

Abstract
Muscarinic acetylcholine receptors are G protein–coupled receptors that respond to acetylcholine and play important signaling roles in the nervous system. There are five muscarinic receptor subtypes (M1R to M5R), which, despite sharing a high degree of sequence identity in the transmembrane region, couple to different heterotrimeric GTP-binding proteins (G proteins) to transmit signals. M1R, M3R, and M5R couple to the Gq/11 family, whereas M2R and M4R couple to the Gi/o family. Here, we present and compare the cryo–electron microscopy structures of M1R in complex with G11 and M2R in complex with GoA. The M1R-G11 complex exhibits distinct features, including an extended transmembrane helix 5 and carboxyl-terminal receptor tail that interacts with G protein. Detailed analysis of these structures provides a framework for understanding the molecular determinants of G-protein coupling selectivity.
Muscarinic acetylcholine receptors (mAChRs) are family A G protein–coupled receptors (GPCRs) activated by the neurotransmitter acetylcholine. Members of this family play key roles in a variety of physiological functions, including regulation of heart rate, smooth muscle contraction, glandular secretion, and memory formation (1). The mAChR family consists of five highly conserved subtypes (M1R to M5R) that share 64 to 82% sequence identity and 82 to 92% sequence similarity in the transmembrane region. However, mAChR subtypes differ in their tissue distribution and the type of heterotrimeric GTP-binding protein (G protein) that they engage to instigate signaling. Heterotrimeric G proteins consist of Gα, Gβ, and Gγ subunits and are classified by the a subunit, which determines GPCR coupling specificity. mAChR subtypes M2R and M4R preferentially signal through Gi/o proteins. The Gi/o-protein family includes Gi1, Gi2, Gi3, GoA, and GoB. Activation of Gi/o proteins inhibit adenylyl cyclase and decrease intracellular concentrations of adenosine 3′,5′-cyclic monophosphate (cAMP). By contrast, M1R, M3R, and M5R predominantly couple to Gq/11 proteins (Gq and G11), leading to the activation of phospholipase C and the increase of cytosolic Ca2+ (Fig. 1A) (2). None of the muscarinic receptors couple efficiently to Gs proteins that activate adenylyl cyclase. M1R is abundantly expressed in the central nervous system (CNS), whereas M2R is more abundant in the peripheral tissues, including heart and colon (3). Studies of knockout mice show that the M1R plays a role in memory formation, whereas the M2R regulates heart rate (2, 4). The M2R is also a presynaptic autoreceptor in the peripheral parasympathetic nervous system and therefore influences responses of other muscarinic receptor subtypes.
Fig. 1. Overall architectures of M1R-G11iN-scFv16 and M2R-GoAiN-scFv16 complexes.
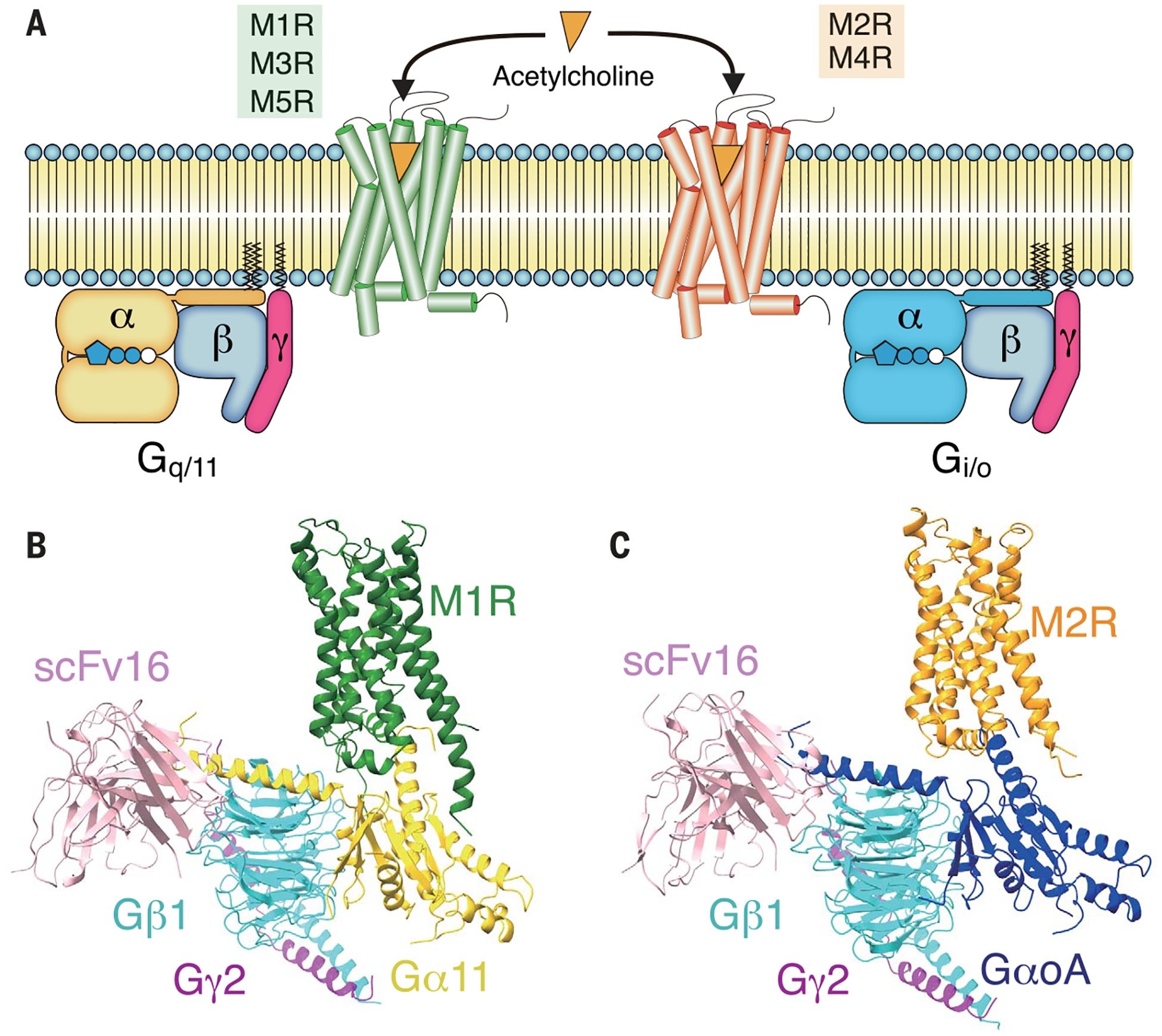
(A) Signaling selectivity among muscarinic receptors. M1R, M3R, and M5R predominantly signal through Gq/11-type G protein, whereas M2R and M4R couple to Gi/o (B and C). Cryo-EM structures of M1R-G11iN-scFv16 and M2R-GoAiN-scFv16 complex. Color code for the proteins is as follows: M1R (green), M2R (orange), Gα11iN (gold), GaoAiN (blue), Gβ1 (cyan), Gg2 (magenta), scFv16 (pink).
Owing to the high sequence homology, yet distinct G-protein preferences, the mAChR subfamily has been used as a model system to study the G-protein selectivity of GPCRs. So far, highresolution structural information on mAChRs has mostly been limited to antagonist-bound inactive states with the exception of an activestate M2R structure stabilized by a G-protein mimetic nanobody bound to a high-affinity agonist and a positive allosteric modulator (PAM) (5–8). The structure of rhodopsin bound to the transducin peptide first revealed interactions between the α5 helix of a G protein and an active GPCR (9). The first intact GPCR–G-protein structure to be reported was that of the β2-adrenergic receptor (β2AR)–Gs complex, determined by x-ray crystallography (10). The structure of the A2A receptor in complex with a modified Gs protein (mini-Gs) was more recently also determined by x-ray crystallography (11). Efforts to understand subtype selectivity on the basis of these structures did not reveal a clear consensus sequence on receptors that recognize the same G protein but concluded that receptors from different subfamilies evolved different mechanisms to activate the same G protein (12). Recently, several family B and family A GPCR structures in complex with either Gs or Gi/o proteins have been determined by single-particle cryo–electron microscopy (cryo-EM). These structures provided further structural insights into G-protein activation and have revealed that the transmembrane helix 6 (TM6) in GPCR-Gs complexes generally undergo a larger outward displacement than in GPCR-Gi/o complexes, resulting in a wider G-protein binding pocket that can accommodate the bulkier C terminus of Gs (13–18). To extend our understanding of coupling specificity to GPCR-Gq/11 complexes, we used single-particle cryo-EM to obtain the structures of active M1R and M2R engaged with heterotrimeric G11 and GoA proteins, two distinct G proteins belonging to the Gq/11 and Gi/o family, respectively. Comparison of these complexes provides structural insights into the activation of the Gq/11 family of G proteins and the basis for the distinct coupling preference of mAChRs for Gq/11 and Gi/o.
Sample preparation and cryo-EM
For this work, we used G11iN and GoAiN, which are modified forms of G11 and GoA, respectively. G11iN is a chimeric G11 in which most of the αN helix is replaced with the equivalent region of Gi1 to impart the ability to bind to scFv16 and stabilize the nucleotide-free GPCR-G protein complex (19) (fig. S1). GoAiN is a modified GoA that has four mutations in the region of the αN helix that binds scFv16 to make this homologous stretch identical to that of Gi1 (fig. S1). We made these mutations in GoA because scFv16 was originally developed against Gi1, even though wild-type GoA also binds scFv16 (19). Hereafter, we refer to them as G11 and GoA, because these modifications do not affect interactions with their respective receptors. M1R-G11-scFv16 and M2R-GoA-scFv16 complexes were formed from purified components and stabilized by the addition of apyrase to remove residual guanosine 5′-diphosphate (GDP). The final size-exclusion chromatography profiles and the SDS–polyacrylamide gel electrophoresis show pure and monodisperse samples before being loaded onto grids, blotted, and frozen (figs. S2, A to E, and S3, A to E). The M2R-GoA-scFv16 complex showed severe dissociation upon cryo-EM grid preparation, resulting in 3 to 6% complex particles out of all autopicked projections, compared with ~10% for the M1R-G11-scFv16 complex. Addition of the nonionic detergent octyl β-d-glucopyranoside (β-OG) at a concentration of 0.05% (~0.1 critical micelle concentration) to the M2R-GoA-scFv16 sample before grid application resulted in a modest increase in the number of vitrified intact complexes (7.5~11%) (fig. S3E). Single-particle cryo-EM analysis of these samples enabled us to obtain maps at nominal resolutions of 3.3 and 3.6 Å for M1R-G11-scFv16 and M2R-GoA-scFv16, respectively (Fig. 1, B and C; figs. S2 to S4; and table S1).
Comparison of overall structures
The higher resolution attained for the M1R-G11-scFv16 complex may be the result of more-extensive interactions between M1R and G11, including those involving intracellular loop 2 (ICL2) and ICL3 and the C terminus of M1R, as discussed later. The overall structure of the active M1R is similar to that of the active conformation of M2R (fig. S5A), with root mean square deviation (RMSD) values of 1.55 Å for the whole complex, 0.94 Å when comparing receptors alone, and 0.84 Å when comparing G proteins alone. The conformations of critical residues for the receptor activation such as D3.49R3.50Y3.51, N7.49P7.50xxY7.53, and P5.50I/V3.40F6.44 motifs [super-scripts indicate Ballesteros-Weinstein numbering for GPCRs (20)] are also similar between active conformations of M1R and M2R (fig. S5B), suggesting that the activation mechanism is shared between these muscarinic receptors that prefer different G-protein partners. When comparing inactive (7) and active states of M1R, we observe the outward displacement of TM6 that is characteristic of receptor activation. The TM6 movement is accompanied by a small rotation of the helix, as well as a tilt of TM5 toward TM6. These structural changes allow the C-terminal helix of the a subunit of the G protein to engage the receptor core (Fig. 2).
Fig. 2. Comparison between inactive and active M1R.
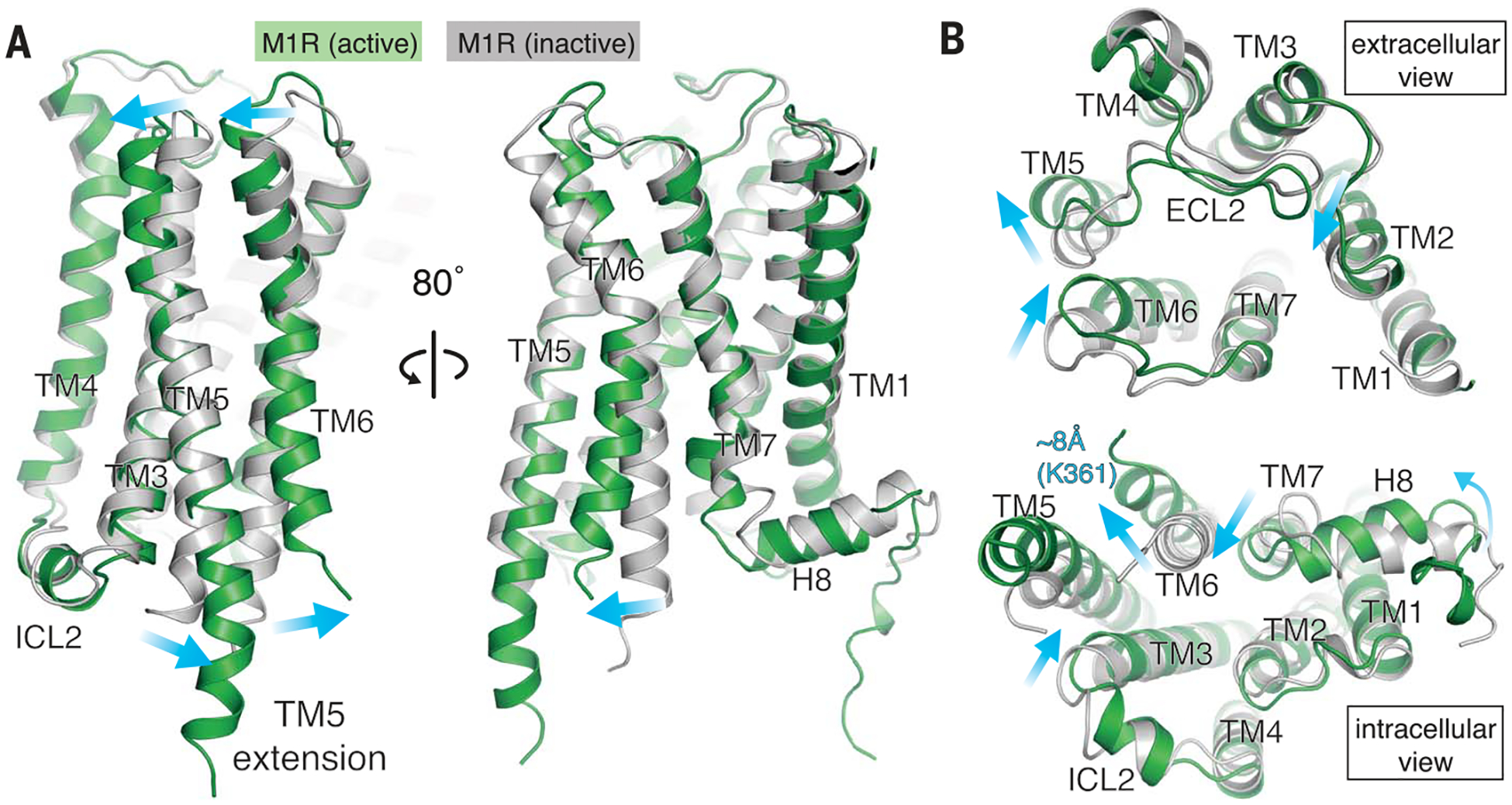
(A) Comparison of M1R between inactive (PDB code 5CXV) and active form viewed from the side. (B) Extracellular view (top), and intracellular view (bottom) of superposed M1Rs. Active and inactive M1R are colored in green and gray, respectively. Conformational changes are shown with blue arrows.
On the extracellular side of the M1R, the extracellular loop 2 (ECL2), TM5, and TM6 reorganize, with TM6 coming closer to ECL2 by 3.9 Å (Tyr179-Ser388) and causing a contraction of the extracellular vestibule (Fig. 2B). The same reorganization takes place in M2R, as seen previously in the nanobody-stabilized active state (5, 8), although it was not observed in active β2AR or μ-opioid receptor (μOR) (10, 15). The extracellular vestibule in muscarinic receptors is a well-documented binding site for allosteric modulators (21, 22), and its contraction may be a key feature for cooperativity with PAMs (23). Indeed, LY2119620, a positive allosteric modulator for M2R, was found to bind to this contracted vestibule (8). Our M2R-GoA-scFv16 cryo-EM map shows a distinct density corresponding to LY2119620 (figs. S4D and S5C). Although we included VU0357017, a PAM for M1R (24), in the M1R-G11-scFv16 sample for cryo-EM, no obvious density corresponding to this PAM was observed at the common allosteric vestibule or other regions of the receptor.
Comparison of the iperoxo binding pocket in M1R and M2R
Even though iperoxo is a highly efficacious agonist for both M1R and M2R, its affinity for M2R is 10- to 100-fold higher than for M1R, depending on how it is measured (25). Although the amino acids that form the iperoxo binding pocket are well resolved in the M1R cryo-EM map (fig. S6A), the density for the ligand does not allow for its unambiguous docking into the binding pocket. This suggests some degree of flexibility in the ligand or more than one binding pose. Our computational docking studies identified a range of closely related poses that both fit well to the experimental map and also received the highest docking scores (fig. S6B). Every pose formed the strong cation–π interactions observed in the crystal structure of M2R; however, there were a range of rotations around the alkyne axis and several positions for the heterocyclic group. Notably, the heterocyclic group is unable to occupy a geometry that makes ideal hydrogen bonds to the protein, which is likely a major contributing factor to the range of docked structures with similar scores. While there may be additional reasons for the poorly resolved density for the ligand in the experimental map, these docking results are consistent with a flexible and dynamic ligand. Although the residues coordinating iperoxo are identical and the side-chain conformations between M1R and M2R are similar (8) (fig. S6C), the surface-accessible volume of the orthosteric binding pocket in M1R is significantly larger than that of the M2R, with 73.7 A3 in the M1R and 55.0 A3 in the M2R [calculated by CASTp server (26)] (table S2). This might contribute to the lower affinity of iperoxo for M1R as compared to M2R (25). In addition, our M1R construct has an ICL3 truncation and an unintentional N110Q mutation, but these modifications do not affect the binding affinity of iperoxo (table S3).
Structure of the nucleotide-free G11
Nucleotide-free G11 in M1R-G11 shares common structural changes with other nucleotide-free G-protein subtypes characteristic of the nucleotide-free GPCR-engaged state (10, 15), including separation of the a-helical domain (AHD) from the Ras-like domain (23, 27, 28) (fig. S7A).
As observed for other GPCR-engaged G proteins, the α5 helix of Gα11 undergoes rotational and translational movement upon GDP release, as highlighted by the conserved side-chain positions of Phe341, Phe336, and Phe376 in Gαq/11, Gαi1/oA, and Gαs, respectively (Fig. 3). This conformational change accompanies the reorganization of the β6-α5 loop, the β1-α1 loop (P-loop), and the a1 helix (Fig. 3). The displacement of the a1 helix toward the GDP binding site and the β6-α5 loop away from the pocket likely contributes to GDP dissociation. The side chain of Gln58 in the α1 helix forms a hydrogen bond with the backbone carbonyl of Ala331 in the β6-α5 loop (fig. S7B). The interaction between the α1 helix and the β6-α5 loop is retained in both GDP-bound and GDP/AlF4-bound states of Gq (29, 30); thus, it is likely that the conformational changes of these elements and the α5 helix take place in a concerted manner. The importance of this interaction is revealed in a mutational study of Gi1, where alanine substitution of the equivalent residue showed a high basal nucleotide exchange (31).
Fig. 3. Structural comparison of G proteins.
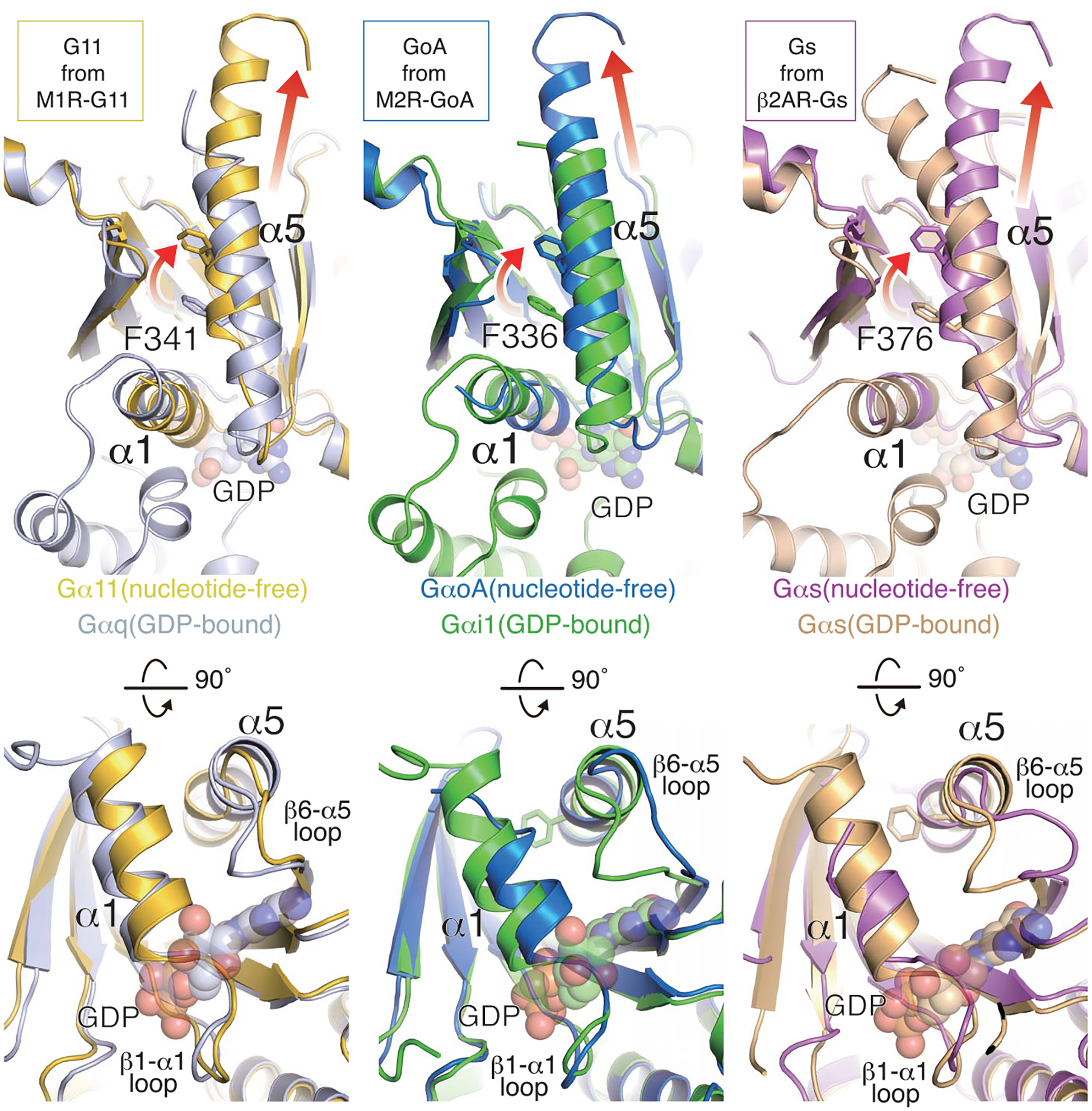
Structural changes upon receptor engagement and nucleotide release in Gαq/11, Gαi1, and Gαs. The GDP-bound structure is superimposed onto the nucleotide-free state for each family member. Rotation and translation of the α5 helix is shown in the upper panels, and a1 helix and β6-α5 loop displacements are shown in the lower panels. The PDB code for each structure is as follows: GDP-bound Gαq (3AH8), GDP-bound Gαi1 (1GP2), nucleotide-free Gαi1 (6DDE), GDP-bound Gs (6EG8), nucleotide-free Gs (3SN6).
Comparison of the receptor G-protein binding interfaces of the M1R-G11 and M2R-GoA complexes
Although the overall structure of M1R-G11 is similar to those of M2R-Go and other GPCR–G-protein complexes, we observe several distinct features. Comparison of the structures of M1R-G11 and M2R-GoA shows a clear difference in the orientation of G11 and GoA relative to the receptor (Fig. 4A). The α5 helix and Ras domain of G11 are rotated ~15° away from receptor TM5 compared to those of GoA. Other GPCR-Gi/o complexes have a similar rotational placement to GoA in M2R-GoA (fig. S8). This rotational shift of the α5 helix in M1R-G11 may be due to more-extensive interactions between the ICL2 of M1R and G11 com pared with the ICL2 of M2R and Go (and other GPCR-Gi/o complexes) (Fig. 4B).
Fig. 4. Comparison of the structures of M1R-G11 and M2R-GoA.
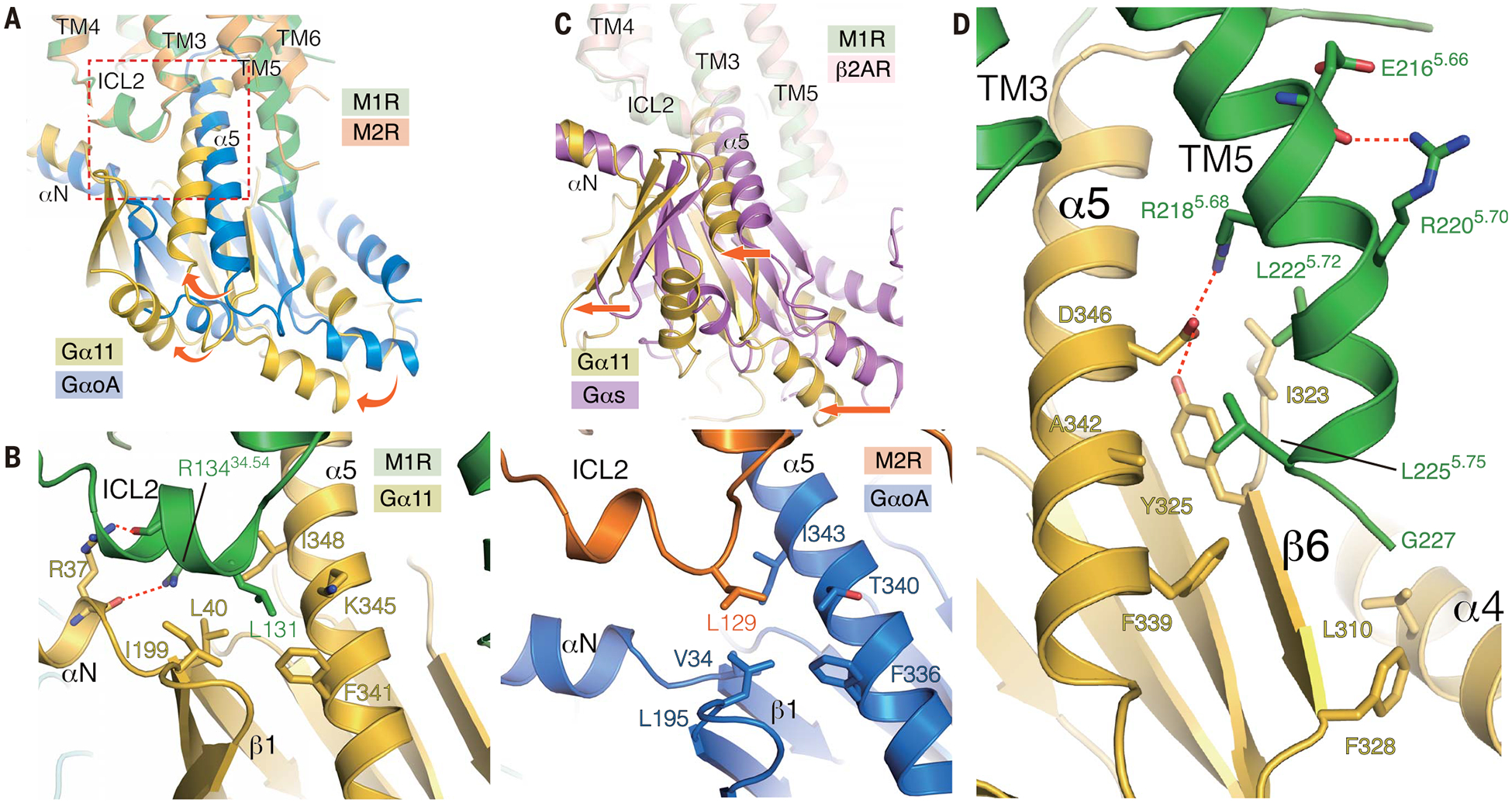
(A) Superposition of M1R-G11 and M2R-GoA complexes with the alignment based on the receptor. Rotational shift from GoA to G11 is depicted with curved arrows. Enlarged area in the panel (B) is shown as a broken rectangle. (B) View of ICL2 interface between the G protein on M1R-G11 (left) and M2R-GoA (right). (C) Superposition of β2AR-Gs and M1R-G11 complexes with the alignment based on the receptor. A translational shift is shown by straight arrows. (D) The extended helical structure from TM5 interacts with G11; interface residues are depicted as sticks.
In the M1R-G11 structure, Arg13434.54 in ICL2 interacts with Arg37 located at the junction between helix αN and strand α1 of G11 (Fig. 4B). The side chain of Arg13434.54 in ICL2 forms a hydrogen bond with the backbone carbonyl of Arg37 in the αN-β1 junction, whereas the side chain of Arg37 forms a hydrogen bond with the backbone carbonyl of Arg13434.54 in ICL2. Arg13434.54 in ICL2 is conserved among Gq/11-coupling muscarinic receptors and has been shown to contribute to the Gq/11 coupling (32). The equivalent position is proline in Gi/o-coupling muscarinic receptors (fig. S9). In the chimeric G11iN used in this study, the junction is made between Lys29 of Gai1 and Ala36 of Gα11, so that observed interactions are between the M1R and G11 amino acids, and not Gαi1 amino acids.
Leu131 in ICL2 of M1R is buried in a hydrophobic groove formed by the αN-β1 junction, the β2-β3 loop, and the α5 helix of Gα11 (Fig. 4B). The same hydrophobic interaction is conserved in other GPCR/G-protein complexes and has been reported to be crucial for the efficient nucleotide exchange reaction in multiple GPCRs, including M1R, M3R, and β2AR (33, 34). Leu131 is buried in this hydrophobic pocket of G11 as deeply as Phe139 of β2AR is buried in a similar pocket in Gs. Reflecting its importance, mutation of L131 in M1R to Ala, Asn, or Asp leads a loss of Gq coupling, whereas coupling is preserved in L131M and L131F mutations (34). This interaction does not appear to be as important for Gi/o coupling. The CB1, which has Leu at the homologous position, couples promiscuously to Gi/o and Gs. Mutation of this Leu to Ala leads to loss of coupling to Gs, but not to Gi (35). Consistent with these studies, the homologous L129 in the M2R forms only weak hydrophobic interactions with L195 in the β2-β3 loop and T340 in the α5 helix of GoA. Indeed, mutation of L131A in M1R reduces coupling with G11 to 17% of wild type, whereas the equivalent mutant L129A in M2R reduces coupling to GoA to 49% of wild type (fig. S10).
Recent structural studies have revealed that the outward displacement of TM6 is generally smaller in Gi/o-coupled GPCRs (e.g., rhodopsin, μOR) (15, 16) than in Gs-coupled GPCRs (e.g., β2AR, GLP1R) (10, 13, 14). The C terminus of the α5 helix of Gas has larger side chains compared to Gi/o, and the large displacement of TM6 may be required to accommodate these side chains. This suggests that the extent of TM6 displacement is one of the determinants of G protein coupling preference (36). When compared to the β2AR-Gs complex, the α5 helix of Gα11 adopts a straight conformation similar to that of Gas (Fig. 3), but is translationally shifted ~3 Å closer to TM1-TM2. As a result, the entire G11 protein is shifted in the same direction (Fig. 4C). Like Gαs, Gα11 harbors bulky side chains at the terminal residues. The translational shift, however, enables M1R to accommodate Gα11 despite the smaller outward displacement of TM6 relative to Gs-coupled GPCRs.
One of the most distinct features of the M1R-G11 complex is the additional 2.5 helical turns of TM5 extending inward toward the β6 strand and the α4 helix of Gα11 (Fig. 4D and fig. S5A). This helical extension is not observed in the M2R-Go complex, even though the M2R construct used in this study has a longer ICL3, and has also not been observed in other GPCR-Gi/o complexes reported to date. An extended TM5 has been observed in the β2AR-Gs complex, where it runs in a straight conformation and makes mostly polar contacts with Gs (10) (fig. S11A). By contrast, interactions between the TM5 extension of M1R and G11 are largely formed by hydrophobic interactions made between the inner surface of the TM5 and a hydrophobic patch in Gα11, but includea salt-bridge (Fig. 4D). The equivalent region in Gs and Gi/o is either modestly or highly negatively charged (fig. S11B), and these charged or polar properties in Gs or Gi/o would disfavor the interaction with hydrophobic residues in the TM5 extension of M1R.
Although this extended TM5 cannot be formed in all Gq/11-coupled receptors owing to shorter ICL3s (37), it is notable that an extended TM5 is also found in the structure of squid rhodopsin in its ground state. Unlike vertebrate rhodopsins that couple with transducin, a Gi/o-family member, invertebrate rhodopsins predominantly couple with Gq/11-type G protein to transduce the photosensing signal (38). Those invertebrate rhodopsins have a longer ICL3 compared to their vertebrate counterparts. The crystal structure of a squid rhodopsin shows an extended TM5 that bends inward (39), and structural superposition with the active M1R shows similarity between these two Gq/11-coupling GPCRs (fig. S11C). The inward bend begins at the same position and the inward surface of the extended TM5 shares hydro- phobic properties similar to those of the extended TM5 in the M1R-G11 complex.
Interactions between the M1R C terminus and G11
In the M1R-G11 complex, we observe 11 residues of the C terminus of M1R after H8 that extend into a groove formed by the Ras domain and the Gβ. A similar C-terminal extension has not been observed in other GPCR–G protein complexes reported thus far. Although the density for the extension is weaker than for other regions of the complex, a continuous backbone density runs from H8 helix along the Gβ subunit at the Gα/Gβ interface, suggesting the presence of a weak interaction between the C terminus of M1R and the Gβ subunit (Fig. 5A). It has been shown that in cells, M3R forms a precoupling assembly specifically with Gq/11, and this complex formation is independent of the ligand but dependent on the polybasic cluster at the C terminus of M3R (40). This polybasic cluster is conserved among Gq/11-coupling muscarinic receptors (fig. S12A). Although the Gβ interface with the Gα subunit is negatively charged across all G-protein subtypes, the Gα subunit of Gs and Gi1 subtypes have a highly positively charged interface, whereas the Gαq/11 surface is neutral (fig. S12B). This characteristic surface property may enable GPCRs that have a polybasic C-terminal cluster to engage Gq/11-type G proteins more efficiently (fig. S12B). Consistent with a functional role for this interaction, a mutant M1R in which most of the polybasic residues in the C-tail were replaced with alanines showed lower affinity for G11 (Fig. 5B). In this assay, G11 coupling to wild-type and mutant M1R was monitored by observing the ability of G11 to enhance the affinity of the M1R for the agonist iperoxo, which then displaces the antagonist N-methyl scopolamine (NMS) from the M1R (Fig. 5B). A C-terminal polybasic sequence is widely found among Gq/11-coupling GPCRs, as previously reported (40) and illustrated in fig. S13, suggesting that this mechanism is conserved in Gq/11-type GPCRs and might contribute to coupling specificity.
Fig. 5. Interaction of the M1R C terminus with G-protein α/β interface.
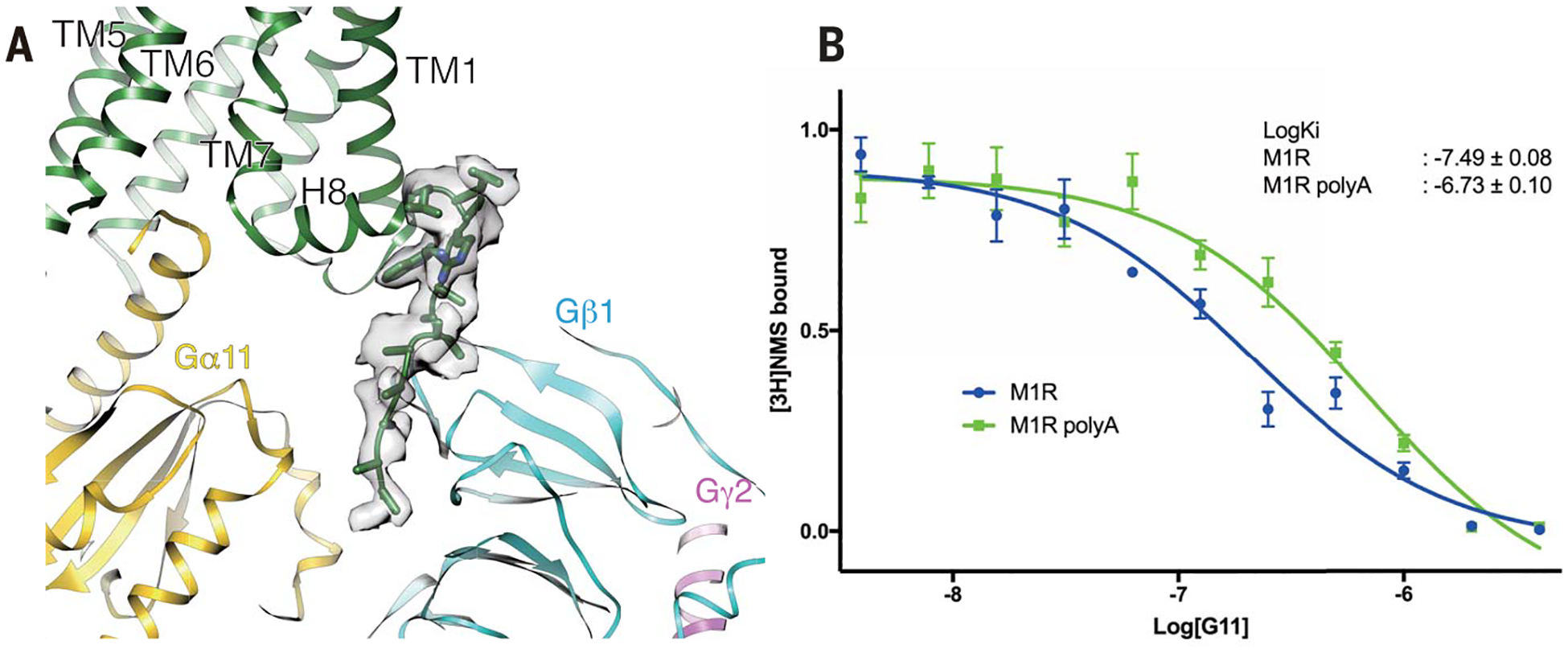
(A) Density of the C terminus of M1R positioned at the interface between Gα11 and Gβ subunit. (B) Titration of G11 in the competition ligand-binding assay using iperoxo and [3H]NMS as probes. Coupling of M1R to G11 increases the affinity for iperoxo, which displaces [3H]NMS. G11 couples to the poly(A) mutant of the C-terminal basic region of M1R with lower affinity. Data points represent the mean ± SEM of three experiments performed in triplicate.
Contributions of receptor TM5-TM6 and the Gα helix α5 to G protein–coupling selectivity
Muscarinic receptors have been used as a model system to study the structural basis of GPCR/G protein coupling selectivity because they share high sequence similarity in the transmembrane region but still show distinct preference for their G-protein signaling pathway. Our structures reveal substantial differences in the interactions between receptors and their cognate G protein for M1R-G11 and M2R-Go complexes. Moreover, of the 19 amino acids involved in interactions between M1R and G11, only 12 are conserved in M2R (fig. S9); however, the structures do not reveal which of these interactions are the most important for coupling specificity. The G-protein preference for muscarinic receptors has been extensively studied by using chimeric receptors and site-directed mutagenesis (32, 41–53). Although most of these comparisons have been made with M2R and M3R, of the 19 amino acids involved in interactions between M1R and G11, 18 are identical in the M3R ((fig. S9). Notably, many of these studies used chimeric G proteins to facilitate signaling studies. For example, it has previously been shown that the last five amino acids of G-protein α subunits are the major determinants of coupling specificity for some GPCRs (51, 54, 55). Thus, replacing the last five amino acids of Gq with the last five amino acids of Go, Gi/o-coupled M2R receptor could stimulate inositol phosphate turnover (48, 52), which is a more robust assay than inhibition of adenylyl cyclase by Gi/o.
Chimeric receptors studies showed that the major determinants of Gi/o and Gq/11 coupling specificity were found in ICL3 (41, 42, 44, 49), but ICL3 chimeras that included ICL2 from M3R could enhance coupling of M2R to Gq/11 (32). Through more refined chimeric and site-directed mutagenesis, it was possible to identify specific residues in ICL2, TM5, and TM6 that were important for M3R coupling to Gq (shown with an asterisk in fig. S9) (46–48, 51). These residues are identical in M1R (fig. S9). Among them, Tyr2555.62 in M3R (equivalent to Tyr2125.62 in M1R) has been identified as a critical residue for efficient Gq/11 coupling (46). Mutagenesis of this position to alanine abolishes Gq/11 coupling, whereas substitution to other aromatic residue shows efficient signaling indistinguishable from that of the wild-type (46, 47). Site-directed mutagenesis showed that A6.33A6.34xxL6.37S6.37 are also critical determinants for Gq/11 coupling to M3R, whereas V6.33T6.34xxI6.37L6.37 are important for coupling of M2R to Gi/o (x denotes any amino acid residue) (figs. S9 and S14) (32, 48).
Unexpectedly, of the five residues in TM5 and TM6 identified as being critical for Gq/11 coupling, only two interact directly with G11: A6.33 and L6.37 form van der Waals interactions with the highly conserved L(–2) (α5 helix numbering starts with –1 from the terminal residue) of G11. Tyr2125.62 does not interact with G11, but rather with Ala6.33 and Ala6.34 (Fig. 6). This interaction likely stabilizes TM6 at a position that is optimal for engaging G11 (Fig. 6 and fig. S14). In M2R, S5.62 forms van der Waals interactions with T6.34 and I6.37, whereas only V6.33 forms interactions with the highly conserved L(−2) and L(−7) of GoA (Fig. 6 and fig. S14). Thus, among the five residues shown by mutagenesis studies to be important for coupling specificity, most of the interactions occur between TM5 and TM6 and may be critical for stabilizing these TM segments for optimal interactions with the C terminus of the G protein (Fig. 6 and fig. S14).
Fig. 6. Comparison of α5/TM5/TM6 interactions in M1R-G11 and M2R-GoA complexes.
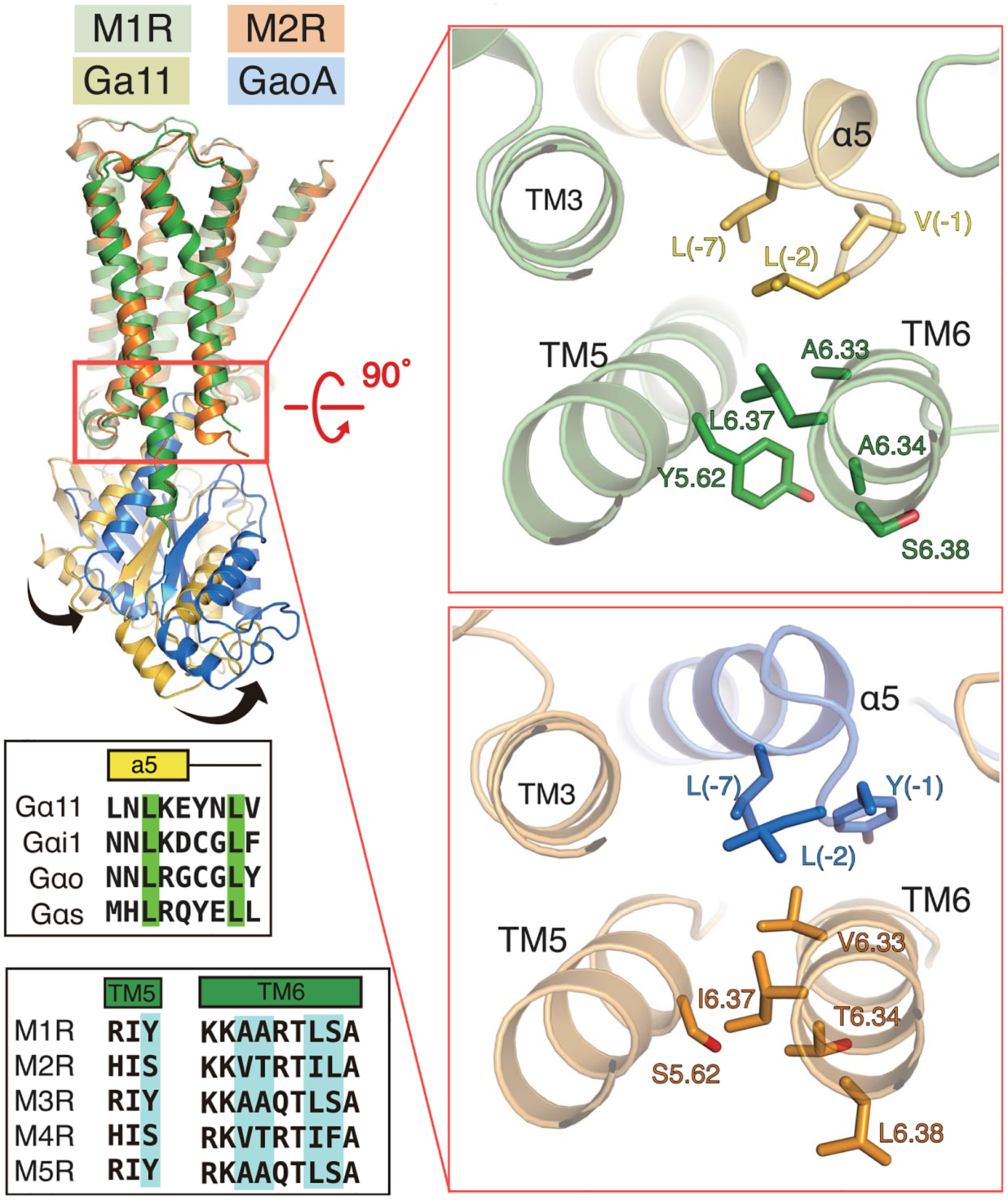
The right panels show a rotated view of the α5/TM5/TM6 interface for M1R-G11 and M2R-GoA complexes indicated in the red box on the left panel. Residues in M1R and M2R that have been indicated by mutagenesis to be important for G-protein coupling specificity are shown as sticks on the receptor structure and are highlighted in blue in the sequence alignments of TM5 and TM6 (lower left).
The interactions between M2R and GoA and between M1R and G11 provide insights into the coupling specificity in muscarinic receptors. Although the M1R amino acids that interact with G11 are conserved in M3R and M5R, they are not conserved in other Gq/11 coupled receptors (fig. S15). Similarly, M2R amino acids that interact with GoA are conserved in M4R but not in other GPCR–Gi/o complexes (fig. S15). These observations suggest a possible intermediate complex that might play a more important role in G-protein coupling specificity. Evidence for such an intermediate state has been observed in single-molecule studies for the β2AR-Gs complex (56). A transient intermediate complex will be difficult to capture by crystallography or cryo-EM and may require the creative application of other biophysical methods. Furthermore, the G proteins used in our study are chimeric proteins, and although these chimeric junctions are not involved in interactions with the receptor, we cannot exclude the possibility that these protein modifications could have a functional or structural impact.
Conclusions
Here, we report the structures of M1R-G11 and M2R-GoA complexes obtained by cryo-EM. Although the overall architecture is similar between M1R-G11 and M2R-GoA, the orientation of the G protein relative to the receptor is distinct for these complexes. Notable features in the M1R-G11 complex are an extended helix from TM5 that makes more extensive interactions with the G protein, and a polybasic cluster in the C terminus of M1R that interacts with Gα/Gβ subunit interface. This C-terminal interaction is likely involved in preassembly of the complex prior to agonist binding for Gq/11-coupled receptors. These structures highlight the importance of the local organization of the TM5/TM6/α5 interface in G-protein selectivity determination in mAChRs.
Supplementary Material
ACKNOWLEDGMENTS
Funding: The work was supported by NIH grant R01GM083118 (B.K.K.) and the Mathers Foundation (G.S. and B.K.K.). B.K.K. is a Chan Zuckerberg Biohub investigator.
Footnotes
Competing interests: B.K.K. is a cofounder of and consultant for ConfometRx. The remaining authors declare no competing interests.
Data and materials availability: The atomic coordinates for M1R/G11iN/scFv16 and M2R/GoAiN/scFv16 have been deposited in the Protein Data Bank with the accession codes 6OIJ and 6OIK, respectively. The EM maps for M1R/G11iN/scFv16 and M2R/GoAiN/scFv16 have been deposited in EMDB with the codes EMD-20078 and EMD-20079, respectively.
SUPPLEMENTARY MATERIALS
science.sciencemag.org/content/364/6440/552/suppl/DC1
Materials and Methods
Figs. S1 to S16
Tables S1 to S3
REFERENCES AND NOTES
- 1.Wess J, Annu. Rev. Pharmacol. Toxicol 44, 423–450 (2004). [DOI] [PubMed] [Google Scholar]
- 2.Langmead CJ, Watson J, Reavill C, Pharmacol. Ther 117, 232–243 (2008). [DOI] [PubMed] [Google Scholar]
- 3.Lebois EP, Thorn C, Edgerton JR, Popiolek M, Xi S, Neuropharmacology 136, 362–373 (2018). [DOI] [PubMed] [Google Scholar]
- 4.Thomsen M, Sørensen G, Dencker D, Neuropharmacology 136, 411–420 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Haga K et al. , Nature 482, 547–551 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kruse AC et al. , Nature 482, 552–556 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Thal DM et al. , Nature 531, 335–340 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kruse AC et al. , Nature 504, 101–106 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Choe H-W et al. , Nature 471, 651–655 (2011). [DOI] [PubMed] [Google Scholar]
- 10.Rasmussen SGF et al. , Nature 477, 549–555 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Carpenter B, Nehmé R, Warne T, Leslie AGW, Tate CG, Nature 536, 104–107 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Flock T et al. , Nature 545, 317–322 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang Y et al. , Nature 546, 248–253 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liang Y-L et al. , Nature 546, 118–123 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Koehl A et al. , Nature 558, 547–552 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kang Y et al. , Nature 558, 553–558 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Draper-Joyce CJ et al. , Nature 558, 559–563 (2018). [DOI] [PubMed] [Google Scholar]
- 18.García-Nafría J, Nehmé R, Edwards PC, Tate CG, Nature 558, 620–623 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Maeda S et al. , Nat. Commun 9, 3712 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ballesteros JA, Weinstein H, in Receptor Molecular Biology, Sealfon SC, Ed. (Academic Press, 1995), vol. 25 of Methods in Neurosciences, pp. 366–428. [Google Scholar]
- 21.Jensen AA, Spalding TA, Eur. J. Pharm. Sci 21, 407–420 (2004). [DOI] [PubMed] [Google Scholar]
- 22.Dror RO et al. , Nature 503, 295–299 (2013). [DOI] [PubMed] [Google Scholar]
- 23.Dror RO et al. , Science 348, 1361–1365 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Digby GJ et al. , J. Neurosci 32, 8532–8544 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schrage R et al. , Biochem. Pharmacol 90, 307–319 (2014). [DOI] [PubMed] [Google Scholar]
- 26.Tian W, Chen C, Lei X, Zhao J, Liang J, Nucleic Acids Res. 46, W363–W367 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Van Eps N et al. , Proc. Natl. Acad. Sci. U.S.A 115, 2383–2388 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kaya AI et al. , J. Biol. Chem 289, 24475–24487 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nishimura A et al. , Proc. Natl. Acad. Sci. U.S.A 107, 13666–13671 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tesmer VM, Kawano T, Shankaranarayanan A, Kozasa T, Tesmer JJG, Science 310, 1686–1690 (2005). [DOI] [PubMed] [Google Scholar]
- 31.Kapoor N, Menon ST, Chauhan R, Sachdev P, Sakmar TP, J. Mol. Biol 393, 882–897 (2009). [DOI] [PubMed] [Google Scholar]
- 32.Blin N, Yun J, Wess J, J. Biol. Chem 270, 17741–17748 (1995). [DOI] [PubMed] [Google Scholar]
- 33.Moro O, Lameh J, Högger P, Sadée W, J. Biol. Chem 268, 22273–22276 (1993). [PubMed] [Google Scholar]
- 34.Moro O, Shockley MS, Lameh J, Sadée W, J. Biol. Chem 269, 6651–6655 (1994). [PubMed] [Google Scholar]
- 35.Chen XP et al. , Br. J. Pharmacol 161, 1817–1834 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rose AS et al. , J. Am. Chem. Soc 136, 11244–11247 (2014). [DOI] [PubMed] [Google Scholar]
- 37.Pándy-Szekeres G et al. , Nucleic Acids Res. 46 (D1), D440–D446 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Koyanagi M, Terakita A, Photochem. Photobiol 84, 1024–1030 (2008). [DOI] [PubMed] [Google Scholar]
- 39.Murakami M, Kouyama T, Nature 453, 363–367 (2008). [DOI] [PubMed] [Google Scholar]
- 40.Qin K, Dong C, Wu G, Lambert NA, Nat. Chem. Biol 7, 740–747 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kubo T et al. , FEBS Lett. 241, 119–125 (1988). [DOI] [PubMed] [Google Scholar]
- 42.Wess J, Brann MR, Bonner TI, FEBS Lett. 258, 133–136 (1989). [DOI] [PubMed] [Google Scholar]
- 43.Lechleiter J et al. , EMBO J. 9, 4381–4390 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wess J, Bonner TI, Dörje F, Brann MR, Mol. Pharmacol 38, 517–523 (1990). [PubMed] [Google Scholar]
- 45.Blüml K, Mutschler E, Wess J, Proc. Natl. Acad. Sci. U.S.A 91, 7980–7984 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Blüml K, Mutschler E, Wess J, J. Biol. Chem 269, 402–405 (1994). [PubMed] [Google Scholar]
- 47.Blüml K, Mutschler E, Wess J, J. Biol. Chem 269, 11537–11541 (1994). [PubMed] [Google Scholar]
- 48.Liu J, Conklin BR, Blin N, Yun J, Wess J, Proc. Natl. Acad. Sci. U.S.A 92, 11642–11646 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Singer-Lahat D, Liu J, Wess J, Felder CC, FEBS Lett. 386, 51–54 (1996). [DOI] [PubMed] [Google Scholar]
- 50.Liu J, Blin N, Conklin BR, Wess J, J. Biol. Chem 271, 6172–6178 (1996). [DOI] [PubMed] [Google Scholar]
- 51.Kostenis E, Gomeza J, Lerche C, Wess J, J. Biol. Chem 272, 23675–23681 (1997). [DOI] [PubMed] [Google Scholar]
- 52.Kostenis E, Conklin BR, Wess J, Biochemistry 36, 1487–1495 (1997). [DOI] [PubMed] [Google Scholar]
- 53.Wess J et al. , Life Sci. 60, 1007–1014 (1997). [DOI] [PubMed] [Google Scholar]
- 54.Conklin BR, Farfel Z, Lustig KD, Julius D, Bourne HR, Nature 363, 274–276 (1993). [DOI] [PubMed] [Google Scholar]
- 55.Conklin BR et al. , Mol. Pharmacol 50, 885–890 (1996). [PubMed] [Google Scholar]
- 56.Gregorio GG et al. , Nature 547, 68–73 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.