

Abstract
Background
Hypothermia has been associated with therapeutic benefits including reduced mortality and better neurologic outcomes in survivors of cardiac arrest. However, undesirable side effects have been reported in patients undergoing coronary interventions. Using a large animal model of temperature management, we aimed to describe how temperature interferes with the coronary vasculature.
Methods and Results
Coronary hemodynamics and endothelial function were studied in 12 pigs at various core temperatures. Left circumflex coronary artery was challenged with intracoronary nitroglycerin, bradykinin, and adenosine at normothermia (38°C) and mild hypothermia (34°C), followed by either rewarming (38°C; n=6) or moderate hypothermia (MoHT; 32°C, n=6). Invasive coronary hemodynamics by Doppler wire revealed a slower coronary blood velocity at 32°C in the MoHT protocol (normothermia 20.2±11.2 cm/s versus mild hypothermia 18.7±4.3 cm/s versus MoHT 11.3±5.3 cm/s, P=0.007). MoHT time point was also associated with high values of hyperemic microvascular resistance (>3 mm Hg/cm per second) (normothermia 2.0±0.6 mm Hg/cm per second versus mild hypothermia 2.0±0.8 mm Hg/cm per second versus MoHT 3.4±1.6 mm Hg/cm per second, P=0.273). Assessment of coronary vasodilation by quantitative coronary analysis showed increased endothelium‐dependent (bradykinin) vasodilation at 32°C when compared with normothermia (normothermia 6.96% change versus mild hypothermia 9.01% change versus MoHT 25.42% change, P=0.044). Results from coronary reactivity in vitro were in agreement with angiography data and established that endothelium‐dependent relaxation in MoHT completely relies on NO production.
Conclusions
In this porcine model of temperature management, 34°C hypothermia and rewarming (38°C) did not affect coronary hemodynamics or endothelial function. However, 32°C hypothermia altered coronary vasculature physiology by slowing coronary blood flow, increasing microvascular resistance, and exacerbating endothelium‐dependent vasodilatory response.
Keywords: coronary physiology, endothelium, microcirculation, NO, therapeutic hypothermia
Subject Categories: Coronary Circulation, Endothelium/Vascular Type/Nitric Oxide, Hemodynamics, Physiology, Vascular Biology
Clinical Perspective
What Is New?
Coronary hemodynamics are influenced by temperature. Moderate hypothermia slows coronary blood velocity and augments microvascular resistance in healthy coronary vasculature.
Moderate hypothermia alters coronary endothelium function. By increasing NO, hypothermia modifies endothelium‐dependent vasodilatory potential in healthy coronary arteries.
What Are the Clinical Implications?
In patients undergoing targeted‐temperature management protocols, hypothermia‐driven alterations of coronary hemodynamics could hamper physiology‐guided coronary assessments of healthy vasculature.
At 32°C, NO‐mediated endothelium hyperreactivity could lead to an overestimation of endothelial‐dependent coronary vasodilatory capacity.
Introduction
For centuries, pathophysiological features of hypothermia have been studied, and used, for multiple clinical purposes.1 In recent decades, the influence of hypothermia on metabolic reactions and biologic processes has emerged as a promising therapeutic tool to endure ischemic injury.2 In particular, therapeutic hypothermia (TH) has been explored to ameliorate brain injury associated with ischemia and reperfusion after cardiac arrest.3 In this context, TH applied in survivors of out‐of‐hospital cardiac arrest (OHCA) was initially related to remarkable improvement in neurologic outcomes and mortality.4, 5 As the use of TH has spread into clinical practice and new clinical trials have been performed, uncertainties about the advantages of TH have emerged6, 7 along with the report of undesirable side effects.8 Vascular and blood disorders play a central role in TH‐related side effects and further study is needed to better understand the implications and limitations of TH in clinical practice.9 Relevant alterations in coronary vasculature require special consideration, as coronary syndromes are related to the majority of OHCA.10, 11 In this context, slow blood flow,12 endothelial dysfunction13 and thrombotic events including stent thrombosis8, 14 have been reported, giving rise to controversy about TH use in patients undergoing percutaneous coronary intervention (PCI).15
Coronary‐specific regulatory mechanisms16 and the presence of multiple variables affecting coronary vessels in patients with OHCA complicate efforts to elucidate the role and independent contribution of TH in the reported side effects. Based on this scenario, we aimed to study the pure effects of mild hypothermia (MiHT; 34°C), moderate hypothermia (MoHT; 32°C), and rewarming (38°C) on coronary hemodynamics and endothelial function in a large animal model of whole‐body temperature management.
Methods
The data that support the findings of this study are available from the corresponding author upon reasonable request.
All procedures were conducted following the European Directive 2010/63/UE and Spanish RD 53/2013 regulations related to the Guide for the Care and Use of Laboratory Animals. The study protocol was approved by the Animal Experimentation Ethics Committee of the University of Barcelona (approval reference number: DAAM 7842).
Study Design
Twelve Landrace x Large White female pigs (32.5±3.6 kg) were used to study the influence of temperature management on coronary hemodynamics and endothelial function. Animals were distributed in 2 protocols according to temperature challenge: normothermia (38°C) and MiHT (34°C), followed by either rewarming (38°C) (rewarming protocol; n=6) or MoHT (32°C) (MoHT protocol; n=6). At each temperature time point, coronary hemodynamics (blood velocity and pressure) and in vivo endothelial‐independent and ‐dependent vasodilatory function were studied, at rest and after intracoronary drug challenges. After completing each temperature protocol, undisturbed coronary arteries were collected for in vitro vascular reactivity study (Figure 1).
Figure 1.
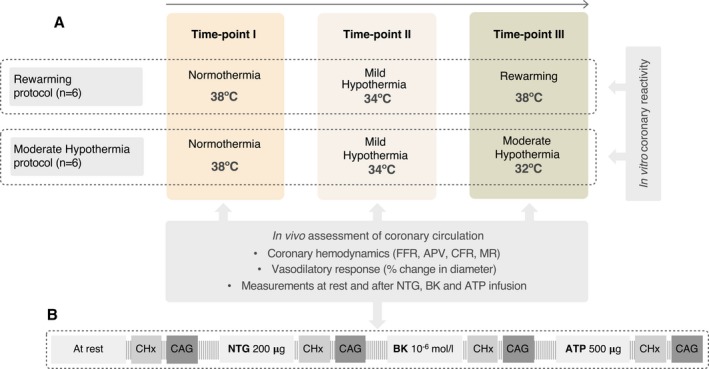
Flow chart illustrating study design with the assessments performed at each temperature time point (A) and the protocol of intracoronary drug administration (B). APV indicates average peak velocity; CAG, coronary angiography; CFR, coronary flow reserve; CHx, coronary hemodynamics; FFR fractional flow reserve; MR, microvascular resistance.
Animal Preparation and Coronary Catheterization
Fasting animals were sedated with intramuscular ketamine (6 mg/kg,), xylazine (3.5 mg/kg,) and midazolam (0.15 mg/kg). The marginal ear vein was cannulated and a bolus of sodium thiopental (5 mg/kg) was administered for anesthesia induction, then animals were orotracheally intubated. Volume‐controlled ventilation settings were modified according to animal weight to maintain normocapnia and normoxia. Anesthesia was maintained with continuous intravenous propofol (10 mg/kg per minute) and analgesia was provided with fentanyl (0.005–0.01 mg/kg per hour). Heart electric activity was assessed with 3‐lead ECG. Right femoral artery was surgically exposed and an 8F introducer was inserted for blood pressure measurement and access to coronary catheterization. At each temperature time point, left circumflex artery (LCx) was cannulated with a 6F guiding catheter using standard over‐the‐wire technique. The LCx was chosen for in vivo coronary assessments as its anatomy is well preserved in animals and has a larger diameter, compared with the left anterior descending artery, in the breed of pigs used in this study. Heparin was used to maintain appropriate anticoagulation throughout the experiment. A bolus of 9000 IU was administered before coronary catheterization, followed by repeated boluses of 3000 IU each hour. The right external jugular vein and right femoral vein were also surgically instrumented. Jugular access was used for rapid liquid infusions, while the femoral vein access was employed for continuous monitoring of the central blood temperature and pressure with a Swan‐Ganz catheter, placed in the inferior cava vein to avoid catheter interference when obtaining coronary angiographies. A small midline abdominal incision was made to visualize and probe the urine bladder with a 23G Foley catheter. Furosemide (bolus of 5 mg) was administered, when needed, to maintain a normal central venous pressure and slight positive fluid balance. Bolus of pancuronium bromide (0.15–0.3 mg/kg) was administered to treat hypothermia‐related shivering episodes. Heart rate, invasive arterial pressures, diuresis, and temperature were continuously monitored and recorded every 15 minutes throughout the experiment.
Whole‐Body Temperature Management
Central blood temperature was managed with a combined approach to induce MiHT and MoHT: superficial cooling (6 ice packs, placed on the thoracoabdominal region, both axillae, and groins) and invasive cooling (4°C crystalloids [0.9% NaCl; 500 mL/h], administered as previously described17). Animals in the rewarming protocol were covered with an external heating blanket over the entire body surface and 38°C crystalloid was administered (0.9% NaCl; 500 mL/h).
Intracoronary Drug Protocol
At each time point, the LCx artery was challenged with intracoronary nitroglycerin (2 mL of 100 μg/mL; rapid bolus) (Solnitrina, Kern Pharma), bradykinin (3 mL of 10−6 mol/L; 1 mL/min) (B3259, Merck KGaA), and ATP (3 mL of 166 μg/mL; rapid bolus) (Atepodín, Medix). Drugs were diluted in 0.9% saline and always administered in the same order, followed by 3 mL 0.9% saline (1 mL/min) washout. To avoid drug interferences, a blanking period based on the half‐life of each drug and previous tests (J. Bobi, et al, unpublished data, 2016) was established between administrations: 2 minutes between nitroglycerin and bradykinin, and 5 minutes between bradykinin and ATP administrations.
In Vivo Assessment of Coronary Hemodynamics
Coronary blood velocity and pressure were obtained from a Doppler flow/pressure wire (ComboWire, Volcano) and visualized in a ComboMap console. After cannulating the LCx with a 6F guiding catheter using standard over‐the‐wire techniques,18 the flow/pressure wire was advanced and retrogradely positioned in order to maximize stability of the Doppler signal through intracoronary drug challenges. At each time point, coronary blood velocity (cm/s) and pressure (mm Hg) were measured at rest and during the peak effect of the administered drugs. Parameters derived from velocity and pressure, including average peak velocity (cm/s), fractional flow reserve, coronary flow reserve, and microvascular resistances (MR; mm Hg/cm per second), were used to assess influence of temperature on coronary hemodynamics (Figure 2). All measurements were recorded and analyzed using dedicated software (ComboMap Software, Volcano Corporation).
Figure 2.
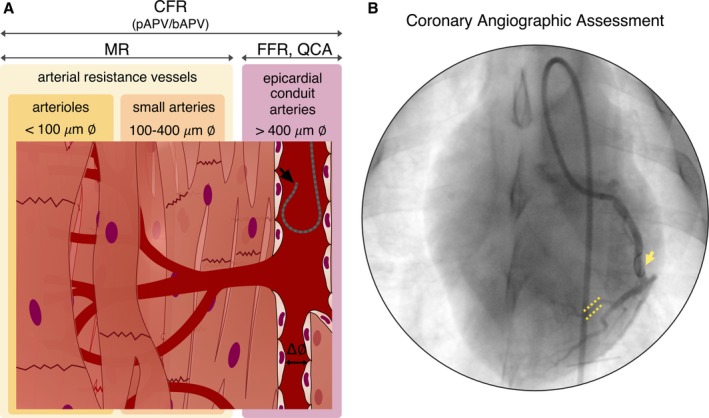
Invasive assessment of coronary hemodynamics by Doppler wire. A, Depiction of each coronary hemodynamic variable studied by Doppler wire (black arrow) and its relevance based on coronary vascular compartments. B, Representative coronary angiography showing the retrograde positioning of the Doppler wire in the left circumflex coronary artery (yellow arrow) and a selected branch used to study drug‐induced vasodilation by measuring the diameter before and after each drug challenge (yellow dotted lines). bAPV indicates basal average peak velocity; CFR, coronary flow reserve; ∅, diameter; FFR, fractional flow reserve; MR, microvascular resistance; pAPV, peak average peak velocity; QCA, quantitative coronary analysis.
In Vivo Coronary Vasodilation Assessed by Quantitative Coronary Angiography
Along with coronary hemodynamics, vasodilatory capacity of coronary arteries was used to study the effects of temperature on coronary endothelium. At each study time point, coronary angiographies of the LCx were obtained at rest and after endothelium‐independent (nitroglycerin) and ‐dependent (bradykinin) drug administration, and at pharmacologically induced hyperemia (ATP). In order to not interfere with the flow and pressure‐derived measurements, radiopaque contrast (Iomeprol 350 mg/mL, Iomeron 350, Bracco) was administered and images were obtained 3 seconds after each drug‐induced peak of flow detected by the Doppler wire. The diameters were then measured using quantitative coronary analysis (QAngio XA, Medis Medical Imaging Systems) by 2 blinded experienced investigators. A nondisturbed and distal‐to‐wire branch (1.0–2.0 mm in diameter) was selected on a baseline image by agreement of both investigators and then measured for all time points and drug challenges.
In Vitro Vascular Reactivity in Temperature‐Preconditioned Coronary Arteries
Immediately after MoHT or rewarming protocols, animals were euthanized, with an overdose of propofol and fentanyl followed by 60 mEq of potassium chloride, and the heart was excised. The left anterior descending coronary artery, which was not disturbed during in vivo coronary assessments, was carefully dissected, cleaned of surrounding tissue, and kept overnight in ice‐cold Leibovitz medium. Coronary rings (4 mm) were mounted in organ bath for isometric recording of tension as previously described.19 Briefly, 2 stainless‐steel holders (100 μm in diameter) were introduced through the arterial lumen and placed in a 20 mL tissue bath containing modified Krebs‐Henseleit (in mmol/L: NaCl 115; KCl 4.6; KH2PO4 1.2; MgCl2 1.2; CaCl2 2.5; NaHCO3 25; glucoses 11.1; EDTA 0.01, pH 7.3–7.4), kept at 37°C and aerated with 95% O2/5% CO2 for isometric force measurements.19 Optimal tension was assessed in preliminary experiments by subjecting arterial segments to different resting tensions and challenging with 100 mmol/L KCl. The vessels were stretched to 10 mN, washed, and allowed to equilibrate for 30 minutes. After equilibration, each ring was precontracted with the thromboxane A2 analogue U46619 (1 μmol/L) and challenged for endothelium‐dependent relaxation with bradykinin (100 pmol/L to 10 μmol/L) and ATP (10 nmol/L to 30 μmol/L). The influence of NO and prostaglandins on endothelium‐dependent relaxation was determined by treating coronary arteries with the nonselective NO synthase inhibitor (N(ω)‐nitro‐L‐arginine methyl ester [l‐NAME], 100 μmol/L) or with the cyclooxygenase inhibitor indomethacin (10 μmol/L). Incubation with inhibitors started 30 minutes before contraction with U46619 and was maintained throughout the experiments. Changes in isometric force were recorded using Chart version 3.4/s software and a MacLab/8e data acquisition system (ADInstruments). Sigmoidal curve fitting was performed on concentration‐response curve data using GraphPad Prism software (version 7.0; GraphPad Software). Maximal vasodilator responses were calculated and expressed relative to the maximal changes from the contraction produced by U46619 in each segment, which was determined as 0% relaxation.
Statistical Analysis
Statistical analysis was performed using SPSS software (version 23.0, IBM) and graphics dots were created on GraphPad Prism (version 5.00 for Windows). All data are represented as mean±SD. Because of interindividual variability of coronary vasculature, a univariate analysis of variance using a general linear model of repeated measures was used to study heart rate, mean arterial pressure, central venous pressure, coronary hemodynamics parameters, the diameter of a selected coronary segment before drug challenge, and diameter changes between temperature time points within each temperature protocol. All 3 time points were included in the analysis. The main effects were compared, when indicated, by Bonferroni post hoc test. Alterations in MR were also studied qualitatively by defining a cutoff value of 3 mm Hg/cm per second for microvascular dysfunction, based on the literature.20 In quantitative coronary analysis, intraclass correlation coefficient was used to assess interobserver reliability on diameter measurements. Changes in in vitro relaxation curves between protocols were studied by 2‐way ANOVA analysis. The area under the curve was calculated from each sigmoidal curve using GraphPad Prism software. The main effects were compared, when indicated, by ANOVA followed by Bonferroni post hoc test. Differences in milligrams of furosemide administration, elapsed time between time points, and fluid balance were assessed by Student t test.
Results
Coronary Catheterization and Temperature Management Protocol
All animals successfully completed both temperature protocols, as well as all intracoronary drug administrations at each temperature time point. Only 1 animal from each protocol presented transitory atrioventricular blockage after intracoronary adenosine (3 mL of 166 μg/mL, bolus). Nonetheless, animals returned to normal sinus rhythm without medical intervention. In both cases, intracoronary adenosine was repeated but dosage was reduced to half dose and no subsequent events were observed.
Core temperature was properly controlled by temperature‐conditioned crystalloid infusions combined with superficial cooling/heating in both protocols. The temperature target at each time point was constantly maintained within a ±0.5°C range during the 1‐hour stabilization and coronary assessments. The time elapsed between temperature time points only differed between the rewarming and MoHT protocols between time points II and III, as more minutes were needed to reach the rewarming target from the MiHT time point, compared with the MoHT target (time points II to III: rewarming 165.0±15.5 min versus MoHT 91.7±20.5 min [P<0.001] and time points II to III: rewarming 165.0±15.5 min versus MoHT 91.7±20.5 min [P<0.001]*). Heart rate behavior differed between protocols, since it was maintained during the rewarming protocol but tended to slow down at time points II and III in the MoHT protocol. Conversely, mean arterial pressure and central venous pressure were maintained over time in both protocols (Table 1). Furosemide requirements to achieve a slight positive fluid balance (rewarming 308.3±428.3 mL versus MoHT 275.0±361.6 mL, P=0.887) were similar in both protocols (rewarming 16.3±3.8 mg versus MoHT 14.1±3.5 mg, P=0.311).
Table 1.
Protocol Variables
| Time Point I | Time Point II | Time Point III | P Value | Bonferroni Multiple Comparisons | |||
|---|---|---|---|---|---|---|---|
| I to II | I to III | II to III | |||||
| Rewarming protocol (n=6) | Normothermia (38°C) | MiHT (34°C) | Rewarming (38°C) | ||||
| Heart rate, beats per min | 93.6±18.3 | 87.7±19.4 | 87.3±21.1 | 0.803 | … | … | … |
| Mean arterial pressure, mm Hg | 95.8±18.7 | 86.5±22.3 | 94.0±11.7 | 0.557 | … | … | … |
| Central venous pressure, mm Hg | 4.7±2.2 | 4.0±2.6 | 4.5±3.3 | 0.733 | … | … | … |
| MoHT protocol (n=6) | Normothermia (38°C) | MiHT (34°C) | MoHT (32°C) | ||||
| Heart rate, beats per min | 117.8±13.8 | 86.0±13.2 | 65.8±22.4 | 0.004 | 0.001 | 0.029 | … |
| Mean arterial pressure, mm Hg | 82.2±15.3 | 89.3±10.0 | 90.2±14.0 | 0.258 | … | … | … |
| Central venous pressure, mm Hg | 4.2±2.3 | 4.7±2.0 | 5.0±1.8 | 0.582 | … | … | … |
MiHT indicates mild hypothermia; MoHT, moderate hypothermia.
Influence of Temperature on Coronary Hemodynamics
The pure effects of temperature on coronary hemodynamics were studied from resting and hyperemic‐derived values. Variables obtained after nitroglycerin and bradykinin administrations were used to assess the impact of endothelium on the modulation of coronary hemodynamics by temperature. Coronary hemodynamics for each temperature time point, at rest and for each drug challenge, are summarized in Table 2.
Table 2.
Coronary Hemodynamics at Each Temperature Time Point
| Time Point I | Time Point II | Time Point III | P Value | Bonferroni Multiple Comparisons | |||
|---|---|---|---|---|---|---|---|
| I to II | I to III | II to III | |||||
| Rewarming protocol (n=6) | Normothermia (38°C) | MiHT (34°C) | Rewarming (38°C) | ||||
| At rest | |||||||
| APV, cm/s | 27.0±11.5 | 18.3±6.2 | 20.8±7.4 | 0.424 | … | … | … |
| MR, mm Hg/cm per s | 4.1±1.3 | 5.0±1.1 | 4.7±1.8 | 0.570 | … | … | … |
| Nitroglycerin | |||||||
| FFR | 0.99±0.08 | 0.97±0.04 | 0.97±0.02 | 0.831 | … | … | … |
| bAPV, cm/s | 25.3±10.4 | 18.2±4.8 | 19.0±5.8 | 0.363 | … | … | … |
| pAPV, cm/s | 53.7±22.2 | 43.8±11.8 | 47.3±8.5 | 0.574 | … | … | … |
| CFR | 2.2±0.7 | 2.6±1.0 | 2.6±0.4 | 0.321 | … | … | … |
| MR mm Hg/cm per s | 2.0±1.0 | 1.9±0.6 | 1.8±0.4 | 0.879 | … | … | … |
| Bradykinin | |||||||
| FFR | 0.94±0.08 | 0.99±0.03 | 0.98±0.02 | 0.512 | … | … | … |
| bAPV, cm/s | 19.0±7.7 | 14.8±5.6 | 12.3±3.8 | 0.052 | … | … | … |
| pAPV, cm/s | 40.1±17.5 | 33.5±11.5 | 37.5±9.4 | 0.483 | … | … | … |
| CFR | 2.2±0.6 | 2.2±0.5 | 3.2±0.8 | 0.125 | … | … | … |
| MR, mm Hg/cm per s | 2.4±1.1 | 2.6±0.9 | 2.6±1.2 | 0.757 | … | … | … |
| ATP | |||||||
| FFR | 0.94±0.05 | 0.96±0.02 | 0.95±0.06 | 0.646 | … | … | … |
| bAPV, cm/s | 14.5±3.8 | 12.3±4.1 | 11.8±3.4 | 0.055 | … | … | … |
| pAPV, cm/s | 57.7±5.9 | 49.7±5.2 | 53.0±8.6 | 0.100 | … | … | … |
| CFR | 4.2±0.9 | 4.5±1.2 | 4.8±0.9 | 0.112 | … | … | … |
| hMR, mm Hg/cm per s | 1.5±0.2 | 1.7±0.4 | 1.6±0.4 | 0.435 | … | … | … |
| MoHT protocol (n=6) | Normothermia (38°C) | MiHT (34°C) | MoHT (32°C) | ||||
| At rest | |||||||
| APV, cm/s | 20.2±11.2 | 18.7±4.3 | 11.3±5.3 | 0.007 | >0.999 | 0.060 | 0.208 |
| MR, mm Hg/cm per s | 4.9±2.2 | 5.9±2.3 | 9.6±5.5 | 0.182 | … | … | … |
| Nitroglycerin | |||||||
| FFR | 1.01±0.26 | 0.95±0.05 | 0.97±0.03 | 0.520 | … | … | … |
| bAPV, cm/s | 17.5±6.6 | 19.1±7.3 | 13.1±6.5 | 0.096 | … | … | … |
| pAPV, cm/s | 32.3±15.6 | 35.3±12.0 | 27.7±4.36 | 0.049 | >0.999 | >0.999 | 0.568 |
| CFR | 1.8±0.5 | 1.9±0.5 | 2.9±1.2 | 0.203 | … | … | … |
| MR, mm Hg/cm per s | 2.8±2.3 | 2.7±1.1 | 3.2±0.6 | 0.418 | … | … | … |
| Bradykinin | |||||||
| FFR | 1.05±0.17 | 0.97±0.34 | 0.97±0.34 | 0.650 | … | … | … |
| bAPV, cm/s | 16.0±5.5 | 12.7±1.4 | 10.0±4.5 | 0.170 | … | … | … |
| pAPV, cm/s | 39.0±20.6 | 33.5±10.2 | 39.0±22.7 | 0.888 | … | … | … |
| CFR | 2.4±0.8 | 2.7±0.8 | 4.5±2.9 | 0.138 | … | … | … |
| MR, mm Hg/cm per s | 2.3±0.9 | 2.6±0.6 | 2.6±1.5 | 0.760 | … | … | … |
| ATP | |||||||
| FFR | 0.97±0.44 | 0.90±0.09 | 0.96±0.04 | 0.205 | … | … | … |
| bAPV, cm/s | 14.0±4.9 | 13.8±4.5 | 9.0±4.0 | 0.119 | … | … | … |
| pAPV, cm/s | 37.7±17.2 | 43.2±14.3 | 32.2±9.9 | 0.226 | … | … | … |
| CFR | 2.6±0.6 | 3.2±0.3 | 3.9±1.2 | 0.181 | … | … | … |
| hMR, mm Hg/cm per s | 2.0±0.6 | 2.0±0.8 | 3.4±1.6 | 0.273 | … | … | … |
APV indicates average peak velocity; bAPV, baseline average peak velocity; CFR, coronary flow reserve; FFR, fractional flow reserve; hMR, hyperemic microvascular resistance; MR, microvascular resistance; MiHT, mild hypothermia; MoHT, moderate hypothermia; pAPV, peak average peak velocity.
Coronary blood velocity before intracoronary drug protocol remained similar across time points in the rewarming protocol, but blood velocity changed significantly in the MoHT protocol, showing a tendency to slow at 32°C compared with normothermia (*normothermia 20.2±11.2 cm/s versus MiHT 18.7±4.3 cm/s versus *MoHT 11.3±5.3 cm/s; P=0.007, *P=0.060). In contrast, hyperemic average peak velocity was similar across time points and hyperemic coronary flow reserve did not change in rewarming and showed a tendency to increase in MoHT (P=0.181).
After nitroglycerin or bradykinin administrations, no relevant differences in hemodynamics were observed between time points in any of the protocols. Only average peak velocity after nitroglycerin decreased among MoHT time points, but this was not associated with a modification of coronary flow reserve after nitroglycerin.
The MR, at rest or hyperemia, did not differ significantly across temperature time points in either experimental protocol. However, MR before any intracoronary drug administration increased at 32°C (normothermia 4.9±22 versus MiHT 5.9±2.3 versus MoHT 9.6±5.5 mm Hg/cm second, P=0.182) and, only in the rewarming protocol, hyperemic MR (hMR) remained below 3 mm Hg/cm per second across time points.
Hyperemic pressure–derived fractional flow reserve was maintained above 0.9 and was similar across different time points in both protocols. The administration of endothelium‐independent and ‐dependent vasodilators did not modify fractional flow reserve at any time point.
Temperature Modulation of In Vivo Coronary Endothelium‐Dependent Vasodilation
Undisturbed branches of LCx of similar diameter (rewarming 1.53±0.17 mm versus MoHT 1.61±0.21 mm, P=0.418) were selected to analyze vasodilatory responses by quantitative coronary analysis. The intraclass correlation coefficient for interobserver measurements of segment diameter was 0.946. The diameters of selected segments at rest did not change significantly between time points in either protocol (Table 3).
Table 3.
Diameters (mm) of Selected Undisturbed Branches of LCx for QCA
| Time Point I | Time Point II | Time point III | P Value | Bonferroni Multiple Comparisons | |||
|---|---|---|---|---|---|---|---|
| I to II | I to III | II to III | |||||
| Rewarming protocol (n=6) | 1.52±0.17 | 1.39±0.18 | 1.40±0.16 | 0.410 | … | … | … |
| MoHT protocol (n=6) | 1.62±0.21 | 1.63±0.25 | 1.44±0.25 | 0.088 | … | … | … |
LCx indicates left circumflex artery; MoHT, moderate hypothermia; QCA, quantitative coronary analysis.
The analysis of change in diameter after intracoronary administration of the endothelium‐independent drug (nitroglycerin) revealed similar coronary vasodilatory behavior across all time points in both the rewarming and MoHT protocols (Figure 3A). However, endothelium‐dependent vasodilation was influenced by hypothermia, as the percent change of coronary diameters after bradykinin administration changed between temperature time points in the MoHT protocol, increasing at 32°C compared with normothermia (*normothermia 7.0±8.9% change versus MiHT 9.0±8.5% change versus *MoHT 24.0±12.1% change; P=0.044, *P=0.025) (Figure 3B). No differences in the change of diameter induced at hyperemia (ATP) were observed in any protocol (Figure 3C).
Figure 3.
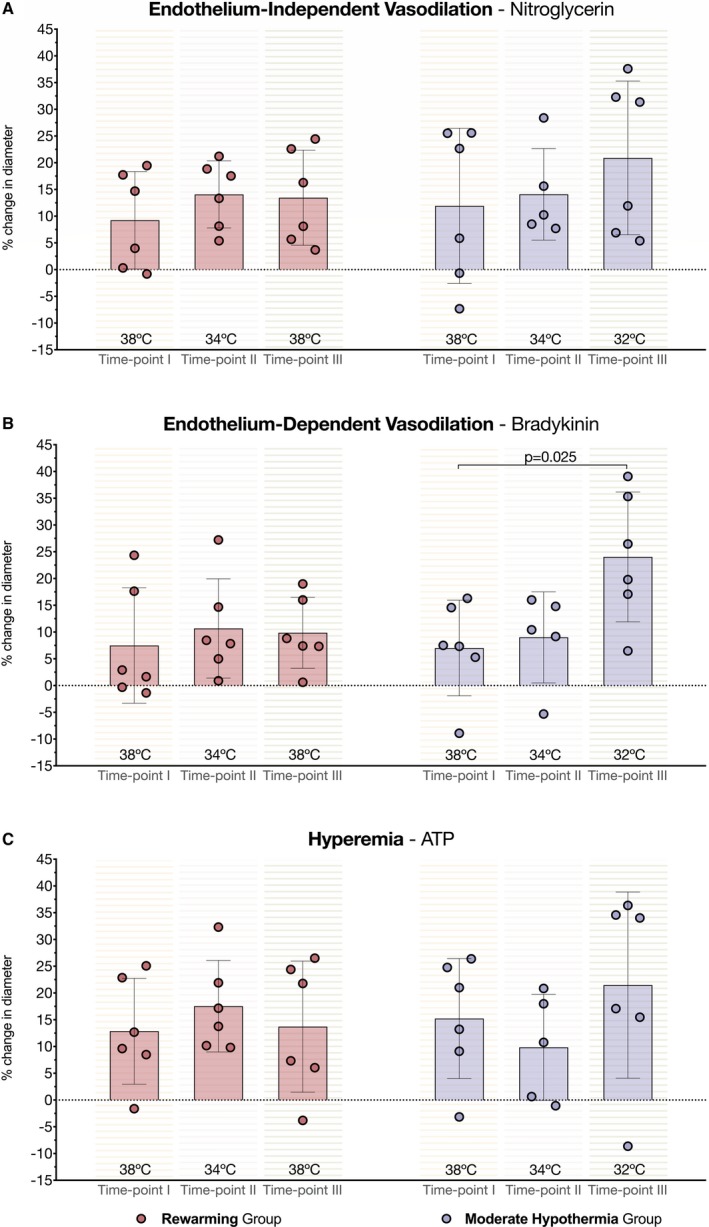
Effects of temperature on coronary vasodilation. A, Left and right parts of the bar chart show similar percent change in diameter after administration of endothelium‐independent vasodilator (nitroglycerin) along rewarming and moderate hypothermia (MoHT) protocols, respectively. B, Right bars (MoHT protocol) show an effect of temperature on endothelium‐dependent vasodilation with a significant increase in the percent change of diameter after administering bradykinin at 32°C time point. Left part of the bar chart (rewarming protocol) shows no differences in coronary vasodilation after bradykinin administration in the rewarming protocol. C, Bar chart depicts no statistical differences in the percent change in diameter at hyperemia (ATP) between time points in either protocol.
Increased Vasorelaxation of Hypothermia‐Preconditioned Coronary Rings Completely Relies on NO
After completing in vivo assessments, the influence of temperature on endothelial function was further determined in vitro in coronary rings isolated from undisturbed left anterior descending arteries. Five left anterior descending arteries from each temperature protocol were successfully harvested to assess vascular reactivity in the organ chamber. Parallel to the findings in vivo, rings from the MoHT protocol (isolated at 32°C) displayed a greater vasodilating potential when challenged with bradykinin, compared with the rewarming protocol (isolated at 38°C). Moreover, MoHT arteries showed increased ATP‐induced relaxation in vitro when compared with rewarming vessels (Figure 4A).
Figure 4.
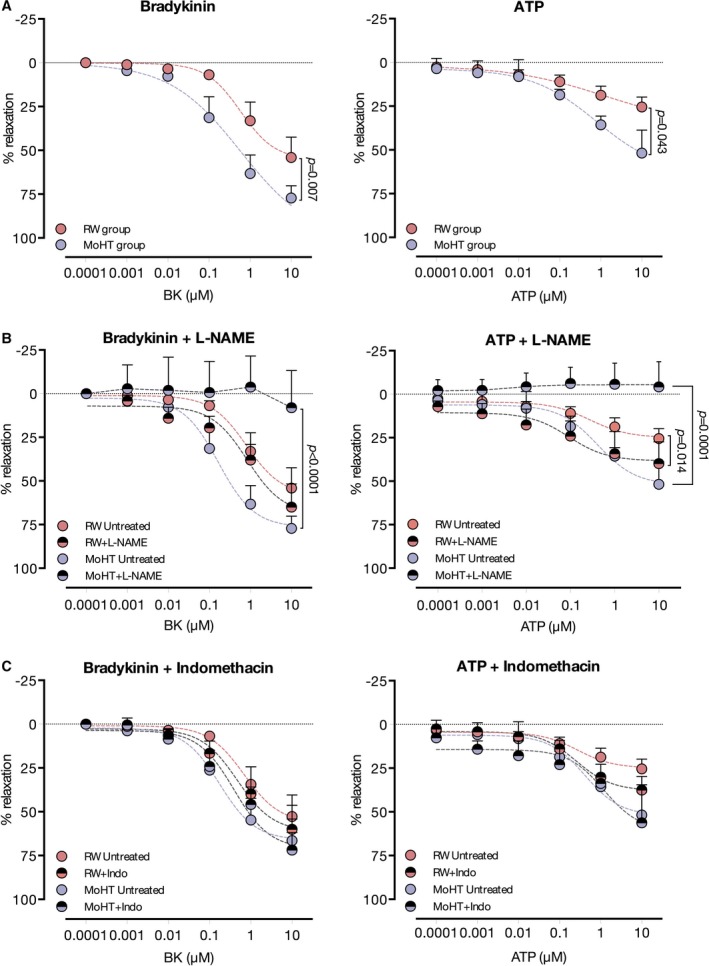
In vitro vasorelaxation of temperature preconditioned coronary rings. A, Left chart depicts increased relaxation of arteries from moderate hypothermia (MoHT), in comparison with the rewarming protocol, when challenged with bradykinin, a pure endothelium‐dependent vasodilator. ATP relaxation curves also showed a greater potential to relax in MoHT rings, compared with rewarming, as shown in the right chart. B, Treatment with the nonselective NO inhibitor, N(ω)‐nitro‐L‐arginine methyl ester (l‐NAME), resulted in a complete suppression of bradykinin‐ (left chart) and ATP‐induced (right chart) relaxation in coronary rings from the MoHT protocol. On the other hand, l‐NAME did not modify the bradykinin relaxation curve (left chart) and augmented the potential of relaxation induced by ATP (right chart) in rings from the rewarming protocol. C, Indomethacin, a cyclooxygenase inhibitor, did not modify either bradykinin‐ (left chart) or ATP‐induced (right chart) relaxation curves in either protocol.
l‐NAME and indomethacin treatments were used to study the magnitude of the contribution of NO and prostaglandins, respectively, to the endothelium‐dependent relaxation in rings from MoHT and rewarming protocols. When the influence of NO was removed by l‐NAME treatment, not only was the augmented relaxation promoted by bradykinin in MoHT rings eliminated but vasorelaxation was completely suppressed. The same pattern of response was observed when stimulating l‐NAME–treated MoHT arteries with ATP. This inhibitory effect was not observed in the rewarming protocol, where rings incubated with or without l‐NAME presented similar relaxation curves when challenged with bradykinin. Surprisingly, when the influence of NO was removed from ATP‐induced relaxation in the rewarming protocol, coronary segments treated with l‐NAME showed a slightly increased vasodilatory response (Figure 4B). On the other hand, treatment with indomethacin did not modify bradykinin‐ and ATP‐induced relaxation patterns in either the MoHT or rewarming segments (Figure 4C).
Discussion
In this large animal study, we analyzed the influence of the modulation of core temperature on coronary hemodynamics and endothelium. Our study found a significant adaptation of coronary vasculature to MoHT (Figure 5), as supported by our main findings: (1) MoHT was associated with a slower coronary blood velocity and an elevated hMR, and (2) MoHT induced a modification of the coronary endothelium behavior by increasing its vasodilatory potential. This was confirmed in vitro by coronary rings isolated at 32°C, which showed an increase in relaxation when challenged with an endothelium‐dependent vasodilator. In addition, in vitro vessel reactivity highlighted the key role of NO in this hypothermia‐induced increased vasodilation. On the other hand, MiHT and rewarming were not associated with any significant change in coronary vasculature.
Figure 5.
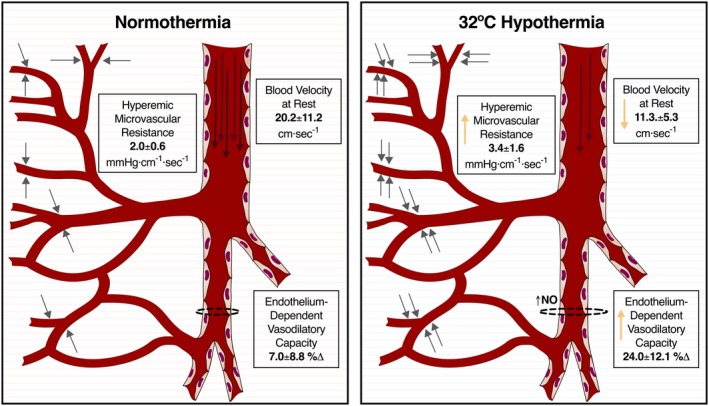
Main effects of moderate hypothermia (MoHT) in coronary vasculature. Briefly, MoHT (right) was associated with slowed coronary velocity (long arrows inside the coronary artery), augmented microvascular resistance (represented as grey short arrows pointing out small vessels), and increased endothelium‐derived vasodilatory capacity (dotted lines around the schematic representation of a coronary branch) associated with greater NO activity in comparison with reference parameters at normothermia (left).
Hemodynamic Adaption of Coronary Vasculature to Hypothermia
The feasibility, safety, and utility of coronary physiology assessment, invasive or not, in coronary syndromes has been reported20, 21, 22, 23 and is gaining impact in daily practice. This physiology‐guided approach to study the coronary vasculature adds relevant information to the visual examination of coronary angiographies, for example, by revealing epicardial vasomotor dysfunction or microvascular disease.24
Coronary blood flow velocity is frequently explored during both diagnostic and therapeutic coronary procedures. For example, the thrombolysis in myocardial infarction frame count is often used to estimate coronary blood flow after primary PCI. The usefulness of coronary blood flow assessment after PCI has been largely demonstrated, as slow flow and therefore high thrombolysis in myocardial infarction frame count values are associated with poor clinical outcomes in myocardial infarction.25, 26 In our study, coronary blood velocity at MoHT decreased to half the normothermic values. In the context of coronary events, different factors, such as incomplete revascularization or microvascular obstruction, might induce slow blood flow. Nevertheless, it is unlikely that these factors would have influenced the slow blood flow at MoHT observed in our experimental model with healthy coronary vessels. More likely, the slow flow could be related to temperature‐mediated changes in myocardium metabolism. In this sense, TH benefits have been attributed, in part, to the temperature‐dependent metabolism slowdown.27 As myocardial metabolism slows with decrease of temperature and oxygen consumption diminishes,28, 29 coronary blood flow might also decrease since coronary oxygen supply is directly related to myocardium oxygen demand.16 In this regard, further studies are needed to better describe the hypothermia‐induced reduction of myocardial metabolism and its influence on coronary blood flow; for example, to investigate whether coronary flow remains slower in hypothermia and, at the same time, with a pharmacologically increased cardiac metabolism. Although the mechanisms behind are not clear yet, hypothermia‐mediated decrease of blood flow is the result of a physiologic adaptation that could have further implications in clinical practice when assessing coronary blood velocity. Furthermore, sluggish coronary flow has been seen in patients with OHCA who have ST‐segment–elevation myocardial infarction undergoing PCI12 and has been proposed to favor prothrombotic states that may increase the risk of thromboembolic events such as stent thrombosis. The higher risk of stent thrombosis in patients with OHCA after PCI remains controversial14, 15, 30, 31 and has recently been attributed to the cardiac arrest pathophysiology8 rather than the actual effects of body temperature.32, 33
Although we observed a reduction in coronary blood velocity in MoHT at rest, it did not notably affect the capacity of coronary vasculature to increase flow (coronary flow reserve) at hyperemia or after the administration of endothelium‐dependent or ‐independent vasodilators. Nonetheless, the peak velocity after intracoronary administration of nitroglycerin was limited in MoHT. A possible explanation for this finding is that the effect of nitroglycerin, an NO donor, is more intense in epicardial coronary arteries compared with the microvasculature.34 The microvascular compartment substantially determines coronary flow35 and, therefore, the elevated MR that we observed at MoHT could be limiting the peak velocity in nitroglycerin‐induced vasodilation. In this context, the importance of the microvascular compartment in ischemic heart disease is gaining attention in interventional cardiology practice. Assessment of the microvascular compartment with hMR or the index of microvascular resistance,36 a thermodilution‐based method, has demonstrated its usefulness as a prognostic tool.20, 21, 22 Indeed, a cutoff value of 3 mm Hg/cm per second of hyperemic MR has been associated with microvascular injury and poor clinical outcomes in myocardial infarction.20 Interestingly, in our protocol, the hMR of nonischemic coronary vasculature exceeded the 3 mm Hg/cm per second cutoff value at MoHT. This finding might be relevant, as it would suggest that MoHT could hinder the diagnosis of microvascular dysfunction by masking normal hMR values, as we also proposed for the hypothermia‐induced slow blood velocity. These results raise further questions since previous preclinical studies have linked TH to reduced microvascular ischemic injury by limiting the no‐reflow areas37 and preserving microvasculature.38 To the best of our knowledge, the earlier studies did not perform direct measurements of coronary hMR; however, it would be interesting to investigate whether in the setting of MI, despite a higher preservation of microvasculature, hMR is also increased in hypothermia. To clarify the influence of hypothermia on coronary hemodynamics, and particularly the changes mediated by the microvascular compartment, further studies using preclinical models of acute myocardial infarction or epicardial stenosis, which more closely resemble the clinical scenario, are needed.
Another key finding of our study is the role of the degree of hypothermia in the described modifications of coronary hemodynamics. The changes in coronary blood flow and microvascular resistance were uniquely observed at 32°C hypothermia. Although we cannot completely rule out that coronary physiology remains unaltered in more extended 34°C hypothermia, our results and previous preclinical data38 suggest it could be reasonable to assume a dose dependency of the impact of hypothermia on coronary vasculature and myocardium.
Role of Endothelium on Coronary Modulation by Temperature
Endothelium is critical in the control of vascular function. In physiological conditions, endothelial cells release vasoactive mediators to maintain vessel homeostasis. In TH, endothelium has been studied to better understand its benefits and undesirable side effects.13 In this context, endothelium dysfunction can be defined as an inappropriate response mediated by a decrease in the production or bioavailability of NO via specific alterations.16 By assessing coronary vasodilation with endothelium‐dependent and ‐independent drugs at different temperatures, we investigated the potential endothelial dysfunction promoted by hypothermia. As a result, we found that coronary endothelium–dependent vasodilatory capacity was modified in hypothermia, but contrary to what would be expected, it showed greater potential to vasodilate at 32°C. Moreover, the vasodilatory function of the studied coronary vessels remained unaltered when challenged with an endothelium‐independent vasodilator at different temperatures, highlighting the importance of endothelium in the hypothermia‐mediated increase in vasodilation. Although the observed results do not meet the criteria of the original definition of endothelial dysfunction, they may indicate a direct influence of MoHT on the coronary endothelium by increasing its reactivity. On the other hand, we cannot discard the possibility that the aforementioned effect of hypothermia on endothelial cells leads to different outcomes in the presence of ischemic events, as previously reported in patients with OHCA and ST‐segment–elevation myocardial infarction who show endothelial impairment.13
Similar to in vivo results, in vitro MoHT‐preconditioned arteries challenged with bradykinin also displayed an endothelial vasodilatory hyperreactivity. Remarkably, the increased vasodilatory effect not only disappeared when adding an NO synthase inhibitor (l‐NAME), but bradykinin‐induced relaxation was completely eliminated. On the other hand, cyclooxygenase inhibition (indomethacin) did not significantly modify bradykinin relaxation properties. In isolated coronary arteries from healthy pigs, the endothelium‐dependent relaxation elicited by bradykinin is only partially dependent on NO or cyclooxygenase‐derived products, and mostly attributable to the endothelium‐derived hyperpolarizing factor.39, 40 Among the candidates, H2O2 has been raised as an endogenous endothelium‐derived hyperpolarizing factor that plays an important role in coronary circulation in animal models41 and humans.42 As we mentioned above, the decrease in temperature affects metabolism and oxygen supply in the myocardium, which may lead to a reduction of reactive oxygen species production, including H2O2.43 Although our study cannot establish a direct correlation, it is plausible that a decrease in H2O2 during MoHT shifts the contribution of endothelium‐derived hyperpolarizing factor/H2O2 to an NO‐mediated endothelium‐dependent vasodilation, which is recovered during rewarming. In our study, only rewarming‐preconditioned coronary rings displayed the above‐mentioned pattern and rings from MoHT protocol showed an apparent shift from endothelium‐derived hyperpolarizing factor–mediated to NO‐dependent vasodilation. These findings were supported by the results obtained from coronary rings challenged with ATP, which dilates porcine coronary arteries in an endothelium‐dependent, NO‐mediated manner.40 Although the higher in vivo vasodilatory capacity at 32°C after ATP administration did not reach statistical significance, MoHT‐preconditioned coronary rings in vitro challenged with ATP displayed greater relaxation compared with vessels from the rewarming protocol. Similar to what was observed with bradykinin, the l‐NAME incubation completely blocked the endothelium‐dependent relaxation in the MoHT protocol. These results suggest the essential role of NO in the regulation of vasculature in hypothermia. In this sense, our data support previous preclinical studies illustrating an upregulated expression of NO in hypothermia.3, 44, 45
Clinical Relevance
Currently, the routine use of urgent coronary angiography and PCI play a pivotal role in the therapeutic strategies of comatose patients following cardiac arrest.11, 46, 47, 48 According to European and American guidelines,49, 50 survivors of OHCA undergoing coronary interventions may be subjected to targeted temperature management protocols to maintain core temperature between 32°C and 36°C during the first 33 hours after cardiac arrest.
Although we did not consider the potential interactions of hypothermia with atherosclerotic coronary arteries, our findings that the coronary hemodynamics of healthy arteries were not the same in hypothermia as in normothermia might have clinical relevance. In particular, 32°C hypothermia could be masking reference values of some parameters used in the assessment of coronary physiology by slowing down the blood flow and increasing hMR, thereby hampering the analysis of these parameters for diagnostic purposes. If confirmed in clinical practice, this could imply a need to adjust the reference values of coronary hemodynamics in hypothermia. Moreover, the NO‐mediated endothelium hyperreactivity observed at 32°C could lead to an overestimation of endothelial‐dependent coronary vasodilatory capacity.
Limitations
The swine model for cardiovascular interventions is well accepted and provides relevant clinical data because of the anatomical and physiological similarities between human and porcine coronary vessels. However, the young age of the animals, the healthy status of the coronary vessels, and the lack of comorbidities represent a limitation when extrapolating results from this model to patients with OHCA undergoing coronary interventions. Another limitation of the study is the physiological temperature of pigs (38°C), which is higher than that of humans (36°C). Nonetheless, this animal model has been used in previous translational studies about hypothermia.38 Last, all of the in vivo assessments were performed after 1 hour of stable targeted temperature; thus, we do not know whether our findings are maintained after longer periods of 32°C targeted temperature management.
Conclusions
We observed that MoHT modified the behavior of coronary vasculature in a large animal model of temperature management. Coronary hemodynamics were modified at 32°C, characterized by slowed blood flow and elevated resistance of microvasculature. In addition, hypothermia altered coronary endothelium function, as evidenced by an exacerbated response to endothelium‐dependent vasodilator mediated by NO. Our findings highlight the relevance of the impact of hypothermia on healthy coronary vasculature and support the design of further studies to determine the magnitude of these interactions in clinical settings.
Sources of Funding
This work was supported by a grant from Fondo de Investigación Sanitario Instituto de Salud Carlos III (FIS PI13/01485) and the CERCA Programme of the Generalitat de Catalunya.
Disclosures
None.
Acknowledgments
We thank Elaine Lilly for English editing. This work was developed at the Centre de Recerca Biomèdica Cellex, Barcelona.
(J Am Heart Assoc. 2020;9:e014035 DOI: 10.1161/JAHA.119.014035.)
References
- 1. Alzaga AG, Cerdan M, Varon J. Therapeutic hypothermia. Resuscitation. 2006;70:369–380. [DOI] [PubMed] [Google Scholar]
- 2. Meybohm P, Gruenewald M, Albrecht M, Zacharowski KD, Lucius R, Zitta K, Koch A, Tran N, Scholz J, Bein B. Hypothermia and postconditioning after cardiopulmonary resuscitation reduce cardiac dysfunction by modulating inflammation, apoptosis and remodeling. PLoS One. 2009;4:e7588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gong P, Hua R, Zhang Y, Zhao H, Tang Z, Mei X, Zhang M, Cui J, Li C. Hypothermia‐induced neuroprotection is associated with reduced mitochondrial membrane permeability in a swine model of cardiac arrest. J Cereb Blood Flow Metab. 2013;33:928–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. HACA Study Group . Mild therapeutic hypothermia to improve the neurologic outcome after cardiac arrest. N Engl J Med. 2002;346:549–556. [DOI] [PubMed] [Google Scholar]
- 5. Bernard SA, Gray TW, Buist MD, Jones BM, Silvester W, Gutteridge G, Smith K. Treatment of comatose survivors of out‐of‐hospital cardiac arrest with induced hypothermia. N Engl J Med. 2002;346:557–563. [DOI] [PubMed] [Google Scholar]
- 6. Kim F, Nichol G, Maynard C, Hallstrom A, Kudenchuk PJ, Rea T, Copass MK, Carlbom D, Deem S, Longstreth WT, Olsufka M, Cobb LA. Effect of prehospital induction of mild hypothermia on survival and neurological status among adults with cardiac arrest a randomized clinical trial. J Am Med Assoc. 2014;311:45–52. [DOI] [PubMed] [Google Scholar]
- 7. Chan PS, Berg RA, Tang Y, Curtis LH, Spertus JA. Association between therapeutic hypothermia and survival after in‐hospital cardiac arrest. J Am Med Assoc. 2016;316:1375–1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Joffre J, Varenne O, Bougouin W, Rosencher J, Jean‐Paul M, Cariou A Stent thrombosis an increased adverse event after angioplasty following resuscitated cardiac arrest. Resuscitation. 2014;85:769–773. [DOI] [PubMed] [Google Scholar]
- 9. Staikou C, Paraskeva A, Drakos E, Anastassopoulou I, Papaioannou E, Donta I, Kontos M. Impact of graded hypothermia on coagulation and fibrinolysis. J Surg Res. 2011;167:125–130. [DOI] [PubMed] [Google Scholar]
- 10. Wijesekera VA, Mullany DV, Tjahjadi CA, Walters DL. Routine angiography in survivors of out of hospital cardiac arrest with return of spontaneous circulation: a single site registry. BMC Cardiovasc Disord. 2014;14:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ko E, Shin JK, Cha WC, Park JH, Lee TR, Yoon H, Lee G, Hwang SY, Shin TG, Sim MS, Jo IJ, Rhee JE, Song KJ, Jeong YK, Do Shin S, Choi JH. Coronary angiography is related to improved clinical outcome of out‐of‐hospital cardiac arrest with initial non‐shockable rhythm. PLoS One. 2017;12:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Regueiro A, Freixa X, Heras M, Penela D, Fernández‐Rodríguez D, Brugaletta S, Martín‐Yuste V, Masotti M, Sabaté M. Impact of therapeutic hypothermia on coronary flow. Int J Cardiol. 2014;172:228–229. [DOI] [PubMed] [Google Scholar]
- 13. Brugaletta S, Scalone G, Dantas AP, Ortega‐Paz L, Garabito M, Roqué M, Martin V, Masotti M, Freixa X, Sabaté M. Endothelial function impairment in STEMI patients with out‐of‐hospital cardiac arrest under therapeutic hypothermia treatment. Int J Cardiol. 2017;232:70–75. [DOI] [PubMed] [Google Scholar]
- 14. Penela D, Magaldi M, Fontanals J, Martin V, Regueiro A, Ortiz JT, Bosch X, Sabaté M, Heras M. Hypothermia in acute coronary syndrome: brain salvage versus stent thrombosis? J Am Coll Cardiol. 2013;61:686–687. [DOI] [PubMed] [Google Scholar]
- 15. Rosillo SO, Lopez‐de‐Sa E, Iniesta AM, de Torres F, Del Prado S, Rey JR, Armada E, Moreno R, López‐Sendón JL. Is therapeutic hypothermia a risk factor for stent thrombosis? J Am Coll Cardiol. 2013;63:939–940. [DOI] [PubMed] [Google Scholar]
- 16. Goodwill AG, Dick GM, Kiel AM, Tune JD. Regulation of coronary blood flow. Compr Physiol. 2017;7:321–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Larsson IM, Wallin E, Rubertsson S. Cold saline infusion and ice packs alone are effective in inducing and maintaining therapeutic hypothermia after cardiac arrest. Resuscitation. 2010;81:15–19. [DOI] [PubMed] [Google Scholar]
- 18. Bobi J, Solanes N, Fernández‐Jiménez R, Galán‐Arriola C, Dantas AP, Fernández‐Friera L, Gálvez‐Montón C, Rigol‐Monzó E, Agüero J, Ramírez J, Roqué M, Bayés‐Genís A, Sánchez‐González J, García‐Álvarez A, Sabaté M, Roura S, Ibáñez B, Rigol M. Intracoronary administration of allogeneic adipose tissue‐derived mesenchymal stem cells improves myocardial perfusion but not left ventricle function, in a translational model of acute myocardial infarction. J Am Heart Assoc. 2017;6:e005771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Costa TJ, Ceravolo GS, Santos RA, De Oliveira MA, Araújo PX, Giaquinto LR, Tostes RC, Akamine EH, Fortes ZB, Dantas AP, Carvalho MH. Association of testosterone with estrogen abolishes the beneficial effects of estrogen treatment by increasing ROS generation in aorta endothelial cells. Am J Physiol Hear Circ Physiol. 2015;723–732. [DOI] [PubMed] [Google Scholar]
- 20. Waard GA, Fahrni G, De Wit D, Kitabata H, Williams R, Patel N, Teunissen PF, Van De Ven PM, Umman S, Knaapen P, Perera D, Akasaka T, Sezer M, Kharbanda RK, Van Royen N. Hyperaemic microvascular resistance predicts clinical outcome and microvascular injury after myocardial infarction. Heart. 2018;104:127–134. [DOI] [PubMed] [Google Scholar]
- 21. Teunissen PF, De Waard GA, Hollander MR, Robbers LF, Danad I, Biesbroek PS, Amier RP, Echavarría‐Pinto M, Quirós A, Broyd C, Heymans MW, Nijveldt R, Lammertsma AA, Raijmakers PG, Allaart CP, Lemkes JS, Appelman YE, Marques KM, Bronzwaer JG, Horrevoets AJ, Van Rossum AC, Escaned J, Beek AM, Knaapen P, Van Royen N. Doppler‐derived intracoronary physiology indices predict the occurrence of microvascular injury and microvascular perfusion deficits after angiographically successful primary percutaneous coronary intervention. Circ Cardiovasc Interv. 2015;8:1–11. [DOI] [PubMed] [Google Scholar]
- 22. Kitabata H, Kubo T, Ishibashi K, Komukai K, Tanimoto T, Ino Y, Kashiwagi M, Ozaki Y, Shiono Y, Shimamura K, Orii M, Hirata K, Tanaka A, Imanishi T, Akasaka T. Prognostic value of microvascular resistance index immediately after primary percutaneous coronary intervention on left ventricular remodeling in patients with reperfused anterior acute ST‐segment elevation myocardial infarction. JACC Cardiovasc Interv. 2013;6:1046–1054. [DOI] [PubMed] [Google Scholar]
- 23. Smits PC, Abdel‐Wahab M, Neumann F‐J, Boxma‐de Klerk BM, Lunde K, Schotborgh CE, Piroth Z, Horak D, Wlodarczak A, Ong PJ, Hambrecht R, Angerås O, Richardt G, Omerovic E. Fractional flow reserve‐guided multivessel angioplasty in myocardial infarction. N Engl J Med. 2017;376:1234–1244. [DOI] [PubMed] [Google Scholar]
- 24. Díez‐delhoyo F, Gutiérrez‐Ibañes E, Sanz‐Ruiz R, Vázquez‐Álvarez ME, González Saldívar H, Rivera Juárez A, Sarnago F, Martínez‐Sellés M, Bermejo J, Soriano J, Elízaga J, Fernández‐Avilés F. Prevalence of microvascular and endothelial dysfunction in the nonculprit territory in patients with acute myocardial infarction. Circ Cardiovasc Interv. 2019;12:e007257. [DOI] [PubMed] [Google Scholar]
- 25. Haager PK, Christott P, Heussen N, Lepper W, Hanrath P, Hoffmann R. Prediction of clinical outcome after mechanical revascularization in acute myocardial infarction by markers of myocardial reperfusion. J Am Coll Cardiol. 2003;41:532–538. [DOI] [PubMed] [Google Scholar]
- 26. Karmpaliotis D, Turakhia MP, Kirtane AJ, Murphy SA, Kosmidou I, Morrow DA, Giugliano RP, Cannon CP, Antman EM, Braunwald E, Gibson CM. Sequential risk stratification using TIMI risk score and TIMI flow grade among patients treated with fibrinolytic therapy for ST‐segment elevation acute myocardial infarction. Am J Cardiol. 2004;94:1113–1117. [DOI] [PubMed] [Google Scholar]
- 27. Nolan JP, Morley PT, Vanden Hoek TL, Hickey RW, Kloek WGJ, Billi J, Böttiger BW, Okada K, Reyes C, Shuster M, Steen PA, Weil MH, Wenzel V, Carli P, Atkins D. Therapeutic hypothermia after cardiac arrest. An advisory statement by the Advanced Life Support Task Force of the International Liaison Committee on Resuscitation. Resuscitation. 2003;57:231–235. [DOI] [PubMed] [Google Scholar]
- 28. Chitwood WR, Sink JD, Hill RC, Wechsler AS, Sabiston DC. The effects of hypothermia on myocardial oxygen consumption and transmural coronary blood flow in the potassium‐arrested heart. Ann Surg. 1979;190:106–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Matschke K, Knaut M, Kanig R, Mrowietz C, Hiebl B, Jung F. Influence of systemic hypothermia on the myocardial oxygen tension during extracorporeal circulation: comparative study in German Landrace pigs. Clin Hemorheol Microcirc. 2012;52:115–122. [DOI] [PubMed] [Google Scholar]
- 30. Jiménez‐Brítez G, Freixa X, Flores‐Umanzor E, San Antonio R, Caixal G, Garcia J, Hernandez‐Enriquez M, Andrea R, Regueiro A, Masotti M, Brugaletta S, Martin V, Sabaté M. Out‐of‐hospital cardiac arrest and stent thrombosis: ticagrelor versus clopidogrel in patients with primary percutaneous coronary intervention under mild therapeutic hypothermia. Resuscitation. 2017;114:141–145. [DOI] [PubMed] [Google Scholar]
- 31. García J, Jiménez‐Brítez G, Flores‐Umanzor E, Mendieta G, Freixa X, Sabaté M. Eventos trombóticos y hemorrágicos después de una intervención coronaria percutánea tras parada cardiaca extrahospitalaria con y sin hipotermia terapéutica. Rev Española Cardiol. 2019;72:433–435. [DOI] [PubMed] [Google Scholar]
- 32. Shah N, Chaudhary R, Mehta K, Agarwal V, Garg J, Freudenberger R, Jacobs L, Cox D, Kern KB, Patel N. Therapeutic hypothermia and stent thrombosis: a nationwide analysis. JACC Cardiovasc Interv. 2016;9:1801–1811. [DOI] [PubMed] [Google Scholar]
- 33. Casella G, Carinci V, Cavallo P, Guastaroba P, Pavesi PC, Pallotti MG, Sangiorgio P, Barbato G, Coniglio C, Iarussi B, Gordini G, Di Pasquale G. Combining therapeutic hypothermia and emergent coronary angiography in out‐of‐hospital cardiac arrest survivors: optimal post‐arrest care for the best patient. Eur Hear J Acute Cardiovasc Care. 2015;4:579–588. [DOI] [PubMed] [Google Scholar]
- 34. Duncker DJ, Bache RJ. Regulation of coronary blood flow during exercise. Physiol Rev. 2008;88:1009–1086. [DOI] [PubMed] [Google Scholar]
- 35. Duncker DJ, Kollerb A, Merkusa D, Canty JMJ. Regulation of coronary blood flow in health and ischemic heart disease. Prog Cardiovasc Dis. 2015;27:215–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Fearon WF, Kobayashi Y. Invasive assessment of the coronary microvasculature: the index of microcirculatory resistance. Circ Cardiovasc Interv. 2017;10:1–11. [DOI] [PubMed] [Google Scholar]
- 37. Hale SL, Herring MJ, Kloner RA. Delayed treatment with hypothermia protects against the no‐reflow phenomenon despite failure to reduce infarct size. J Am Heart Assoc. 2013;2:e004234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Dash R, Mitsutake Y, Pyun WB, Dawoud F, Lyons J, Tachibana A, Yahagi K, Matsuura Y, Kolodgie FD, Virmani R, McConnell MV, Illindala U, Ikeno F, Yeung A. Dose‐dependent cardioprotection of moderate (32°C) versus mild (35°C) therapeutic hypothermia in porcine acute myocardial infarction. JACC Cardiovasc Interv. 2018;11:181–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Matoba T, Shimokawa H, Morikawa K, Kubota H, Kunihiro I, Urakami‐Harasawa L, Mukai Y, Hirakawa Y, Akaike T, Takeshita A. Electron spin resonance detection of hydrogen peroxide as an endothelium‐derived hyperpolarizing factor in porcine coronary microvessels. Arterioscler Thromb Vasc Biol. 2003;23:1224–1230. [DOI] [PubMed] [Google Scholar]
- 40. Durand MJ, Gutterman DD. Diversity in mechanisms of endothelium‐dependent vasodilation in health and disease. Microcirculation. 2013;20:239–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Yada T, Shimokawa H, Hiramatsu O, Kajita T, Shigeto F, Goto M, Ogasawara Y, Kajiya F. Hydrogen peroxide, an endogenous endothelium‐derived hyperpolarizing factor, plays an important role in coronary autoregulation in vivo. Circulation. 2003;107:1040–1045. [DOI] [PubMed] [Google Scholar]
- 42. Liu Y, Bubolz AH, Mendoza S, Zhang DX, Gutterman DD. H2O2 is the transferrable factor mediating flow‐induced dilation in human coronary arterioles. Circ Res. 2011;108:566–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Polderman KH. Mechanisms of action, physiological effects, and complications of hypothermia. Crit Care Med. 2009;37:S186–S202. [DOI] [PubMed] [Google Scholar]
- 44. Mochizuki T, Yu S, Katoh T, Aoki K, Sato S. Cardioprotective effect of therapeutic hypothermia at 34°C against ischaemia/reperfusion injury mediated by PI3K and nitric oxide in a rat isolated heart model. Resuscitation. 2012;83:238–242. [DOI] [PubMed] [Google Scholar]
- 45. Shao ZH, Sharp WW, Wojcik KR, Li CQ, Han M, Chang WT, Ramachandran S, Li J, Hamann KJ, Vanden Hoek TL. Therapeutic hypothermia cardioprotection via Akt‐ and nitric oxide‐mediated attenuation of mitochondrial oxidants. Am J Physiol Circ Physiol. 2010;298:H2164–H2173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Nolan JP, Berg RA, Bernard S, Bobrow BJ, Callaway CW, Cronberg T, Koster RW, Kudenchuk PJ, Nichol G, Perkins GD, Rea TD, Sandroni C, Soar J, Sunde K, Cariou A. Intensive care medicine research agenda on cardiac arrest. Intensive Care Med. 2017;1–12. [DOI] [PubMed] [Google Scholar]
- 47. Akin M, Sieweke JT, Zauner F, Garcheva V, Tongers J, Napp LC, Friedrich L, Treptau J, Bahntje MU, Flierl U, Sedding DG, Bauersachs J, Schäfer A. Mortality in patients with out‐of‐hospital cardiac arrest undergoing a standardized protocol including therapeutic hypothermia and routine coronary angiography: experience from the HACORE registry. JACC Cardiovasc Interv. 2018;11:1811–1820. [DOI] [PubMed] [Google Scholar]
- 48. Yannopoulos D, Bartos JA, Aufderheide TP, Callaway CW, Deo R, Garcia S, Halperin HR, Kern KB, Kudenchuk PJ, Neumar RW, Raveendran G, Null N. The evolving role of the cardiac catheterization laboratory in the management of patients with out‐of‐hospital cardiac arrest: a scientific statement from the American Heart Association. Circulation. 2019;139:e1–e23. [DOI] [PubMed] [Google Scholar]
- 49. Soar J, Nolan JP, Böttiger BW, Perkins GD, Lott C, Carli P, Pellis T, Sandroni C, Skrifvars MB, Smith GB, Sunde K, Deakin CD, Koster RW, Monsieurs KG, Nikolaou NI. European resuscitation council guidelines for resuscitation 2015. section 3. adult advanced life support. Resuscitation. 2015;95:100–147. [DOI] [PubMed] [Google Scholar]
- 50. Neumar RW, Shuster M, Callaway CW, Gent LM, Atkins DL, Bhanji F, Brooks SC, De Caen AR, Donnino MW, Ferrer JME, Kleinman ME, Kronick SL, Lavonas EJ, Link MS, Mancini ME, Morrison LJ, O'Connor RE, Samson RA, Schexnayder SM, Singletary EM, Sinz EH, Travers AH, Wyckoff MH, Hazinski MF. Part 1: executive summary: 2015 American Heart Association guidelines update for cardiopulmonary resuscitation and emergency cardiovascular care. Circulation. 2015;132:S315–S367. [DOI] [PubMed] [Google Scholar]