

Abstract
β-Cell antigen recognition by autoreactive T cells is essential in type 1 diabetes (T1D) pathogenesis. Recently, insulin hybrid peptides (HIPs) were identified as strong agonists for CD4 diabetogenic T cells. Here, using BDC2.5 transgenic and NOD mice, we investigated T-cell recognition of the HIP2.5 epitope, which is a fusion of insulin C-peptide and chromogranin A (ChgA) fragments, and compared it with the WE14 and ChgA29–42 epitopes. We measured in situ two-dimensional affinity on individual live T cells from thymus, spleen, pancreatic lymph nodes, and islets before and after diabetes. Although preselection BDC2.5 thymocytes possess higher affinity than splenic BDC2.5 T cells for all three epitopes, peripheral splenic T cells maintained high affinity only to the HIP2.5 epitope. In polyclonal NOD mice, a high frequency (∼40%) of HIP2.5-specific islet T cells were identified at both prediabetic and diabetic stages comprising two distinct high- and low-affinity populations that differed in affinity by 100-fold. This high frequency of high- and low-affinity HIP2.5 T cells in the islets potentially represents a major risk factor in diabetes pathogenesis.
Introduction
Type 1 diabetes (T1D) is a complex T cell–mediated autoimmune disease. Consistent with the requirement of CD4 T cells for T1D development, HLA-DR4 and DQ8 (human) and I-Ag7 (murine) are key genetic risk factors (1,2). Despite great progress in T1D research, we still do not understand how CD4 T cells break tolerance to β-cell autoantigens and cause disease. Numerous MHC II–restricted epitopes have been identified from various islet proteins such as insulin and chromogranin A (ChgA), among others (3,4). Many corresponding diabetogenic CD4 T-cell receptors (TCRs) have been cloned, including the well-characterized BDC2.5 TCR that has been made into a transgenic (Tg) mouse model (5). Insulin and ChgA have surfaced as two prime autoantigen targets in NOD mice, since a point mutation in the insulin B chain (replacing a tyrosine at position 16 with an alanine, Y16A) (6) or deficiency of ChgA (ChgA−/−) (7) protects NOD mice from diabetes. Although ChgA−/− confers complete protection, the dominant epitope of T1D has remained controversial. Islet cells from wild-type NOD mice are potent stimulators of the BDC2.5 clone but completely lose stimulation on the ChgA−/− background (7). Two antigenic ChgA peptides have been identified, namely, WE14 (ChgA358–371) and ChgA29–42, but both are only minimally stimulatory to BDC2.5 Tg cells. More recently, a dominant strong agonist for the BDC2.5 TCR was discovered as a hybrid insulin peptide (HIP) fusion between an insulin C-peptide fragment and the MHC anchor residues from the WE14 (ChgA358–362) peptide (8). A series of HIPs were identified to be agonistic peptides for previously cloned CD4 TCRs from diabetic NOD mice. Importantly, HIPs also exist in humans (8). Therefore, the HIPs potentially represent an important new class of posttranslationally modified β-cell autoantigens that offer an exciting explanation for the protection from T1D found in ChgA-deficient mice.
A fundamental premise of T1D research is that β-cell autoantigen recognition by diabetogenic T cells initiates and perpetuates the disease. A clear understanding of autoantigen recognition may hold the key to the ultimate goal of specifically depleting or restoring tolerance of diabetogenic T cells and at the same time sparing the functioning T-cell repertoire. However, there is limited quantitative information on the binding kinetics (e.g., affinity) of the TCR–peptide-MHC (pMHC) interaction as opposed to the downstream less direct measures of functional readouts such as expression of activation markers and cytokine secretion. Tetramer staining is a popular assay for enumeration of β-cell autoantigen-specific T-cell populations, but it provides a measure of avidity and interacts with only the highest affinity TCRs. MHC class II tetramer binding therefore underestimates the number of antigen-specific T cells by as much as 10-fold (9,10). The TCR/CD3 protein complex is embedded in the two-dimensional (2D) cell membrane, as is their pMHC ligand. Recently, we and others applied novel 2D techniques to measure TCR-pMHC binding kinetics on live T cells and demonstrated a dramatic role of the cellular environment in T-cell antigen recognition (11–15); more importantly, such in situ 2D TCR kinetics, notably 2D affinity, correlates with T-cell functional responses and T-cell fate (13–17).
Here, we tracked 2D affinity of the insulin/ChgA HIP2.5 epitope recognized by the BDC2.5 transgenic T cells and polyclonal NOD CD4 T cells during thymocyte development and peripheral activation upon disease onset and compared it to two previously identified ChgA epitopes (i.e., WE14 and ChgA29–42). Our results showed that the HIP2.5 peptide possesses much higher affinity than WE14 and ChgA29–42. This was especially true in the periphery as the HIP2.5 epitope maintained high affinity in the spleen and islets. Our sensitive 2D assay also revealed a much higher percentage (∼40%) of HIP2.5-specific CD4 T cells than did HIP2.5 tetramer staining in NOD islets both before and after diabetes, demonstrating that there are many more HIP2.5-reactive T cells than previously realized (8).
Research Design and Methods
Mice
NOD and BDC2.5 TCR Tg mice were bred in the Emory University and University of Utah animal facilities and used between 5 and 20 weeks of age, with 9 weeks of age being the average age used. BDC2.5 retrogenic (Rg) mice were generated in the animal facility at St. Jude Children’s Research Hospital, as previously described, using retroviral producers encoding these TCRs (18). For BDC2.5 Rg mice, reconstitution was verified by flow cytometric analysis 4–5 weeks after bone marrow transplant, and healthy mice that exhibited engraftment and expression of the desired TCR were used for experiments. Mouse diabetes incidence was monitored weekly by testing for the presence of glucose in the urine by Diastix (Bayer, Elkhart, IN). Mice that tested positive by Diastix were further tested with a Breeze2 glucometer (Bayer) for elevated blood glucose levels and were considered diabetic if their blood glucose was >400 mg/dL. All experiments were performed in accordance with Institutional Animal Care and Use Committee protocols at Emory University, University of Utah, St. Jude Children’s Research Hospital, and Baylor College of Medicine.
Reagents and Antibodies
Cell culture media R10 was RPMI-1640 supplemented with 10% heat-inactivated FBS, 2 mmol/L l-glutamine, 2 × 10−5 mol/L 2-mercaptoethanol, 10 mmol/L HEPES buffer, and 50 μg/mL gentamicin sulfate. All media components were from Mediatech except FBS (Gibco) and 2-mercaptoethanol (Sigma-Aldrich). Experimental additive solution 45 (EAS45) was prepared as described, sterile filtered, and frozen in aliquots at −20°C until needed (19). Antibodies and clones used were RT1B phycoerythrin (PE) (clone Ox-6, detects I-Ag7), CD4 allophycocyanin (APC) (RM4-5), Vα2 FITC (B20.1), Vβ4 PE (KT4), TCRβ PE (H57-597), CD25 APC (clone PC61), CD69 BV650 (clone H1.2F3), and PD1 PE-cy7 (clone 29F.1A12) (BD Pharmingen). All peptides (HIP2.5, LQTLALWSRMD; mimotope 1 [Mim1], AHHPIWARMDA; ChgA29–42, DTKVMKCVLEVISD; and WE14, WSRMDQLAKELTAE) were synthesized on a Prelude peptide synthesizer (Gyro Protein Technologies) using fluorenylmethyloxycarbonyl chemistry and resuspended in molecular-grade water to 3 mmol/L and sterile filtered through a 0.22-μm syringe filter.
T-Cell Proliferation
Single-cell suspensions of BDC2.5 Tg splenocytes were labeled with CellTrace Violet (Thermo Fisher Scientific) according to the manufacturer’s instructions. Labeled BDC2.5 T cells were plated into a 96-well flat-bottom plate at 6 × 105 cells/well with a series of indicated peptide concentrations in complete RPMI media and incubated at 37°C with 5% CO2. After 24 and 72 h of incubation, T cells were analyzed by flow cytometry.
T-Cell Isolation
A CD53-based negative selection method was used to isolate double-positive preselection thymocytes from thymus (20). CD4-positive selection MACS kit from Miltenyi Biotec was used to isolate CD4 T cells from spleen and pancreatic lymph nodes. When indicated, CD4-enriched T cells were sorted based on Vα2 staining. For pancreatic T cells, pancreata of female NOD mice were perfused by injecting 3 mL collagenase IV (Worthington, Lakewood, NJ) through the bile duct, harvested, and placed in 2 mL collagenase IV. Pancreata were incubated at 37°C for 30 min, after which they were washed once with 10 mL 5% FBS/Hanks’ balanced salt solution (HBSS) and resuspended in 10 mL 5% FBS/HBSS. Islets were handpicked and incubated at 37°C for 15 min in 1 mL cell dissociation buffer (Invitrogen, Carlsbad, CA) and further dissociated by vortexing. Cells were then washed in 10 mL 5% FBS/HBSS and directly analyzed by flow cytometry or sorted based on CD4+CD5+ for 2D affinity measurements. Tetramer analysis was performed by gating on single cells within the lymphocyte gate, followed by gating on B220-CD4+CD3+ T cells. Double staining with HIP2.5 PE and APC tetramers was used to improve staining specificity. CLIP tetramer was used as a negative control.
2D Affinity Measurement
The micropipette adhesion frequency assay has been described previously (21). Peptide:I-Ag7 monomers (National Institutes of Health Tetramer Core Facility) were coupled to biotinylated human red blood cells (RBCs) via biotin-streptavidin chemistry, which serves as a surrogate antigen-presenting cell. In a typical test cycle, a coated RBC aspirated onto a glass micropipette is mechanically controlled (by a piezoelectric actuator) to approach, contact, and retract from a CD4 T cell held by an opposing static pipette. This test cycle is repeated 50 times for each RBC–T-cell pair. An adhesion event is visually identified by stretching of the RBC membrane upon its retraction. Adhesion frequency (Pa) was then calculated as the number of adhesion events divided by total cycle number. The frequency of antigen-specific cells was determined for the entire population by dividing number of cells with Pa greater than the cutoff for nonspecific binding (Pa > 0.1) by the total number of cells tested. The effective 2D affinity AcKa was calculated using the following equation:
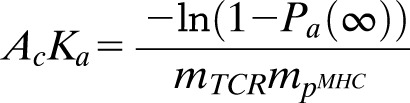 |
where Pa(∞) is the adhesion frequency at an equilibrium contact time (≥2 s) and mTCR and mpMHC are the densities of TCR and pMHC, respectively. TCR surface density was calculated with a PE Fluorescence Quantitation Kit from BD Pharmigen with TCR stained by either Vβ4 PE for BDC2.5 T cells or TCRβ for polyclonal CD4 T cells and with pMHC (I-Ag7) by RT1B PE.
Statistical Analysis
GraphPad Prism 6 (GraphPad Software) was used to determine significance using unpaired two-tailed t tests. Sample means were deemed significantly different if P < 0.05, P < 0.01, P < 0.001, or P < 0.0001.
Data and Resource Availability
The data sets generated during and/or analyzed during the current study are available from the corresponding author upon reasonable request.
Results
Splenic Tg BDC2.5 T Cells Recognize the ChgA-Related Autoantigens With a Broad Range of Affinity That Correlates With Their Functional Responses
The micropipette adhesion frequency assay uses a pMHC-decorated RBC to directly visualize pMHC-to-TCR binding on individual live T cells via deformation of the flexible RBC (14). By varying the concentration of pMHC molecules coated on the RBC, one can ensure that adhesion events between the RBC and the T cell are infrequent and mediated by a small number of pMHC:TCR bonds, allowing derivation of 2D affinity (22). Densities of TCR and pMHC are defined components used to calculate TCR affinity for pMHC. Using this assay, we quantified 2D affinity for WE14, ChgA29–42, and HIP2.5 epitopes for splenic BDC2.5 T cells (Fig. 1). WE14 was the first discovered ChgA epitope for the BDC2.5 TCR. It, however, lacks two major anchor residues at positions 1 and 4 for the sole MHC II I-Ag7 allele in NOD mice and is only minimally antigenic (23). ChgA29–42, with a full binding motif of I-Ag7, was a second ChgA epitope reported to activate BDC2.5 T cells at high concentrations (24). We also included analysis of a high-potency Mim1 (25) as a positive control (5.6 × 10−4 μm4) and an insulin epitope as a negative control, which confirmed antigen specificity of the BDC2.5 system with no binding (Fig. 1B). Among the four antigenic peptides, the HIP2.5 possesses the highest 2D affinity (1.8 × 10−3 μm4), almost 100 times greater than WE14 and 3 times greater than the mimotope. The HIP2.5 and Mim1 epitopes were both of sufficiently high affinities (>10−4 μm4), which we have defined as the approximate cutoff for detection by tetramers for the BDC2.5 TCR. WE14 has the lowest 2D affinity (3.0 × 10−6 μm4), with ChgA29–42 being marginally higher (Fig. 1A). These average affinities would be below what is needed for the respective peptide:MHC tetramers to detect BDC2.5 T cells (9,10,26). In addition to affinity, our adhesion frequency assay also provides information on the population of responding cells in terms of the number of responding cells and the range of response, as each symbol represents a separate T cell. All of the T cells (100%) possessed affinity for the HIP2.5 and Mim1 epitopes compared with only ∼50% of tested T cells bound to either WE14 or ChgA29–42 (Fig. 1C).
Figure 1.

BDC2.5 Tg splenic T cells recognize autoantigens with a broad range of affinity. Purified BDC2.5 splenic CD4 splenic T cells were interrogated for interaction with WE14, ChgA29–42, HIP2.5, and Mim1 presented by I-Ag7, from which 2D affinity and antigen-specific frequency were derived. A: 2D affinities of indicated antigens. B: Adhesion frequency for all individual cells tested. Some cells did not bind to RBCs coated with the WE14 or ChgA29–42 epitope presented by I-Ag7, demonstrating that not all BDC2.5 T cells are specific for the two weak antigens. The insulin epitope control shows the specificity of the 2D assay. Each point in panels A and B represents an individual cell. For each epitope, data were pooled from multiple experiments, with each experiment corresponding to one mouse (WE14, experiment number n = 6; ChgA29–42, n = 14; HIP2.5, n = 2; Mim1, n = 3). C: Corresponding frequency of antigen (Ag)-specific BDC2.5 CD4 T cells for experiments in panels A and B. Each point represents an individual experiment. In all of the panels, lines and error bars represent mean and SEM. **P < 0.01, ****P < 0.0001.
To examine the impact of 2D affinity on T-cell responses, we measured functional outcomes of Tg BDC2.5 splenic T cells toward the four peptides, focusing on early activation markers and proliferation. Using cell trace violet to track T-cell proliferation, we found that even the lowest HIP2.5 peptide concentration achieved complete cell division (greater than six divisions), although with increased peptide doses, more cells divided (Fig. 2). Therefore, the strength of HIP2.5 stimulation appears to only determine rapidity and simultaneity but not a binary decision of proliferation. In addition, proliferation does not equate to upregulation of activation markers such as CD25, CD69, and PD1; rather, the strength of stimulation plays a dominant role in their upregulation. The same proliferation characteristics were observed for the Mim1 epitope, whereas neither ChgA29–42 nor WE14 induced proliferation or increased surface marker expression (Fig. 3). These data demonstrated that 2D affinity for ChgA29–42 and WE14 was too low to fully activate BDC2.5 Tg cells in our in vitro experiments.
Figure 2.

The HIP2.5 peptide is agonistic for the BDC2.5 Tg splenic T cells. BDC2.5 splenocytes were labeled with cell trace violet (CTV), followed by ex vivo activation by pulsing with the HIP2.5 peptide at the indicated concentrations. Data from a representative mouse on day 3 after HIP2.5 stimulation are shown for proliferation (A–F) and surface marker expression of CD25 (G–L), CD69 (M–R), and PD1 (S–X).
Figure 3.

BDC2.5 Tg splenic T cells mount strong responses to the HIP2.5 epitope but no observable responses for the WE14 and ChgA29–42 epitopes. A–E: Dose-response curves of BDC2.5 splenic T cells to WE14, ChgA29–42, HIP2.5, and Mim1 at day 1 poststimulation by corresponding peptides. The HIP2.5 peptide elicited similarly strong responses to the mimotope in TCR downregulation (A), upregulation of CD25 (B), CD69 (C), and PD1 (D), and proliferation (E). MFI, mean fluorescence intensity. F–H: Comparison of T-cell responses between day 1 and day 3 poststimulation. Data shown are for 100 μmol/L peptide concentration (no antigen as a control). Lines and error bars represent mean and SEM from three experiments (with three, three, and two mice per experiment). For CD25, CD69, and PD1, there was no statistical difference between any pair of no antigen, WE14, and ChgA29–42 conditions or between Mim1 and HIP2.5 conditions; in contrast, there was a significant difference between any one of no antigen, WE14, and ChgA29–42 conditions and either Mim1 or HIP condition. ****P < 0.0001.
BDC2.5 Thymocytes Display Increased Affinity for Ligand
One major function of thymocyte selection is to purge high-affinity T-cell clones for self-antigens to avoid autoimmunity. To test whether such selection pressure may alter autoantigen recognition by BDC2.5 T cells for ChgA29–42 and WE14, we compared 2D affinity for CD4+CD8+ double-positive (DP) preselection BDC2.5 thymocytes to splenic T cells. The affinity of Tg preselection BDC2.5 thymocytes (3.5 × 10−4 μm4) for ChgA29–42 is >20 times higher than that of splenic T cells (Fig. 4A). This higher affinity is at the same level as that of a model foreign reactive CD4 SMARTA clone interacting with an lymphocytic choriomeningitis virus (LCMV) epitope (26). Surprisingly, preselection BDC2.5 thymocytes have even higher 2D affinity for the WE14 epitope (5.6 × 10−4 μm4), ∼200-fold higher than that of splenic cells (Fig. 4B). This would suggest that selective pressure decreases BDC2.5 TCR affinity for ChgA during development. To further support the notion that thymic selection alters how BDC2.5 TCR recognizes antigen, we tracked the frequency of endogenous Vα TCR chains. Because of limited availability of antibodies targeting TCR Vα chains, we tracked Vα2-expressing T cells (27). Indeed, the percentage of cells expressing Vα2 increased 40-fold from the Tg preselection thymocytes to Tg splenic T cells (DP 0.36% vs. single-positive [SP] 14%) (Fig. 4C and D). However, Tg splenic BDC2.5 T cells have the same affinity for WE14 or ChgA29–42 whether from the FACS-sorted Vα2+ or Vα2− counterparts (Fig. 4A and B), indicating that other TCRαs might contribute. To eliminate endogenous TCRα-chain contributions, we analyzed the affinity for both ChgA29–42 and WE14 using Rg splenic BDC2.5 T cells on a Rag−/− background. In this case, the affinity of splenic Rg T cells was similar to that of preselection Tg BDC2.5 T cells (Fig. 4A and B).
Figure 4.

Splenic BDC2.5 Tg T cells recognize the ChgA29–42 and the WE14 epitopes with lower 2D affinity than preselection thymocytes. A and B: 2D affinity of DP (CD4+CD8+) preselection, splenic SP (CD4+CD8−), splenic SP Vα2+ and Vα2− BDC2.5 Tg, and splenic SP BDC2.5 Rg T cells for the ChgA29–42 (A) and WE14 (B) epitopes. Each point in panels A and B represents one cell. For each condition, data were pooled from multiple experiments, with each experiment corresponding to one mouse (WE14, Tg DP, experiment number n = 6; WE14, Tg SP, n = 6; WE14, Rg SP, n = 2; ChgA29–42, Tg DP, n = 6; ChgA29–42, Tg SP, n = 14; ChgA29–42, Rg SP, n = 3). C: Representative flow plots of Vα2 (plotted with Vβ4) expression in the preselection and splenic BDC2.5 Tg T cells from panel D. D: Vα2 expression is much higher on splenic Tg BDC2.5 T cells than preselection thymocytes. Each point in panel D represents one mouse. The lines and error bars in panels A, B, and D represent mean and SEM. ****P < 0.0001.
The decrease in BDC2.5 affinity for antigen(s) was also observed for the HIP2.5 and Mim1 epitopes, but the decrease in affinity from DP to splenic T cells was less. The 2D affinity of preselection BDC2.5 thymocytes for HIP2.5 was approximately fivefold higher than splenic T cells (1.1 × 10−2 μm4 vs. 1.8 × 10−3 μm4, respectively) (Fig. 5A). 2D affinity for the Mim1 epitope followed the same pattern as the HIP2.5 epitope (Fig. 5B–D). Compared with ChgA29–42 and WE14, TCR affinity for the Mim1 and HIP2.5 epitopes was maintained at a higher level after thymic development. Unlike ChgA, neither of these epitopes is likely expressed in the thymus.
Figure 5.

BDC2.5 Tg T cells recognize the HIP2.5 and Mim1 epitopes with high 2D affinity. A and C: 2D affinity of preselection DP, splenic SP, and ex vivo activated BDC2.5 Tg T cells for the HIP2.5 and the Mim1 epitopes, respectively. The 2D affinity of the BDC2.5 Tg SP splenic T cells is much lower than that of preselection DP T cells, whereas 2D affinity partially recovered after ex vivo activation. B and D: Adhesion frequency for all individual cells tested. In all panels, each point represents an individual cell. Lines and error bars represent mean and SEM. For each condition, data were pooled from multiple experiments, with each experiment corresponding to one mouse (HIP2.5, preselection thymocyte, n = 2; HIP2.5, splenic, n = 2; HIP2.5, activated, n = 2; Mim1, preselection thymocyte, n = 6; Mim1, splenic, n = 3; Mim1, activated, n = 3). *P < 0.05, ****P < 0.0001.
HIP2.5 Reacts With a High Percentage of Polyclonal NOD CD4 T Cells With High 2D Affinity From Pre- to Post-diabetes
We next used the micropipette adhesion assay to probe the islets of NOD mice for HIP2.5-reactive polyclonal T cells. We found that 40% of the islet-derived T cells possessed affinity for the HIP2.5 epitope at the prediabetic time point (Fig. 6A, total HIP2.5-specific data). The high frequency of islet CD4 HIP2.5-reactive T cells is ∼20 times higher than tetramer staining (Fig. 6C–E). Although the frequency of HIP2.5-specific CD4 T cells trended higher in post-diabetic islets, the difference was not statistically significant. In contrast, splenic CD4 T cells did not show HIP2.5 specificity (Fig. 6B). This is consistent with the expected frequency of antigen-reactive T cells in the periphery as opposed to the site of autoimmune disease. Of note, the HIP2.5-reactive polyclonal islet CD4 T-cell populations clearly exhibited a bimodal distribution of high or low 2D affinity separated by three orders of magnitude for both pre- and post-diabetic conditions (Fig. 6F). The high affinity of polyclonal TCRs for the HIP2.5 epitope was higher at both pre- (1.1 × 10−2 μm4) and post-diabetes (0.9 × 10−2 μm4) compared with the BDC2.5 T cells (1.8 × 10−3 μm4). The bimodal distribution was surprising, as previous analysis of polyclonal populations has identified a normal distribution (28), which can be seen for the ChgA29–42 and insulin mimotope p8G-reactive T cells (29) (Fig. 6F). Approximately 20% of the pancreatic lymph node–derived T cells were specific to ChgA29–42 epitope, but all were of lower affinity (Fig. 6F). Similarly, CD4 T cells specific for the insulin p8G epitope were predominantly low affinity (96%) at the prediabetic stage, although high-affinity binders were enriched (15%) after mice became diabetic. Overall, ∼50% of islet CD4 T cells were p8G specific with the micropipette assay. Collectively, our data showed that there are substantial populations of high- and low-affinity CD4 T cells for the HIP2.5 epitope before disease onset and that they are maintained during overt diabetes.
Figure 6.

Bimodal distribution of HIP2.5 affinity for polyclonal NOD T cells. A: The frequency of HIP2.5-specific cells in the islets of prediabetic and diabetic NOD mice, where the line is the mean value and each dot represents data from one experiment (one mouse). In addition to the frequency of total HIP2.5-specific T cells, antigen specificity was also reported for high– and low–2D affinity T cells for the HIP2.5 defined in panel F. B: NOD splenic CD4 T cells do not bind to the HIP2.5 epitope. Enriched splenic CD4 T cells from three prediabetic NOD mice were tested for HIP2.5 reactivity, but none was a binder. C and D: HIP2.5 tetramer staining of prediabetic NOD islet CD4 T cells. Panel C shows representative flow plots (CLIP as a control). PE- and APC-conjugated tetramers were both used to improve staining specificity. Each point in panel D represents results from one mouse. E: Micropipette adhesion assay detects more antigen-specific CD4 islet T cells than tetramer staining. Filled bar shows the percentage of HIP2.5-specific CD4 islet T cells from prediabetic mice detected by micropipette (from panel F) (white bar by tetramer staining [from panel D]). F: Effective 2D affinities for HIP2.5 (open symbols, data pooled from three mice pre- and three mice post-diabetes) and p8G (half-filled symbols, data pooled from two mice pre- and two mice post-diabetes) were calculated for the islet polyclonal CD4 T cells, where lines represent the mean and error bars the SEM and each dot represents an individual cell. Also shown are ChgA29–42 affinities for polyclonal CD4 T cells in the pancreatic lymph node (filled symbols). *P < 0.05, **P < 0.01.
Discussion
In T1D, CD4 T-cell recognition of β-cell autoantigen is a key event in disease pathology. Among identified β-cell–derived CD4 epitopes, HIP2.5 is a potential prime target of murine CD4 diabetogenic T cells that triggers and perpetuates the disease. Recent identification of HIPs in the islets of healthy hosts (e.g., BALB/c mice and control donors) (30) suggests that HIP epitopes alone are not sufficient to cause diabetes. As one of the highest genetic risk factors in T1D is MHC II, presentation of the HIP peptides may be required for activation or regulation of HIP-reactive T cells. In this study, we have characterized how Tg and polyclonal mouse CD4 autoimmune T cells recognize the HIP2.5 epitope. Using the high sensitivity of the micropipette adhesion frequency assay, we quantified affinity and frequency of HIP2.5-specific murine diabetogenic T cells. We found that the HIP2.5 epitope (presented by IAg7) binds to transgenic BDC2.5 T cells with high affinity and behaves as a strong agonist. Furthermore, in NOD islets, a high frequency of high-affinity HIP2.5-specific T cells (20%) exist in the prediabetic phase and are maintained during diabetes; in comparison, high-affinity p8G-specific T cells were few in the prediabetic phase but enriched after diabetes (15%). Such high frequency of high-affinity autoreactive CD4 T cells for single epitopes is surprising and is ∼10 times higher than that reported by tetramer staining (8,29) (Fig. 6). In addition, there is a high frequency of low-affinity autoreactive CD4 T cells in the islets. Together, we found that ∼40%, 50%, and 20% of CD4 T cells in NOD islets are specific for the HIP2.5, p8G, and ChgA29–42 epitopes, respectively. Considering other insulin hybrid peptides (8,31) and many previously identified self-peptides from both insulin and ChgA (4,32), we expect most if not all islet T cells to interact with local antigens. These high percentages of reactive T cells would also suggest increased levels of cross-reactivity between T cells.
The affinity profile of polyclonal CD4 T cells in the islet for the HIP2.5 epitope was different from BDC2.5 T cells, showing a clear bimodal distribution, with high-affinity binders representing nearly 50% of the total HIP2.5-specific population. The high frequency of strong HIP2.5 binders implies an absence of negative selection in the thymus, a common problem for autoantigens derived from posttranslational modifications (33). We also recently reported high-affinity TCRs for hybrid HLA-DQ8 molecules (34). This bimodal response to HIP2.5 from polyclonal T cells is also very different from other systems we have analyzed. Using a well-characterized dominant epitope from myelin oligodendrocyte glycoprotein (MOG), we previously demonstrated that the dynamic range in affinity of polyclonal central nervous system–infiltrating CD4 T cells covered a broad continuum that spans at least 3 logs (9), which can be described by a single normal distribution with lower mean (∼10−5 μm4). As expected from thymic negative selection, the peripheral CD4 T cells are predominantly of low affinity (like that of splenic BDC2.5 interacting with the WE14 and ChgA29–42 epitopes). The importance and cause of the bimodal distribution to HIP2.5 remain to be determined. Of interest, we have postulated, based on our analysis of myelin-specific T cells, that high-affinity together with low-affinity T cells are key components of autoimmune disease (9,35). This model would be compatible with the suggestion of high-affinity clones to initiate disease and lower-affinity TCRs to perpetuate an anti-self response.
In Bettini et al. (36), we reported the 2D affinity for a panel of murine CD4 clones specific for the insulin B-chain epitope InsB9–23 and found that 2D affinity correlated with ex vivo T-cell activation responses. The dynamic range of 2D affinity for the seven TCR panel was limited to 1 log, with all TCRs having relatively high affinity (ranging from 10−4 to 10−3μm4). Of interest, there was no obvious correlation among these high–2D affinity TCRs with diabetes onset or incidence in Rg mice. This study did not include any low-affinity T cells for the insulin antigens, which likely differs from the polyclonal setting (Fig. 6).
WE14 and ChgA29–42 peptides are recognized by the BDC2.5 TCR in peripheral T cells with affinity that is two orders of magnitude lower than the HIP2.5 epitope; however, in the thymus, preselection BDC2.5 thymocytes bind to ChgA29–42:I-Ag7 or WE14:I-Ag7 with much higher affinity. Thus, it seems that affinity for self-ChgA29–42 and WE14 epitopes was downregulated during thymic development, possibly as a means to limit self-reactivity in the periphery. In contrast, the affinity of the HIP2.5 epitope remains high for diabetogenic CD4 T cells in the periphery; in fact, it is still five times higher than that of typical foreign reactive SMARTA T cells for an LCMV epitope (26). Therefore, our data support a hypothesis of ChgA but not HIP2.5 expression in the thymus (37,38) allowing for high affinity for the HIP2.5 epitope in the periphery to eventually trigger disease.
It is thought that the frequency and kinetics of diabetogenic T cells specific for β-cell autoantigens determine disease outcome. The first study documenting a link between TCR antigen recognition and diabetes progression used a mimetope (called NRP-A7) of the CD8 NY8.3 TCR (specific for an islet-specific glucose-6-phosphatase epitope presented by Kd) to quantify tetramer staining of polyclonal islet T cells and track their frequency (39). As diabetes progressed, tetramer avidity and the percentage of tetramer-positive cells progressively increased within expanded islet T cells in vitro. However, results from later studies are mixed. Some reports on NOD mice and patient blood samples are supportive, showing higher frequency of autoantigen-specific T cells correlated with worsened disease (40) or diabetes progression (41–45). Even in these cases, there are usually significant overlaps between compared groups. Yet, other studies did not reveal a significant difference of frequency for autoimmune T cells between healthy control subjects and patients with diabetes (46,47). A recent study making use of tetramers for analysis showed an increase in the frequency of HIP2.5-reactive T cells as diabetes progressed (31), which was not observed in the current study. The discrepancy might be due to the different tracking methods used. Our 2D assay has been proven to be much more sensitive than tetramer staining and has consistently been able to identify more antigen-specific T cells (9,28,48). It is possible that tetramer staining may have missed a subpopulation of HIP2.5-reactive T cells, leading to an underestimation of HIP2.5-specific T-cell frequency. Therefore, considering all available data, whether tetramer-derived frequency of autoantigen-specific T cells can serve as a reliable biomarker for disease progression is still an unresolved issue in T1D.
In summary, we have determined the affinity and frequency of CD4 T cells specific to HIP2.5 and related ChgA autoantigens. For BDC2.5 T cells, ChgA29–42 and WE14 affinity is greatly downregulated in the periphery during thymic development, yet peripheral BDC2.5 T cells maintain high affinity for the HIP2.5 epitope. The high-affinity and optimal functional responses (Figs. 1–3) highlight HIP2.5 as a factor in BDC2.5 T1D. Importantly, we also observed a higher frequency of high-affinity polyclonal T cells reactive to HIP2.5 in NOD islets than has previously been reported. An interesting future avenue of research would be to investigate the underlying regulatory mechanism of affinity downregulation for the ChgA29–42 and WE14 epitopes and apply such understanding to modify autoimmune CD4 T-cell recognition of posttranslationally modified β-cell autoantigens, such as HIP2.5, as a novel intervention method in treating diabetes.
Article Information
Acknowledgments. The authors thank Laurel Ann Lawrence (Emory University) and Linda Morrison (University of Utah) for managing the mouse colony and screening of mice, Dr. Rustom Antia and Dr. Veronika Zarnitsyna (Emory University) for their expertise and guidance in statistical analysis, Dr. Cheng Zhu and his laboratory (Georgia Institute of Technology), Dr. Joseph Sabatino (Emory University) for technical support and advice with the micropipette adhesion frequency assay, and Hiran Thyagarajan, Mike Faust, Gavile Cathy, and Douglas Cornwall (University of Utah) for careful reading of the manuscript. The authors also thank the National Institutes of Health Tetramer Core Facility (Emory University) for providing all of the pMHC monomers and tetramers used in this study.
Funding. Training support from National Institutes of Health (NIH) National Institute of Diabetes and Digestive and Kidney Diseases (F31-DK-089932) was provided for J.D.H. This work was supported by NIH National Institute of Diabetes and Digestive and Kidney Diseases grant DK-089125 (D.A.V. and M.B.), National Institute of Allergy and Infectious Diseases grant AI-125301 (M.B.), National Institute of Neurological Disorders and Stroke grant NS-071518 (B.D.E.), and American Diabetes Association grant 1-09-IN-16 (B.D.E.).
Duality of Interest. No potential conflicts of interest relevant to this article were reported.
Author Contributions. B.L., J.D.H., and B.D.E. designed the experiments. B.L., J.D.H., and E.M.K. obtained 2D affinity data. B.L., J.D.H., and D.M.W. obtained T-cell functional data. B.L., J.D.H., and B.D.E. designed the experiments. B.L., J.D.H., and B.D.E. wrote the manuscript. D.A.V. and M.B. contributed to writing the manuscript. B.D.E. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Footnotes
B.L. and J.D.H. contributed equally.
References
- 1.Anderson MS, Bluestone JA. The NOD mouse: a model of immune dysregulation. Annu Rev Immunol 2005;23:447–485 [DOI] [PubMed] [Google Scholar]
- 2.DiMeglio LA, Evans-Molina C, Oram RA. Type 1 diabetes. Lancet 2018;391:2449–2462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Roep BO, Peakman M. Antigen targets of type 1 diabetes autoimmunity. Cold Spring Harb Perspect Med 2012;2:a007781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McGinty JW, Marré ML, Bajzik V, Piganelli JD, James EA. T cell epitopes and post-translationally modified epitopes in type 1 diabetes. Curr Diab Rep 2015;15:90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Katz JD, Wang B, Haskins K, Benoist C, Mathis D. Following a diabetogenic T cell from genesis through pathogenesis. Cell 1993;74:1089–1100 [DOI] [PubMed] [Google Scholar]
- 6.Nakayama M, Abiru N, Moriyama H, et al. Prime role for an insulin epitope in the development of type 1 diabetes in NOD mice. Nature 2005;435:220–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Baker RL, Bradley B, Wiles TA, et al. Cutting edge: nonobese diabetic mice deficient in chromogranin A are protected from autoimmune diabetes. J Immunol 2016;196:39–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Delong T, Wiles TA, Baker RL, et al. Pathogenic CD4 T cells in type 1 diabetes recognize epitopes formed by peptide fusion. Science 2016;351:711–714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sabatino JJ Jr., Huang J, Zhu C, Evavold BD. High prevalence of low affinity peptide-MHC II tetramer-negative effectors during polyclonal CD4+ T cell responses. J Exp Med 2011;208:81–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Martinez RJ, Andargachew R, Martinez HA, Evavold BD. Low-affinity CD4+ T cells are major responders in the primary immune response. Nat Commun 2016;7:13848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu B, Chen W, Evavold BD, Zhu C. Accumulation of dynamic catch bonds between TCR and agonist peptide-MHC triggers T cell signaling. Cell 2014;157:357–368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sibener LV, Fernandes RA, Kolawole EM, et al. Isolation of a structural mechanism for uncoupling T cell receptor signaling from peptide-MHC binding. Cell 2018;174:672–687.e27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Adams JJ, Narayanan S, Liu B, et al. T cell receptor signaling is limited by docking geometry to peptide-major histocompatibility complex. Immunity 2011;35:681–693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huang J, Zarnitsyna VI, Liu B, et al. The kinetics of two-dimensional TCR and pMHC interactions determine T-cell responsiveness. Nature 2010;464:932–936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huppa JB, Axmann M, Mörtelmaier MA, et al. TCR-peptide-MHC interactions in situ show accelerated kinetics and increased affinity. Nature 2010;463:963–967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu B, Zhong S, Malecek K, et al. 2D TCR-pMHC-CD8 kinetics determines T-cell responses in a self-antigen-specific TCR system. Eur J Immunol 2014;44:239–250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.DiToro D, Winstead CJ, Pham D, et al. Differential IL-2 expression defines developmental fates of follicular versus nonfollicular helper T cells. Science 2018;361:eaao2933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bettini ML, Bettini M, Nakayama M, Guy CS, Vignali DA. Generation of T cell receptor-retrogenic mice: improved retroviral-mediated stem cell gene transfer. Nat Protoc 2013;8:1837–1840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dumaswala UJ, Wilson MJ, José T, Daleke DL. Glutamine- and phosphate-containing hypotonic storage media better maintain erythrocyte membrane physical properties. Blood 1996;88:697–704 [PubMed] [Google Scholar]
- 20.Hong J, Ge C, Jothikumar P, et al. A TCR mechanotransduction signaling loop induces negative selection in the thymus. Nat Immunol 2018;19:1379–1390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jiang N, Huang J, Edwards LJ, et al. Two-stage cooperative T cell receptor-peptide major histocompatibility complex-CD8 trimolecular interactions amplify antigen discrimination. Immunity 2011;34:13–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chesla SE, Selvaraj P, Zhu C. Measuring two-dimensional receptor-ligand binding kinetics by micropipette. Biophys J 1998;75:1553–1572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stadinski BD, Delong T, Reisdorph N, et al. Chromogranin A is an autoantigen in type 1 diabetes. Nat Immunol 2010;11:225–231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nikoopour E, Sandrock C, Huszarik K, et al. Cutting edge: vasostatin-1-derived peptide ChgA29-42 is an antigenic epitope of diabetogenic BDC2.5 T cells in nonobese diabetic mice. J Immunol 2011;186:3831–3835 [DOI] [PubMed] [Google Scholar]
- 25.Stratmann T, Martin-Orozco N, Mallet-Designe V, et al. Susceptible MHC alleles, not background genes, select an autoimmune T cell reactivity. J Clin Invest 2003;112:902–914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rosenthal KM, Edwards LJ, Sabatino JJ Jr., et al. Low 2-dimensional CD4 T cell receptor affinity for myelin sets in motion delayed response kinetics. PLoS One 2012;7:e32562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gonzalez A, Katz JD, Mattei MG, Kikutani H, Benoist C, Mathis D. Genetic control of diabetes progression. Immunity 1997;7:873–883 [DOI] [PubMed] [Google Scholar]
- 28.Hood JD, Zarnitsyna VI, Zhu C, Evavold BD. Regulatory and T effector cells have overlapping low to high ranges in TCR affinities for self during demyelinating disease. J Immunol 2015;195:4162–4170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Crawford F, Stadinski B, Jin N, et al. Specificity and detection of insulin-reactive CD4+ T cells in type 1 diabetes in the nonobese diabetic (NOD) mouse. Proc Natl Acad Sci U S A 2011;108:16729–16734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wiles TA, Powell R, Michel R, et al. Identification of hybrid insulin peptides (HIPs) in mouse and human islets by mass spectrometry. J Proteome Res 2019;18:814–825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Baker RL, Jamison BL, Wiles TA, et al. CD4 T cells reactive to hybrid insulin peptides are indicators of disease activity in the NOD mouse. Diabetes 2018;67:1836–1846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Eissa N, Hussein H, Hendy GN, Bernstein CN, Ghia JE. Chromogranin-A and its derived peptides and their pharmacological effects during intestinal inflammation. Biochem Pharmacol 2018;152:315–326 [DOI] [PubMed] [Google Scholar]
- 33.Doyle HA, Mamula MJ. Autoantigenesis: the evolution of protein modifications in autoimmune disease. Curr Opin Immunol 2012;24:112–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chow IT, Gates TJ, Papadopoulos GK, et al. Discriminative T cell recognition of cross-reactive islet-antigens is associated with HLA-DQ8 transdimer–mediated autoimmune diabetes. Sci Adv 2019;5:eaaw9336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Martinez RJ, Evavold BD. Lower affinity T cells are critical components and active participants of the immune response. Front Immunol 2015;6:468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bettini M, Blanchfield L, Castellaw A, et al. TCR affinity and tolerance mechanisms converge to shape T cell diabetogenic potential. J Immunol 2014;193:571–579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Soultanova A, Panneck AR, Rafiq A, Kummer W. Terminally differentiated epithelial cells of the thymic medulla and skin express nicotinic acetylcholine receptor subunit α 3. BioMed Res Int 2014;2014:757502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Panneck AR, Rafiq A, Schütz B, et al. Cholinergic epithelial cell with chemosensory traits in murine thymic medulla. Cell Tissue Res 2014;358:737–748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Amrani A, Verdaguer J, Serra P, Tafuro S, Tan R, Santamaria P. Progression of autoimmune diabetes driven by avidity maturation of a T-cell population. Nature 2000;406:739–742 [DOI] [PubMed] [Google Scholar]
- 40.Mariño E, Richards JL, McLeod KH, et al. Gut microbial metabolites limit the frequency of autoimmune T cells and protect against type 1 diabetes. Nat Immunol 2017;18:552–562 [DOI] [PubMed] [Google Scholar]
- 41.Oling V, Marttila J, Ilonen J, et al. GAD65- and proinsulin-specific CD4+ T-cells detected by MHC class II tetramers in peripheral blood of type 1 diabetes patients and at-risk subjects. J Autoimmun 2005;25:235–243 [DOI] [PubMed] [Google Scholar]
- 42.Yu W, Jiang N, Ebert PJ, et al. Clonal deletion prunes but does not eliminate self-specific αβ CD8(+) T lymphocytes. Immunity 2015;42:929–941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kronenberg D, Knight RR, Estorninho M, et al. Circulating preproinsulin signal peptide-specific CD8 T cells restricted by the susceptibility molecule HLA-A24 are expanded at onset of type 1 diabetes and kill β-cells. Diabetes 2012;61:1752–1759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Standifer NE, Burwell EA, Gersuk VH, Greenbaum CJ, Nepom GT. Changes in autoreactive T cell avidity during type 1 diabetes development. Clin Immunol 2009;132:312–320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kracht MJ, van Lummel M, Nikolic T, et al. Autoimmunity against a defective ribosomal insulin gene product in type 1 diabetes. Nat Med 2017;23:501–507 [DOI] [PubMed] [Google Scholar]
- 46.Velthuis JH, Unger WW, Abreu JR, et al. Simultaneous detection of circulating autoreactive CD8+ T-cells specific for different islet cell-associated epitopes using combinatorial MHC multimers. Diabetes 2010;59:1721–1730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Danke NA, Yang J, Greenbaum C, Kwok WW. Comparative study of GAD65-specific CD4+ T cells in healthy and type 1 diabetic subjects. J Autoimmun 2005;25:303–311 [DOI] [PubMed] [Google Scholar]
- 48.Blanchfield L, Sabatino JJ Jr., Lawrence L, Evavold BD. NFM cross-reactivity to MOG does not expand a critical threshold level of high-affinity T cells necessary for onset of demyelinating disease. J Immunol 2017;199:2680–2691 [DOI] [PMC free article] [PubMed] [Google Scholar]