

Neurodegenerative diseases are an ever-increasing burden in an aging society. Currently no cure is available for any of these diseases and treatment is based on managing symptoms. Despite many candidate therapeutics demonstrating promise in animal models, none has yet shown efficacy in human trials. It is self-evident that humans are different from the animals used to model our diseases, especially models that have been highly manipulated to generate a disease in an animal that does not naturally have such a disease. These differences are likely the reason for the failures of drug candidates in human trials but, until recently, human models of neurodegenerative diseases were lacking. The development of the human cerebral organoid model, by differentiating three-dimensional human neuronal tissue from pluripotent stem cells, represents a significant advance in studying human brain diseases. Cerebral organoids have been used to model Alzheimer’s disease, Parkinson’s disease, Down’s syndrome dementia and we have now shown they can be infected with human prions creating a new model of human prion diseases.
Prion diseases are fatal, transmissible, neurodegenerative diseases that affect humans and animals. The disease can arise spontaneously, have genetic origins, or be acquired due to exposure to contaminated materials. The most common form of human prion disease is sporadic Creutzfeldt-Jakob disease (CJD), occurring in 1–2 people per million per year. Progression of prion diseases results in protease resistant prion protein (PrP) deposition, astrogliosis, and neuronal loss with spongiosis. The molecular mechanism underlying these diseases is the conformational restructuring of PrP from a monomeric, primarily alpha-helical protein, to an abnormally folded, typically protease resistant, beta-sheet rich oligomer/fibril (PrPSc). Understanding the role of prions in the infection, transmission, and mechanism of disease has required the advent of animal and cell culture models.
Animal models for human prion disease have been instrumental to our understanding of the disease; they demonstrate many of the pathological features found in human disease including PrP deposition, protease resistant PrP, spongiform change, astrogliosis and death. However, animal models are low-throughput and, as for other neurodegenerative diseases, promising therapeutics tested in animals have failed to translate into effective therapies in human disease. Cell culture models allow for a high-throughput platform to test infection, treatments, and fundamental questions of prion biology. Many cell culture models have been developed to study prion diseases, allowing for acute infection or faithful (“stable”) propagation of various prions over many passages (Priola, 2018). Traditionally, however, human cell lines and cell lines expressing human PrP have not allowed for stable prion infection and do not display cytopathic signs. Primary culture systems, such as the cerebellar organotypic cultured slice system, better recapitulate more aspects of prion disease but are restricted to animal tissues. Together, this has been a source of difficulty when studying human prion diseases.
To date, only one report of successfully infecting an immortalized cell line with human prions has been described (Ladogana et al., 1995). This study, published in 1995, described infecting CJD into sub-clones of the neuroblastoma cell line Sh-SY5Y. However, there have been no subsequent studies using this model in nearly 15 years.
Aside from the above study, a cellular model for human prion disease was not reported until 2017 when Krejciova et al. (2017) successfully infected astrocytes, derived from human induced pluripotent stem cells (Hu-iPSCs), with sporadic and variant CJD. This model was a significant advancement in the modeling of human prion diseases and allowed for infection, faithful subtype reproduction, and sub-passage of PrPSc prions.
Another benefit to Hu-iPSC derived cell lines is that they can be patient specific. Fibroblasts are isolated from a living donor, reprogrammed into Hu-iPSCs and used to generate various types of cells. This technique is particularly useful for studying genetic variants. For example, Matamoros-Angles et al. (2018) generated Hu-iPSC derived neurons and spherical neural masses (SNMs) using fibroblasts isolated from individuals with a predisposition to prion disease due to the genetic mutation Y218N in the PRNP gene, which causes Gerstmann-Straussler-Scheinker syndrome. SNMs are multipotent masses of precursor cells that can be matured into two-dimensional neurons with a limited glial population. With these systems, select pathological changes such as astrogliosis and hyperphosphorylation of tau were observed in the Y218N cultures. However, this model failed to replicate PrPSc production and deposition seen in vivo (Matamoros-Angles et al., 2018). Furthermore, neither neurons or SNMs generated from Hu-iPSCs with nor without the Y218N mutation were able to propagate infection by inoculation with human PrPSc, from either sporadic CJD or Y218N GSS. Additionally, these systems are both monolayer cultures and do not provide for the study of three-dimentional neuronal responses to infection or larger network cross-talk analysis. Finally, one general difficulty with genetic neurodegenerative diseases, such as the above-mentioned study, is that following differentiation, disease progression may take a longer time to manifest than the culture system will allow. While these models enable critical “pre-clinical” studies, they do not yet fully recapitulate the end stage of disease.
As seen with the Hu-iPSC derived astrocytes, neurons, and SNMs in the previously mentioned study, Hu-iPSC technology has progressed a great deal in recent years. Specifically, Hu-iPSCs have now been used to generate three-dimensional human brain tissues called cerebral organoids (Lancaster and Knoblich, 2014; Renner et al., 2017). This model displays self-organization of brain regions such as the cerebral cortex and generates a neuronal network organization similar to that seen in the developing brain. The organoids eventually populate with mature cortical neuronal subtypes, including mature pyramidal cells, astrocytes and oligodendrocytes. This is the closest in vitro model of the human brain that has yet been described (Lancaster and Knoblich, 2014; Renner et al., 2017; Marton et al., 2019). These organoids can survive in culture for months to years and develop spontaneous network activity as well as regional specification (Lancaster and Knoblich, 2014; Yakoub, 2019).
Organoids have already shown robust potential to model developmental diseases such as microcephaly following infection with Zika virus (Lancaster et al., 2013; Salick et al., 2017). Additionally, their longevity in vitro allows researchers to study disorders that develop over a longer time period such as tau hyperphosphorylation with amyloid deposition seen in Alzheimer’s disease and Down’s syndrome dementia (Gonzalez et al., 2018). This provides a new platform for studying human prion infection in a human brain analog.
Similar to the previously mentioned Hu-iPSC derived models, organoids can be derived from specific patients. Although afflicted with some of the drawbacks as other genetic models, iPSCs containing mutations in the PRNP gene can be used to generate organoids with a pre-disposition to prion disease. And while these genetically predisposed organoids have yet to show that they can fully develop the disease (Gonzalez et al., 2018), they can significantly aid in our understanding of the early pre-clinical stages and the mechanisms that may ultimately lead to disease.
Recently, our group determined that human cerebral organoids are capable of reproducing human prion infection and various aspects of pathogenesis (Groveman et al., 2019). When subjected to human PrPSc in the form of brain homogenates from sporadic CJD, the organoids demonstrate uptake, clearance, and re-emergence of prion self-seeding activity as well as de novo propagation with deposition of fully glycosylated protease resistant PrPSc. Furthermore, organoids differentiated from the same underlying iPSCs may have the ability to differentiate distinct sCJD subtypes based on their biological properties. This exciting new platform for studying human prion disease infection opens the door to understanding the pathologies caused by different human prion subtypes.
Organoids provide the opportunity to study changes over the course of the disease that other prion models cannot. For example, general health of the tissue and degree of cell death can be easily monitored to track disease progression. Cytokine profiles can also be studied for insight into how the disease influences neuroinflammation, as well as understanding cross-talk between different cell types as the disease progresses. Indeed, our group has shown that certain changes in cytokine profiles in the organoids mimic was has been seen in human disease (Groveman et al., 2019).
The cerebral organoid system is not without its limitations. As a model for prion disease, we found astrogliosis and spongiosis to be indistinguishable between the infected and control organoids. This is likely due to the age of the organoids as earlier timepoints of infection showed some evidence of these cytopathic signs being disease associated. Further studies will help determine if these disease-associated changes can be seen reliably. More generally, cerebral organoids have a distinct level of heterogeneity, they are not vascularized, and they lack non-neuronally derived cells such as epithelial cells and microglia. Many studies are now looking to address these deficiencies to generate an ever-closer model to bona fide human brain tissue. Furthermore, organoid technology is continually advancing such that organoids can be differentiated corresponding to different brain regions with different neuronal populations. The information on disease processes that could be gleaned by combining such advances with the prion infection model is exciting to say the least.
Cerebral organoids may hold the key for many of the outstanding questions still plaguing the prion research community. Organoids provide a unique three-dimensional model with which to study the infection and spread of prions from cell to cell and throughout the entire brain. Unlike more commonly used monocultures (Figure 1), organoids can be used to determine how different neuronal subtypes interact with each other to cause the myriad of pathologies observed in clinical cases as well as what role other common cell types found within the mature brain, such as oligodendrocytes and astrocytes, play in disease incubation and pathology. This enhanced interaction and cellular crosstalk found in a three-dimensional model provides a distinct advantage over two-dimensional models and, coupled with the longevity of this system without need for subsequent passage, is likely an important factor for successful infection with human prions. Further, our model will hopefully enable researchers to observe any shared pathological mechanisms across prion disorders thereby elucidating the common mechanisms of cell death which could most readily be targeted (Figure 1). Ultimately, cerebral organoids provide a promising platform for therapeutic drug discovery in a far more relevant human tissue background, which may finally overcome the failures of therapeutic compounds previously tested only in animal systems.
Figure 1.
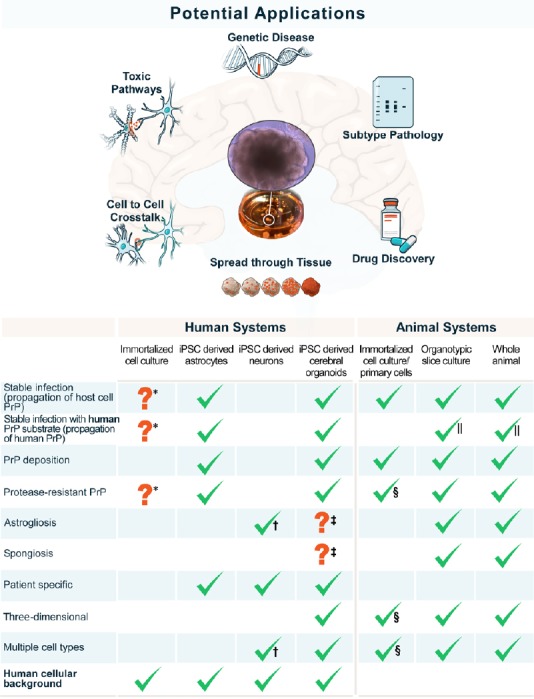
Applications and attributes of cerebral organoids as a prion disease model.
Schematic showing the potential investigations that could be carried out using human cerebral organoids (upper) and direct comparison of the disease features recapitulated by organoids with other systems used for the investigation of human prion disease (lower). *: Only found in one study; †: limited astroglial population; ‡: spongiosis and gliosis were observed but could not be discerned from aging; §: if differentiated from neural stem cell cultures; ||: transgenic mice expressing human PrP. iPSC: Induced pluripotent stem cells; PrP: prion protein.
The authors would like to thank Austin Athman and Anita Mora for their graphical assistance.
This work was supported in part by the Intramural Research Program of the NIH, NIAID (to CLH).
Footnotes
Copyright license agreement: The Copyright License Agreement has been signed by all authors before publication.
Plagiarism check: Checked twice by iThenticate.
Peer review: Externally peer reviewed.
C-Editors: Zhao M, Li JY; T-Editor: Jia Y
References
- 1.Gonzalez C, Armijo E, Bravo-Alegria J, Becerra-Calixto A, Mays CE, Soto C. Modeling amyloid beta and tau pathology in human cerebral organoids. Mol Psychiatry. 2018;23:2363–2374. doi: 10.1038/s41380-018-0229-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Groveman BR, Foliaki ST, Orru CD, Zanusso G, Carroll JA, Race B, Haigh CL. Sporadic Creutzfeldt-Jakob disease prion infection of human cerebral organoids. Acta Neuropathol Commun. 2019;7:90. doi: 10.1186/s40478-019-0742-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Krejciova Z, Alibhai J, Zhao C, Krencik R, Rzechorzek NM, Ullian EM, Manson J, Ironside JW, Head MW, Chandran S. Human stem cell-derived astrocytes replicate human prions in a PRNP genotype-dependent manner. J Exp Med. 2017;214:3481–3495. doi: 10.1084/jem.20161547. J Exp Med 214:3481-3495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ladogana A, Liu Q, Geng Xi Y, Pocchiari M. Proteinase-resistant protein in human neuroblastoma cells infected with brain material from Creutzfeldt-Jakob patient. Lancet. 1995;345:594–595. doi: 10.1016/s0140-6736(95)90508-1. [DOI] [PubMed] [Google Scholar]
- 5.Lancaster MA, Knoblich JA. Generation of cerebral organoids from human pluripotent stem cells. Nat Protoc. 2014;9:2329–2340. doi: 10.1038/nprot.2014.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lancaster MA, Renner M, Martin CA, Wenzel D, Bicknell LS, Hurles ME, Homfray T, Penninger JM, Jackson AP, Knoblich JA. Cerebral organoids model human brain development and microcephaly. Nature. 2013;501:373–379. doi: 10.1038/nature12517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Marton RM, Miura Y, Sloan SA, Li Q, Revah O, Levy RJ, Huguenard JR, Pasca SP. Differentiation and maturation of oligodendrocytes in human three-dimensional neural cultures. Nat Neurosci. 2019;22:484–491. doi: 10.1038/s41593-018-0316-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Matamoros-Angles A, Gayosso LM, Richaud-Patin Y, di Domenico A, Vergara C, Hervera A, Sousa A, Fernandez-Borges N, Consiglio A, Gavin R, Lopez de Maturana R, Ferrer I, Lopez de Munain A, Raya A, Castilla J, Sanchez-Pernaute R, Del Rio JA. iPS cell cultures from a Gerstmann-Straussler-Scheinker patient with the Y218N PRNP mutation recapitulate tau pathology. Mol Neurobiol. 2018;55:3033–3048. doi: 10.1007/s12035-017-0506-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Priola SA. Cell biology of prion infection. Handb Clin Neurol. 2018;153:45–68. doi: 10.1016/B978-0-444-63945-5.00003-9. [DOI] [PubMed] [Google Scholar]
- 10.Renner M, Lancaster MA, Bian S, Choi H, Ku T, Peer A, Chung K, Knoblich JA. Self-organized developmental patterning and differentiation in cerebral organoids. EMBO J. 2017;36:1316–1329. doi: 10.15252/embj.201694700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Salick MR, Wells MF, Eggan K, Kaykas A. Modelling zika virus infection of the developing human brain in vitro using stem cell derived cerebral organoids. J Vis Exp. 2017 doi: 10.3791/56404. doi: 103791/56404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yakoub AM. Cerebral organoids exhibit mature neurons and astrocytes and recapitulate electrophysiological activity of the human brain. Neural Regen Res. 2019;14:757–761. doi: 10.4103/1673-5374.249283. [DOI] [PMC free article] [PubMed] [Google Scholar]