

Abstract
The contribution of an impaired astrocytic K+ regulation system to epileptic neuronal hyperexcitability has been increasingly recognized in the last decade. A defective K+ regulation leads to an elevated extracellular K+ concentration ([K+]o). When [K+]o reaches peaks of 10–12 mM, it is strongly associated with seizure initiation during hypersynchronous neuronal activities. On the other hand, reactive astrocytes during a seizure attack restrict influx of K+ across the membrane both passively and actively. In addition to decreased K+ buffering, aberrant Ca2+ signaling and declined glutamate transport have also been observed in astrogliosis in epileptic specimens, precipitating an increased neuronal discharge and induction of seizures. This review aims to provide an overview of experimental findings that implicated astrocytic modulation of extracellular K+ in the mechanism of epileptogenesis.
Keywords: AQP4, Ca2+ signaling, cell volume, connexins, epilepsy, K+ buffering, miRNA, NKCC, pannexin, TBI
Introduction
Over 50 million people are suffering from epilepsy globally (Ngugi et al., 2010; Thurman et al., 2011; Losi et al., 2012), which amounts to 1% of the world population (Witcher and Ellis, 2012; Baldin et al., 2014). A seizure, as a fundamental component of epilepsy, is an uncontrolled sudden attack of behavioral change invoked by neuronal hyperexcitability. For decades, the underlying mechanism of seizure has baffled neuroscientists. As an increasing number of evidences surface through researches, the cardinal role of astrocytes in this perplexing condition has come to unfold and be recognized. Astrocytes are now believed to contribute to seizure both independently and concertedly.
As the most abundant sub-type of glial cells, astrocytes consist of one third of the brain. Their leaflet-like processes envelope the majority of the central nervous system (CNS) synapses, and the end-feet contribute to and retain the integrity of blood brain barrier. Astrocytes were formerly believed to provide energy for neurons and function primarily as metabolic and physical support in the CNS. Resent research has advanced our knowledge to the next level and shown that astrocytes also exert a pivotal function in neuronal homeostasis and modulation (Seifert and Steinhauser, 2013; Verkhratsky et al., 2017). Through potassium buffering (Kofuji and Newman, 2004) and agonist-induced Ca2+ wave (Wang et al., 2013), astrocytes can actively regulate ionic homeostasis, control neuronal excitability (Kofuji and Newman, 2004), maintain the blood-brain barrier integrity, sustain cellular water distribution, modulate amino acid neurotransmitter metabolism, and provide energy and nutrient support to neurons (Verkhratsky et al., 2014). These processes all contribute to a stable internal osmotic environment that normal cerebral activities rely on. These new findings along with other features render astrocyte a vital target in the field of epilepsy treatment through neuromodulation. In addition, recent epilepsy research has discovered genetic mutations of voltage-gated Na+, Ca2+ and K+ channels (Carmignoto and Haydon, 2012) and inhibitory synaptic transmission, which have been identified to be involved in the purported idiopathic epilepsy (McNamara et al., 2006). Herein, we reviewed the current concepts of astrocytic modulation of K+ under seizures.
Search Strategy and Selection Criteria
We conducted a literature search of the electronic databases, including PubMed/MEDLINE, Google Scholar from 1990 to 2019. The following terms were used accordingly: astrocytic potassium modulation; astrocytes calcium signaling; astrocytic epilepsy models; astrocytic cell volume; astrocyte and traumatic brain injury; traumatic brain injury and seizure. We have also reviewed reference lists of obtained publications. Literature that is not published in English has not been considered.
Increase of K+ Induces Paroxysmal Oscillation
The neuronal excitability in the CNS is heavily regulated by the K+ channel and extracellular potassium concentration (Kofuji and Newman, 2004). The opening of the K+ channel after a single action potential can result in a transient increase in the extracellular K+ concentration that amounts to nearly 1 mM above the resting potential; whereas that of an intensive neuronal firing discharge can create an increase of nearly 5 mM above the resting potential. An increased K+ derived from the latter scenario, i.e., an intense neuronal stimulation, predispose neurons to develop epileptiform discharges (Timofeev et al., 2002). In epileptogenesis studies, a peak of 10–12 mM in the [K+]o has been observed during an epileptic episode with hyper-synchronous neuronal activities (Walz, 2000; Carmignoto and Haydon, 2012). Although the association between [K+]o concentration and epilepsies has been well recognized and substantiated, it remains unclear whether the increase of [K+]o is the culprit of a seizure attack, or the result of the intense epileptic electrical discharge.
Grafstein (1956) hypothesized that K+ released during intense neuron discharge accumulated in the interstitial space, which in turn led to depolarization of neurons and spike inactivation. A combination of intrinsic conductance and elevated K+ can be sufficient to initiate 2–3 Hz oscillations. K+ modulation can influence immensely the maximal conductance of some depolarizing currents and the excitability of neurons (Timofeev et al., 2002; Bazhenov et al., 2004). Spontaneous local increase of neuronal discharge may rise K+ concentration; through the shift of reversal potential of K+ currents, elevated K+ can produce synchronized neuronal bursts (Bazhenov et al., 2004). A recent study has observed periodic bursting caused by increase of K+ in a cortical network model and a cerebellar Purkinje neuron circuit (Wang et al., 2012a). In vitro, bursting firing activities resulted from a simultaneous increase in extracellular K+ and decrease in Ca2+ concentrations have been recorded from granule cells of the dentate gyrus (Bazhenov et al., 2004). In external stimulation-initiated seizure, excessive electrical discharge provoked outward flow of K+ exceeding the capacity of the glial potassium buffering system – resulting in periodic bursting. Intriguingly, glial cells obtained from immediate biopsied tissue of the seizure focus from patients inflicted with intractable mesial-temporal lobe epilepsy have impaired K+ buffering (Rangroo Thrane et al., 2013), which is modulated by astrocyte.
Current Understanding of Astrocytic Modulation of K+
The [K+]o is continuously modulated by K+ pumps and K+ currents in neurons and astrocytes. Accumulation of K+ in the extracellular space can cause depolarization of neurons, further disturb neuronal signaling. Astrocytes play an essential role in maintaining extracellular K+ at a compatible level and continuing normal neuronal function (Seifert and Steinhauser, 2013). Astrocytic transmembrane modulation of K+ conductance exceeds all other conductance (Seifert et al., 2018). In vitro, external stimulation-induced elevation of K+ are accompanied with K+ accumulation in astrocytes, which reflects their role in potassium regulation (Larsen et al., 2014). The K+ level is mediated by the collaborative functions of K+ pumps and glial buffering mechanism. Undoubtably, increase of K+ will ensue from failure of either function, eventually invoking paroxysmal bursting.
Astrocytes are linked with abundant gap junctions that are permeable to K+. This provides an expansive network that facilitates a swift K+ redistribution through areas of different neuronal activities levels (Walz, 2000). As a major mechanism of K+ clearance, spatial buffering was initially reported by Orkand in 1960 (Kofuji and Newman, 2004). Briefly, spatial buffering theory asserts that the positive charged K+ enters and travels through the cytoplasm, it then exits again as positive charged K+ at a location distant from the active neurons. The overall effect is that, instead of accumulating intracellularly, K+ would be passively shipped from an extracellular space of a high K+ concentration to that of a low K+ concentration, in the absence of an increase of intracellular K+ concentration (Walz, 2000; Bellot-Saez et al., 2017).
Spatial buffering involves K+ influx possibility facilitated through astrocytic Kir4.1 K+ channels (Steinhauser et al., 2012; Elorza-Vidal et al., 2019), which are expressed in astrocytes surrounding both synapses and blood vessels in the brain. Consistently, astrocyte-specific knockout of Kir4.1 results in seizure activity and premature death (Djukic et al., 2007; Stewart et al., 2010; Tong et al., 2014). Brain-derived neurotrophic factor (BDNF) has been found to be expressed in astrocytes besides neurons. By forming a tripartite synapse, it can regulate neuronal activity. Interestingly, evidence has shown that Kir4.1 channel modulate the expression of BDNF in astrocytes (Miklic et al., 2004). Apart from attenuated K+ buffering and increased extracellular K+, dysfunctional astrocytes with inhibited Kir4.1 channel promotes BDNF expression, which triggers elevated neuronal discharges (Ohno et al., 2018). This has been speculated to contribute to epileptogenesis (Mukai et al., 2018).
At the same time, K+ leak channels also have prominent impact in this process as well as the high resting K+ conductance. The two-pore-domain K+ channels are the first to be reported of the potassium leak channels family. Although their fundamental roles in the generation of membrane potential were well recognized at the dawn of electrophysiology (Minieri et al., 2013), the molecular entities responsible for the background K+ currents, however, have remained obscure for nearly half a century. In 1995, K+ channels with two-pore-domain in tandem were successfully cloned and expressed with their functionality preserved. From 1996 to 2003, 14 new members joined the family of background K+ leak channels that were further divided into six subfamilies (TWIK, TREK, TASK, TALK, THIK, and TRESK) according to the sequence similarity and functional resemblance. All 15 types of K+ leak channels are encoded by different genes, yet sharing the same general molecular architecture that upholds characteristic features of relative time- and voltage-independence and K+ selectivity (Talley et al., 2003). The two-pore-domain channels are regulated by a variety of biological and chemical stimuli, intracellular and extracellular pH regulated by carbon dioxide, physiological fluctuation of oxygen tension and osmolarity, least but not last membrane stretch (Lesage et al., 2000).
Besides spatial buffering, additional mechanisms of potassium regulation have found to contribute to extracellular K+ homeostasis as well. Astrocytes intercellular communication is mainly fulfilled by gap junction channels (GJCs) consisted of connexin-based channels (Rovegno and Sáez, 2018). During synchronization of neuronal discharges, astrocytic GJCs expedite the clearance of extracellular K+. Studies investigating the role of Na+, K+-ATPase in the process of clearing K+ from extracellular space revealed that when extracellular K+ concentration was raised to 10 mM, a net increase of intracellular K+ concentration could reach up to 25 mM (Walz and Wuttke, 1999). This amplitude has been observed in astrocytes both in vivo and in vitro across studies (Ransom et al., 2000; Xiong and Stringer, 2000; Wang et al., 2012b). Disturbance of K+-Na+ exchange in the membrane promoted by ouabain-induced inhibition of Na+, K+-ATPase activity, on the other hand, has yielded an instantaneous decrease in the K+ concentration (Walz and Wuttke, 1999). In addition, there is a lack of difference in the diffusion coefficients for the extracellular K+ between normal and gliotic cortex (Walz and Wuttke, 1999). These altogether indicate that brain Na+, K+-ATPase also mediates the potassium clearing process in the extracellular space.
Loss of intracellular K+ to the extracellular space in neurons is recovered mainly through the activities of neuronal Na+, K+-ATPase. Glial cells, as described previously, employ various mechanisms to achieve a transient intracellular K+ accumulation, or redistribution through spatial buffering. Post-stimulus recovery of activity-dependent increase in [K+]o can be attributed to the activities of Na+, K+-ATPase in both neurons and glial cell- with the glial uptake contributing to the early stage of rapid fall in [K+]o, and neuronal uptake prevailing in the late stage of slow decrease in [K+]o (Ransom et al., 2000). During an extensive and intense neuronal stimulation, bulk K+ would accumulate in the extracellular space. Recovery of [K+]o mediated by Na+, K+-ATPase activities can be exceedingly slow and inefficient. Diffusion of K+ is anticipated to occur. In this circumstance, the Kir4.1 potassium channels are presumed to be greatly involved in regulating extracellular clearance of K+, either by spatial buffering or temporary storage (Meeks and Mennerick, 2007).
Effects of Extracellular K+ on Intracellular Cl–
The intracellular Na+ concentration of astrocyte ranges from 10 to 15mM (Rose and Karus, 2013). This is evidently inadequate for a 1:1 or 3:2 exchange of Na+ by K+ while the Na+, K+-ATPase functions at a high uptake rate. A transmembrane Na+ cycle has been proposed by Walz to explain the supplementation of intracellular Na+ (Rose and Karus, 2013). This cycle is mainly operated and fulfilled by the Na+, K+-ATPase and Na+-K+-Cl– co-transporter-1 (NKCC1) (Amadeo et al., 2018). As extracellular K+ is pumped into the intracellular space by Na+, K+-ATPase, intracellular Na+ will be exchanged to the extracellular space. This maneuver creates an electrochemical gradient of sodium across the membrane, which in turn provides the energy required by the NKCC to actively transport Na+, K+, and Cl– into the cell body with a stoichiometry of 1Na:1K:2Cl (Haas and Forbush, 2000).
While NKCC1 actively replenishes intracellular Na+ by transporting Na+ into the cells, it simultaneously creates a continuing influx of Cl–, which is believed to contribute to the active intracellular astrocytes Cl– accumulation (Liang and Huang, 2017). Active accumulation of Cl– has been demonstrated with GABA currents or Cl– substitution experiments (Walz and Wuttke, 1999), and observed in cortical astrocytes (Rangroo Thrane et al., 2013). The astrocyte intracellular Cl– concentration approximates 20–40 mM (Walz and Wuttke, 1999), with an average resting Cl– values around 30 mM. The inhibition of the NKCC1 induced with 1 uM bumetanide has reduced this resting Cl– by 50% in astrocyte (Su et al., 2000).
ClC-2, a voltage-gated chloride channel (CLC), is mainly expressed in the endfeet of astrocytes (Poroca et al., 2017). Upon depolarization, glial cell adhesion molecule (GlialCAM) will bind and modify CIC-2 to form a transmembrane complex, which then produce an influx of Cl– to counterbalance the excess K+ concentration (Elorza-Vidal et al., 2019). This compensatory mechanism can only occur under depolarization and may be required in certain high neuronal activity conditions (Estevez et al., 2018). A ClC-2 suppressed vacuolizing phenotype has been observed in the Kir4.1 ablated vacuolization (Blanz et al., 2007). This indicated that ClC-2 also contributes to the associated influx of Cl– in the process of K+ siphoning in addition to NKCC1 (Blanz et al., 2007; Sirisi et al., 2017; Elorza-Vidal et al., 2019).
Regulation of Astrocytic Cell Volume and Extracellular Space
Potassium level may also serve as an important modulator of cell volume and extracellular space (Larsen et al., 2014). Elevation of extracellular K+ concentration activates both active and passive uptake of K+ through Na+, K+-ATPase and other ion channels on astrocyte membrane. Rapid reversible swelling of astrocyte occurs subsequently with a correspondent decrease in extracellular space up to 30% (Risher et al., 2009). In contrast, decline of cell volume and enlargement of the extracellular space are associated with a fall in extracellular K+ concentration (Dallwig et al., 2000).
Astrocyte is particularly sensitive to the change in the extracellular fluid osmolarity. As extracellular osmolarity decreases following excessive uptake of K+, subsequent influx of water ensues and leads to astrocytic swelling. The inflow of water from extracellular space was speculated to be mediated through aquaporin-4 (AQP4) channel, coupled with the water-impermeable potassium selective K+ channels (Simard and Nedergaard, 2004; Mack and Wolburg, 2013). However, this previous popular coupling theory has recently been overturned (Haj-Yasein et al., 2011). In substitute, NKCC has been identified to modulate stimulus-evoked cell swelling in astrocytes (MacVicar et al., 2002), as cell shrinkage stimulates NKCC activity of astrocyte while NKCC inhibition reduces astrocyte swelling and K+ uptake. Following swelling of glial cells, a corrective process aiming for cell volume recovery will be enacted through extrusion of intracellular osmolytes consisting of K+ and Cl– and organic molecules including poly amines, taurine, GABA, glutamate and glycine. In addition, alternations of cell volume set off signal pathways that trigger swelling-activated channels (Kimelberg et al., 2006), which regulate the cell volume in return.
Volume-regulated anion channel (VRAC) is widely expressed in various types of cell membranes including astrocytes. Under cell swelling, VRACs are activated and transport anions and excitatory mediators outwards (Osei-Owusu et al., 2018). The exact role of VRAC is still largely under investigation. However, it has been postulated that the K+ spatial buffering triggers astrocytes swelling and lead to the activation of VRAC on astrocytic membranes, which facilitate the release of excitatory mediators and result in neuronal excitability (Lutter et al., 2017; Elorza-Vidal et al., 2019).
Astrocytic Ca2+ Signaling and Modulation
In the last two decades, it has been observed that astrocyte can transmit Ca2+ signal (Nedergaard, 1994; Parpura et al., 1994). Meanwhile, an increase in cytosolic Ca2+ has been documented in astrocytes in responses to synaptic neurotransmissions (Fiacco and McCarthy, 2004; Halassa et al., 2009). Neuroscientists have come to an agreement that astrocytic Ca2+ transients are of great importance in signal transmission between neurons and glial cells. The underlying mechanism, however, has yet to be unraveled.
Agonists such as glutamate and GABA combine with Gq-coupled plasma membrane receptors, which activate phospholipase C. Activated phospholipase C then hydrolyzes phosphatidylinositol 4,5-bisphosphate into the second messengers inositol 1,,4,5-trisphosphate and diacylglycerol, which trigger endoplasmic reticulum to release calcium into the cytosol (Bosanac et al., 2002; Dickson et al., 2013; Sakuragi et al., 2017). This provides the majority of Ca2+ in the astrocytic soma. In the astrocytic processes, however, ion channel mediated influx of Ca2+ consists nearly half of the Ca2+ transients. Increase of astrocytic Ca2+ in turn provokes release of astrocytic gliotransmitters that mediate neuronal excitability. Modern computerized model has also depicted that astrocytes synchronize an extensive network of neurons through a spaciotemporal characteristic of calcium dynamics. Our recent study has demonstrated the activation of Na+, K+-ATPase by astrocytic Ca2+ wave (Figure 1) induced by agonists, such as ATP and glutamate (data not published). This supplemented the current knowledge and understanding of second messenger Ca2+ regulated Na+, K+-ATPase activities.
Figure 1.
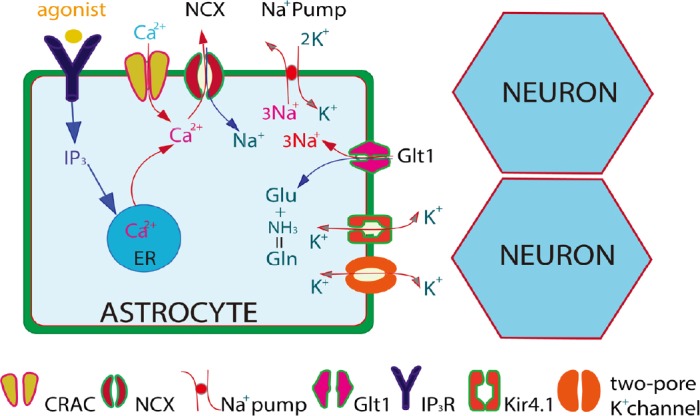
Agonist-induced Ca2+ signaling in astrocytes might work through lowering extracellular K+ to affect synaptic activities.
Agonist-induced astrocytic Gq-linked receptors trigger release of intracellular IP3 and mobilize Ca2+ endoplasmic reticulum (ER) Ca2+. Intracellular Ca2+ is transported outwards across the membrane by the Na+,Ca2+ exchanger (NCX) at the expense of Na+ influx. In turn, elevation of cytosolic Na+ activates the Na+,K+-ATPase, leading to a decrease in extracellular K+. The effect of Ca2+ wave on glutamate clearance is unclear. The figure is modified from Wang et al. (2013). CRAC: Ca2+ release activated Ca2+.
Multiple pathological conditions including ischemia, hypoglycemia, and seizures have demonstrated a decrease in extracellular Ca2+. It has been well established that clinical hypocalcemia is closely associated with seizure activities (Han et al., 2015). When extracellular Ca2+ concentration falls to the range of 0.001–0.1 mM, it will constantly evoke glial signaling (Tsumura et al., 2010). Decreased extracellular Ca2+ may mediate and increase neuronal excitability through four ways (Pan and Stringer, 1997): 1) subdue inhibitory interneurons functions by blocking neurotransmitters release; 2) suppress the afterhyperpolarization by decreasing the Ca2+-sensitive K+ conductance; 3) increase excitability by modulating surface charge density; 4) repress the normal function of the Na+, K+-ATPase. Both K+ and Ca2+ can, either directly or indirectly, affect neurons and generate bursting. It has been hypothesized that increase in [K+]o could be a primary factor in inducing low extracellular Ca2+ burst initiation and propagation (Ghai et al., 2000).
Modulation of the extracellular Ca2+ can be attributed to the Na+-Ca2+ exchanger, a bidirectional transporter that is activated by high intracellular Na+. Contingent on the electrochemical gradient of the substrate, Na+-Ca2+ exchanger promotes the electrogenic exchange of 3 or 4 Na+ for 1 Ca2+ (Tsumura et al., 2010), controls the resting Ca2+, and mediates the clearance of Ca2+ during agonist activation (DiPolo and Beauge, 2006; Reyes et al., 2012). Submerging hippocampal slice in a high K+ (over 5 mM) and low Ca2+ (below 0.5 mM) solution produced non-synaptic epileptiform activities (Feng and Durand, 2004). Similar epileptiform activities can be provoked in rat with 6.5 mM K+ and 5 mM EGTA induced low hippocampal extracellular Ca2+. This low-Ca2+ epilepsy model possesses characteristics of a focal hippocampal seizure, including a focal origin, local spread, a progressive increase in synchronicity, and post-event refractoriness (Ghai et al., 2000). Moreover, in vitro and in situ studies (Ghai et al., 2000) have shown that seizure associated neuronal activity increment results in increased K+ concentration and lowered Ca2+ levels in brain tissue. Initiation of epilepsy may arise from multiple pathways involving a concurrent disruption of both Ca2+ dependant astrocyte-neuro communication and K+ clearance mechanism (Li et al., 2019).
Effects of K+ on Astrocytic Modulation of Glutamate
Astrocyte plays a pivotal role in mediating extracellular glutamate concentration. The astrocyte processes are enriched with high affinity glutamate transporters, mainly GLT-1 transporter. This renders astrocyte the ability of transporting extracellular glutamate across the membrane into the intracellular space. Without astrocytic modulation, glutamate as a neurotransmitter can be build up in the extracellular space and reach an excitotoxic level to the CNS. Astrocyte prevents this from happening by maintaining a low extracellular glutamate concentration (Coulter and Eid, 2012; Gimenez-Cassina et al., 2012).
Astrocytic glutamate transporter harnesses the energy of electrochemical gradient of sodium produced by activity of Na+, K+-ATPase, moves one glutamate with three Na+ into the glial cell. Impairment of Na+, K+-ATPase function by extremely high extracellular K+ could impede the reuptake of glutamate. Once inside the astrocyte, glutamate is converted to glutamine astrocyte-specific glutamine synthetase enzyme (Coulter and Eid, 2012), which then will be released to the extracellular space to fuel neurons. Glutamine synthetase and glutamate transporters are key catalyzers in glutamate metabolism cycle.
Astrocyte reacts to CNS epileptic changes comprehensively. Studies have shown an upregulation of glutamine synthetase in reactive astrocytes, as well as a reduction of both glutamine synthetases and glutamate transporters in epileptic hippocampus (Proper et al., 2002; Coulter and Eid, 2012). Astrocyte can potentially modify and extend peri-synaptic processes after detecting copious glutamate released by synapses during a seizure attack (Coulter and Eid, 2012).
Traumatic Brain Injury, Astrocyte, and Seizures
An increased astrocyte activity, or astrocytosis, is believed to be activated by mechanical stress in brain injury, followed by a cascade of complex astrocytic signaling (Karve et al., 2016; Cheng et al., 2019). Astrocytosis entails changes in the number of astrocytes, astrocyte morphology, and gene expression. David et al. (2009) proposed that the disruption of blood brain barrier caused extravasation of albumin leading to modulation of astrocytic gene expression including downregulation of Kir4.1. This would reduce the K+ buffering and result in an accumulation of extracellular K+, followed by frequency-dependent facilitation of excitatory postsynaptic potentials and epileptiform firing. Astrocytes have a substantial increase of ATP sensitive potassium channels in brain contusion specimens (Kir6.2), indicating astrocytes are involved in potassium modulation in TBI (Castro et al., 2018).
Uncontrolled release of tremendous amount of excitatory amino acids after a severe traumatic brain injury (TBI) further disturbs the ion hemostasis, along with a loss of K+ conductance, eventually results in an increase in extracellular K+ concentration. As discussed previously, astrocytes exert homeostatic functions involving the balance of various ions including K+, Cl–, Ca2+. K+ efflux is specifically associated with excessive neuronal activity. An upregulation of NKCC and AQP4 in the astrocyte end-feet have also been observed in TBI, indicating that astrocytes are also involved in the TBI related brain edema (Lo Pizzo et al., 2013; Wang et al., 2017; Palazzo et al., 2019). Extracellular K+ clearance through the mechanism of K+ spatial buffering mediated by astrocytes is critical for homeostatic recovery (Du et al., 2016; Amadeo et al., 2018; Lykke et al., 2019). In the event of TBI, disruption of normal astrocyte function could lead to an persistent increase of extracellular K+ triggering paroxysmal neuronal activities, followed by abnormal neuronal phenomena including seizures (Topolnik et al., 2003). This might be the underlying mechanism of posttraumatic epileptogenesis.
Dysfunctional Astrocytes and Epilepsy
It is indisputable that astrocytes involve extensively in epileptogenesis. Investigators have developed and studied numerous epileptic models with a particular focus on astrocyte. In its expansive role of regulating ion homeostasis, astrocytes mediate extracellular K+ concentration through a concerted mechanism incorporating seamlessly K+ channels including Kir4.1 and Kir6.2 (ATP sensitive), AQP4 water channels, NKCC, and GJCs (Kadala et al., 2015; Haj-Mirzaian et al., 2019). Specimen study from patients suffering temporal lobe epilepsy and epilepsy models have revealed dysfunctional astrocytes with altered expression of Kir4.1, AQP4 and connexin-based channels (Steinhauser et al., 2016; Dossi et al., 2018). Studies in computerized model, knock out animal epilepsy model, and epileptic patients have revealed that extracellular K+ accumulation as a consequence of dysfunctional astrocytes further prompts seizure like neuronal discharges (Grigorovsky and Bardakjian; Lu et al., 2019; Patel et al., 2019). Pannexins, a recently discovered innexins sharing similar topology with connexins, also contribute to GJCs. Pannexin-1 (Panx1) channels open in response to high extracellular K+ and release ATP, glutamate to the extracellular space firing up a series of excitatory neuronal activities that commence a seizure attack in animal models (Dossi et al., 2018; Rovegno and Sáez, 2018; Aquilino et al., 2019).
MicroRNAs (miRNAs), a group of non-coding small RNAs, have been discovered to regulate gene expression at a post-transcriptional level. Epileptic models have shown that many members of miRNAs found in the astrocytes are associated with pathophysiology of epilepsy. Korotkov et al. suggested that miRNA-132 in particular repress astrocytic expression of pro-epileptogenic factors (Korotkov et al., 2019). Zheng et al. (2019) demonstrated suppression of astrocytic activation in mouse epilepsy model by inhibiting miRNA-103a and improving epileptic pathology. Downregulation of A-type potassium currents by miRNAs in epileptic mice (Tiwari et al., 2019) has also been observed, and A-type K+ currents contribute to neuronal excitability and in-vitro epileptic activity (Wu et al., 2015; Niespodziany et al., 2019).
Astrocytes’ wide implication in epileptogenesis have undoubtedly provided a reservoir of targets, such as pannexin and miRNAs, that hold great potential for instigating innovative interventions to epilepsy.
Conclusion
Traditionally known as a mere supportive subtype of glial cell, astrocyte has proven to neuroscientists its imperative role in regulating extracellular K+ level, mediating and maintaining an internal K+ homeostasis (Wang et al., 2012b). Astrocytes fulfill this by employing two major systems: passive redistribution of K+ via spatial buffering, and active K+ uptake via Na+, K+-ATPase. The electrochemical gradient of sodium across the membrane produced in this process will provide energy to NKCC and glutamate transporters, which facilitate the reuptake of Cl– and glutamate from extracellular space.
Astrocytes respond to a number of external stimulations and changes by activation of Ca2+ signaling. However, the significance of astrocytic Ca2+ signaling remains largely unclear. Our studies indicated that agonist-induced increase in cytosolic Ca2+ modulates the astrocytic Na+, K+-ATPase activity, and furthermore neuronal excitability (Figure 2). Astrocytes diligently modulate epileptic activities owing primarily to their contribution to microenvironmental homeostasis and expansive integration within the neural network (Simard and Nedergaard, 2004).
Figure 2.
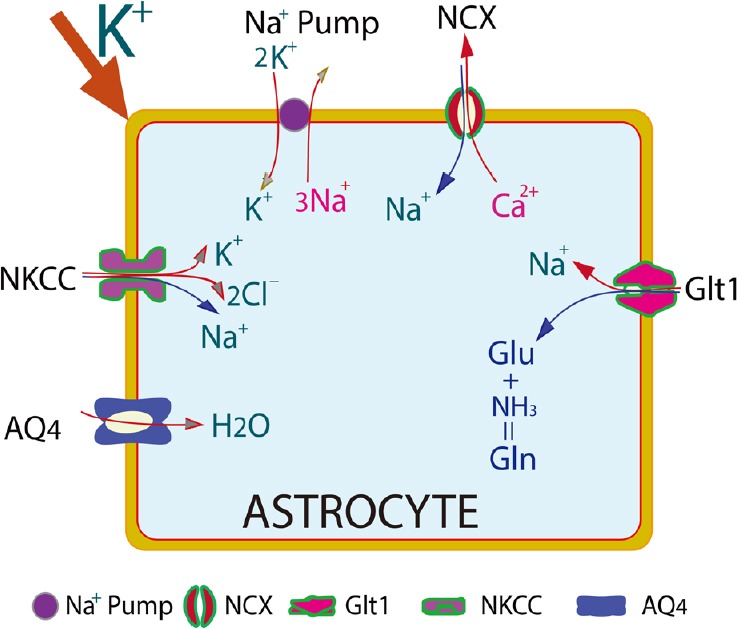
High extracellular K+ might act as a signaling in affecting many ion channels and transporters to induce seizures.
High [K+]o activates Na+,K+-ATPase, which pumps K+ into the astrocytes and Na+ out of the astrocytes. Na+ will be transported back into the cell through Na+,Ca2+ exchanger (NCX), or Na+,K+,2Cl– co-transporter (NKCC), accompanied by Ca2+ and Cl–, resulting in high intracellular Ca2+ and Cl–. High intracellular Na+ might further block the uptake of glutamate from extracellular space. Aquaporin-4 (AQP4) is intimately involved in post-traumatic edema. Its astrocytic expression has also been altered in epileptic models.
As a fundamental element of astrocyte physiological functions, astrocytic modulation of potassium homeostasis will undoubtably attain mounting attention in the intense search of new targets for epilepsy treatment (Crunelli and Carmignoto, 2013).
Footnotes
Conflicts of interest: The authors declare no conflicts of interest.
Financial support: This work was supported partially by Helen Vosburg McCrillus Plummer and Robert Edward Lee Plummer, Jr. Chair in Neurosurgery (to JHH) and by NIH-R01-NS-067435 (to JHH).
Copyright license agreement: The Copyright License Agreement has been signed by all authors before publication.
Plagiarism check: Checked twice by iThenticate.
Peer review: Externally peer reviewed.
Funding: This work was supported partially by Helen Vosburg McCrillus Plummer and Robert Edward Lee Plummer, Jr. Chair in Neurosurgery (JHH) and by NIH-R01-NS-067435 (to JHH).
C-Editors: Zhao M, Li JY; T-Editor: Jia Y
References
- 1.Amadeo A, Coatti A, Aracri P, Ascagni M, Iannantuoni D, Modena D, Carraresi L, Brusco S, Meneghini S, Arcangeli A, Pasini ME, Becchetti A. Postnatal changes in K(+)/Cl(-) cotransporter-2 expression in the forebrain of mice bearing a mutant nicotinic subunit linked to sleep-related epilepsy. Neuroscience. 2018;386:91–107. doi: 10.1016/j.neuroscience.2018.06.030. [DOI] [PubMed] [Google Scholar]
- 2.Aquilino MS, Whyte-Fagundes P, Zoidl G, Carlen PL. Pannexin-1 channels in epilepsy. Neurosci Lett. 2019;695:71–75. doi: 10.1016/j.neulet.2017.09.004. [DOI] [PubMed] [Google Scholar]
- 3.Baldin E, Hauser WA, Buchhalter JR, Hesdorffer DC, Ottman R. Yield of epileptiform electroencephalogram abnormalities in incident unprovoked seizures: a population-based study. Epilepsia. 2014;55:1389–1398. doi: 10.1111/epi.12720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bazhenov M, Timofeev I, Steriade M, Sejnowski TJ. Potassium model for slow (2-3 Hz) in vivo neocortical paroxysmal oscillations. J Neurophysiol. 2004;92:1116–1132. doi: 10.1152/jn.00529.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bellot-Saez A, Kekesi O, Morley JW, Buskila Y. Astrocytic modulation of neuronal excitability through K(+) spatial buffering. Neurosci Biobehav Rev. 2017;77:87–97. doi: 10.1016/j.neubiorev.2017.03.002. [DOI] [PubMed] [Google Scholar]
- 6.Blanz J, Schweizer M, Auberson M, Maier H, Muenscher A, Hubner CA, Jentsch TJ. Leukoencephalopathy upon disruption of the chloride channel ClC-2. J Neurosci. 2007;27:6581–6589. doi: 10.1523/JNEUROSCI.0338-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bosanac I, Alattia JR, Mal TK, Chan J, Talarico S, Tong FK, Tong KI, Yoshikawa F, Furuichi T, Iwai M, Michikawa T, Mikoshiba K, Ikura M. Structure of the inositol 1, 4,5-trisphosphate receptor binding core in complex with its ligand. Nature. 2002;420:696–700. doi: 10.1038/nature01268. [DOI] [PubMed] [Google Scholar]
- 8.Carmignoto G, Haydon PG. Astrocyte calcium signaling and epilepsy. Glia. 2012;60:1227–1233. doi: 10.1002/glia.22318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Castro L, Noelia M, Vidal-Jorge M, Sanchez-Ortiz D, Gandara D, Martinez-Saez E, Cicuendez M, Poca MA, Simard JM, Sahuquillo J. Kir6, 2, the pore-forming subunit of ATP-sensitive K(+) channels, is overexpressed in human posttraumatic brain contusions. J Neurotrauma? 2, the pore-forming subunit of ATP-sensitive K(+) channels, is overexpressed in human posttraumatic brain contusions. J Neurotrauma. 2018 doi: 10.1089/neu.2017.5619. doi: 101089/neu20175619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cheng X, Wang J, Sun X, Shao L, Guo Z, Li Y. Morphological and functional alterations of astrocytes responding to traumatic brain injury. J Integr Neurosci. 2019;18:203–215. doi: 10.31083/j.jin.2019.02.110. [DOI] [PubMed] [Google Scholar]
- 11.Coulter DA, Eid T. Astrocytic regulation of glutamate homeostasis in epilepsy. Glia. 2012;60:1215–1226. doi: 10.1002/glia.22341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Crunelli V, Carmignoto G. New vistas on astroglia in convulsive and non-convulsive epilepsy highlight novel astrocytic targets for treatment. J Physiol. 2013;591:775–785. doi: 10.1113/jphysiol.2012.243378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dallwig R, Vitten H, Deitmer JW. A novel barium-sensitive calcium influx into rat astrocytes at low external potassium. Cell Calcium. 2000;28:247–259. doi: 10.1054/ceca.2000.0153. [DOI] [PubMed] [Google Scholar]
- 14.David Y, Cacheaux LP, Ivens S, Lapilover E, Heinemann U, Kaufer D, Friedman A. Astrocytic dysfunction in epileptogenesis: Consequence of altered potassium and glutamate homeostasis? J Neurosci. 2009;29:10588–10599. doi: 10.1523/JNEUROSCI.2323-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dickson EJ, Falkenburger BH, Hille B. Quantitative properties and receptor reserve of the IP3 and calcium branch of Gq-coupled receptor signaling. J Gen Physiol. 2013;141:521–535. doi: 10.1085/jgp.201210886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.DiPolo R, Beauge L. Sodium/calcium exchanger: influence of metabolic regulation on ion carrier interactions. Physiol Rev. 2006;86:155–203. doi: 10.1152/physrev.00018.2005. [DOI] [PubMed] [Google Scholar]
- 17.Djukic B, Casper KB, Philpot BD, Chin LS, McCarthy KD. Conditional knock-out of Kir4, 1 leads to glial membrane depolarization, inhibition of potassium and glutamate uptake, and enhanced short-term synaptic potentiation. J Neurosci. 2007;27:11354–11365. doi: 10.1523/JNEUROSCI.0723-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dossi E, Blauwblomme T, Moulard J, Chever O, Vasile F, Guinard E, Le Bert M, Couillin I, Pallud J, Capelle L, Huberfeld G, Rouach N. Pannexin-1 channels contribute to seizure generation in human epileptic brain tissue and in a mouse model of epilepsy. Sci Transl Med. 2018 doi: 10.1126/scitranslmed.aar3796. doi:101126/scitranslmedaar3796. [DOI] [PubMed] [Google Scholar]
- 19.Du M, Li J, Wang R, Wu Y. The influence of potassium concentration on epileptic seizures in a coupled neuronal model in the hippocampus. Cogn Neurodyn. 2016;10:405–414. doi: 10.1007/s11571-016-9390-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Elorza-Vidal X, Gaitán-Peñas H, Estévez R. Chloride channels in astrocytes: Structure, roles in brain homeostasis and implications in disease. Int J Mol Sci. 2019 doi: 10.3390/ijms20051034. doi:103390/ijms20051034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Estevez R, Elorza-Vidal X, Gaitan-Penas H, Perez-Rius C, Armand-Ugon M, Alonso-Gardon M, Xicoy-Espaulella E, Sirisi S, Arnedo T, Capdevila-Nortes X, Lopez-Hernandez T, Montolio M, Duarri A, Teijido O, Barrallo-Gimeno A, Palacin M, Nunes V. Megalencephalic leukoencephalopathy with subcortical cysts: A personal biochemical retrospective. Eur J Med Genet. 2018;61:50–60. doi: 10.1016/j.ejmg.2017.10.013. [DOI] [PubMed] [Google Scholar]
- 22.Feng Z, Durand DM. Suppression of excitatory synaptic transmission can facilitate low-calcium epileptiform activity in the hippocampus in vivo. Brain Res. 2004;1030:57–65. doi: 10.1016/j.brainres.2004.09.063. [DOI] [PubMed] [Google Scholar]
- 23.Fiacco TA, McCarthy KD. Intracellular astrocyte calcium waves in situ increase the frequency of spontaneous AMPA receptor currents in CA1 pyramidal neurons. J Neurosci. 2004;24:722–732. doi: 10.1523/JNEUROSCI.2859-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ghai RS, Bikson M, Durand DM. Effects of applied electric fields on low-calcium epileptiform activity in the CA1 region of rat hippocampal slices. J Neurophysiol. 2000;84:274–280. doi: 10.1152/jn.2000.84.1.274. [DOI] [PubMed] [Google Scholar]
- 25.Gimenez-Cassina A, Martinez-Francois JR, Fisher JK, Szlyk B, Polak K, Wiwczar J, Tanner GR, Lutas A, Yellen G, Danial NN. BAD-dependent regulation of fuel metabolism and K(ATP) channel activity confers resistance to epileptic seizures. Neuron. 2012;74:719–730. doi: 10.1016/j.neuron.2012.03.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Grafstein B. Mechanism of spreading cortical depression. J Neurophysiol. 1956;19:154–171. doi: 10.1152/jn.1956.19.2.154. [DOI] [PubMed] [Google Scholar]
- 27.Grigorovsky V, Bardakjian BL. Effects of astrocytic mechanisms on neuronal hyperexcitability. Conf Proc IEEE Eng Med Biol Soc. 2014;2014:4880–4883. doi: 10.1109/EMBC.2014.6944717. [DOI] [PubMed] [Google Scholar]
- 28.Haas M, Forbush B. The Na-K-Cl cotransporter of secretory epithelia. Annu Rev Physiol. 2000;62:515–534. doi: 10.1146/annurev.physiol.62.1.515. [DOI] [PubMed] [Google Scholar]
- 29.Haj-Mirzaian A, Ramezanzadeh K, Afshari K, Mousapour P, Abbasi N, Haj-Mirzaian A, Nikbakhsh R, Haddadi NS, Dehpour AR. Activation of ATP-sensitive K-channel promotes the anticonvulsant properties of cannabinoid receptor agonist through mitochondrial ATP level reduction. Epilepsy Behav. 2019;93:1–6. doi: 10.1016/j.yebeh.2019.01.025. [DOI] [PubMed] [Google Scholar]
- 30.Haj-Yasein NN, Vindedal GF, Eilert-Olsen M, Gundersen GA, Skare O, Laake P, Klungland A, Thoren AE, Burkhardt JM, Ottersen OP, Nagelhus EA. Glial-conditional deletion of aquaporin-4 (Aqp4) reduces blood-brain water uptake and confers barrier function on perivascular astrocyte endfeet. Proc Natl Acad Sci U S A. 2011;108:17815–17820. doi: 10.1073/pnas.1110655108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Halassa MM, Fellin T, Haydon PG. Tripartite synapses: roles for astrocytic purines in the control of synaptic physiology and behavior. Neuropharmacology. 2009;57:343–346. doi: 10.1016/j.neuropharm.2009.06.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Han P, Trinidad BJ, Shi J. Hypocalcemia-induced seizure: demystifying the calcium paradox. ASN Neuro. 2015 doi: 10.1177/1759091415578050. doi:101177/1759091415578050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kadala A, Verdier D, Morquette P, Kolta A. Ion homeostasis in rhythmogenesis: The interplay between neurons and astroglia. Physiology. 2015;30:371–388. doi: 10.1152/physiol.00023.2014. [DOI] [PubMed] [Google Scholar]
- 34.Karve IP, Taylor JM, Crack PJ. The contribution of astrocytes and microglia to traumatic brain injury. Br J Pharmacol. 2016;173:692–702. doi: 10.1111/bph.13125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kimelberg HK, Macvicar BA, Sontheimer H. Anion channels in astrocytes: biophysics, pharmacology, and function. Glia. 2006;54:747–757. doi: 10.1002/glia.20423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kofuji P, Newman EA. Potassium buffering in the central nervous system. Neuroscience. 2004;129:1045–1056. doi: 10.1016/j.neuroscience.2004.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Korotkov A, Broekaart DWM, Banchaewa L, Pustjens B, van Scheppingen J, Anink JJ, Baayen JC, Idema S, Gorter JA, van Vliet EA, Aronica E. microRNA-132 is overexpressed in glia in temporal lobe epilepsy and reduces the expression of pro-epileptogenic factors in human cultured astrocytes. Glia. 2019 doi: 10.1002/glia.23700. doi:101002/glia23700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Larsen BR, Assentoft M, Cotrina ML, Hua SZ, Nedergaard M, Kaila K, Voipio J, MacAulay N. Contributions of the Na(+)/K(+)-ATPase, NKCC1, and Kir4, 1 to hippocampal K(+) clearance and volume responses. Glia. 2014;62:608–622. doi: 10.1002/glia.22629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lesage F, Maingret F, Lazdunski M. Cloning and expression of human TRAAK, a polyunsaturated fatty acids-activated and mechano-sensitive K(+) channel. FEBS Lett. 2000;471:137–140. doi: 10.1016/s0014-5793(00)01388-0. [DOI] [PubMed] [Google Scholar]
- 40.Li Q, Li QQ, Jia JN, Liu ZQ, Zhou HH, Mao XY. Targeting gap junction in epilepsy: Perspectives and challenges. Biomed Pharmacother. 2019;109:57–65. doi: 10.1016/j.biopha.2018.10.068. [DOI] [PubMed] [Google Scholar]
- 41.Liang B, Huang JH. Elevated NKCC1 transporter expression facilitates early post-traumatic brain injury seizures. Neural Regen Res. 2017;12:401–402. doi: 10.4103/1673-5374.202939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lo Pizzo M, Schiera G, Di Liegro I, Di Liegro CM, Pal J, Czeiter E, Sulyok E, Doczi T. Aquaporin-4 distribution in control and stressed astrocytes in culture and in the cerebrospinal fluid of patients with traumatic brain injuries. Neurol Sci. 2013;34:1309–1314. doi: 10.1007/s10072-012-1233-4. [DOI] [PubMed] [Google Scholar]
- 43.Losi G, Cammarota M, Carmignoto G. The role of astroglia in the epileptic brain. Front Pharmacol. 2012;3:132. doi: 10.3389/fphar.2012.00132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lu J, Huang H, Zeng Q, Zhang X, Xu M, Cai Y, Wang Q, Huang Y, Peng Q, Deng L. Hippocampal neuron loss and astrogliosis in medial temporal lobe epileptic patients with mental disorders. J Integr Neurosci. 2019;18:127–132. doi: 10.31083/j.jin.2019.02.16. [DOI] [PubMed] [Google Scholar]
- 45.Lutter D, Ullrich F, Lueck JC, Kempa S, Jentsch TJ. Selective transport of neurotransmitters and modulators by distinct volume-regulated LRRC8 anion channels. J Cell Sci. 2017;130:1122–1133. doi: 10.1242/jcs.196253. [DOI] [PubMed] [Google Scholar]
- 46.Lykke K, Assentoft M, Horlyck S, Helms HC, Stoica A, Toft-Bertelsen TL, Tritsaris K, Vilhardt F, Brodin B, MacAulay N. Evaluating the involvement of cerebral microvascular endothelial Na(+)/K(+)-ATPase and Na(+)-K(+)-2Cl(-) co-transporter in electrolyte fluxes in an in vitro blood-brain barrier model of dehydration. J Cereb Blood Flow Metab. 2019;39:497–512. doi: 10.1177/0271678X17736715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mack AF, Wolburg H. A novel look at astrocytes: aquaporins, ionic homeostasis, and the role of the microenvironment for regeneration in the CNS. Neuroscientist. 2013;19:195–207. doi: 10.1177/1073858412447981. [DOI] [PubMed] [Google Scholar]
- 48.MacVicar BA, Feighan D, Brown A, Ransom B. Intrinsic optical signals in the rat optic nerve: role for K(+) uptake via NKCC1 and swelling of astrocytes. Glia. 2002;37:114–123. doi: 10.1002/glia.10023. [DOI] [PubMed] [Google Scholar]
- 49.McNamara JO, Huang YZ, Leonard AS. Molecular signaling mechanisms underlying epileptogenesis. Sci STKE. 2006;2006:re12. doi: 10.1126/stke.3562006re12. [DOI] [PubMed] [Google Scholar]
- 50.Meeks JP, Mennerick S. Astrocyte membrane responses and potassium accumulation during neuronal activity. Hippocampus. 2007;17:1100–1108. doi: 10.1002/hipo.20344. [DOI] [PubMed] [Google Scholar]
- 51.Miklic S, Juric DM, Carman-Krzan M. Differences in the regulation of BDNF and NGF synthesis in cultured neonatal rat astrocytes. Int J Dev Neurosci. 2004;22:119–130. doi: 10.1016/j.ijdevneu.2004.03.001. [DOI] [PubMed] [Google Scholar]
- 52.Minieri L, Pivonkova H, Caprini M, Harantova L, Anderova M, Ferroni S. The inhibitor of volume-regulated anion channels DCPIB activates TREK potassium channels in cultured astrocytes. Br J Pharmacol. 2013;168:1240–1254. doi: 10.1111/bph.12011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mukai T, Kinboshi M, Nagao Y, Shimizu S, Ono A, Sakagami Y, Okuda A, Fujimoto M, Ito H, Ikeda A, Ohno Y. Antiepileptic drugs elevate astrocytic Kir4, 1 expression in the rat limbic region. Front Pharmacol. 2018;9:845. doi: 10.3389/fphar.2018.00845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nedergaard M. Direct signaling from astrocytes to neurons in cultures of mammalian brain cells. Science. 1994;263:1768–1771. doi: 10.1126/science.8134839. [DOI] [PubMed] [Google Scholar]
- 55.Ngugi AK, Bottomley C, Kleinschmidt I, Sander JW, Newton CR. Estimation of the burden of active and life-time epilepsy: a meta-analytic approach. Epilepsia. 2010;51:883–890. doi: 10.1111/j.1528-1167.2009.02481.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Niespodziany I, Mullier B, Andre VM, Ghisdal P, Jnoff E, Moreno-Delgado D, Swinnen D, Sands Z, Wood M, Wolff C. Discovery of a small molecule modulator of the Kv1, 1/Kvbeta1 channel complex that reduces neuronal excitability and in vitro epileptiform activity. CNS Neurosci Ther. 2019;25:442–451. doi: 10.1111/cns.13060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ohno Y, Kinboshi M, Shimizu S. Inwardly rectifying potassium channel Kir4, 1 as a novel modulator of BDNF expression in astrocytes. Int J Mol Sci? 1 as a novel modulator of BDNF expression in astrocytes. Int J Mol Sci. 2018 doi: 10.3390/ijms19113313. doi: 103390/ijms19113313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Osei-Owusu J, Yang J, Vitery MDC, Qiu Z. Molecular biology and physiology of volume-regulated anion channel (VRAC) Curr Top Membr. 2018;81:177–203. doi: 10.1016/bs.ctm.2018.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Palazzo C, Buccoliero C, Mola MG, Abbrescia P, Nicchia GP, Trojano M, Frigeri A. AQP4ex is crucial for the anchoring of AQP4 at the astrocyte end-feet and for neuromyelitis optica antibody binding. Acta Neuropathol Commun. 2019;7:51. doi: 10.1186/s40478-019-0707-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pan E, Stringer JL. Role of potassium and calcium in the generation of cellular bursts in the dentate gyrus. J Neurophysiol. 1997;77:2293–2299. doi: 10.1152/jn.1997.77.5.2293. [DOI] [PubMed] [Google Scholar]
- 61.Parpura V, Basarsky TA, Liu F, Jeftinija K, Jeftinija S, Haydon PG. Glutamate-mediated astrocyte-neuron signalling. Nature. 1994;369:744–747. doi: 10.1038/369744a0. [DOI] [PubMed] [Google Scholar]
- 62.Patel DC, Tewari BP, Chaunsali L, Sontheimer H. Neuron-glia interactions in the pathophysiology of epilepsy. Nat Rev Neurosci. 2019;20:282–297. doi: 10.1038/s41583-019-0126-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Poroca DR, Pelis RM, Chappe VM. ClC channels and transporters: Structure, physiological functions, and implications in human chloride channelopathies. Front Pharmacol. 2017;8:151. doi: 10.3389/fphar.2017.00151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Proper EA, Hoogland G, Kappen SM, Jansen GH, Rensen MG, Schrama LH, van Veelen CW, van Rijen PC, van Nieuwenhuizen O, Gispen WH, de Graan PN. Distribution of glutamate transporters in the hippocampus of patients with pharmaco-resistant temporal lobe epilepsy. Brain. 2002;125:32–43. doi: 10.1093/brain/awf001. [DOI] [PubMed] [Google Scholar]
- 65.Rangroo Thrane V, Thrane AS, Wang F, Cotrina ML, Smith NA, Chen M, Xu Q, Kang N, Fujita T, Nagelhus EA, Nedergaard M. Ammonia triggers neuronal disinhibition and seizures by impairing astrocyte potassium buffering. Nat Med. 2013;19:1643–1648. doi: 10.1038/nm.3400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ransom CB, Ransom BR, Sontheimer H. Activity-dependent extracellular K+ accumulation in rat optic nerve: the role of glial and axonal Na+ pumps. J Physiol. 2000;522:427–442. doi: 10.1111/j.1469-7793.2000.00427.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Reyes RC, Verkhratsky A, Parpura V. Plasmalemmal Na+/Ca2+ exchanger modulates Ca2+-dependent exocytotic release of glutamate from rat cortical astrocytes. ASN Neuro. 2012 doi: 10.1042/AN20110059. doi:101042/AN20110059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Risher WC, Andrew RD, Kirov SA. Real-time passive volume responses of astrocytes to acute osmotic and ischemic stress in cortical slices and in vivo revealed by two-photon microscopy. Glia. 2009;57:207–221. doi: 10.1002/glia.20747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Rose CR, Karus C. Two sides of the same coin: sodium homeostasis and signaling in astrocytes under physiological and pathophysiological conditions. Glia. 2013;61:1191–1205. doi: 10.1002/glia.22492. [DOI] [PubMed] [Google Scholar]
- 70.Rovegno M, Sáez JC. Role of astrocyte connexin hemichannels in cortical spreading depression. Biochim Biophys Acta Biomembr. 2018;1860:216–223. doi: 10.1016/j.bbamem.2017.08.014. [DOI] [PubMed] [Google Scholar]
- 71.Sakuragi S, Niwa F, Oda Y, Mikoshiba K, Bannai H. Astroglial Ca(2+) signaling is generated by the coordination of IP3R and store-operated Ca(2+) channels. Biochem Biophys Res Commun. 2017;486:879–885. doi: 10.1016/j.bbrc.2017.03.096. [DOI] [PubMed] [Google Scholar]
- 72.Seifert G, Steinhauser C. Neuron-astrocyte signaling and epilepsy. Exp Neurol. 2013;244:4–10. doi: 10.1016/j.expneurol.2011.08.024. [DOI] [PubMed] [Google Scholar]
- 73.Seifert G, Henneberger C, Steinhauser C. Diversity of astrocyte potassium channels: An update. Brain Res Bull. 2018;136:26–36. doi: 10.1016/j.brainresbull.2016.12.002. [DOI] [PubMed] [Google Scholar]
- 74.Simard M, Nedergaard M. The neurobiology of glia in the context of water and ion homeostasis. Neuroscience. 2004;129:877–896. doi: 10.1016/j.neuroscience.2004.09.053. [DOI] [PubMed] [Google Scholar]
- 75.Sirisi S, Elorza-Vidal X, Arnedo T, Armand-Ugon M, Callejo G, Capdevila-Nortes X, Lopez-Hernandez T, Schulte U, Barrallo-Gimeno A, Nunes V, Gasull X, Estevez R. Depolarization causes the formation of a ternary complex between GlialCAM, MLC1 and ClC-2 in astrocytes: implications in megalencephalic leukoencephalopathy. Hum Mol Genet. 2017;26:2436–2450. doi: 10.1093/hmg/ddx134. [DOI] [PubMed] [Google Scholar]
- 76.Steinhauser C, Seifert G, Bedner P. Astrocyte dysfunction in temporal lobe epilepsy: K+ channels and gap junction coupling. Glia. 2012;60:1192–1202. doi: 10.1002/glia.22313. [DOI] [PubMed] [Google Scholar]
- 77.Steinhauser C, Grunnet M, Carmignoto G. Crucial role of astrocytes in temporal lobe epilepsy. Neuroscience. 2016;323:157–169. doi: 10.1016/j.neuroscience.2014.12.047. [DOI] [PubMed] [Google Scholar]
- 78.Stewart TH, Eastman CL, Groblewski PA, Fender JS, Verley DR, Cook DG, D’Ambrosio R. Chronic dysfunction of astrocytic inwardly rectifying K+ channels specific to the neocortical epileptic focus after fluid percussion injury in the rat. J Neurophysiol. 2010;104:3345–3360. doi: 10.1152/jn.00398.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Su G, Haworth RA, Dempsey RJ, Sun D. Regulation of Na(+)-K(+)-Cl(-) cotransporter in primary astrocytes by dibutyryl cAMP and high [K(+)](o) Am J Physiol Cell Physiol. 2000;279:C1710–1721. doi: 10.1152/ajpcell.2000.279.6.C1710. [DOI] [PubMed] [Google Scholar]
- 80.Talley EM, Sirois JE, Lei Q, Bayliss DA. Two-pore-Domain (KCNK) potassium channels: dynamic roles in neuronal function. Neuroscientist. 2003;9:46–56. doi: 10.1177/1073858402239590. [DOI] [PubMed] [Google Scholar]
- 81.Thurman DJ, et al. Standards for epidemiologic studies and surveillance of epilepsy. Epilepsia. 2011;7:2–26. doi: 10.1111/j.1528-1167.2011.03121.x. [DOI] [PubMed] [Google Scholar]
- 82.Timofeev I, Bazhenov M, Sejnowski T, Steriade M. Cortical hyperpolarization-activated depolarizing current takes part in the generation of focal paroxysmal activities. Proc Natl Acad Sci U S A. 2002;99:9533–9537. doi: 10.1073/pnas.132259899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Tiwari D, Brager DH, Rymer JK, Bunk AT, White AR, Elsayed NA, Krzeski JC, Snider A, Schroeder Carter LM, Danzer SC, Gross C. MicroRNA inhibition upregulates hippocampal A-type potassium current and reduces seizure frequency in a mouse model of epilepsy. Neurobiol Dis. 2019;130:104508. doi: 10.1016/j.nbd.2019.104508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Tong X, Ao Y, Faas GC, Nwaobi SE, Xu J, Haustein MD, Anderson MA, Mody I, Olsen ML, Sofroniew MV, Khakh BS. Astrocyte Kir4. 1 ion channel deficits contribute to neuronal dysfunction in Huntington’s disease model mice. Nat Neurosci. 2014;17:694–703. doi: 10.1038/nn.3691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Topolnik L, Steriade M, Timofeev I. Hyperexcitability of intact neurons underlies acute development of trauma-related electrographic seizures in cats in vivo. Eur J Neurosci. 2003;18:486–496. doi: 10.1046/j.1460-9568.2003.02742.x. [DOI] [PubMed] [Google Scholar]
- 86.Tsumura M, Okumura R, Tatsuyama S, Ichikawa H, Muramatsu T, Matsuda T, Baba A, Suzuki K, Kajiya H, Sahara Y, Tokuda M, Momose Y, Tazaki M, Shimono M, Shibukawa Y. Ca2+ extrusion via Na+-Ca2+ exchangers in rat odontoblasts. J Endod. 2010;36:668–674. doi: 10.1016/j.joen.2010.01.006. [DOI] [PubMed] [Google Scholar]
- 87.Verkhratsky A, Nedergaard M, Hertz L. Why are astrocytes important? Neurochem Res. 2014;40:389–401. doi: 10.1007/s11064-014-1403-2. [DOI] [PubMed] [Google Scholar]
- 88.Verkhratsky A, Zorec R, Parpura V. Stratification of astrocytes in healthy and diseased brain. Brain Pathol. 2017;27:629–644. doi: 10.1111/bpa.12537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Walz W. Role of astrocytes in the clearance of excess extracellular potassium. Neurochem Int. 2000;36:291–300. doi: 10.1016/s0197-0186(99)00137-0. [DOI] [PubMed] [Google Scholar]
- 90.Walz W, Wuttke WA. Independent mechanisms of potassium clearance by astrocytes in gliotic tissue. J Neurosci Res. 1999;56:595–603. doi: 10.1002/(SICI)1097-4547(19990615)56:6<595::AID-JNR5>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 91.Wang F, Xu Q, Wang W, Takano T, Nedergaard M. Bergmann glia modulate cerebellar Purkinje cell bistability via Ca2+-dependent K+ uptake. Proc Natl Acad Sci U S A. 2012a;109:7911–7916. doi: 10.1073/pnas.1120380109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Wang F, Smith NA, Xu Q, Goldman S, Peng W, Huang JH, Takano T, Nedergaard M. Photolysis of caged Ca2+ but not receptor-mediated Ca2+ signaling triggers astrocytic glutamate release. J Neurosci. 2013;33:17404–17412. doi: 10.1523/JNEUROSCI.2178-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Wang F, Smith NA, Xu Q, Fujita T, Baba A, Matsuda T, Takano T, Bekar L, Nedergaard M. Astrocytes modulate neural network activity by Ca(2)+-dependent uptake of extracellular K+ Sci Signal. 2012b;5:ra26. doi: 10.1126/scisignal.2002334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Wang F, Wang X, Shapiro LA, Cotrina ML, Liu W, Wang EW, Gu S, Wang W, He X, Nedergaard M, Huang JH. NKCC1 up-regulation contributes to early post-traumatic seizures and increased post-traumatic seizure susceptibility. Brain Struct Funct. 2017;222:1543–1556. doi: 10.1007/s00429-016-1292-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Witcher MR, Ellis TL. Astroglial networks and implications for therapeutic neuromodulation of epilepsy. Front Comput Neurosci. 2012;6:61. doi: 10.3389/fncom.2012.00061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Wu KC, Kuo CS, Chao CC, Huang CC, Tu YK, Chan P, Leung YM. Role of voltage-gated K(+) channels in regulating Ca(2+) entry in rat cortical astrocytes. J Physiol Sci. 2015;65:171–177. doi: 10.1007/s12576-015-0356-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Xiong ZQ, Stringer JL. Sodium pump activity, not glial spatial buffering, clears potassium after epileptiform activity induced in the dentate gyrus. J Neurophysiol. 2000;83:1443–1451. doi: 10.1152/jn.2000.83.3.1443. [DOI] [PubMed] [Google Scholar]
- 98.Zheng P, Bin H, Chen W. Inhibition of microRNA-103a inhibits the activation of astrocytes in hippocampus tissues and improves the pathological injury of neurons of epilepsy rats by regulating BDNF. Cancer Cell Int. 2019;19:109. doi: 10.1186/s12935-019-0821-2. [DOI] [PMC free article] [PubMed] [Google Scholar]