and thrombin (0.025 U/mL) or collagen (0.25 μg/mL) for 5 minutes. Platelet lysates were resolved in 10% SDS–PAGE and assessed for p‐IκBα. The actin blot is from stripped membranes of the p‐IκBα blot. Histograms represent the median of blot data, expressed in optical density (n=4, median±IQR). *P<0.01 vs. control (Kruskal–Wallis followed by Dunn's post hoc test). ASA indicates acetylsalicylic acid; IQR, interquartile range; NF‐κB, nuclear factor kappa B; sCD40L, soluble CD40L.
Figure 3.
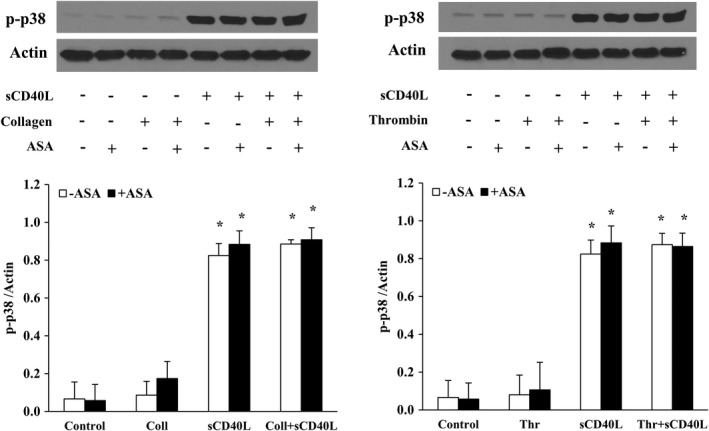
ASA does not affect p38‐MAPK activation by sCD40L. Washed human platelets (1000×106/mL) were pretreated or not with ASA (30 μmol/L) for 10 minutes, then stimulated with sCD40L (1000 ng/mL) and thrombin (0.025 U/mL) or collagen (0.25 μg/mL) for 5 minutes. Platelet lysates were resolved in 10% SDS–PAGE and assessed for p‐p38‐MAPK. The actin blot is from stripped membranes of the p‐p38‐MAPK blot. Histograms represent the median of blot data, expressed in optical density (n=4, median±IQR). *P<0.01 vs control (Kruskal–Wallis followed by Dunn's post hoc test). ASA indicates acetylsalicylic acid; IQR, interquartile range; p38‐MAPK, p38 mitogen‐activated protein kinase; sCD40L, soluble CD40L.
Figure 4.
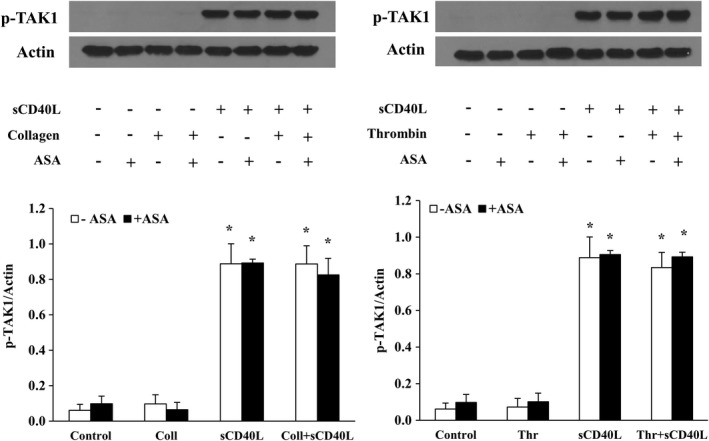
ASA does not affect TAK1 activation by sCD40L. Washed human platelets (1000×106/mL) were pretreated or not with ASA (30 μmol/L) for 10 minutes, then stimulated with sCD40L (1000 ng/mL) and thrombin (0.025 U/mL) or collagen (0.25 μg/mL) for 5 minutes. Platelet lysates were resolved in 10% SDS–PAGE and assessed for p‐TAK1. Actin blot is from stripped membranes of p‐TAK1 blot. Histograms represent the median of blot data, expressed in optical density (n=4, median±IQR). *P<0.01 vs. control (Kruskal–Wallis followed by Dunn's post hoc test). ASA indicates acetylsalicylic acid; IQR, interquartile range; sCD40L, soluble CD40L; TAK1, transforming growth factor‐β–activated kinase 1.
ASA Reduces the Effect of CD40L on Platelet Aggregation
sCD40L alone was unable to induce platelet aggregation (Figure 5A), but potentiated platelet aggregation in combination with suboptimal doses of collagen (Figure 5B) or thrombin (Figure 5C). Interestingly, the effect of sCD40L in potentiating platelet aggregation was completely reversed by ASA in response to a suboptimal dose of collagen (Figure 5B). However, the potentiating impact of sCD40L on platelet aggregation was significantly reduced by ≈50% by ASA when sCD40L‐pretreated platelets were stimulated with a suboptimal dose of thrombin (Figure 5C). Furthermore, only the effect of ASA in response to collagen and sCD40L was associated with complete inhibition of platelet shape change (Figure 5B, red and green aggregation traces) that precedes the onset of the aggregation process.
Figure 5.
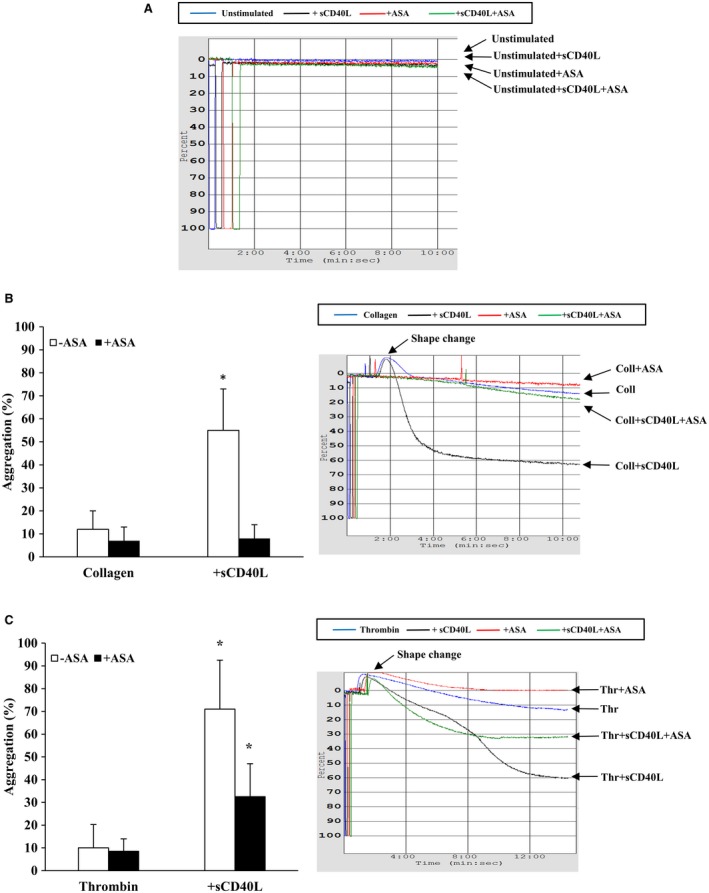
Effects of sCD40L and ASA on platelet aggregation in response to suboptimal doses of agonists. Washed human platelets (250×106/mL) were left untreated or pretreated with ASA (30 μmol/L) for 5 minutes at 37°C. In (A) sCD40L (1000 ng/mL) was used as the platelet agonist, whereas in B and C, platelets were preincubated with sCD40L or not for 30 minutes. Platelet aggregation was induced with suboptimal doses of collagen (0.25 μg/mL, B) or thrombin (0.025 U/mL, C). Histograms represent medians of aggregation percentages (n=25, median±IQR). *P<0.01 vs other treatments (Kruskal–Wallis followed by Dunn's post hoc test). ASA indicates acetylsalicylic acid; IQR, interquartile range; sCD40L, soluble CD40L.
Involvement of MLC in ASA Action
To gain insight into the observed effect of ASA on platelet shape change in response to collagen, we assessed the phosphorylation of MLC, a regulator of the dynamic remodeling of the actin cytoskeleton required for shape change.37, 38, 39 As shown in Figure 6, ASA inhibits MLC phosphorylation induced by a suboptimal aggregating dose of collagen in sCD40L‐treated platelets. However, in sCD40L‐treated platelet stimulated with a suboptimal dose of thrombin, ASA had no significant effect. To understand the impact of such findings on platelet function, we used ML7, a selective inhibitor of MLC kinase, in platelet aggregation tests. A concentration of 50 μmol/L of ML7 was able to abolish MLC activation in platelets (Figure S2). ML7 inhibited platelet aggregation and shape change when platelets were stimulated with suboptimal doses of collagen (Figure 7A) or thrombin (Figure 7B), thereby preventing the potentiating action of sCD40L on platelet aggregation. Finally, similar to ASA, ML7 had no effect on the phosphorylation of IκBα, p38‐MAPK, and TAK1 (Figure 8).
Figure 6.
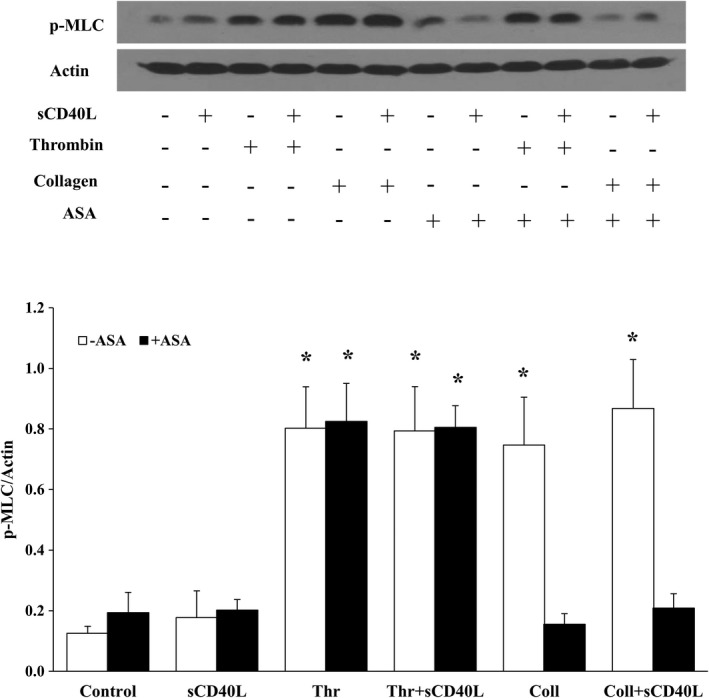
ASA inhibits MLC phosphorylation in platelets stimulated with collagen but not thrombin. Washed human platelets (1000×106/mL) were pretreated or not with ASA (30 μmol/L) for 10 minutes, then stimulated with sCD40L (1000 ng/mL). Aggregation was triggered with suboptimal doses of thrombin (0.025 U/mL) or collagen (0.25 μg/mL). Platelet lysates were resolved in 15% SDS–PAGE and assessed for p‐MLC. The MLC blot is from stripped membranes of the p‐MLC blot. Histograms represent the median of blot data, expressed in optical density (n=4, median±IQR). *P<0.01 vs. control (Kruskal–Wallis followed by Dunn's post hoc test). ASA indicates acetylsalicylic acid; IQR, interquartile range; MLC, myosin light chain; sCD40L, soluble CD40L.
Figure 7.
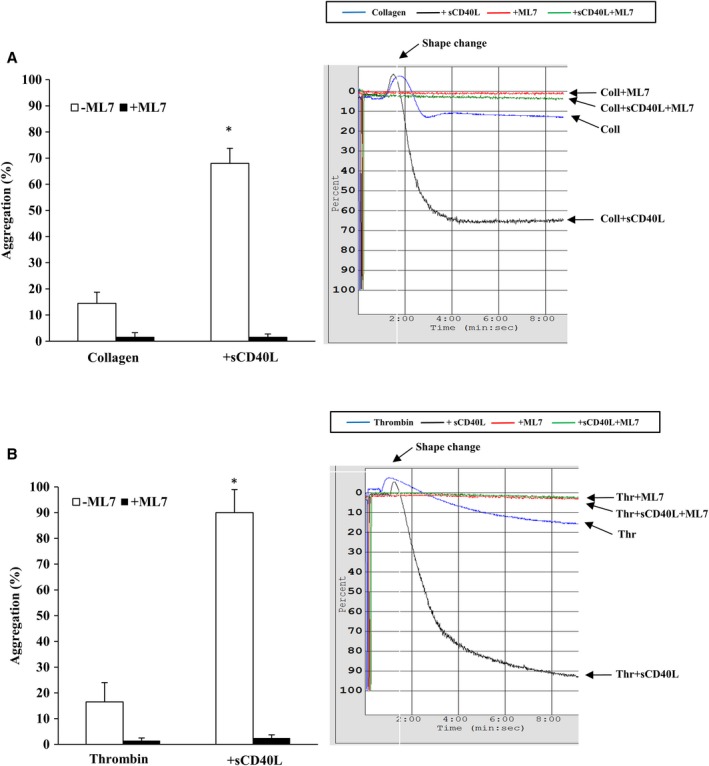
Effect of ML7 on the potentiating action of sCD40L on platelet aggregation. Washed human platelets (250×106 platelets/mL) were left untreated or pretreated with ML7 (50 μmol/L) for 5 minutes at 37°C. They were preincubated with sCD40L or not (1000 ng/mL) for 30 minutes. Platelet aggregation was induced with suboptimal doses of collagen (0.25 μg/mL, A) or thrombin (0.025 U/mL, B). Histograms represent medians of aggregations (n=9, median±IQR). *P<0.01 vs. other treatments (Kruskal–Wallis followed by Dunn's post hoc test). IQR indicates interquartile range; ML7, selective inhibitor of myosin light chain kinase; sCD40L, soluble CD40L.
Figure 8.
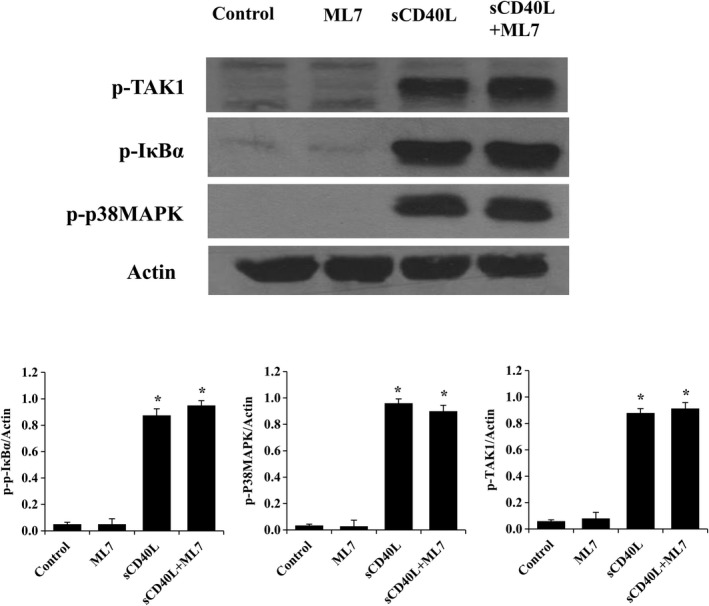
ML7 has no effect on sCD40L signaling. Washed human platelets (1000×106/mL) were pretreated or not with ML7 (50 μmol/L) for 5 minutes, then left untreated or stimulated with sCD40L (1000 ng/mL) for 5 minutes. Platelet lysates were resolved in 10% SDS–PAGE and assessed for p‐TAK1 and stripped for p‐IκBα, p‐p38, and actin. Histograms represent the median of blot data, expressed in optical density (n=5, median±IQR). *P<0.01 vs. control (Kruskal–Wallis followed by Dunn's post hoc test). IQR indicates interquartile range; ML7, selective inhibitor of myosin light chain kinase; sCD40L, soluble CD40L; TAK1, transforming growth factor‐β–activated kinase 1.
Discussion
In this study, we hypothesized that elevated levels of sCD40L, like those found in the circulation of coronary patients, attenuate the efficacy of antiplatelet drugs. As such, combating the influence of sCD40L on platelet activity might improve antiplatelet efficacy. To address our hypothesis, we assessed the effects of ASA on sCD40L‐induced signaling, secretion, and aggregation of unstimulated and primed platelets with suboptimal concentrations of physiological agonists, thrombin, and collagen. We found that ASA is highly effective in completely reversing the potentiating effect of sCD40L on platelet aggregation in response to collagen. However, the response of sCD40L in response to thrombin was only partially reduced. Such effect was independent of sCD40L signaling via NF‐κB, p38‐MAPK or TAK1, but related to inhibition of MLC phosphorylation and platelet shape change in response to collagen of sCD40L‐primed platelets.
The priming role of sCD40L, which significantly potentiated the aggregation in accordance with our previous studies,30, 31, 33 was completely reversed by ASA when platelets were stimulated with collagen. However, sCD40L was able to partially but significantly overcome the inhibitory effect of ASA when aggregation was induced with suboptimal doses of thrombin. This could be explained by the ability of ASA to inhibit the effects of collagen on platelets, which relies principally on thromboxane A2,40 whereas thrombin is known to be a potent platelet activator41, 42 independently of thromboxane A2. Indeed, despite dual therapies with antagonists of ADP receptors and ASA, a large number of patients with acute coronary syndrome experience recurrent major cardiovascular ischemic events attributable to markers of thrombin activation remaining high after an acute coronary syndrome.43, 44 This suggests sustained platelet activation through a thrombin‐mediated mechanism.45 Interestingly, the effect of ASA was related to inhibition of collagen‐induced platelet shape changes, which frequently occur even when platelets are exposed to minimal degrees of activation that cannot result in aggregation or adhesion, such as suboptimal concentrations of platelet agonists or shear stress.46 The effect of ASA on platelet shape changes correlated with its effect on MLC phosphorylation, a key protein player in the process.37, 38, 39 ASA pretreatment also inhibited shape change in response to a suboptimal concentration of collagen, but not thrombin, and in the presence or absence of CD40L.This indicates that ASA attenuates collagen signaling through inhibition of MLC. This was further confirmed with ML7, a selective inhibitor of MLC kinase, whereby we observed that MLC inhibition does not interfere with sCD40L signaling. Furthermore, ML7 was able to inhibit the potentiating action of sCD40L in response to suboptimal doses of collagen and thrombin. In fact, MLC is major player in platelet activation and aggregation.47, 48 However, ASA had no effect on MLC activation in thrombin‐stimulated platelets, which translated to maintained shape change and sCD40L‐potentiation action in the aggregation process. This could be explained by the ability of thrombin to induce shape change in a thromboxane A2–independent manner. By measuring thromboxane B2 secretion, we observed that sCD40L is capable of enhancing thromboxane A2 secretion in response to suboptimal doses of agonists. Thus, CD40L, aside from being able to enhance platelet alpha‐granule release,24 can also amplify thromboxane A2 secretion. This observation is consistent with the results of a previous study where other platelet primers also enhanced thromboxane A2 secretion.34 Interestingly, following inhibition with ASA, sCD40L maintains its ability to trigger platelet aggregation in response to suboptimal doses of thrombin indicating that the priming action of sCD40L is independent of thromboxane A2. In fact, one of the proposed mechanisms by which ASA resistance develops is via activation of alternative signaling pathways that are thromboxane A2 independent.49 Also, our results demonstrate that this resistance may be explained by the fact that ASA does not affect sCD40L signaling. Indeed, ASA seems to have an indirect effect on preventing sCD40L potentiation action in platelet aggregation; as shown, unlike thrombin, which induces thromboxane A2–independent aggregation, collagen's action, which is thromboxane A2 dependent, is inhibited by ASA.50 Furthermore, it has been reported that ASA inhibits sCD40L release in collagen‐stimulated platelets of patients undergoing ASA treatments but does not affect the release of sCD40L when platelets of those patients are stimulated with a protease‐activated receptor agonist, which acts similarly to thrombin.51, 52 In addition, it has been shown that, in response to a protease‐activated receptor agonist, ASA does not influence the release of platelet inflammatory mediators (P‐selectin and sCD40L) in patients with acute stroke, while it does in healthy volunteers.53 In our in vitro study, the use of sCD40L‐stimulated platelets reflects a pathological state, similar to that observed in coronary or stroke patients, where ASA is less effective in response to thrombin. Our results with thromboxane A2 production corroborate previous findings obtained with sCD40L, as ASA inhibits the release of thromboxane A2 in collagen‐ but not thrombin‐stimulated platelets. This may also explain why the potentiation of aggregation in sCD40L‐primed platelets was inhibited after collagen but not thrombin stimulation. Our previous work showed that sCD40L signaling in platelets is transduced through 2 pathways: the CD40/TAK1/NF‐κB pathway31, 33 and the Ras‐related C3 botulinum toxin substrate 1/p38‐MAPK.30 These pathways remained activated when ASA‐treated platelets were stimulated with sCD40L, indicating that sCD40L‐mediated resistance to ASA is dependent on TAK1, NF‐κB, and p38‐MAPK.
To translate our findings to a clinical setting, our results suggest that patients undergoing antiplatelet therapy that blocks thromboxane A2 formation are not totally protected from the platelet priming impact of sCD40L, whose elevated levels might reduce the efficacy of such therapies. Thus, revealing the priming mechanisms of the CD40L/CD40 axis in platelet function may help to identify molecular targets in its signaling pathways that may emerge as innovative preventive treatments in coronary patients with elevated circulating levels of sCD40L. The major clinical outcome is to improve the management of atherothrombotic events in patients with coronary artery disease that are nonresponsive or less responsive to conventional antiplatelet therapies. This will add significant value to the healthcare system beyond current prevention programs and existing traditional therapies in coronary artery disease. Indeed, prospective studies may be planned in patients with metabolic syndrome at high risk of coronary artery disease and presenting with high levels of circulating sCD40L. In addition, it would be also beneficial to surmise finding a subgroup of patients who are nonresponsive or less responsive to antiplatelet therapies and whose platelets can be manipulated in vitro with an experimental treatment that interferes with the sCD40L/CD40 axis. The receptor CD40, its adaptor protein tumor necrosis factor receptor‐associated factor 2, the downstream effectors TAK1/NF‐κB, and Ras‐related C3 botulinum toxin substrate/p38‐MAPK have been involved in the sCD40L/CD40 axis. However, the specific pathways that can be targeted pharmacologically in platelets without affecting other physiological functions are yet to be identified and characterized in further studies.
We recognize that this study has some limitations that should be mentioned, as previously described.33 sCD40L is released in humans in vivo in thrombotic conditions, although it attains circulating concentrations lower than those required in vitro to prime platelets. Indeed, at least 100 ng/mL of sCD40L was necessary to induce platelet NF‐κB activation, whereas the levels of sCD40L in the circulating blood are ≈1 to 5 ng/mL and increase 10‐fold up to 50 ng/mL in patients with diabetes mellitus or thrombotic events.54, 55 However, the level of sCD40L is predicted to be higher at the site of vascular injury. Indeed, the environment that forms in the gaps between aggregated platelets and the injured vessel wall might allow attaining active higher localized concentrations56 than those found in the circulation.
In conclusion, ASA does not affect platelet sCD40L signaling but prevents its effect on thromboxane A2 secretion and platelet aggregation in response to collagen, via a mechanism implying inhibition of platelet shape change and MLC phosphorylation. Elevated levels of sCD40L in the blood of coronary patients may attenuate ASA efficiency. Targeting sCD40L axis in platelets may, therefore, have a therapeutic potential in patients with elevated levels of CD40L and who are nonresponsive or less responsive to ASA.
Sources of Funding
This study was supported by the Research Foundation of the Montreal Heart Institute and the Heart and Stroke Foundation of Canada.
Disclosures
None.
Supporting information
Figure S1. Dose‐response curve of platelet aggregation in response to collagen and thrombin.
Figure S2. Washed human platelets (1000×106/mL) were treated with several concentrations of ML7 for 5 minutes at 37°C, and then stimulated with 1000 ng/mL of sCD40L for 10 minutes.
Acknowledgments
The authors thank all blood donors and the nursing department of the Montreal Heart Institute bio‐bank for their valuable technical support in blood sampling.
(J Am Heart Assoc. 2020;9:e013396 DOI: 10.1161/JAHA.119.013396.)
References
- 1. Thomas MR, Storey RF. The role of platelets in inflammation. Thromb Haemost. 2015;114:449–458. [DOI] [PubMed] [Google Scholar]
- 2. Willoughby S, Holmes A, Loscalzo J. Platelets and cardiovascular disease. Eur J Cardiovasc Nurs. 2002;1:273–288. [DOI] [PubMed] [Google Scholar]
- 3. Lozano R, Naghavi M, Foreman K, Lim S, Shibuya K, Aboyans V, Abraham J, Adair T, Aggarwal R, Ahn SY, Alvarado M, Anderson HR, Anderson LM, Andrews KG, Atkinson C, Baddour LM, Barker‐Collo S, Bartels DH, Bell ML, Benjamin EJ, Bennett D, Bhalla K, Bikbov B, Bin Abdulhak A, Birbeck G, Blyth F, Bolliger I, Boufous S, Bucello C, Burch M, Burney P, Carapetis J, Chen H, Chou D, Chugh SS, Coffeng LE, Colan SD, Colquhoun S, Colson KE, Condon J, Connor MD, Cooper LT, Corriere M, Cortinovis M, de Vaccaro KC, Couser W, Cowie BC, Criqui MH, Cross M, Dabhadkar KC, Dahodwala N, De Leo D, Degenhardt L, Delossantos A, Denenberg J, Des Jarlais DC, Dharmaratne SD, Dorsey ER, Driscoll T, Duber H, Ebel B, Erwin PJ, Espindola P, Ezzati M, Feigin V, Flaxman AD, Forouzanfar MH, Fowkes FG, Franklin R, Fransen M, Freeman MK, Gabriel SE, Gakidou E, Gaspari F, Gillum RF, Gonzalez‐Medina D, Halasa YA, Haring D, Harrison JE, Havmoeller R, Hay RJ, Hoen B, Hotez PJ, Hoy D, Jacobsen KH, James SL, Jasrasaria R, Jayaraman S, Johns N, Karthikeyan G, Kassebaum N, Keren A, Khoo JP, Knowlton LM, Kobusingye O, Koranteng A, Krishnamurthi R, Lipnick M, Lipshultz SE, Ohno SL, Mabweijano J, MacIntyre MF, Mallinger L, March L, Marks GB, Marks R, Matsumori A, Matzopoulos R, Mayosi BM, McAnulty JH, McDermott MM, McGrath J, Mensah GA, Merriman TR, Michaud C, Miller M, Miller TR, Mock C, Mocumbi AO, Mokdad AA, Moran A, Mulholland K, Nair MN, Naldi L, Narayan KM, Nasseri K, Norman P, O'Donnell M, Omer SB, Ortblad K, Osborne R, Ozgediz D, Pahari B, Pandian JD, Rivero AP, Padilla RP, Perez‐Ruiz F, Perico N, Phillips D, Pierce K, Pope CA III, Porrini E, Pourmalek F, Raju M, Ranganathan D, Rehm JT, Rein DB, Remuzzi G, Rivara FP, Roberts T, De Leon FR, Rosenfeld LC, Rushton L, Sacco RL, Salomon JA, Sampson U, Sanman E, Schwebel DC, Segui‐Gomez M, Shepard DS, Singh D, Singleton J, Sliwa K, Smith E, Steer A, Taylor JA, Thomas B, Tleyjeh IM, Towbin JA, Truelsen T, Undurraga EA, Venketasubramanian N, Vijayakumar L, Vos T, Wagner GR, Wang M, Wang W, Watt K, Weinstock MA, Weintraub R, Wilkinson JD, Woolf AD, Wulf S, Yeh PH, Yip P, Zabetian A, Zheng ZJ, Lopez AD, Murray CJ, AlMazroa MA, Memish ZA. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet. 2012;380:2095–2128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cheng‐Ching E, Samaniego EA, Naravetla BR, Zaidat OO, Hussain MS. Update on pharmacology of antiplatelets, anticoagulants, and thrombolytics. Neurology. 2012;79:S68–S76. [DOI] [PubMed] [Google Scholar]
- 5. Antithrombotic Trialists’ Collaboration . Collaborative meta‐analysis of randomised trials of antiplatelet therapy for prevention of death, myocardial infarction, and stroke in high risk patients. BMJ. 2002;324:71–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Elwood PC, Hughes C, O'Brien JR. Platelets, aspirin, and cardiovascular disease. Postgrad Med J. 1998;74:587–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Antithrombotic Trialists’ Collaboration , Baigent C, Blackwell L, Collins R, Emberson J, Godwin J, Peto R, Buring J, Hennekens C, Kearney P, Meade T, Patrono C, Roncaglioni MC, Zanchetti A. Aspirin in the primary and secondary prevention of vascular disease: collaborative meta‐analysis of individual participant data from randomised trials. Lancet. 2009;373:1849–1860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Roth GJ, Majerus PW. The mechanism of the effect of aspirin on human platelets. I. Acetylation of a particulate fraction protein. J Clin Invest. 1975;56:624–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Floyd CN, Ferro A. Mechanisms of aspirin resistance. Pharmacol Ther. 2014;141:69–78. [DOI] [PubMed] [Google Scholar]
- 10. Halushka MK, Walker LP, Halushka PV. Genetic variation in cyclooxygenase 1: effects on response to aspirin. Clin Pharmacol Ther. 2003;73:122–130. [DOI] [PubMed] [Google Scholar]
- 11. Cambria‐Kiely JA, Gandhi PJ. Aspirin resistance and genetic polymorphisms. J Thromb Thrombolysis. 2002;14:51–58. [DOI] [PubMed] [Google Scholar]
- 12. Szczeklik A, Undas A, Sanak M, Frolow M, Wegrzyn W. Relationship between bleeding time, aspirin and the PlA1/A2 polymorphism of platelet glycoprotein IIIa. Br J Haematol. 2000;110:965–967. [DOI] [PubMed] [Google Scholar]
- 13. Andrioli G, Minuz P, Solero P, Pincelli S, Ortolani R, Lussignoli S, Bellavite P. Defective platelet response to arachidonic acid and thromboxane A(2) in subjects with Pl(A2) polymorphism of beta(3) subunit (glycoprotein IIIa). Br J Haematol. 2000;110:911–918. [DOI] [PubMed] [Google Scholar]
- 14. Goodman T, Sharma P, Ferro A. The genetics of aspirin resistance. Int J Clin Pract. 2007;61:826–834. [DOI] [PubMed] [Google Scholar]
- 15. Ross R. Atherosclerosis–an inflammatory disease. N Engl J Med. 1999;340:115–126. [DOI] [PubMed] [Google Scholar]
- 16. Hansson GK, Libby P. The immune response in atherosclerosis: a double‐edged sword. Nat Rev Immunol. 2006;6:508–519. [DOI] [PubMed] [Google Scholar]
- 17. Dichiara J, Bliden KP, Tantry US, Chaganti SK, Kreutz RP, Gesheff TB, Kreutz Y, Gurbel PA. Platelet function measured by VerifyNow identifies generalized high platelet reactivity in aspirin treated patients. Platelets. 2007;18:414–423. [DOI] [PubMed] [Google Scholar]
- 18. Macchi L, Christiaens L, Brabant S, Sorel N, Allal J, Mauco G, Brizard A. Resistance to aspirin in vitro is associated with increased platelet sensitivity to adenosine diphosphate. Thromb Res. 2002;107:45–49. [DOI] [PubMed] [Google Scholar]
- 19. Stolla MC, Li D, Lu L, Woulfe DS. Enhanced platelet activity and thrombosis in a murine model of type I diabetes are partially insulin‐like growth factor 1‐dependent and phosphoinositide 3‐kinase‐dependent. J Thromb Haemost. 2013;11:919–929. [DOI] [PubMed] [Google Scholar]
- 20. Lupia E, Bosco O, Mariano F, Dondi AE, Goffi A, Spatola T, Cuccurullo A, Tizzani P, Brondino G, Stella M, Montrucchio G. Elevated thrombopoietin in plasma of burned patients without and with sepsis enhances platelet activation. J Thromb Haemost. 2009;7:1000–1008. [DOI] [PubMed] [Google Scholar]
- 21. Angiolillo DJ, Fernandez‐Ortiz A, Bernardo E, Ramirez C, Sabate M, Jimenez‐Quevedo P, Hernandez R, Moreno R, Escaned J, Alfonso F, Banuelos C, Costa MA, Bass TA, Macaya C. Platelet function profiles in patients with type 2 diabetes and coronary artery disease on combined aspirin and clopidogrel treatment. Diabetes. 2005;54:2430–2435. [DOI] [PubMed] [Google Scholar]
- 22. Gresele P, Falcinelli E, Momi S. Potentiation and priming of platelet activation: a potential target for antiplatelet therapy. Trends Pharmacol Sci. 2008;29:352–360. [DOI] [PubMed] [Google Scholar]
- 23. Aukrust P, Muller F, Ueland T, Berget T, Aaser E, Brunsvig A, Solum NO, Forfang K, Froland SS, Gullestad L. Enhanced levels of soluble and membrane‐bound CD40 ligand in patients with unstable angina. Possible reflection of T lymphocyte and platelet involvement in the pathogenesis of acute coronary syndromes. Circulation. 1999;100:614–620. [DOI] [PubMed] [Google Scholar]
- 24. Inwald DP, McDowall A, Peters MJ, Callard RE, Klein NJ. CD40 is constitutively expressed on platelets and provides a novel mechanism for platelet activation. Circ Res. 2003;92:1041–1048. [DOI] [PubMed] [Google Scholar]
- 25. Hassan GS, Merhi Y, Mourad WM. CD154 and its receptors in inflammatory vascular pathologies. Trends Immunol. 2009;30:165–172. [DOI] [PubMed] [Google Scholar]
- 26. Hassan GS, Merhi Y, Mourad W. CD40 ligand: a neo‐inflammatory molecule in vascular diseases. Immunobiology. 2012;217:521–532. [DOI] [PubMed] [Google Scholar]
- 27. Lutgens E, Gorelik L, Daemen MJ, de Muinck ED, Grewal IS, Koteliansky VE, Flavell RA. Requirement for CD154 in the progression of atherosclerosis. Nat Med. 1999;5:1313–1316. [DOI] [PubMed] [Google Scholar]
- 28. van Kooten C, Banchereau J. CD40‐CD40 ligand. J Leukoc Biol. 2000;67:2–17. [DOI] [PubMed] [Google Scholar]
- 29. Henn V, Slupsky JR, Grafe M, Anagnostopoulos I, Forster R, Muller‐Berghaus G, Kroczek RA. CD40 ligand on activated platelets triggers an inflammatory reaction of endothelial cells. Nature. 1998;391:591–594. [DOI] [PubMed] [Google Scholar]
- 30. Yacoub D, Hachem A, Theoret JF, Gillis MA, Mourad W, Merhi Y. Enhanced levels of soluble CD40 ligand exacerbate platelet aggregation and thrombus formation through a CD40‐dependent tumor necrosis factor receptor‐associated factor‐2/Rac1/p38 mitogen‐activated protein kinase signaling pathway. Arterioscler Thromb Vasc Biol. 2010;30:2424–2433. [DOI] [PubMed] [Google Scholar]
- 31. Hachem A, Yacoub D, Zaid Y, Mourad W, Merhi Y. Involvement of nuclear factor kappaB in platelet CD40 signaling. Biochem Biophys Res Commun. 2012;425:58–63. [DOI] [PubMed] [Google Scholar]
- 32. Mussbacher M, Salzmann M, Brostjan C, Hoesel B, Schoergenhofer C, Datler H, Hohensinner P, Basilio J, Petzelbauer P, Assinger A, Schmid JA. Cell type‐specific roles of NF‐kappaB linking inflammation and thrombosis. Front Immunol. 2019;10:85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kojok K, Akoum SE, Mohsen M, Mourad W, Merhi Y. CD40L priming of platelets via NF‐kappaB activation is CD40‐ and TAK1‐dependent. J Am Heart Assoc. 2018;7:e009636 DOI: 10.1161/JAHA.118.009636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Blair TA, Moore SF, Hers I. Circulating primers enhance platelet function and induce resistance to antiplatelet therapy. J Thromb Haemost. 2015;13:1479–1493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Meinders M, Kulu DI, van de Werken HJ, Hoogenboezem M, Janssen H, Brouwer RW, van Ijcken WF, Rijkers EJ, Demmers JA, Kruger I, van den Berg TK, Suske G, Gutierrez L, Philipsen S. Sp1/Sp3 transcription factors regulate hallmarks of megakaryocyte maturation and platelet formation and function. Blood. 2015;125:1957–1967. [DOI] [PubMed] [Google Scholar]
- 36. Yacoub D, Theoret JF, Villeneuve L, Abou‐Saleh H, Mourad W, Allen BG, Merhi Y. Essential role of protein kinase C delta in platelet signaling, alpha IIb beta 3 activation, and thromboxane A2 release. J Biol Chem. 2006;281:30024–30035. [DOI] [PubMed] [Google Scholar]
- 37. Siess W. Molecular mechanisms of platelet activation. Physiol Rev. 1989;69:58–178. [DOI] [PubMed] [Google Scholar]
- 38. Klages B, Brandt U, Simon MI, Schultz G, Offermanns S. Activation of G12/G13 results in shape change and Rho/Rho‐kinase‐mediated myosin light chain phosphorylation in mouse platelets. J Cell Biol. 1999;144:745–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lokeshwar VB, Bourguignon LY. The involvement of Ca2+ and myosin light chain kinase in collagen‐induced platelet activation. Cell Biol Int Rep. 1992;16:883–897. [DOI] [PubMed] [Google Scholar]
- 40. Best LC, Holland TK, Jones PB, Russell RG. The interrelationship between thromboxane biosynthesis, aggregation and 5‐hydroxytryptamine secretion in human platelets in vitro. Thromb Haemost. 1980;43:38–40. [PubMed] [Google Scholar]
- 41. Furie B, Furie BC. Mechanisms of thrombus formation. N Engl J Med. 2008;359:938–949. [DOI] [PubMed] [Google Scholar]
- 42. Davi G, Patrono C. Platelet activation and atherothrombosis. N Engl J Med. 2007;357:2482–2494. [DOI] [PubMed] [Google Scholar]
- 43. Loeffen R, van Oerle R, Leers MP, Kragten JA, Crijns H, Spronk HM, Ten Cate H. Factor XIa and thrombin generation are elevated in patients with acute coronary syndrome and predict recurrent cardiovascular events. PLoS One. 2016;11:e0158355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Manten A, de Winter RJ, Minnema MC, ten Cate H, Lijmer JG, Adams R, Peters RJ, van Deventer SJ. Procoagulant and proinflammatory activity in acute coronary syndromes. Cardiovasc Res. 1998;40:389–395. [DOI] [PubMed] [Google Scholar]
- 45. de Souza Brito F, Tricoci P. Novel anti‐platelet agents: focus on thrombin receptor antagonists. J Cardiovasc Transl Res. 2013;6:415–424. [DOI] [PubMed] [Google Scholar]
- 46. Shin EK, Park H, Noh JY, Lim KM, Chung JH. Platelet shape changes and cytoskeleton dynamics as novel therapeutic targets for anti‐thrombotic drugs. Biomol Ther (Seoul). 2017;25:223–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Toth‐Zsamboki E, Oury C, Cornelissen H, De Vos R, Vermylen J, Hoylaerts MF. P2X1‐mediated ERK2 activation amplifies the collagen‐induced platelet secretion by enhancing myosin light chain kinase activation. J Biol Chem. 2003;278:46661–46667. [DOI] [PubMed] [Google Scholar]
- 48. Hashimoto Y, Sasaki H, Togo M, Tsukamoto K, Horie Y, Fukata H, Watanabe T, Kurokawa K. Roles of myosin light‐chain kinase in platelet shape change and aggregation. Biochim Biophys Acta. 1994;1223:163–169. [DOI] [PubMed] [Google Scholar]
- 49. Topcuoglu MA, Arsava EM, Ay H. Antiplatelet resistance in stroke. Expert Rev Neurother. 2011;11:251–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lewis GP, Watts IS. Prostaglandin endoperoxides, thromboxane A2 and adenosine diphosphate in collagen‐induced aggregation of rabbit platelets. Br J Pharmacol. 1982;75:623–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Nannizzi‐Alaimo L, Alves VL, Phillips DR. Inhibitory effects of glycoprotein IIb/IIIa antagonists and aspirin on the release of soluble CD40 ligand during platelet stimulation. Circulation. 2003;107:1123–1128. [DOI] [PubMed] [Google Scholar]
- 52. Chandler AB, Earhart AD, Speich HE, Kueter TJ, Hansen J, White MM, Jennings LK. Regulation of CD40L (CD154) and CD62P (p‐selectin) surface expression upon GPIIb‐IIIa blockade of platelets from stable coronary artery disease patients. Thromb Res. 2010;125:44–52. [DOI] [PubMed] [Google Scholar]
- 53. Lukasik M, Dworacki G, Michalak S, Kufel‐Grabowska J, Golanski J, Watala C, Kozubski W. Aspirin treatment influences platelet‐related inflammatory biomarkers in healthy individuals but not in acute stroke patients. Thromb Res. 2011;128:e73–e80. [DOI] [PubMed] [Google Scholar]
- 54. Varo N, Vicent D, Libby P, Nuzzo R, Calle‐Pascual AL, Bernal MR, Fernandez‐Cruz A, Veves A, Jarolim P, Varo JJ, Goldfine A, Horton E, Schonbeck U. Elevated plasma levels of the atherogenic mediator soluble CD40 ligand in diabetic patients: a novel target of thiazolidinediones. Circulation. 2003;107:2664–2669. [DOI] [PubMed] [Google Scholar]
- 55. Fong SW, Few LL, See Too WC, Khoo BY, Nik Ibrahim NN, Yahaya SA, Yusof Z, Mohd Ali R, Abdul Rahman AR, Yvonne‐Tee GB. Systemic and coronary levels of CRP, MPO, sCD40L and PlGF in patients with coronary artery disease. BMC Res Notes. 2015;8:679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Brass LF, Zhu L, Stalker TJ. Novel therapeutic targets at the platelet vascular interface. Arterioscler Thromb Vasc Biol. 2008;28:s43–s50. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Dose‐response curve of platelet aggregation in response to collagen and thrombin.
Figure S2. Washed human platelets (1000×106/mL) were treated with several concentrations of ML7 for 5 minutes at 37°C, and then stimulated with 1000 ng/mL of sCD40L for 10 minutes.