

Abstract
Among collagen members in the collagen superfamily, type XIX collagen has raised increasing interest in relation to its structural and biological roles. Type XIX collagen is a Fibril-Associated Collagen with Interrupted Triple helices member, one main subclass of collagens in this superfamily. This collagen contains a triple helix composed of three polypeptide segments aligned in parallel and it is associated with the basement membrane zone in different tissues. The molecular structure of type XIX collagen consists of five collagenous domains, COL1 to COL5, interrupted by six non-collagenous domains, NC1 to NC6. The most relevant domain by which this collagen exerts its biological roles is NC1 domain that can be cleavage enzymatically to release matricryptins, exerting anti-tumor and anti-angiogenic effect in murine and human models of cancer. Under physiological conditions, type XIX collagen expression decreases after birth in different tissues although it is necessary to keep its basal levels, mainly in skeletal muscle and hippocampal and telencephalic interneurons in brain. Notwithstanding, in amyotrophic lateral sclerosis, altered transcript expression levels show a novel biological effect of this collagen beyond its structural role in basement membranes and its anti-tumor and anti-angiogenic properties. Type XIX collagen can exert a compensatory effect to ameliorate the disease progression under neurodegenerative conditions specific to amyotrophic lateral sclerosis in transgenic SOD1G93A mice and amyotrophic lateral sclerosis patients. This novel biological role highlights its nature as prognostic biomarker of disease progression in and as promising therapeutic target, paving the way to a more precise prognosis of amyotrophic lateral sclerosis.
Keywords: anti-tumor and anti-angiogenic properties, C1 domain, COL19A1 gene and protein levels, compensatory effect, FACIT collagens, hippocampal interneurons, matricryptins, multiplexins, NC1 domain, regenerative response, skeletal muscle, type XIX collagen
The Collagen Superfamily, a Backward Glance in the Nature of Type XIX Collage
Since it was discovered in 1992, type XIX collagen has raised interest among members of Fibril-Associated Collagens with Interrupted Triple helices (FACIT) family. FACIT collagens belong to the collagen superfamily that encompasses 28 members numbered with Roman numerals in vertebrates from I to XXVIII (Ricard et al., 2011; Oudart et al., 2017) (Table 1). A novel epidermal collagen, type XXIX collagen is the last collagen identified albeit its gene was identical to the COL6A5 gene, and the α1 (XXIX) chain corresponds to the α5 (VI) chain (Ricard et al., 2011). All the collagen members share in common three polypeptide chains known as chains. These α chains can vary in size from 600 to more than 3000 amino acid residues, and they are characterized by one or more polypeptide segments with multiple Gly-Xaa-Yaa sequence repeats, often proline for Xaa and hydroxyproline for Yaa, respectively. To date, 42 genes coding for 46 different α chains have been identified in this collagen superfamily which represents for about 30% of the total protein weight in mammals and they are also present in all invertebrate phyla (Brown and Timpl, 1995; Oudart et al., 2017). All these family members share in common a triple helix composed of three polypeptide segments aligned in parallel and their exclusive localization in extracellular matrices (Brown and Timpl, 1995). The main fact is that most of the α chains can contain several non-triple helical protein modules, which confers greater module diversity than any other protein family since they are unique to this family or they can be shared by other extracellular proteins (Brown and Timpl, 1995).
Table 1.
Collagen members of the collagen superfamily
| HGNC ID (gene) | Approved symbol | Approved name | Previous symbols | Synonyms | Chromosome |
|---|---|---|---|---|---|
| HGN02197 | COL1A1 | collagen type 1 alpha 1 chain | 014 | 17q21.33 | |
| HGNC:2198 | COL1A2 | collagen type 1 alpha 2 chain | 014 | 7q21.3 | |
| HGNC:2200 | COL2A1 | collagen type II alpha 1 chain | SE DC, AO M | STL1 | 12ql3.11 |
| HGNC2201 | COL3A1 | collagen type III alpha 1 chain | EDS4A | 2q32.2 | |
| HGNC2202 | COL4A1 | collagen type IV alpha 1 chain | 13q34 | ||
| HGNC:2203 | COL4A2 | collagen type IV alpha 2 chain | FU222S9,DKFZp686ll4213 | 13q34 | |
| HGNC2204 | COL4A3 | collagen type IV alpha 3 chain | 2q36.3 | ||
| HGNC2206 | COL4A4 | collagen type IV alpha 4 chain | CA44 | 2q36.3 | |
| HGNC:2207 | COL4A5 | collagen type IV alpha S chain | ASLN.ATS | Xq22.3 | |
| HGNC2208 | COL4A6 | collagen type IV alpha 6 chain | Xq22.3 | ||
| HGNC2209 | COL5A1 | collagen type V alpha 1 chain | 9q34.3 | ||
| HGNC:2210 | COL5A2 | collagen type V alpha 2 chain | 2q32.2 | ||
| HGNC:14864 | COL5A3 | collagen type V alpha 3 chain | 19pl3.2 | ||
| HGNC2211 | COL6A1 | collagen type VI alpha 1 chain | 21q22.3 | ||
| HGN02212 | COL6A2 | collagen type VI alpha 2 chain | 21q22.3 | ||
| HGNC2213 | COL6A3 | collagen type VI alpha 3 chain | 2q37.3 | ||
| HGN033484 | COL6A4P1 | collagen type VI alpha 4 pseudogene 1 | DVWA | VWA6,DIVA,COL6A4,COL6A4P | 3p25.1 |
| HGNC:38501 | COL6A4P2 | collagen type VI alpha 4 pseudogene 2 | COL6A4 | 3q22.1 | |
| HGNC:26674 | COL6A5 | collagen type VI alpha 5 chain | COL29A1 | FU 35880, VWA4 | 3q22.1 |
| HGNC:27023 | COL6A6 | collagen type VI alpha 6 chain | 3q22.1 | ||
| HGNC2214 | COL7A1 | collagen type VII alpha 1 chain | EBDCT,EBD1,EBR1 | 3p21.31 | |
| HGNC2215 | COL8A1 | collagen type VIII alpha 1 chain | C30rf7 | MGC9568 | 3ql2.1 |
| HGNC2216 | COL8A2 | collagen type VIII alpha 2 chain | FECD | PPCD,FECD1,PPCD2 | lp34.3 |
| HGNC:2217 | COL9A1 | collagen type IX alpha 1 chain | 6ql3 | ||
| HGNC2218 | COL9A2 | collagen type IX alpha 2 chain | EDM2 | MED | lp34.2 |
| HGNC2219 | COL9A3 | collagen type IX alpha 3 chain | IDD,MED,EDM3,FU90759,DJ885L7.4.1 | 20ql3.33 | |
| HGNC2185 | COLlOAl | collagen type X alpha 1 chain | 6q22.1 | ||
| HGNC2186 | COL11A1 | collagen type XI alpha 1 chain | COLL6.DFNA37 | STL2,C011A1 | lp21.1 |
| HGNC2187 | COL11A2 | collagen type XI alpha 2 chain | DFNA13,DFNB53 | HKES | 6p21.32 |
| HGNC2188 | COL12A1 | collagen type XII alpha 1 chain | COL12A1L | 6ql3-ql4.1 | |
| HGNC:2190 | COL13A1 | collagen type XIII alpha 1 chain | 10q22.1 | ||
| HGNC2191 | COL14A1 | collagen type XIV alpha 1 chain | UND | 8q24.12 | |
| HGNG2192 | COL1SA1 | collagen type XV alpha 1 chain | 9q22.33 | ||
| HGNC:2193 | COL16A1 | collagen type XVI alpha 1 chain | lp35.2 | ||
| HGNC2194 | COL17A1 | collagen type XVII alpha 1 chain | BPAG2 | BP180 | 10q25.1 |
| HGNC2195 | COL18A1 | collagen type XVIII alpha 1 chain | KNO | KS,KN01 | 21q22.3 |
| HGNC2196 | COL19A1 | collagen type XIX alpha 1 chain | 6ql3 | ||
| HGNC14670 | COL20A1 | collagen type XX alpha 1 chain | KIAA1510 | 20ql3.33 | |
| HGNC:17025 | COL21A1 | collagen type XXI alpha 1 chain | 6pl2.1 | ||
| HGNC:22989 | COL22A1 | collagen type XXII alpha 1 chain | 8q24.23-q24.3 | ||
| HGNC:22990 | COL23A1 | collagen type XXIII alpha 1 chain | DKFZp434K0621 | 5q35.3 | |
| HGNC:20821 | COL24A1 | collagen type XXIV alpha 1 chain | lp22.3 | ||
| HGNC:18603 | COL25A1 | collagen type XXV alpha 1 chain | 4q25 | ||
| HGNC:18038 | COL26A1 | collagen type XXVI alpha 1 chain | EMID2 | Emu2,EMI6 | 7q22.1 |
| HGNC:22986 | COL27A1 | collagen type XXVII alpha 1 chain | KIAA1870,MGC11337, FU 11895 | 9q32 | |
| HGNC:22442 | COL28A1 | collagen type XXVIII alpha 1 chain | 7p21.3 |
Adapted from HUGO Gene Nomenclature Committee, 2019.
This diversity in protein modules has hindered an easy classification of collagens in different subfamilies that seem to be grouped in three main categories. One homogeneous group is fibrillary collagens that are involved in the formation of major cross-striated fibrils. These fibrillary collagens share a triple helical segment of approximately 1000 amino acids residues following a perfect Gly-Xaa-Yaa sequence repeat over its entire length. Structurally, they contain a C-terminal propeptide or CF module and an N-terminal propeptide or VC/TN module, which are both released by proteases together with a few smaller triple helical and non-triple helical segments to form fibrils. Notwithstanding, the heterogeneous group of non-fibrillar collagens share triple helical segments of variable length, some of them subdivided by larger non-collagenous segments that contains Gly-Xaa or Xaa-Yaa sequence. They also share non-collagenous modules such as the VA module of the von Willebrand factor, the C4 module present in collagen IV and the F3 module present in fibronectin. Contrary to fibrillary collagens, most of these modules are not released by processing and become part of the globular domain. These non-fibrillar collagens can associate with fibrillary collagens or they can form separate networks. Finally, the FACIT family, multiplexins and the membrane collagen subfamilies that contain several triple-helical domains can vary in the length of their triple-helical segments albeit they share the frequent interruption of these segments by large non-triple helical regions and they present N- and/or C-terminal noncollagenous domains that could represent globular structures (Brown and Timpl, 1995; Ricard et al., 2011).
In particular, types IX, XII, XIV, XVI, XX, XXI, XXII form the FACIT family to which type XIX collagen also belongs to. These collagens are involved in the integrity and stability of the extracellular matrix, modulating the formation and size of the collagen fibrils and controlling cellular organization in the extracellular matrix (Oudart et al., 2017). Their specific localization in some tissues and the fine regulation of their expression make FACIT family members key targets to understand functional alterations in different cells and organs. In fact, type XIX collagen was discovered from a human rhabdomyosarcoma cell line (CCL-36) (Brown and Timpl, 1995; Oudart et al., 2017). The precise functions of this collagen still remain unclear although it is known that it is involved in the formation and maintenance of the extracellular matrix, especially during the embryonic stage. In contrast, the biochemical characterization and the chromosomal location of type XIX collagen has been deeply analyzed.
This review includes our experience in the field of type XIX collagen related to amyotrophic lateral sclerosis (ALS), as well as articles retrieved using electronic search of the PubMed database. Searching criteria included articles describing the following subjects: “type XIX collagen, COL19A1, skeletal muscle, neurodegeneration, amyotrophic lateral sclerosis, structure, properties”. Articles that were published up to April 2019 were included.
Molecular Characterization of Type XIX Collagen
The human type XIX collagen (COL19A1) gene that is mapped on human chromosome 6 in the q12-q14 region contains 55 exons in a full length of 250 kb. This gene encodes for a protein that exhibits a molecular structure based on three α1 chains in a 400 kDa homotrimer fashion. A total of 3426 amino acids are present in these α1 chains, 1142 amino acids per chain with a molecular mass of 112 kDa with a 23 residue signal peptide (Inoguchi et al., 1995). The structure in each chain follows the same distribution based on multidomains: 5 collagenous domains, COL1 to COL5, interrupted by 6 non-collagenous domains, NC1 to NC6, which consist of 19, 44, 23, 31, 20 and 291 amino acids, respectively (Oudart et al., 2017) (Figure 1A). The high homology observed between some exons encoding the first four non-collagenous and collagenous domains of the collagen α1 (XIX) chain and the collagen α2 (IX) chain suggest that an ancestral gene could encode the different FACITα chains (Gerecke et al., 1997; Khaleduzzaman et al., 1997; Oudart et al., 2017). Furthermore, there is also high sequence homology between the human and murine collagen α1 (XIX) chains (82% identity), especially in the C-terminal side in the NC1 domain that is essential for the conformational and functional roles of the protein and it contains a plasmin cleavage site (Sumiyoshi et al., 1997; Oudart et al., 2017).
Figure 1.

Molecular structure of type XIX collagen and gene levels variations from embryonic to adult stages.
(A) Structural organization of type XIX collagen protein. Five collagenous domains designated by COL1 to COL5, and six non-collagenous domains designated by NC1 to NC6 complete the molecular structure of the protein encoded by type XIX collagen. Fourteen cysteine residues located in NC1, NC4 and NC6 domains play a relevant role in the formation and stabilization of the triple helix. (B) Variations of transcript levels of Col19a1 in mice. Col19a1 gene levels are essential in the regulation of muscle differentiation in skeletal muscle and they decrease from the embryonic to the adult stage, while in the hippocampus their levels increase ten-fold higher in the adult stage to maintain synapse formation in hippocampal interneurons. TSP: Thrombospondin domain.
Interestingly, the coding region of type XIX collagen gene is quite short in relation to the length of the mRNA because of the long 3′untranslated region, which spans more than 5 kb (Inoguchi et al., 1995). A second relevant feature of this gene is based on the fact that 3 microsatellites were identified in 50 unrelated individuals in a Japanese cohort. These microsatellites consisted of cytosine-adenine repeated sequences located 1.9 kb upstream of exon 3, 120 bp upstream of exon 6 and 1.0 kb downstream of exon 12, suggesting that they could be useful for linkage study of heritable connective tissue disorders since the combined heterozygosity was found 0.82 (Khaleduzzaman et al., 1997).
The amino acid sequence of type XIX collagen shares strong similarities to other FACIT members, such as types IX, XII, XIV, XVI and XXII collagens. Indeed, types IX and XII collagens are located in the same chromosomal localization (Oudart et al., 2017). The NC6 domain is the one that shows the strongest homologies with NC4, NC2 and NC11 α1 chains domains of types IX, XII, XIV and XVI collagens, respectively, and contains an heparin binding site. The NC2 domain is involved in the trimerization of α1 (XIX) chain as well as in the formation of disulfide inter-chain bonds that connect COL1 and NC1 domains (Boudko et al., 2008, 2010). These disulfide bonds are promoted by the 14 cysteine residues that enable the formation of both inter and intra-chain disulfide bonds, enhancing the stabilization of the triple helix and increasing the thermal transition temperature of the triple helix. Moreover, the presence of a disulfide bond at the end of the last collagen domain prevents the C-terminal end of the collagen triple helix from fraying (Boudko et al., 2008). Electron microscopy confirmed a rod-like structure of 240 nm in the type XIX collagen with a globular domain at the N-terminal region (Myers et al., 2003). This rod-like structure can change when temperature rises above 27°C into a semi flexible three arm star-like chain, in which the single coil peptide chain is extended by the high segment density surrounding the linking domain (Terao et al., 2015). This dynamic property could be explained by the involvement of the interruption sequence G5G in the middle of Gly-Xaa-Yaa repeats and in relation to hydrogen bonding topology, torsion angles and helical parameters (Xu et al., 2018).
Considering that it contains a thrombospondin like N-terminal domain in NC6 and it is not associated with fibres, type XIX collagen has also been described as a member of the multiplexin family, to which types XV and XVIII collagens belongs to (Myers et al., 1997; Sumiyoshi et al., 2001; Ricard et al., 2011). Additionally, the localization of type XIX collagen in the basement membrane zone of different organs resembles the one observed in other multiplexins. Albeit these similarities, type XIX collagen as a whole is a unique collagen member that can play different roles depending on its localization.
A Wide Range of Expression Patterns and Functions in Type XIX Collage
Type XIX collagen is structural part of the basement membrane, a specialized extracellular matrix in vertebrates and some invertebrates. These membranes are present in epithelia, connective tissue, blood vessel walls, axons, adipocytes and muscle tissue. Furthermore, they present a double functionality: one the one side, these membranes preserve the architecture of the tissues, and on the other side, they can regulate biological functions in touch with other cells by means of adapter proteins. In this sense, they can play a role in cell migration, adhesion, differentiation, proliferation and chemotaxis (Oudart et al., 2017).
Albeit type XIX collagen was firstly identified in rhabdomyosarcoma cDNA clones, immunohistochemical analysis provided a wider distribution of this collagen in different human and murine tissues, such as breast, colon, kidney, liver, placenta, prostate, skeletal muscle, brain (cerebral cortex and hippocampus), skin and spleen (Sumiyoshi et al., 2001; Myers et al., 2003; Su et al., 2010; Oudart et al., 2017). Notwithstanding, the transcript and protein levels of type XIX collagen (COL19A1) not only vary from the embryonic to the adult stage in some tissues but they also decrease gradually after birth, except for the brain in which Col19a1 gene expression is ten-fold higher in adult mice respect to the embryonic state (Sumiyoshi et al., 1997).
In mice, the Col19a1 gene expression is transient and confined to muscle cells and it is localized in limbs, tongue and the smooth muscle layers of the stomach and esophagus of the developing embryo (Sumiyoshi et al., 2001). In particular, it is detected in the embryonic state at 9th day of gestation and its expression gradually declines concomitantly to the increase in myogenin gene activation, suggesting that the presence of type XIX collagen at early embryonic stages is relevant for the muscle tissue differentiation (Sumiyoshi et al., 2001) (Figure 1B). These findings are in accordance with the kind of cells in which type XIX collagen was initially discovered. Rhabdomyosarcoma cell lines were used as a model to analyze the embryogenesis of skeletal muscle and differentiation of mesenchymal cells. Indeed, some cells of these lines produce type XIX collagen and they can induce myoblast differentiation under culture conditions, highlighting the main role of this type of collagen in muscle differentiation and its association with a subpopulation of cells that synthesize embryonic skeletal muscle-specific proteins (Myers et al., 1999). Additionally, knockout Col19a1 mice exhibited muscle alterations related to extracellular matrix disruption and failed in the activation of myogenic regulatory factors, confirming the role of Col19a1 gene in muscle development (Sumiyoshi et al., 2004).
Apart from muscle tissue, Col19a1 is also present in brain, especially in some populations of hippocampal interneurons that express neuropeptide Y, somatostatin and calbindin. Contrary to muscle tissue, Col19a1 expression is higher in adult brain than in the embryonic state (Figure 1B) and it is necessary for the formation of hippocampal synapses and assembly of inhibitory nerve terminals by means of a matricryptin, a fragment that derives from Col19a1 gene in a proteolytic process, revealing a novel paracrine mechanism that can regulate the assembly of these synapses (Su et al., 2010, 2016). In addition, the lack of Col19a1 gene in mice exerted a decrease of inhibitory synapses and in the hippocampus and subiculum, which are mainly involved in memory tasks (Su et al., 2010). Furthermore, loss of Col19a1 enhances degradation of perineuronal nets in telencephalic interneurons of collagen XIX-deficient mice, which suggests that the lack of this collagen could contribute to complex brain disorders (Su et al., 2017). In line with this, Col19a1 has also a role in axon guidance in a zebrafish model, suggesting its involvement in axonal migration (Hilario et al., 2010).
In human, COL19A1 shows a wider distribution during the embryonic stage rather than in the adult stage. In the former stage it is localized in specific regions such as vasculature, neural and muscular basal membrane zone of skeletal muscle, spleen, skin, kidney, colon, prostate and in the hippocampus in the brain (Oudart et al., 2013, 2017). Interestingly, during the last decade some relevant studies have analyzed the connection of type XIX collagen with specific disorders or diseases, such as breast cancer and melanoma and ALS. As a matter of fact, the susceptibility of type XIX collagen to proteolysis or abnormal levels of synthesis could be involved in different pathological states.
Proteolysis of Type XIX Collagen Exerts an Anti-Angiogenic and Anti-Tumor Effects
The first studies that revealed a biological function of type XIX collagen analyzed the role of this collagen in tumor progression (Oudart et al., 2013, 2015, 2016; Monboisse et al., 2014). The molecular basis relies on the fact that NC1 domain of type XIX collagen can suffer enzymatic proteolysis and release matricryptins or matrikines which resides in the NC1 domain of this collagen and other network-forming collagens (type IV collagen) or multiplexin collagens (types XV and XVIII collagens) (Ricard et al., 2011; Oudart et al., 2017). These matricryptins derived from type XIX collagen can induce a decrease in vascular endothelial growth factor expression in an in vivo murine model of melanoma and in a xenograft model of human melanoma (Ramont et al., 2007). Additionally, these matricryptins in the NC1 domain of type XIX collagen can interact with integrin receptors to inhibit the phosporylation of the focal adhesion kinase (FAK)/phosphoinositide 3-kinase/protein kinase B/mammalian target of rapamycin pathway. Since this pathway is activated to promote the proliferation, migration and angiogenesis of many cancers, such as breast cancer or melanoma, its inhibition by the C-terminal of NC1 domain in type XIX collagen confers anti-tumor and anti-angiogenic properties to this collagen member, providing new insights for the development of more promising anti-cancer therapeutic strategies (Toubal et al., 2010; Oudart et al., 2016). These anti-tumor properties can be favored by the proteolytic effect of plasmin, which is an important enzyme involved in tumor invasion and it can promote the release of a matricryptin from this collagen in a conformation-dependent fashion (Oudart et al., 2015).
Interestingly, as the two sides of a coin, the FAK molecular pathway can also promote a beneficial effect in skeletal muscle under physiological conditions beyond its role in cancer. The FAK pathway has a relevant role in the myogenesis, muscle homeostasis and in the modulation of the oxidative capacity of the muscle fiber (Graham et al., 2015). In particular, under pathological conditions such as muscular dystrophies or spinal cord injury, FAK expression is significantly reduced in the skeletal muscle, while satellite cells and myoblast differentiation are activated as a compensatory response for the muscle regeneration (Sakuma et al., 2004; Graham et al., 2015). Contrary to this activation, in transgenic SOD1G93A mice, one of the best characterized murine model of ALS, we found that the functions of skeletal muscle satellite cells are impaired in mutant superoxide 1 (SOD1) myogenic cell cultures from these mice. More in depth, a significant downregulation of myogenic regulatory factors, such as myogenin, was observed at the presymptomatic stages of the disease in these mutant SOD1 cell cultures, suggesting that the compensatory response to ameliorate disease progression is altered from early stages in this mouse model (Manzano et al., 2013). In addition, transgenic SOD1G93A mice that exhibited increasing mRNA levels of Col19a1 and myogenin along disease progression also showed a shorter lifespan (Calvo et al., 2012). This suggests that Col19a1 mRNA levels could be increased as a compensatory response to activatemuscle regeneration in mice in which the disease progressed faster. Therefore, since ALS is characterized by a progressive muscle weakness in a similar way as in muscular dystrophy pathologies, our hypothesis is based on the fact that the activation of Col19a1 gene expression could also exert a compensatory response, probably by means of the FAK pathway, to ameliorate muscle damage in the mice that would not be able to activate so efficiently the muscle regeneration mechanisms.
Abnormal Levels of Synthesis of Type XIX Collagen Reveal Its Nature as a Prognostic Biomarker in Amyotrophic Lateral Sclerosis
The detection and measurement of COL19A1 transcript and protein levels in different tissues under pathological conditions can shed light on its role as a prognostic and/or diagnostic biomarker for human diseases. The first study that gathered different methodological approaches to measure type XIX collagen was proposed by Oudart and coworkers (Oudart et al., 2013). This study provided evidence that this collagen can be detected and measured in different human tissue extracts and biological fluids, such as serum, amniotic fluid or cord blood. In the cases in which the diagnosis and prognosis of the disease is poor, as in ALS, the identification of potential biomarkers of disease in different tissues is a valuable tool to monitor the disease, even under treatment conditions, and to acquire a better understanding of its progression and ethiopathogenic origin.
Understanding amyotrophic lateral sclerosis
ALS is a rare and neurodegenerative disease, in which degeneration and death of motor neurons takes place gradually (Calvo et al., 2014; Al-Chalabi et al., 2016). In accordance with the Awaji criteria, both the upper motor neurons and the lower motor neurons degenerate or die in ALS, and as a consequence communication between neuron and muscle is lost, prompting progressive muscle weakening and the appearance of fasciculations. In the later stages of the disease, patients become paralyzed and up to 50% of patients demonstrate mild to moderate cognitive or behavioural impairment (Ringholz et al., 2005). The majority of ALS cases are sporadic (SALS) where 5–10% of cases correspond to familial ALS (FALS). FALS follows a predominantly autosomal dominant pattern, while in SALS mutations in several modifier genes have been identified and linked to its pathogenesis. To date, the most known mutations that produce the typical adult onset ALS phenotype are related to the SOD1 gene, Tar DNA-binding protein gene (TARDBP) (previously known as TDP-43), DNA/RNA-binding protein called fused in sarcoma (FUS), translocation in liposarcoma (TLS), and the most recent hexanucleotide repeat expansion in oC9ORF72 (Andersen et al., 2011; DeJesús Hernández et al., 2011; Jones et al., 2013; Al-Chalabi et al., 2016).
From a diagnostic point of view, the evaluation of the first steps of ALS diagnosis is difficult since there is a wide range of motor neuron diseases that share its common and heterogeneous symptoms (Burgunder et al., 2011). Regarding ALS, the dysfunction and loss of both upper and lower motor neurons together with gradual spasticity may be present in the weakened limb, affecting manual dexterity and gait although there should be no autonomic, sensory, or cognitive involvement (Vucic and Kiernan, 2009). Therefore, the need of identifying reliable diagnostic or prognostic biomarkers in ALS is an increasing field of research.
ALS disease in patients is highly heterogeneous although the selective degeneration and loss of motor neurons is present in all the cases. Two main hypotheses have been proposed to explain this selective neurodegeneration. Firstly, the “dying-forward” hypothesis postulates that glutamate cytotoxicity can disrupt cortical motor neurons, favouring the neurodegeneration in an anterograde way to the motor neurons from the anterior horn (Eisen and Weber, 2001). Secondly, the “dying-back” hypothesis suggests that the degeneration of motor neurons starts in the neuromuscular junction and it spreads to the soma of the neurons (Kiernan et al., 2011). In support of the latter hypothesis, the early degeneration of the neuromuscular junction comes before the loss of neurons of the spinal cord from transgenic SOD1G93A mice (Martineau et al., 2018). Albeit many molecular mechanisms have been described altered in the pathology of ALS, the main mechanisms that prompt the disease still remain unknown. Deregulations of RNA and protein processing, glutamate cytotoxicity, mitochondrial dysfunction, oxidative stress, microglia dysfunction, neuroinflammation and failure in the axonal transport are the most related mechanisms to ALS pathogenesis (Figure 2).
Figure 2.
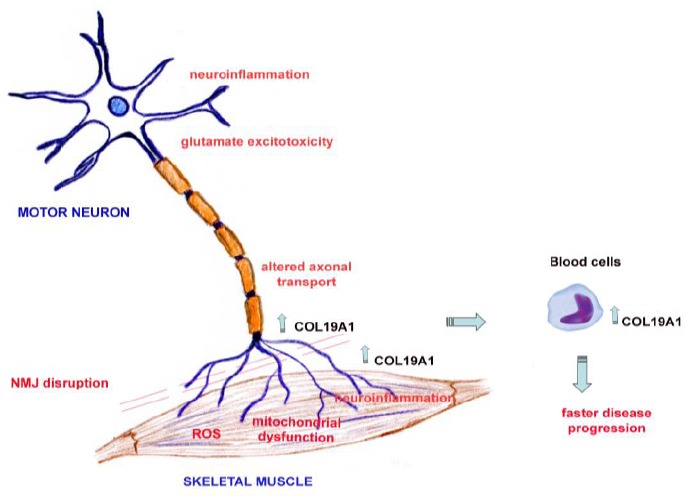
Schematic representation of the potential role of COL19A1 under neurodegenerative conditions in ALS.
In ALS, our hypothesis is that COL19A1 could try to counteract altered molecular mechanisms described in motor neurons and skeletal muscle that drive NMJ disruption. Adapted from Calvo et al., 2014. ALS: Amyotrophic lateral sclerosis; NMJ: neuromuscular junction; ROS: reactive oxygen species.
The understanding of mutations in SOD1, TARDBP, FUS, ATXN2, TAF15, hnRNPA1, hnRNPA2 B1, MATR3, EWSR1, TIA1, SETX, ANG and C9orf72 genes has open a new field of knowledge in ALS related to transcription process, alternative splicing and axonal transport of mRNA and biogenesis of microRNAs (Butti and Patten, 2019). In the case of TARDBP and FUS, mutant proteins translocate from the nucleus to the cytoplasm and this translocation could favour an abnormal processing of their target RNAs. In addition, the GGGGCC repeat expansion of C9orf72 could promote RNA toxicity by sequestering RNA binding proteins. Mutations in other genes such as VCP, OPTN, TBK1, VAPB, UBQLN2 and SQSTM1/p62 could enable the formation of misfolded protein aggregates. These aggregates might finally induce autophagy and proteasome dysfunction (Chang et al., 2013).
Glutamate cytotoxicity in neurons enhanced by an overstimulation of glutamate receptors can trigger a deregulation of calcium homeostasis and an accumulation of reactive oxygen species that in turn unbalance the mitochondrial homeostasis and the endosomal and vesicle transport. Therefore, the oxidative stress finally induced in motor neurons could enhance the vulnerability of these cells, as it has been described in both animal models and ALS patients (Magrane et al., 2014; Hardiman et al., 2017). Additionally, mitochondrial dysfunction could also prompt an insufficient ATP production and a failure in the axonal transport albeit mutations in DCTN, PFN1, TUBA4A, NEFH genes are also closely related to alterations in the axonal transport (Hardiman et al., 2017).
Another key mechanism that contributes to the ALS pathogenesis is the alteration of the microglial function. The activation of microglia could drive increased levels of reactive oxygen species and proinflammatory cytokines, and consequently a neuroinflammatory response, especially during the late stages of the disease (Beers and Appel, 2019). Neuroinflammation plays a relevant role in the neuronal damage and during the disease progression. This molecular mechanism early starts as an anti-inflammatory process to facilitate the production of neurotrophic factors and anti-inflammatory cytokines, while in later stages, it becomes an inflammatory process, enhancing the activation of inflammatory cytokines and a faster disease progression (Hooten et al., 2015).
Potential role of type XIX collagen in amyotrophic lateral sclerosis
We have previously identified five genes, Mef2c, Gsr, Calm1, Snx10 and Col19a1, as prognostic biomarkers of longevity in a mouse model of ALS, the transgenic SOD1G93A mice (Calvo et al., 2012). More in depth, increased Col19a1 gene expression levels in serial skeletal muscle biopsies correlated to a shorter survival in this animal model, suggesting that this collagen could exert a compensatory response in the skeletal muscle of the animals along the disease progression. Indeed, Col19a1 gene could exert a regenerative response to the muscle damage under muscle hypertrophy conditions, inhibiting the FAK molecular pathway, similarly as previously described in tumors (Oudart et al., 2017). Moreover, COL19A1 transcript levels have been also validated as prognostic biomarkers of disease progression in ALS patients (Calvo et al., 2019). In a multicenter study, involving three different cohorts of participants and a total of 148 ALS patients, we could validate COL19A1 transcript and protein levels as prognostic biomarkers of a faster progression of the disease in skeletal muscle biopsies and blood samples. In both skeletal muscle and blood, increased levels of COL19A1 indicated a worse functional state of the patients and a faster disease progression. These findings are in accordance with the observations in the animal model of ALS and with previous studies on a different cohort of ALS patients (Shtilbans et al., 2011). These suggest that COL19A1 could represent a compensatory stimulus to counteract the denervation processes along disease progression, in which the neuromuscular junctions are damaged and induce finally the motor neuron and skeletal muscle degeneration and loss. In this sense, the higher level of COL19A1 in blood or skeletal muscle could be indicative of a larger destabilization of motor neuron terminals, contributing to the neurodegenerative progression in ALS. As previously mentioned, the presence of type XIX collagen at early embryonic stages is relevant for the muscle tissue differentiation and its expression decreases to the adult stage in normal conditions (Sumiyoshi et al., 2001). However, under neurodegenerative conditions in ALS we have observed an increase of COL19A1 transcript levels in skeletal muscle tissue from animal models and ALS patients (Calvo et al., 2012, 2019) and this could possibly indicate that the increasing levels of COL19A1 in ALS is an attempt to induce a regeneration of the tissue by means of activating myogenic regulatory factors in the muscle, probably thorough the FAK pathway (Myers et al., 1999; Sumiyoshi et al., 2001). Additionally, an increase of COL19A1 transcript levels in blood might resemble the degeneration in the skeletal muscle, suggesting a faster progression of the disease (Zhou et al., 2016) (Figure 2). Consequently, COL19A1 levels could indicate a stimulus to restore the connection between motor neurons and skeletal muscle through the neuromuscular junction disruption. In this sense, the faster the disease progression is, the greater the COL19A1 response is induced.
These findings provide new evidence about the role of COL19A1 as a prognostic biomarker of ALS. A better understanding of the key role of this relatively unknown collagen in ALS could facilitate novel therapeutic strategies focused on the maintenance of the main affected tissues in ALS. However, our hypothesis is that although increased COL19A1 is indicative of a faster disease progression and a worsening of the functional state in ALS. Further studies will elucidate if this type of collagen could be also a direct cause in ALS, promoting a direct target for a therapeutic approach in ALS. To date, COL19A1 can be a suitable biomarker to facilitate functional monitoring of ALS patients in future clinical trials.
Future Considerations
Type XIX collagen is one of the FACIT member of collagen superfamily that it is associated with the basement membrane zone in different tissues. During the last two decades, growing knowledge about its structure at gene and protein level, its localization and its regenerating properties has been acquired in mammals, albeit it is still considered as one of the most peculiar collagen member. Under physiological conditions, type XIX collagen expression decreases after birth in different tissues although it is expressed in basal conditions, mainly in skeletal muscle and hippocampal and telencephalic interneurons in brain. Notwithstanding, under physiopathological conditions such as tumors and neurodegenerative diseases like ALS, conformational changes in its molecular structure or altered transcript expression levels show the biological effect of this collagen beyond its structural role in basement membranes. In such different scenarios, type XIX collagen exerts a regenerative and compensatory effect to slow tumor progression or to compensate the disease progression under neurodegenerative conditions specific to ALS. Precisely, this last biological role enables it to be considered a reliable prognostic biomarker in a neurodegenerative disease of unknown origin as ALS, paving the way to a better stratification of ALS patients in clinical trials and to more efficient therapeutic strategies to ameliorate the disease progression.
Footnotes
Conflicts of interest: The authors declare no conflicts of interest.
Financial support: This work was supported by Institutode Salud Carlos III (Grant PI17/00949) and Fondo Europeode Desarrollo Regional (FEDER) “Una manera de hacer Europa” from the European Union, Centrode Investigación Biomédicaen Redsobre Enfermedades Neurodegenerativas (CIBERNED-612), Fundación FEDER (Federación Españolade Enfermedades Raras), Consolidated Groupsfrom Gobiernode Aragón.
Copyright license agreement: The Copyright License Agreement has been signed by all authors before publication.
Plagiarism check: Checked twice by iThenticate.
Peer review: Externally peer reviewed.
Open peer reviewer: Colin Barnstable, University of Pennsylvania, USA.
Funding: This work was supported by Institutode Salud Carlos III (Grant PI17/00949) and Fondo Europeode Desarrollo Regional (FEDER) “Una manera de hacer Europa” from the European Union, Centrode Investigación Biomédicaen Redsobre Enfermedades Neurodegenerativas (CIBERNED-612), Fundación FEDER (Federación Españolade Enfermedades Raras), Consolidated Groupsfrom Gobiernode Aragón.
P-Reviewer: Barnstable C; C-Editors: Zhao M, Li JY; T-Editor: Jia Y
References
- 1.Al-Chalabi A, Hardiman O, Kiernan MC, Chiò A, Rix-Brooks B, van den Berg LH. Amyotrophic lateral sclerosis: moving towards a new classification system. Lancet Neurol. 2016;15:1182–1194. doi: 10.1016/S1474-4422(16)30199-5. [DOI] [PubMed] [Google Scholar]
- 2.Andersen PM, Al-Chalabi A. Clinical genetics of amyotrophic lateral sclerosis: what do we really know? Nat Rev Neurol. 2011;7:603–615. doi: 10.1038/nrneurol.2011.150. [DOI] [PubMed] [Google Scholar]
- 3.Beers DR, Appel SH. Immune dysregulation in amyotrophic lateral sclerosis: mechanisms and emerging therapies. Lancet Neurol. 2019;18:211–220. doi: 10.1016/S1474-4422(18)30394-6. [DOI] [PubMed] [Google Scholar]
- 4.Boudko SP, Engel J, Bachinger HP. Trimerization and triple helix stabilization of the collagen XIX NC2 domain. J Biol Chem. 2008;283:34345–34351. doi: 10.1074/jbc.M806352200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boudko SP, Zientek KD, Vance J, Hacker JL, Engel J, Bächinger HP. The NC2 domain of collagen IX provides chain selection and heterotrimerization. J Biol Chem. 2010;285:23721–23731. doi: 10.1074/jbc.M110.128405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brown JC, Timpl R. The collagen superfamily. Int Arch Allergy Immunol. 1995;107:484–490. doi: 10.1159/000237090. [DOI] [PubMed] [Google Scholar]
- 7.Burgunder JM, Schöls L, Baets J, Andersen P, Gasser T, Szolnoki Z, Fontaine B, Van Broeckhoven C, Di Donato S, De Jonghe P, Lynch T, Mariotti C, Spinazzola A, Tabrizi SJ, Tallaksen C, Zeviani M, Harbo HF, Finsterer J EFNS. EFNS guidelines for the molecular diagnosis of neurogenetic disorders: motoneuron, peripheral nerve and muscle disorders. Eur J Neurol. 2011;18:207–217. doi: 10.1111/j.1468-1331.2010.03069.x. [DOI] [PubMed] [Google Scholar]
- 8.Butti Z, Patten SA. RNA Dysregulation in Amyotrophic Lateral Sclerosis. Front Genet. 2019;9:712. doi: 10.3389/fgene.2018.00712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Calvo AC, Atencia G, Torre P, Roy JF, Galiana A, Jua’rez A, Cano JM, Marti’n MA, Moreno L, Larrode’ P, Cordero P, Gala’n L, Mora J, Mun~oz JL, Mun~oz MJ, Zaragoza P, Pegoraro E, Soraru’ G, Mora M, Lunetta C, et al. Collagen XIX alpha 1 improves prognosis in amyotrophic lateral sclerosis. Aging Dis. 2019;10:278–292. doi: 10.14336/AD.2018.0917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Calvo AC, Manzano R, Atencia-Cibreiro G, Oliva’n S, Mun~oz MJ, Zaragoza P, Cordero-Vázquez P, Esteban-Pérez J, García-Redondo A, Osta R. Genetic biomarkers for ALS disease in transgenic SOD1(G93A) mice. PLoS One. 2012;7:e32632. doi: 10.1371/journal.pone.0032632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Calvo AC, Manzano R, Mendonça DM, Muñoz MJ, Zaragoza P, Osta R. Amyotrophic lateral sclerosis: a focus on disease progression. Biomed Res Int. 2014;2014:925101. doi: 10.1155/2014/925101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chang HY, Hou SC, Way TD, Wong CH, Wang IF. Heat-shock protein dysregulation is associated with functional and pathological TDP-43 aggregation. Nat Commun. 2013;4:2757. doi: 10.1038/ncomms3757. [DOI] [PubMed] [Google Scholar]
- 13.DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, Nicholson AM, Finch NA, Flynn H, Adamson J, Kouri N, Wojtas A, Sengdy P, Hsiung GY, Karydas A, Seeley WW, Josephs KA, Coppola G, Geschwind DH, Wszolek ZK, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron. 2011;72:245–256. doi: 10.1016/j.neuron.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Eisen A, Weber M. The motor cortex and amyotrophic lateral sclerosis. Muscle Nerve. 2001;24:564–573. doi: 10.1002/mus.1042. [DOI] [PubMed] [Google Scholar]
- 15.Gerecke DR, Olson PF, Koch M, Knoll JH, Taylor R, Hudson DL, Champliaud MF, Olsen BR, Burgeson RE. Complete primary structure of two splice variants of collagen XII, and assignment of alpha 1(XII) collagen (COL12A1), alpha 1(IX) collagen (COL9A1), and alpha 1(XIX) collagen (COL19A1) to human chromosome 6q12–q13. Genomics. 1997;41:236–242. doi: 10.1006/geno.1997.4638. [DOI] [PubMed] [Google Scholar]
- 16.Graham ZA, Gallagher PM, Cardozo CP. Focal adhesion kinase and its role in skeletal muscle. J Muscle Res Cell Motil. 2015;36:305–315. doi: 10.1007/s10974-015-9415-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hardiman O, Al-Chalabi A, Chio A, Corr EM, Logroscino G, Robberecht W, Shaw PJ, Simmons Z, van den Berg LH. Amyotrophic lateral sclerosis. Nat Rev Dis Primers. 2017;3:17071. doi: 10.1038/nrdp.2017.71. [DOI] [PubMed] [Google Scholar]
- 18.Hilario JD, Wang C, Beattie CE. Collagen XIXa1 is crucial for motor axon navigation at intermediate targets. Development. 2010;137:4261–4269. doi: 10.1242/dev.051730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hooten KG, Beers DR, Zhao W, Appel SH. Protective and toxic neuroinflammation in amyotrophic lateral sclerosis. Neurotherapeutics. 2015;12:364–375. doi: 10.1007/s13311-014-0329-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Inoguchi K, Yoshioka H, Khaleduzzaman M, Ninomiya Y. The mRNA for alpha 1(XIX) collagen chain, a new member of FACITs, contains a long unusual 3′ untranslated region and displays many unique splicing variants. J Biochem. 1995;117:137–146. doi: 10.1093/oxfordjournals.jbchem.a124700. [DOI] [PubMed] [Google Scholar]
- 21.Jones AR, Woollacott I, Shatunov A, Cooper-Knock J, Buchman V, Sproviero W, Smith B, Scott KM, Balendra R, Abel O, McGuffin P, Ellis CM, Shaw PJ, Morrison KE, Farmer A, Lewis CM, Leigh PN, Shaw CE, Powell JF, Al-Chalabi A. Residual association at C9orf72 suggests an alternative amyotrophic lateral sclerosis-causing hexanucleotide repeat. Neurobiol Aging. 2013;34:2234e1–7. doi: 10.1016/j.neurobiolaging.2013.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Khaleduzzaman M, Sumiyoshi H, Ueki Y, Inoguchi K, Ninomiya Y, Yoshioka H. Structure of the human type XIX collagen (COL19A1) gene, which suggests it has arisen from an ancestor gene of the FACIT family. Genomics. 1997;45:304–312. doi: 10.1006/geno.1997.4921. [DOI] [PubMed] [Google Scholar]
- 23.Kiernan MC, Vucic S, Cheah BC, Turner MR, Eisen A, Hardiman O, Burrell JR, Zoing MC. Amyotrophic lateral sclerosis. Lancet. 2011;377:942–955. doi: 10.1016/S0140-6736(10)61156-7. [DOI] [PubMed] [Google Scholar]
- 24.Magrane J, Cortez C, Gan WB, Manfredi G. Abnormal mitochondrial transport and morphology are common pathological denominators in SOD1 and TDP43 ALS mouse models. Hum Mol Genet. 2014;23:1413–1424. doi: 10.1093/hmg/ddt528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Manzano R, Toivonen JM, Calvo AC, Oliva’n S, Zaragoza P, Rodellar C, Montarras D, Osta R. Altered in vitro proliferation of mouse SOD1-G93A skeletal muscle satellite cells. Neurodegener Dis. 2013;11:153–164. doi: 10.1159/000338061. [DOI] [PubMed] [Google Scholar]
- 26.Martineau E, Di Polo A, Vande Velde C, Robitaille R. Dynamic neuromuscular remodeling precedes motor-unit loss in a mouse model of ALS. Elife. 2018;7:e41973. doi: 10.7554/eLife.41973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Monboisse JC, Oudart JB, Ramont L, Brassart-Pasco S, Maquart FX. Matrikines from basement membrane collagens: a new anti-cancer strategy. Biochim Biophys Acta. 2014;1840:2589–2598. doi: 10.1016/j.bbagen.2013.12.029. [DOI] [PubMed] [Google Scholar]
- 28.Myers JC, Li D, Amenta PS, Clark CC, Nagaswami C, Weisel JW. Type XIX collagen purified from human umbilical cord is characterized by multiple sharp kinks delineating collagenous subdomains and by intermolecular aggregates via globular, disulfide-linked, and heparin-binding amino termini. J Biol Chem. 2003;278:32047–32057. doi: 10.1074/jbc.M304629200. [DOI] [PubMed] [Google Scholar]
- 29.Myers JC, Li D, Bageris A, Abraham V, Dion AS, Amenta PS. Biochemical and immunohistochemical characterization of human type XIX defines a novel class of basement membrane zone collagens. Am J Pathol. 1997;151:1729–1740. [PMC free article] [PubMed] [Google Scholar]
- 30.Myers JC, Li D, Rubinstein NA, Clark CC. Up-regulation of type XIX collagen in rhabdomyosarcoma cells accompanies myogenic differentiation. Exp Cell Res. 1999;253:587–598. doi: 10.1006/excr.1999.4642. [DOI] [PubMed] [Google Scholar]
- 31.Oudart JB, Brassart-Pasco S, Luczka E, Dupont-Deshorgue A, Bellon J, Boudko SP, Bachinger HP, Monboisse JC, Maquart FX, Ramont L. Analytical methods for measuring collagen XIX in human cell cultures, tissue extracts, and biological fluids. Anal Biochem. 2013;437:111–117. doi: 10.1016/j.ab.2013.03.007. [DOI] [PubMed] [Google Scholar]
- 32.Oudart JB, Brassart-Pasco S, Vautrin A, Sellier C, Machado C, Dupont- Deshorgue A, Brassart B, Baud S, Dauchez M, Monboisse JC, Harakat D, Maquart FX, Ramont L. Plasmin releases the anti-tumor peptide from the NC1 domain of collagen XIX. Oncotarget. 2015;6:3656–3668. doi: 10.18632/oncotarget.2849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Oudart JB, Doue’ M, Vautrin A, Brassart B, Sellier C, Dupont-Deshorgue A, Monboisse JC, Maquart FX, Brassart-Pasco S, Ramont L. The anti-tumor NC1 domain of collagen XIX inhibits the FAK/PI3K/Akt/mTOR signaling pathway through αvβ3 integrin interaction. Oncotarget. 2016;7:1516–1528. doi: 10.18632/oncotarget.6399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Oudart JB, Monboisse JC, Maquart FX, Brassart B, Brassart-Pasco S, Ramont L. Type XIX collagen: A new partner in the interactions between tumor cells and their microenvironment. Matrix Biol. 2017;57-58:169–177. doi: 10.1016/j.matbio.2016.07.010. [DOI] [PubMed] [Google Scholar]
- 35.Ramont L, Brassart-Pasco S, Thevenard J, Deshorgue A, Venteo L, Laronze JY, Pluot M, Monboisse JC, Maquart FX. The NC1 domain of type XIX collagen inhibits in vivo melanoma growth. Mol Cancer Ther. 2007;6:506–514. doi: 10.1158/1535-7163.MCT-06-0207. [DOI] [PubMed] [Google Scholar]
- 36.Ricard-Blum S. The collagen family. Cold Spring Harb Perspect Biol. 2011;3:a004978. doi: 10.1101/cshperspect.a004978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ringholz GM, Appel SH, Bradshaw M, Cooke NA, Mosnik DM, Schulz PE. Prevalence and patterns of cognitive impairment in sporadic ALS. Neurology. 2005;65:586–590. doi: 10.1212/01.wnl.0000172911.39167.b6. [DOI] [PubMed] [Google Scholar]
- 38.Sakuma K, Nakao R, Inashima S, Hirata M, Kubo T, Yasuhara M. Marked reduction of focal adhesion kinase, serum response factor and myocyte enhancer factor 2C, but increase in RhoA and myostatin in the hindlimb dy mouse muscles. Acta Neuropathol. 2004;108:241–249. doi: 10.1007/s00401-004-0884-5. [DOI] [PubMed] [Google Scholar]
- 39.Shtilbans A, Choi SG, Fowkes ME, Khitrov G, Shahbazi M, Ting J Zhang W, Sun Y, Sealfon SC, Lange DJ. Differential gene expression in patients with amyotrophic lateral sclerosis. Amyotroph Lateral Scler. 2011;12:250–256. doi: 10.3109/17482968.2011.560946. [DOI] [PubMed] [Google Scholar]
- 40.Su J, Gorse K, Ramirez F, Fox MA. Collagen XIX is expressed by interneurons and contributes to the formation of hippocampal synapses. J Comp Neurol. 2010;518:229–253. doi: 10.1002/cne.22228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Su J, Chen J, Lippold K, Monavarfeshani A, Carrillo GL, Jenkins R, Fox MA. Collagen-derived matricryptins promote inhibitory nerve terminal formation in the developing neocortex. J Cell Biol. 2016;212:721–736. doi: 10.1083/jcb.201509085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Su J, Cole J, Fox MA. Loss of interneuron-derived collagen XIX leads to a reduction in perineuronal nets in the mammalian telencephalon. ASN Neuro. 2017;9:1–16. doi: 10.1177/1759091416689020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sumiyoshi H, Inoguchi K, Khaleduzzaman M, Ninomiya Y, Yoshioka H. Ubiquitous expression of the alpha1(XIX) collagen gene (Col19a1) during mouse embryogenesis becomes restricted to a few tissues in the adult organism. J Biol Chem. 1997;272:17104–17111. doi: 10.1074/jbc.272.27.17104. [DOI] [PubMed] [Google Scholar]
- 44.Sumiyoshi H, Laub F, Yoshioka H, Ramirez F. Embryonic expression of type XIX collagen is transient and confined to muscle cells. Dev Dyn. 2001;220:155–162. doi: 10.1002/1097-0177(2000)9999:9999<::AID-DVDY1099>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 45.Sumiyoshi H, Mor N, Lee SY, Doty S, Henderson S, Tanaka S, Yoshioka H, Rattan S, Ramirez F. Esophageal muscle physiology and morphogenesis require assembly of a collagen XIX-rich basement membrane zone. J Cell Biol. 2004;166:591–600. doi: 10.1083/jcb.200402054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Terao K, Mizuno K, Bächinger HP. Conformational change from rigid rod to star: a triple-helical peptide with a linker domain at the C-terminal end. J Phys Chem B. 2015;119:3714–3719. doi: 10.1021/jp5129172. [DOI] [PubMed] [Google Scholar]
- 47.Toubal A, Ramont L, Terryn C, Brassart-Pasco S, Patigny D, Sapi J, Monboisse JC, Maquart FX. The NC1 domain of type XIX collagen inhibits melanoma cell migration. Eur J Dermatol. 2010;20:712–718. doi: 10.1684/ejd.2010.1070. [DOI] [PubMed] [Google Scholar]
- 48.Vucic S, Kiernan MC. Pathophysiology of neurodegeneration in familial amyotrophic lateral sclerosis. Curr Mol Med. 2009;9:255–272. doi: 10.2174/156652409787847173. [DOI] [PubMed] [Google Scholar]
- 49.Xu T, Zhou CZ, Xiao J, Liu J. Unique Conformation in a natural interruption sequence of type XIX collagen revealed by its high-resolution crystal structure. Biochemistry. 2018;57:1087–1095. doi: 10.1021/acs.biochem.7b01010. [DOI] [PubMed] [Google Scholar]
- 50.Zhou L, Hinerman JM, Blaszczyk M, Miller JL, Conrady DG, Barrow AD, Chirgadze DY, Bihan D, Farndale RW, Herr AB. Structural basis for collagen recognition by the immune receptor OSCAR. Blood. 2016;127:529–537. doi: 10.1182/blood-2015-08-667055. [DOI] [PMC free article] [PubMed] [Google Scholar]