

Abstract
Heritable factors account for approximately 35% of colorectal cancer (CRC) risk, and almost 30% of the population in the UK have a family history of CRC. The quantification of an individual’s lifetime risk of gastrointestinal cancer may incorporate clinical and molecular data, and depends on accurate phenotypic assessment and genetic diagnosis. In turn this may facilitate targeted risk-reducing interventions, including endoscopic surveillance, preventative surgery and chemoprophylaxis, which provide opportunities for cancer prevention. This guideline is an update from the 2010 British Society of Gastroenterology/Association of Coloproctology of Great Britain and Ireland (BSG/ACPGBI) guidelines for colorectal screening and surveillance in moderate and high-risk groups; however, this guideline is concerned specifically with people who have increased lifetime risk of CRC due to hereditary factors, including those with Lynch syndrome, polyposis or a family history of CRC. On this occasion we invited the UK Cancer Genetics Group (UKCGG), a subgroup within the British Society of Genetic Medicine (BSGM), as a partner to BSG and ACPGBI in the multidisciplinary guideline development process. We also invited external review through the Delphi process by members of the public as well as the steering committees of the European Hereditary Tumour Group (EHTG) and the European Society of Gastrointestinal Endoscopy (ESGE). A systematic review of 10 189 publications was undertaken to develop 67 evidence and expert opinion-based recommendations for the management of hereditary CRC risk. Ten research recommendations are also prioritised to inform clinical management of people at hereditary CRC risk.
Keywords: colorectal cancer, genetic testing, inherited cancers, surveillance, colorectal surgery
Objective
To provide a clear strategy for the management of people at hereditary risk of colorectal cancer (CRC), which includes diagnosis, endoscopic management, prevention and surgical care.
Aims and methods
An estimated 35% of CRC is due to heritable factors,1 with approximately 29% of the UK population having a family history of a first-degree relative (FDR) or second degree relative (SDR) with CRC.2 While highly penetrant syndromes such as Lynch syndrome (LS), familial adenomatous polyposis (FAP) and other polyposis syndromes account for account for only 5–10% of all CRC diagnoses, advances in genetic diagnosis, improvements in endoscopic surgical control, and medical and lifestyle interventions provide opportunities for CRC prevention and effective treatment in susceptible individuals.
The purpose of this guideline is to provide an evidence-based framework for the optimal management of hereditary CRC for clinicians involved in their management, including gastroenterologists, nurse practitioners, physicians, colorectal surgeons, clinical geneticists, genetic counsellors and pathologists. This guideline was commissioned by the Clinical Services and Standards Committee (CSSC) of the British Society of Gastroenterology (BSG), via the colorectal section, and a guideline chair selected. It is an update of the previous iteration of the BSG/Association of Coloproctology of Great Britain and Ireland (ACPGBI) guideline published in 2010 and developed in accordance with the BSG National Institute for Health and Care Excellence (NICE)-compliant guideline process.
The Guideline Development Group (GDG), which included gastroenterologists from the BSG, clinical geneticists from United Kingdom Cancer Genetics Group (UKCGG), colorectal surgeons from the ACPGBI, a pathologist, a genetic counsellor and a patient representative, was selected to ensure wide-ranging expertise across all relevant disciplines. Members of the GDG, and participants in the eDelphi process, completed a Declaration of Conflict of Interests (COI) form which was reviewed and vetted by the BSG.
A scoping meeting was held on 13 October 2017, and in advance of this meeting the GDG was asked to develop key priorities and questions.
The GDG determined that the primary measure of effectiveness of any intervention was a reduction of the lifetime risk of CRC, and the following secondary outcome measures:
Reduction in the incidence of advanced adenomas at colonoscopy
Prevention of CRC
Reduced morbidity related to CRC, or morbidity secondary to complications of surveillance and treatment
Improved identification of hereditary CRC syndromes
Improved pathways from diagnosis to treatment in susceptible populations.
We sought a consistent approach in our assessment of the relative effectiveness of interventions. In principle we agreed that surveillance should only be offered to individuals who remain at higher risk of developing CRC than the general population. As CRC risk is not always clearly defined, as a surrogate we accepted that advanced adenoma yield on surveillance should be approximately double that in susceptible populations compared with the average risk population.
A relative threshold for genetic testing was agreed for people with a 10% or greater probability of having a germline pathogenic variant in a cancer susceptibility gene in accordance with previous UK guidelines.3 4 However the GDG agreed that the arbitrary nature of this threshold meant that it could be modified in cases where objective risk assessment was difficult to attain, and clinicians had sufficient clinical suspicion of risk.
Key questions we sought to cover included the following:
Which aspects of the previous guidelines require updating?
What is the lifetime CRC risk and optimal surveillance for those with a family history of CRC (where LS and polyposis syndromes have been excluded)?
What is the diagnostic yield of genetic testing and/or surveillance for high-risk populations?
What is the optimal gastrointestinal (GI) surveillance for patients at hereditary risk GI cancer?
What is the impact of high-quality endoscopy in patients with known or suspected hereditary cancer syndromes?
Should we develop gene- or gender-specific guidelines for surveillance?
What is the optimal diagnostic assessment and surveillance interval for ‘Lynch-like’ syndrome patients?
How can we improve recognition, diagnosis and treatment of patients at hereditary risk of CRC?
Which diagnostic genetic tests should we offer serrated polyposis syndrome (SPS), multiple colorectal adenoma (MCRA) and early onset CRC (EOCRC) patients (if any)?
When should colonoscopic surveillance for familial risk patients stop, because it is no longer necessary, or because the patient should be referred for surgery?
Which are the optimal surgical approaches in patients with hereditary CRC syndromes?
What is the evidence for chemoprophylaxis in patients who are at hereditary risk of CRC?
What is the evidence for the effect of lifestyle modification on hereditary risk of CRC?
What information do we need to share with our patients at inherited risk of GI cancer?
Twenty-three PICOs (Patients, Interventions, Controls and Outcomes) were developed which considered these questions. The Appraisal of Guidelines for Research and Evaluation (AGREE II) instrument provided a methodological framework.5
A literature search was commissioned externally, with search strategies agreed, and was performed by the Yorkshire Healthcare Consortium, which returned 10 189 publications. Returned abstracts were reviewed for relevance. Additional references were obtained by cross-referencing and by recommendation from the GDG. Relevant published national and international guidelines were also scrutinised. After each round of Delphi, and before the guideline was finalised, the search was repeated and any important studies published since the initial evidence search were incorporated.
A modified electronic Delphi process6 was used to develop and refine statements. Initial draft statements formulated by the writing committee were reviewed by the GDG to allow for modification and to identify additional references. After a preliminary discussion, formal anonymous voting rounds were undertaken using SurveyMonkey. Each statement was scored by each member of the GDG using a five-point scale. We also invited key national and international opinion leaders from the UKCGG steering group, ACPGBI, BSG, the European Hereditary Tumour Group (EHTG) and the European Society of Gastrointestinal Endoscopy (ESGE) to participate in the modified Delphi process. We included additional patient and public involvement in the Delphi process by inviting participants through the national charities Bowel Cancer UK and Lynch Syndrome UK. Consensus required at least 80% agreement, and consensus of over 70% was accepted if the GDG felt a statement was required for clinical practice. Where consensus was not reached, feedback from the GDG members was disseminated after each round to allow members to reconsider their original position.7 Where appropriate, revisions to statements were made and a further voting round was undertaken in second and third rounds. A final (fourth) round of voting for statements where consensus had not been reached for 11 statements was performed within the GDG only.
Surveillance and molecular testing recommendations are summarised in table 1 and table 2 respectively. The GDG also developed 10 research recommendations (online supplementary file 1) which were prioritised by electronic voting.
Table 1.
Summary of surveillance recommendations
| Indication for surveillance | Category | Modality | Age to commence (years) | Interval |
| Family history of CRC | Average risk | National screening | National screening age | As defined by national screening |
| Moderate risk | Colonoscopy | 55 | Post-polypectomy guidelines | |
| High risk* | Colonoscopy | 40 | 5 yearly until age 75 years | |
| Lynch syndrome | MMR gene pathogenic variant carriers | |||
| MLH1 and MSH2 gene carriers | Colonoscopy | 25 | 2 yearly until age 75 years | |
| MSH6 and PMS2 gene carriers | Colonoscopy | 35 | 2 yearly until age 75 years | |
| Stomach, small bowel and pancreas | Not indicated outside a clinical trial | |||
| Lynch-like syndrome | Individuals with deficient MMR tumours without hypermethylation/BRAF pathogenic variant and no pathogenic constitutional pathogenic variant in MMR genes, and no evidence of biallelic somatic MMR gene inactivation (and their unaffected FDRs). | Colonoscopy | 25 | 2 yearly until age 75 years |
| Serrated polyposis syndrome | Affected individuals (WHO 2019) | Colonoscopy | From age of diagnosis | 1–2 yearly until age 75 years |
| FDRs of affected individuals | Colonoscopy | 40 (or 10 years earlier than the index case) | 5 yearly until age 75 years | |
| Multiple colorectal adenomas (MCRAs) | 10 or more adenomas without constitutive pathogenic variants in APC or MUTYH | Colonoscopy | From age of diagnosis | 1–2 yearly until age 75 years |
| Familial adenomatous polyposis (FAP) | APC pathogenic variant carriers | Colonoscopy | 12 to 14 | 1–3 yearly depending on phenotype |
| Gastroscopy and duodenoscopy | 25 | As per Spigelman classification | ||
| Sigmoidoscopy/ pouchoscopy | From time of colectomy | 1–3 yearly depending on phenotype | ||
| Individuals with an FDR with a clinical diagnosis of FAP (ie, “at-risk”) and in whom a constitutional pathogenic variant has not been identified | Colonoscopy | 12 to 14 | 5 yearly until national screening age | |
| Gastroscopy and duodenoscopy | Commence only if clinical diagnosis made of colorectal polyposis phenotype | As per Spigelman classification | ||
| MUTYH-associated polyposis (MAP) | MUTYH gene pathogenic variant carriers | Colonoscopy | 18 to 20 years | Annual |
| Gastroscopy and duodenoscopy | 35 | As per Spigelman classification | ||
| Peutz-Jeghers syndrome (PJS) | STK11 gene pathogenic variant carriers | Upper gastrointestinal endoscopy, colonoscopy and video capsule endoscopy | 8 | see main text |
| Juvenile polyposis syndrome (JPS) | SMAD4 and BMPR1A pathogenic variant carriers | Colonoscopy | 15 | 1–3 yearly depending on phenotype |
| SMAD4 pathogenic variant carriers | Gastroscopy and duodenoscopy | 18 | 1–3 yearly depending on phenotype | |
| BMPR1A pathogenic variant carriers | Gastroscopy and duodenoscopy | 25 | 1–3 yearly depending on phenotype | |
*Amsterdam criteria families where MMR testing is not possible may be offered surveillance as per Lynch syndrome families and/or additional constitutional testing.
CRC, colorectal cancer; FDR, first degree relative; MMR, mismatch repair.
Table 2.
Molecular testing strategies in hereditary colorectal cancer (CRC)
| Risk category | Somatic or constitutive testing | Eligibility | Test |
| Family history of CRC | Somatic | Moderate-risk or high-risk family history | dMMR/pMMR |
| Constitutive | Amsterdam criteria112 families where MMR testing is not possible | Panel testing of affected individuals or unaffected testing | |
| CRC | Somatic | Universal testing | dMMR/pMMR and subsequent testing as defined by NICE DG27 guideline5 7 8 18 112 |
| Early onset CRC (EOCRC) | Constitutive | Diagnosis of CRC at 30 years and under | Panel testing determined by MMR status |
| Lynch-like syndrome | Somatic | dMMR tumours without hypermethylation/BRAF pathogenic variant and no constitutional pathogenic variant in MMR genes | Somatic testing panel |
| Serrated polyposis syndrome | Constitutive/ somatic |
Diagnosis of exclusion | Exclude known predisposition syndromes |
| Multiple colorectal adenoma (MCRAs) | Constitutive | MCRAs under 60 years of age with ≥10 adenomas, or patients over 60 years of age with ≥20 adenomas, or ≥10 with a family history of multiple adenomas or CRC | Gene panel testing |
dMMR, MMR proficient; MMR, mismatch repair; NICE, National Institute for Health and Care Excellence; pMMR, MMR deficient.
gutjnl-2019-319915supp001.pdf (78.1KB, pdf)
The GRADE (Grading of Recommendations, Assessment, Development and Evaluations) tool8 was used to evaluate the strength of evidence and the strength of recommendations made (see executive summary). The GRADE system specifically separates the strength of evidence from the strength of a recommendation. While the strength of a recommendation may often reflect the evidence base, the GRADE system allows for occasions where this is not the case—for example, where it seems good sense to make a recommendation despite the absence of high-quality scientific evidence such as a large randomised controlled trial (RCT).
Executive summary of key recommendations
Service provision, communication and management principles
We recommend that the moderate risk category of family history of CRC (FHCC) is the minimum threshold for referral from primary care (GRADE of evidence: very low; Strength of recommendation: strong)
We recommend that individuals with a FHCC, which meets this referral criteria, be referred to a specialist familial CRC clinic in secondary or tertiary care (GRADE of evidence: low; Strength of recommendation: weak)
We recommend that patients should be referred to a specialist service which includes access to constitutional genetic testing in the presence of either deficient mismatch repair (MMR) (with no evidence of MLH1 promoter methylation or BRAF V600E), or polyposis. (GRADE of evidence: low; Strength of recommendation: strong)
There are insufficient clinical data to develop specific guidance for patients with very rare conditions such as polymerase proofreading associated polyposis (PPAP), or NTHL1-associated polyposis (NAP); therefore, we suggest patients with these syndromes should be referred to multidisciplinary expert centres for clinical management. (GRADE of evidence: low; Strength of recommendation: weak)
We recommend that hospitals which diagnose or manage patients at hereditary CRC risk should ensure clinical pathways to facilitate their care, and processes to monitor the quality of the service. (GRADE of evidence: low; Strength of recommendation: strong)
We recommend that individuals at increased familial CRC risk receive specialist knowledge and are aware of patient/support organisations and discussion with regard to lifestyle and participation in research projects. (GRADE of evidence: very low; Strength of recommendation: strong)
Family history of CRC (FHCC)
We recommend that for all patients referred from primary care for assessment for a FHCC, MMR status should be assessed in tumour tissue from a close affected family member. (GRADE of evidence: moderate; Strength of recommendation: strong)
We recommend that a reported family history of polyposis should be verified by review of histopathology and/or endoscopy reports which confirm the presence of a minimum of 10 adenomas or serrated lesions in a FDR. (GRADE of evidence: low; Strength of recommendation: strong)
We recommend that patients with a moderate familial CRC risk should have a one-off colonoscopy at age 55 years. (GRADE of evidence: moderate; Strength of recommendation: strong)
We recommend that subsequent colonoscopic surveillance should be performed as determined by post-polypectomy surveillance guidelines. (GRADE of evidence: moderate; Strength of recommendation: strong)
We suggest that in high-risk families (a cluster of 3× FDRs with CRC across >1 generation) a 5 yearly colonoscopy should be performed from age 40 years until age 75 years. (GRADE of evidence: moderate; Strength of recommendation: weak)
Prevention and lifestyle modification in familial CRC
We recommend that individuals with LS should be advised that regular use of daily aspirin reduces CRC risk. (GRADE of evidence: moderate; Strength of recommendation: strong)
We suggest that people with LS should be offered research opportunities to take aspirin daily at different dosages. If they decline research participation they may be advised on their choices regarding dose of aspirin, risks and benefits of long-term aspirin use and ensure their medical practitioner is aware of their intake. (GRADE of evidence: low; Strength of recommendation: weak)
There is insufficient evidence of the benefit of chemoprophylaxis in polyposis syndromes. (GRADE of evidence: moderate; Strength of recommendation: strong)
We suggest that individuals at increased familial risk of CRC should be strongly encouraged not to smoke, to maintain a normal body mass index (BMI), to moderate their consumption of red and processed meat, and to exercise regularly. (GRADE of evidence: low; Strength of recommendation: weak)
Quality and advanced endoscopic imaging in colonoscopic surveillance
We recommend that colonoscopy is the gold standard diagnostic and preventative method of surveillance for people with hereditary risk of CRC. (GRADE of evidence: moderate; Strength of recommendation: strong)
We recommend that all surveillance colonoscopies are performed by endoscopists who consistently achieve BSG colonoscopy KPI (key performance indicators) minimum standards, specifically caecal intubation rate, adenoma/polyp detection rate and comfort score. (GRADE of evidence: low; Strength of recommendation: strong)
We suggest high-quality, high-definition white light endoscopy as the preferred modality for colonoscopy surveillance. Chromoendoscopy (virtual or dye-based) does not offer a clear advantage over high definition white light examination for colonoscopic surveillance, apart from in the context of determining the multiple polyp phenotype. (GRADE of evidence: moderate; Strength of recommendation: weak)
We suggest a repeat colonoscopy performed by an expert endoscopist is indicated in the event of a previously failed colonoscopy, with efforts made to both improve patient experience and to ensure procedure completion, given the advantages of colonoscopic surveillance. If colonoscopy is not possible then consider CT colonography. (GRADE of evidence: low; Strength of recommendation: weak)
We suggest that if the bowel preparation for colonoscopy is inadequate or if the examination is incomplete then a repeat colorectal surveillance procedure should be arranged within 3 months. (GRADE of evidence: low; Strength of recommendation: weak)
There is insufficient evidence to recommend other methods of surveillance for those with familial CRC risk such as FIT (faecal immunochemical test), MR or CT colonography. (GRADE of evidence: low; Strength of recommendation: strong)
Lynch syndrome (LS)
We recommend that for all people when first diagnosed with CRC, testing using immunohistochemistry (IHC) for MMR proteins or microsatellite instability is used to identify tumours with deficient DNA MMR, and to guide further sequential testing for LS. (GRADE of evidence: moderate; Strength of recommendation: strong)
We recommend that colonoscopic surveillance should be performed at a 2 yearly interval for all LS patients. (GRADE of evidence: moderate; Strength of recommendation: strong)
We recommend that age of onset of surveillance colonoscopy should be stratified according to the LS-associated gene. We recommend colonoscopy from age 25 years for MLH1 and MSH2 mutation carriers and 35 years for MSH6 and PMS2 mutation carriers. There are insufficient data to support stratifying age of onset of surveillance by gender. (GRADE of evidence: moderate; Strength of recommendation: strong)
We suggest that for LS patients with MLH1 or MSH2 mutations who develop colon cancer or colonic neoplasia not amenable to endoscopic control, the decision to perform segmental versus total/near total colectomy should balance the risks of metachronous cancer, the functional consequences of surgery, the patient’s age and patient’s wishes. (GRADE of evidence: Moderate; Strength of recommendation: strong)
We recommend that for LS patients with MSH6 or PMS2 mutations there is insufficient evidence for oncological benefit of extended colectomy over segmental resection. (GRADE of evidence: low; Strength of recommendation: strong)
We suggest that when abdominal-perineal excision can be avoided, a standard low anterior resection is a reasonable option to treat rectal cancers in LS patients, even though the residual colon is at high-risk of metachronous neoplasia. (GRADE of evidence: low; Strength of recommendation: weak)
We recommend that gastric, small bowel, or pancreatic surveillance in LS patients is only performed in the context of a clinical trial. (GRADE of evidence: low; Strength of recommendation: strong)
We recommend screening for H elicobacter pylori in patients with LS and subsequent eradication therapy if indicated. (GRADE of evidence: low; Strength of recommendation: strong)
Lynch-like syndrome (LLS)
We recommend that deficient MMR tumours without hypermethylation/BRAF mutation and without a germline pathogenic variant in MMR genes should undergo somatic tumour testing with a CRC gene panel. (GRADE of evidence: low; Strength of recommendation: strong)
We recommend that if double somatic MMR pathogenic variants are identified, manage proband and their FDRs based on the FHCC. (GRADE of evidence: low; Strength of recommendation: strong)
We suggest that if no or one somatic mutations are identified, the proband and their FDRs should be managed as per LS. (GRADE of evidence: low; Strength of recommendation: weak)
Early onset CRC (EOCRC)
We recommend that in patients under 30 years of age with dMMR CRC, an LS constitutional panel test should be performed, followed by tumour testing for somatic testing if constitutional testing is negative. (GRADE of evidence: low; Strength of recommendation: strong)
We recommend that in patients under 30 years of age with pMMR CRC, a constitutional CRC multiple gene panel test should be performed. (GRADE of evidence: low; Strength of recommendation: strong)
We suggest that people diagnosed with CRC under age 50 years, where hereditary CRC syndromes have been excluded, undergo standard post-CRC surveillance for 3 years, then continue 5 yearly colonoscopic surveillance until the age they are eligible for national screening. (GRADE of evidence: low; Strength of recommendation: weak)
Serrated polyposis syndrome (SPS)
We recommend a diagnosis of SPS should be made in accordance with the new WHO 2019 criteria for SPS. Since causative gene pathogenic variants for SPS have not been identified, a definitive diagnosis of SPS should be phenotype-driven. (GRADE of evidence: moderate; Strength of recommendation: strong)
Other intestinal polyposis syndromes may present with serrated lesions. If (i) the patient is under 50 or (ii) there are multiple affected individuals within kindred or (iii) there is dysplasia within any of the polyps, then we suggest that other polyposis syndromes should be excluded by gene panel testing before making a definitive diagnosis of SPS. (GRADE of evidence: very low; Strength of recommendation: weak)
We recommend the cumulative number of serrated polyps from all endoscopic examinations should be used when applying the WHO 2019 diagnostic criteria for SPS. (GRADE of evidence: moderate; Strength of recommendation: strong)
We recommend that patients with SPS should have colonoscopic surveillance yearly once the colon has been cleared of all lesions >5 mm in size. If no polyps ≥10 mm in size are identified at subsequent surveillance examinations the interval can be extended to 2 yearly. (GRADE of evidence: moderate; Strength of recommendation: strong)
We recommend all FDRs of patients with SPS on the basis of the new WHO 2019 SPS criteria, one or two should be offered an index colonoscopic screening examination at age 40 years or 10 years before the diagnosis of the index case. (GRADE of evidence: moderate; Strength of recommendation: strong)
We suggest all FDRs of SPS patients have a surveillance examination every 5 years unless polyp burden indicates an examination is required earlier according to post-polypectomy surveillance guidelines. (GRADE of evidence: low; Strength of recommendation: strong)
Multiple colorectal adenoma (MCRA) patients
-
We suggest an individualised approach to germline testing of patients with MCRA (defined as having 10 or more metachronous adenomas). Consider this testing for:
Patients under 60 years of age with lifetime total of ≥10 adenomas; or
-
Patients from 60 years of age with lifetime total of:
≥20 adenomas, or
≥10 adenomas and a FHCC or polyposis
(GRADE of evidence: low; Strength of recommendation: weak)
We suggest that patients with a finding of 10 or more polyps (adenomas or serrated lesions) should, at their next colonoscopy, have a high-quality colonoscopic assessment with pancolonic dye spray in order to define accurately the multiple polyp phenotype. (GRADE of evidence: very low; Strength of recommendation: weak)
We suggest that the endoscopic management of patients with 10 or more metachronous adenomas, without MUTYH or APC gene mutations, should be individualised according to phenotype. (GRADE of evidence: very low; Strength of recommendation: weak)
We suggest annual colonoscopic surveillance for patients with 10 or more metachronous adenomas after the colon has been cleared of all lesions >5 mm in size. If no polyps 10 mm or greater in size are identified at subsequent surveillance examinations the interval can be extended to 2 yearly. (GRADE of evidence: very low; Strength of recommendation: weak)
Familial adenomatous polyposis (FAP)
We recommend that colonic surveillance should normally commence age 12–14 years in those confirmed to have FAP on predictive genetic testing. (GRADE of evidence: low; Strength of recommendation: strong)
We suggest that for those with FAP, intervals between surveillance colonoscopy may be individualised depending on colonic phenotype every 1–3 years. (GRADE of evidence: low; Strength of recommendation: weak)
We suggest that colonoscopy screening is performed for individuals who have an FDR with a clinical diagnosis of FAP (ie, “at-risk”) and in whom an APC mutation has not been identified, starting at age 12–14 years, and should continue on 5 yearly surveillance until either a clinical diagnosis is made and they are then managed as FAP, or they reach the age at which they can enrol in national screening. (GRADE of evidence: very low; Strength of recommendation: weak)
We recommend upper GI surveillance for FAP patients starting at age 25 years. (GRADE of evidence: low; Strength of recommendation: strong)
We suggest that for those considered at risk, where predictive genetic testing is not possible, screening with upper GI endoscopy is not routinely recommended but should be started if/when a clinical diagnosis of FAP is made based on colorectal phenotype. (GRADE of evidence: very low; Strength of recommendation: weak)
We suggest that patients with congenital hypertrophy retinal pigmentation epithelium (CHRPE) be referred for a specialist ophthalmic review. Patients with bilateral and multiple CHRPE lesions should be referred for screening for FAP and considered for genetic testing and colonoscopy. (GRADE of evidence: low; Strength of recommendation: weak)
FAP: Surgery, and desmoid disease
We recommend that for patients with FAP who are undergoing colonoscopic surveillance, relative indications for surgery are: polyps >10 mm in diameter, high grade dysplasia within polyps and a significant increase in polyp burden between screening examinations. (GRADE of evidence: low; Strength of recommendation: strong)
We recommend that absolute indications for immediate colorectal surgery in FAP include: documented or suspected cancer or significant symptoms attributable to the polyposis. (GRADE of evidence: low; Strength of recommendation: strong)
We suggest that FAP patients should be counselled about the risk of postoperative desmoid disease formation. (GRADE of evidence: low; Strength of recommendation: weak)
Consider, for FAP patients before colectomy, determining genotypes or family history of desmoid disease which may be predictive of desmoid formation. (GRADE of evidence: very low; Strength of recommendation: weak)
We suggest that sulindac in combination with high-dose selective oestrogen receptor modulators may be effective in FAP patients with intra-abdominal desmoids and desmoids located at the abdominal wall. (GRADE of evidence: low; Strength of recommendation: weak)
We recommend the role of elective surgery for intra-abdominal desmoids should be restricted to treating secondary effects of the desmoid disease, and this surgery should be performed in expert centres. (GRADE of evidence: low; Strength of recommendation: strong)
MUTYH-associated polyposis (MAP)
We recommend that colorectal surveillance is commenced in MAP commencing age 18–20 years. If surgery is not undertaken then annual surveillance is suggested. (GRADE of evidence: moderate; Strength of recommendation: strong)
We recommend that for monoallelic MUTYH pathogenic variant carriers, the risk of CRC is not sufficiently different to population risk to meet thresholds for screening and routine colonoscopy is not recommended. (GRADE of evidence: moderate; Strength of recommendation: strong)
We suggest that upper GI surveillance should be considered starting at the age of 35 years in MAP. We recommend that the surveillance interval is determined as outlined for FAP. (GRADE of evidence: low; Strength of recommendation: weak).
Peutz-Jeghers syndrome (PJS)
We suggest that in an asymptomatic patient with PJS, GI surveillance by upper GI endoscopy, colonoscopy and video capsule endoscopy commence at age 8 years. We recommend that small bowel surveillance should continue 3 yearly. If baseline colonoscopy and oesophago-gastro-duodenoscopy (OGD) are normal, then they can be safely deferred until age 18 years; however, if polyps are found at baseline examination, then they should be repeated 3 yearly. Earlier investigation of the GI tract should be performed in symptomatic patients. (GRADE of evidence: low; Strength of recommendation: weak)
We suggest elective polypectomy to prevent polyp related complications. Small bowel polyps greater than 1.5–2 cm in size (or smaller if symptomatic) should be considered for elective resection to prevent intussusception. (GRADE of evidence: low; Strength of recommendation: weak)
Juvenile polyposis syndrome (JPS)
We suggest colonoscopic surveillance should commence from the age of 15 years or earlier if symptomatic. The surveillance interval should be 1–3 yearly, personalised according to colorectal phenotype. (GRADE of evidence: low; Strength of recommendation: weak)
We suggest that for those with a confirmed clinical or genetic diagnosis, upper GI endoscopic surveillance should start at the age of 18 years for SMAD4 mutation carriers and 25 years for BMPR1A mutation carriers and those without an identified constitutional. The surveillance interval should be 1–3 yearly, personalised according to upper GI tract phenotype. (GRADE of evidence: low; Strength of recommendation: weak)
We suggest that for those with an FDR with a clinical diagnosis of JPS and in whom a mutation has not been identified, screening of the upper GI tract is not required routinely but should be initiated if/when a clinical diagnosis is made on the basis of colonic phenotype. It may, however, be considered if there is a family history suggestive of hereditary haemorrhagic telangiectasia (HHT), even in the absence of colonic polyps. (GRADE of evidence: low; Strength of recommendation: weak)
We suggest that patients with SMAD4 pathogenic variant should be evaluated for HHT, and that those at risk of, or with a confirmed diagnosis of, HHT are best managed in conjunction with a specialist HHT centre. (GRADE of evidence: low; Strength of recommendation: weak)
Patients with JPS and a microdeletion involving BMPR1A and PTEN are at risk of the clinical manifestations of both JPS and PTEN-hamartoma tumour syndrome (PHTS). We suggest that they should be referred to their local genetics centre for further advice and to coordinate their surveillance needs. (GRADE of evidence: low; Strength of recommendation: weak)
Service provision, communication and management principles
We recommend that moderate risk of FHCC is the minimum threshold for referral from primary care
(GRADE of evidence: very low; Strength of recommendation: strong)
Consensus reached: 100% agreement.
We recommend that individuals with an FHCC, which meets this referral criteria, be referred to a specialist familial CRC clinic in secondary or tertiary care.
(GRADE of evidence: low; Strength of recommendation: weak)
Consensus reached: 82% agreement.
We recommend that patients should be referred to a specialist service which includes access to constitutional genetic testing in the presence of defective MMR (with no evidence of MLH1 promoter methylation or BRAF V600E), or polyposis.
(GRADE of evidence: low; Strength of recommendation: strong)
There are insufficient clinical data to develop specific guidance for patients with very rare conditions such as polymerase proofreading associated polyposis (PPAP), or NTHL1 associated polyposis (NAP), therefore patients with these syndromes should be referred to multidisciplinary expert centres for clinical management.
(GRADE of evidence: low; Strength of recommendation: weak)
Consensus reached: 91% agreement.
We recommend that hospitals that diagnose or manage patients at hereditary CRC risk should ensure clinical pathways to facilitate their care, and processes to monitor the quality of the service.
(GRADE of evidence: low; Strength of recommendation: strong)
Consensus reached: 100% agreement.
We recommend that individuals at increased familial CRC risk receive specialist knowledge and are aware of patient/support organisations and discussion with regard to lifestyle and participation in research projects.
(GRADE of evidence: very low; Strength of recommendation: strong)
Consensus reached: 95% agreement
People at hereditary CRC risk require coordinated, timely and high-quality care to reduce their cancer risk and should have access to a full range of management options that minimise the risk of morbidity and mortality.9 A structured referral pathway may ensure better inter-specialty communication and timely, efficient management of hereditary risk from primary through to tertiary care provision. It also facilitates an audit trail and subsequent monitoring of performance. Patients should have access to a full range of management options that minimise the risk of morbidity and mortality.9
Moreover, studies consistently report that high quality screening and surveillance services result in a reduction of CRC incidence and mortality in individuals with FAP and LS.10 Registries of high-risk patients should be linked to robust quality assurance mechanisms for interventions, such as colonoscopy, with effective recall mechanisms in place to ensure high-risk individuals receive surveillance procedures on schedule.
Awareness of hereditary conditions may be inadequate, resulting in an inconsistent approach to the management of these individuals.9 11 Many patients do not have personalised management strategies and there is a failure to provide adequate follow-up.12 13 Patient advocacy organisations recommend improvements in the detection of pre-cancerous polyps, early diagnosis of CRC, and personalised treatment options for LS individuals, who should be seen by a team of specialists.14 The relevance of genomics is growing in clinical practice, and is increasingly relevant across the cancer multidisciplinary teams, with improving access to constitutional genetic testing.15 Genetic testing and/or counselling may resolve uncertainty about personal and familial cancer risk and provide information to guide and personalise decisions about future health care in anyone with an FHCC.16 17 It has been recommended that a dedicated clinical champion for hereditary CRC should be established in each colorectal multidisciplinary team (MDT) to oversee service coordination and to ensure patient pathways.9 18 The establishment of these champions will be another critical component in establishing equity of care. We recommend the establishment of family cancer specialist services by CRC teams in secondary care to ensure local pathways for patients at hereditary risk of CRC, and which can arrange testing of relatives for MMR status. This service should be supported by commissioners and incorporate a multidisciplinary approach involving geneticists, gastroenterologists and colorectal surgeons with links between secondary and tertiary care. Adherence to surveillance recommendations should be monitored at least annually. We suggest a minimum standard of ≥90% compliance. Non-compliant cases should be reviewed to determine whether reason for deviation from surveillance recommendations was clearly documented and clinically appropriate. Thus patients with a family history of CRC may be managed by their local hospital, and patients who require constitutional gene testing be managed by a tertiary care clinic, for example, in clinical genetics, either locally or regionally.
Family history of CRC (FHCC)
Definitions and terminology
A substantial proportion of the UK population have an FHCC without evidence of an inherited CRC syndrome. These individuals have a moderately increased relative risk (RR) of CRC (2–6 fold) compared with the general population.19 Lifetime CRC risk may be inferred from the age of onset of CRC in affected relatives, and familial aggregation, that is, the number of family members affected with CRC.
This section refers to asymptomatic patients referred for optimal management of a family history of either CRC or multiple polyps. The GDG agreed three categories of familial risk (in the absence of known hereditary CRC syndromes) which were determined according to lifetime CRC risk and the diagnostic yield of colorectal surveillance (box 1). Familial clusters (or aggregations) are of affected family members with CRC who are FDRs of each other. The individual referred for assessment should be an FDR of at least one affected member of such families.
Box 1. Categories of risk in patients with a family history of colorectal cancer (FHCC).
Categories of risk – FHCC
Average risk: No FHCC, or a FHCC which does not fulfil moderate or high-risk categories.
-
Moderate risk FHCC:
One FDR diagnosed with CRC under 50 years, or
Two FDRs (in first degree kinship) diagnosed with CRC at any age, of whom the patient under assessment is an FDR of at least one affected individual.
High risk FHCC: Families with a cluster of at least three affected FDRs with CRC diagnosed at any age, across at least two generations, of whom the patient under assessment is an FDR of at least one affected individual.
CRC, colorectal cancer; FDR, first degree relative.
Patients with average risk include those without an FHCC, or with an FHCC which does not significantly increase their lifetime CRC risk, that is, below the level of the moderate risk population. For the average risk populations surveillance may be effectively managed via national bowel cancer screening programmes. Those people in moderate- or high-risk categories require additional surveillance above and beyond national screening however (figure 1).
Figure 1.

Management of people with a family history of colorectal cancer. BSG, British Society of Gastroenterology; CRC, colorectal cancer; FHCC, family history of colorectal cancer; FDR, first degree relative; MMR, mismatch repair.
Assessment of tumours in the affected relatives of those with an FHCC
We recommend that for all patients referred from primary care for assessment for an FHCC, MMR status should be assessed in tumour tissue from a close affected family member.
(GRADE of evidence: Moderate; Strength of recommendation: strong)
Consensus reached: 82% agreement.
We recommend that a reported family history of polyposis should be verified by review of histopathology and/or endoscopy reports which confirm the presence of a minimum of 10 adenomas or serrated lesions in an FDR.
(GRADE of evidence: low; Strength of recommendation: strong)
Consensus reached: 90% agreement.
Histopathological confirmation of CRC alters management of familial CRC surveillance in 20% of UK patients through verification of a diagnosis of CRC, multiple adenomas or other relevant features.20 Similarly review of endoscopy reports may assist in identification of patients with suspected familial risk such as those with polyposis syndromes.
When LS and Lynch-like tumours are excluded in families, their lifetime risk of CRC decreases. To quantify familial CRC risks associated with MMR deficient (dMMR) or MMR proficient (pMMR) tumours, a UK group analysed 2941 population-based cases of CRC.21 CRC risks in FDRs were strongly associated with dMMR tumours, early-onset disease and more than one affected FDR.
In a study by Bapat et al of 3143 CRC patients, dMMR tumours were associated with increasing numbers of FDRs with CRC (p=0.002); this association disappeared, however, when dMMR cases meeting Amsterdam criteria were removed from the analysis.22
A multicentre international registry based study23 assessed MMR status in 33 496 FDRs of 4853 cases of CRC. In comparison with the FDRs of pMMR CRC cases the FDRs of CRC cases with suspected ‘Lynch-like’ syndrome and with LS had a higher risk of CRC, but not those with dMMR non-LS. There was a greater risk of CRC in FDRs if CRC cases were diagnosed under 50 years of age, or if the tumours had clinicopathological features suggestive of LS.
Surveillance for colorectal neoplasia in those with a moderate risk FHCC
We recommend that patients with a moderate familial CRC risk should have a one-off colonoscopy at the age of 55 years.
(GRADE of evidence: moderate; Strength of recommendation: strong)
Consensus reached: 85% agreement.
We recommend that subsequent colonoscopic surveillance should be performed as determined by post-polypectomy surveillance guidelines.
(GRADE of evidence: moderate; Strength of recommendation: strong)
Consensus reached: 95% agreement.
An important question is whether the adenoma detection rate in those with an FHCC is higher than the detection rate in the general population. Most CRCs develop from adenomas and “advanced” adenomas (AAs, defined as either an adenoma size of at least 10 mm, villous architecture of at least 25%, or high grade dysplasia24) are considered to be the precursors of CRC. The term “advanced neoplasia” (AN) refers to the identification of either AAs or CRC.
The effectiveness and requirement for familial risk surveillance may be best determined by comparing the long-term CRC risk of a defined cohort of at-risk patients not undergoing surveillance with that of the general population. Theoretical relative risks of CRC <2 may be dominated by other genetic or environmental effects (and may require complex and validated risk modelling tools to determine suitability for surveillance).25 Where long-term CRC data are not available, the findings at surveillance may be used as a surrogate means to determine the need for post-polypectomy surveillance, although this method is inferior. In this context, that surveillance procedure may not have been warranted where the AA yield on that surveillance was less than doubled compared with a comparable yield in a control population.
There is a low prevalence of CRC in studies of surveillance in familial risk populations. There are limited data suggesting that metachronous CRC risk may be higher in patients at moderate familial risk versus population risk.26 As AAs are strongly associated with CRC development, AAs may be considered a proxy for CRC risk. In studies of patients with a moderate familial risk there is considerable heterogeneity in the prevalence of AA with a typical prevalence of between 8% and 10%; approximately double that of those without a family history (online supplementary tables 1-GRADE table 1). There is evidence from observational studies that the diagnostic yield of colonoscopy has increased in familial CRC risk cohorts over the past two decades, consistent with improvements in endoscopic technique, equipment and quality assurance.27
gutjnl-2019-319915supp002.pdf (220.8KB, pdf)
In the German bowel cancer screening population (an average risk population) the prevalence of AAs measured between 2003 and 2012 increased from 7.4% to 9.0% among men, and from 4.4% to 5.2% among women.28 29 In meta-analysis,30 the prevalence of adenomas is significantly higher in individuals with an FHCC than in controls (OR 1.7, 95% CI 1.4 to 3.5). Many observational control studies of surveillance colonoscopy for familial risk report a lower prevalence of AAs in the average risk population compared with the data from this German screening cohort. This may be related to lower ages of familial risk populations studied, and that many of the studies pre-date improvements in colonoscopic quality standards.
There is some observational evidence that colonoscopic surveillance mitigates this increased risk. In a German case-control study of CRC patients with an FHCC, those who had had a prior colonoscopy had a lower CRC risk that individuals without a family history who had not undergone colonoscopy. In the E3N French prospective study of 92 078 women, 692 CRCs were diagnosed after a median follow-up of 15.4 years31; women with FHCC who had not had a previous colonoscopy had a 80% higher CRC risk than those without FHCC. In women who had had a previous colonoscopy, CRC risk was similar in women with and without FHCC.
Age and risk of AAs in those with an FHCC
Projected annual transition rates from advanced adenomas to CRC strongly increase with age, with annual transition rates increasing from 2.6% in patients in their 50 s to >5% in their 80s.28 In an influential prospective study from 199432 FDRs of CRC patients had a risk of CRC at the age of 40 years equivalent to that of the average risk population aged 50 years. Notably, this historical study did not exclude patients with LS, and the increased risk conferred was predominantly in individuals with an affected FDR diagnosed under of 45 years.
Age is a strong predictor for adenomas and AAs in both familial risk individuals and controls in a series of observational studies. The incidence of AAs in patients aged 40–49 years in a surveillance cohort is equivalent to the general population,27 33 although the age of diagnosis of CRC in the affected FDR is not a predictor of risk of AAs. While adenomas but not AAs are more common in these studies, this may reflect the natural history of the adenoma to carcinoma sequence, whereby patients derive more benefit from surveillance colonoscopy from the age of 50 onwards, due to resection of AAs.
The subdivision of FHCC risk into those with 2× FDRs who were affected over or under 60 years of age is not associated with any difference in the diagnostic yield on colonoscopic surveillance.34 Although this age criterion was used in previous iterations of this guideline35 to subdivide into “low-moderate” and “high-moderate”risk, no evidence was identified to support this differentiation.
Several studies of cohorts of patients with moderate FHCC have demonstrated a negligible incidence in AAs in colonoscopic surveillance before the age of 50 years, but an increased risk after this age.36–41 There is little evidence in case-control series of significant differences in AA incidence between patients with one FDR diagnosed under the age of 50 years, and those families with a cluster of two FDRs diagnosed at any age.
The prevalence of AAs under the age of 50 years is not significantly increased in patients under surveillance for FHCC compared with the average risk population. This supports commencing colonoscopic surveillance at the age of 50–55 years for those at moderate familial risk. As the incidence of CRC is increasing in younger patients, this age recommendation may need to be reviewed in future guideline iterations—pending further relevant data specifically in those with a FHCC.
Surveillance in patients with a moderate familial risk of CRC
To exclude LS in those with an FHCC, a close affected relative’s tumour should undergo MMR tumour testing. A pathology review of the relative’s tumour should also be undertaken to ensure that there is no evidence of multiple polyps. After this risk assessment a decision about colonoscopic intervention should be considered.
The risk of AAs in surveillance colonoscopy (ie, after index/screening procedure) is largely determined by the presence of advanced neoplasia at the index procedure.
In a surveillance programme from St Mark’s Hospital, London, with well-organised recall of high and moderate risk families, AAs and cancer were more common in families who fulfilled Amsterdam criteria compared with those at moderate risk (on initial colonoscopy 5.7% and 0.9%, respectively).42 In families with moderate risk, advanced pathology was particularly uncommon under the age of 45 (1.1% and 0%) and on follow-up colonoscopy if AAs were absent initially (1.7% and 0.1%). With colonoscopic surveillance the incidence of CRC was substantially lower (80% in families with moderate risk (p=0.00004) and 43% in families with LS (p=0.06)) than the expected incidence in the absence of surveillance.
Registry data from other populations also suggest a benefit in selected moderate familial risk populations undergoing surveillance colonoscopy. These studies also confirm an association of AAs at the index procedure with advanced neoplasia in subsequent surveillance procedures (GRADE online supplementary tables 1-GRADE table 1). In a Swedish moderate familial risk cohort43 the risk of future AAs was associated with the prevalence of advanced lesions at the screening colonoscopy (multivariate analysis OR 5.22, 9% CI 2.3 to 9.94). It is of interest that adenomas and advanced lesions were not associated with the same risk factors: family history was predictive of advanced adenomas but not adenomas at the index screening colonoscopy.
The FACTS (Familial CRC Surveillance) randomised controlled trial compared intervals of surveillance in familial CRC.44 Individuals aged between 45 and 65 years with moderate familial CRC risk, where LS had been largely excluded, were randomly assigned to either a colonoscopy at 6 years or a colonoscopy at 3 and 6 years. Intention-to-treat analysis showed no significant difference in the proportion of patients with AAs (the primary outcome measure) at the first follow-up examination at 6 years (6.9%) versus 3 years (3.5%). The presence of AAs at the index colonoscopy was the only significant predictor for the presence of AAs at first follow-up (OR 5.2, 95% CI 1.6 to 16.87). Thus a 6 yearly interval was non-inferior to a 3 yearly surveillance interval, with the exception being that an AA at index colonoscopy predicts further advanced neoplasia at 3 years.
Surveillance for colorectal neoplasia in those with a high familial risk of CRC
We suggest that in high-risk families (a cluster of 3× FDRs with CRC across > 1 generation) a 5 yearly colonoscopy should be performed from the age of 40 years until the age of 75.
(GRADE of evidence: low; Strength of recommendation: weak)
Consensus reached: 86% agreement.
Families who fulfil Amsterdam criteria but who do not have evidence of dMMR do not share the same cancer incidence as families with LS (ie, hereditary MMR deficiency).45 46 Relatives in such families were found to have a lower incidence of CRC than those in families with LS, and incidence was not increased for other cancers. These families should not be described or counselled as having LS. To facilitate distinguishing these entities, the designation of “familial CRC type X” was suggested by Lindor et al to describe this type of familial aggregation of CRC.45
In a prospective surveillance study of a high familial risk population32 there was no significant difference in the prevalence of AAs in LS individuals versus FCC-X individuals. However on follow-up there were no incident cancers in the FCC-X group versus 4.4% CRC in Lynch patients, indicating lower risk of interval CRC in FCC-X despite equivalent AA risk.
In a prospective pooled cohort study of 1585 patients from eight international centres37 families were classified as FCC type X if they fulfilled the original Amsterdam criteria and late onset (LOFCC) if they fulfilled the Amsterdam criteria apart from not having a cancer diagnosed aged under 50. The results for FCC type X and LOFCC were very similar. At baseline, 22 prevalent asymptomatic CRCs were diagnosed, 120 (7.6%) individuals had high-risk adenomas and 225 (14.2%) simple adenomas. On follow-up high-risk adenomas were detected in 92 (8.7%) and multiple adenomas were detected in 20 (1.9%) individuals, from approximately 35 years of age onwards. Again the presence of AA at index colonoscopy was predictive of advanced neoplasia at subsequent procedures—33% of patients with an AA at index colonoscopy had an AA or cancer on follow-up.
This study by Mesher et al 37 indicated patients at high familial CRC risk should be managed similarly with 5 yearly colonoscopies undertaken from between 30 and 40 years of age with more intensive surveillance in individuals developing multiple or high-risk adenomas.
Amsterdam criteria families, where MMR testing of a CRC from an affected individual is not possible, may be offered surveillance as per LS. However such patients should be reviewed by a specialist service who may consider alternative testing strategies such as panel testing of affected individuals, or unaffected testing.
Prevention and lifestyle modification in familial CRC
We recommend that individuals with LS should be advised that regular use of daily aspirin reduces CRC risk.
(GRADE of evidence: moderate; Strength of recommendation: strong)
Consensus reached: 90% agreement.
Long-term data from the CAPP2 RCT suggests that aspirin reduces this risk by approximately half as compared with placebo.47 The benefits of regular aspirin intake take at least 3 to 5 years to become evident. Taking aspirin for less than 2 years’ duration does not seem to confer any benefit in reducing the incidence of cancer, or increasing survival in patients with LS.47 48
We recommend that people with LS should be offered research opportunities to take aspirin daily at different dosages. If they decline research participation they may be advised on their choices regarding dose of aspirin, risks and benefits of long-term aspirin use and ensure their medical practitioner is aware of their intake.
(GRADE of evidence: low; Strength of recommendation: strong)
Consensus reached: 90% agreement.
There is uncertainty about the optimum dosage of aspirin to recommended to individuals with LS. There is some evidence that long-term intake of daily 600 mg aspirin can reduce the risk of all cancers including CRC in LS from the CAPP2 randomised control trial.47 There is no other high quality evidence for any other dose of aspirin/ length of treatment but there are studies ongoing (CAPP3 trial49) which aim to identify optimum dosage. Evidence for the optimum dose will inform awareness and education among health professionals to mitigate the reluctance to prescribe higher doses of aspirin within primary care.50 In the interim clinicians may consider 150 mg aspirin in the context of LS outside of a clinical trial, with 300 mg doses in those with a BMI above 25 kg/m2.
There is insufficient evidence of the benefit of chemoprophylaxis in polyposis syndromes.
(GRADE of evidence: moderate; Strength of recommendation: strong)
Consensus reached: 100% agreement.
Non-steroidal anti-inflammatory drugs (NSAIDs) have been the most commonly studied chemoprophylaxis agents in patients with FAP, predominantly for lower GI tract disease, with some RCTs. Aspirin and tiracoxib have been found to be ineffective,51–53 although sulindac, celecoxib and rofecoxib have been demonstrated to reduce adenoma burden in the short term.54–57 A number of small series provide further support for these drugs and have also shown benefit from topical indomethacin.58 However long-term cancer prevention as an end point has not been adequately addressed.59 60 In the largest cohort of 54 patients, published in abstract form, 10% developed cancer while on chemoprophylaxis.61
Other classes of agents have also been assessed, such as omega 3 fish oils, but again the results are only short term with reduction in polyp size and number as the main endpoint.62
Celecoxib and the combination of sulindac and erlotinib have been reported as being beneficial63 64 for those with FAP and advanced duodenal disease; this was a short-term study using polyp number and size as the primary endpoint. A cohort study reported outcomes of the use of Eviendep, which was observed to reduce polyp number and size.65 However, no studies have demonstrated an effect on duodenal cancer prevention in FAP.
Cyclooxygenase-2 (COX-2) expression may be increased in JPS.66 67 There exists a theoretical potential benefit in the use of selective COX-2 inhibitors in JPS or PJS,66–68 but to date there are no trials demonstrating efficacy.
We recommend that individuals at increased familial risk of CRC should be strongly encouraged not to smoke, to maintain a normal BMI, to moderate their consumption of red and processed meat, and to exercise regularly.
(GRADE of evidence: low; Strength of recommendation: moderate)
Consensus reached: 81% agreement.
Diet and lifestyle factors are well established as significant contributors to up to half of all CRCs.69 70 A systematic review of epidemiological studies investigating the associations between nutritional factors, FHCC and CRC risk71 suggests that combinations of FHCC and higher consumption of alcoholic beverages, red or processed meat, or overweight/obesity increases the risk of CRC. There is evidence that LS individuals who smoke (particularly males with MLH1 mutations) have an increased risk of CRC. Data suggest current smokers are at significant increased risk of CRC irrespective of the age of initiation of smoking. Risk in former smokers decreased with each non-smoking year.72–75 CAPP2 study data from 29 month follow-up also indicate that overweight individuals with LS were more likely to develop CRC than those normal/underweight.76–79 Though modifiable environmental risk factors such as weight and exercise80 are common to both sporadic and familial CRC, individuals with familial risk may benefit from discussion about modifiable factors in order to potentially reduce their level of risk.81 82 There is emerging evidence of the benefit of targeted lifestyle modification in those with an FHCC.83
Quality and advanced endoscopic imaging in colonoscopic surveillance
We recommend that colonoscopy is the gold standard diagnostic and preventative method of surveillance for people with a hereditary risk of CRC.
(GRADE of evidence: moderate; Strength of recommendation: strong)
Consensus reached: 100% agreement.
We recommend that all surveillance colonoscopies are performed by endoscopists who consistently achieve BSG colonoscopy KPI minimum standards, specifically caecal intubation rate, adenoma/polyp detection rate and comfort score.
(GRADE of evidence: low; Strength of recommendation: strong)
Consensus reached: 94% agreement.
We suggest a repeat colonoscopy performed by an expert endoscopist is indicated in the event of a previously failed colonoscopy, with efforts made to both improve patient experience and to ensure procedure completion, given the advantages of colonoscopic surveillance. If colonoscopy is not possible then consider CT colonography.
(GRADE of evidence: low; Strength of recommendation: weak)
Consensus reached: 90% agreement.
We suggest that if the bowel preparation for colonoscopy is inadequate or if the examination is incomplete then a repeat colorectal surveillance procedure should be arranged within 3 months.
(GRADE of evidence: moderate; Strength of recommendation: weak)
Consensus reached: 95% agreement.
We suggest high-quality, high-definition white light endoscopy as the preferred modality for colonoscopy surveillance. Chromoendoscopy (virtual or dye-based) does not offer a clear advantage over high definition white light examination for colonoscopic surveillance, apart from in the context of determining the multiple polyp phenotype.
(GRADE of evidence: moderate; Strength of recommendation: weak)
Consensus reached: 89% agreement.
High quality colonoscopy has been recognised as a core element of successful cancer prevention in sporadic patients.84 There are limited data that this may also be relevant to cancer prevention in LS.85 Therefore colonoscopic quality indicators in endoscopists performing surveillance in LS patients should at least reach if not exceed the KPIs required for sporadic colonoscopy, using validated measures, in particular caecal intubation rate, adenoma/polyp detection rate and, given that patients may require serial colonoscopic procedures, comfort score.86 87
Colonoscopy is less effective for cancer prevention if the procedure is not complete to the caecum or the bowel preparation is inadequate. Where caecal intubation is not achieved, a repeat examination with an expert colonoscopist is appropriate. Inadequate bowel preparation reduces adenoma and advanced adenoma detection rates.88 89 Inadequate preparation at initial colonoscopy led to a threefold increase in miss rate in adenomas 5 mm or smaller.90 Therefore, repeat colonoscopy within 3 months seems appropriate for individuals at high familial risk.
Advanced imaging techniques have been proposed to help reduce missed lesions, especially small and flat lesions. More non-polypoid lesions are found in LS compared with sporadic patients.91 Chromoendoscopy, both dye-based and virtual, was recommended in recent ESGE guidelines on colonoscopic surveillance in LS.92
Tandem studies with chromoendoscopy show a consistent benefit (online supplementary 1 GRADE table 2)93–98; however, a study comparing a second pass with chromoendoscopy to a second white light pass did not show improved adenoma detection.94 Meta-analysis of data in sporadic patients shows an OR for at least one neoplastic lesion of 1.53 (95% CI 1.31 to 1.79).99 Real world cohort data comparing white light to chromoendoscopy provides some support for improved detection in high familial risk patients (39% LS) (15/24 (63%) adenomas with chromoendoscopy versus 15/77 (19%) white light endoscopy).100 A recent large, multicentre, Spanish randomised parallel group study, using high definition endoscopes and with high adenoma-detecting endoscopists, did not demonstrate a significant increase in adenoma detection with chromoendoscopy in 256 Lynch patients (OR 1.34, 95% CI 0.79 to 2.28).98 A similar sized multi-centre parallel group RCT study in the Netherlands had similar results using chromoendoscopy in the proximal colon (overall adenoma detection rate (ADR) 33% vs 27% with white light endoscopy).97 At 2 year follow-up there was no difference in ADR between the two groups, but there were four cancers in the chromoendoscopy group versus one in the white light endoscopy group. Simply combining the results for ADR of these two studies gives a risk ratio of 1.23 (95% CI 0.94 to 1.60, p=0.14; Fisher exact, n=497; our calculation) suggesting a limited clinical benefit in terms of ADR for considerable extra effort, which may not translate into improved cancer prevention.
Virtual chromoendoscopy, narrow band imaging (NBI, second generation, Olympus) and I-SCAN (Pentax) have shown some benefit in tandem studies (online supplementary tables 1-GRADE table 3); however, this improved detection is not consistent with meta-analysis data from sporadic patients,101 and NBI performed less well than chromoendoscopy in a cohort, tandem study.102 In a study comparing different forms of colonoscopic imaging, chromoendoscopy was superior to white light colonoscopy, autofluorescence imaging, and narrow-band imaging for detection of diminutive colorectal lesions in adenomatous polyposis.103
Advanced colonoscopic imaging can assist in making a diagnosis of polyposis by revealing additional lesions required to meet diagnostic criteria: diagnoses of adenomatous polyposis may be missed if dye spray is not used,104 and there is similar evidence from the CONSCOP study that the identification of serrated polyps is enhanced through the use of pancolonic dye spray.105
In summary, a high quality, high definition white light colonoscopic examination, by an endoscopist who meets all colonoscopy KPIs, seems adequate for LS and high familial risk patients, with the exception of those with multiple polyps where chromoendoscopy may help define the phenotype.
Non-invasive surveillance methods for people with FHCC
There is insufficient evidence to recommend other methods of surveillance for those with familial CRC risk such as FIT, MR or CT colonography
(GRADE of evidence: low; Strength of recommendation: strong)
Consensus reached: 95% agreement.
In meta-analysis of 12 published studies of patients at increased risk of CRC (predominantly familial risk), the average sensitivity of FIT for advanced neoplasia was 48% (95% CI 39% to 57%) and the average specificity was 93%.106 A subgroup analysis of patients with familial risk only was performed, and the sensitivity for CRC was 86% and for advanced neoplasia 46%. Thus, although FIT may be close to equivalence to colonoscopy for the detection of CRC, AAs would be missed by surveillance with FIT alone.
Patients with an FHCC no longer on colonoscopic surveillance should participate in national bowel cancer screening programmes designed for the average risk population. Some indirect evidence suggests that FIT or other forms of stool testing methods alongside colonoscopy may potentially be a useful adjunct in surveillance in patients after discharge from colonoscopic surveillance.107
Other methods such as colon capsule108 or MR colonography109 lack efficacy in this population. Although there is some evidence of the efficacy of CT colonography,110 it is not clear how effective CT may be in the identification of serrated or non-polypoid lesions. Repeated CT scanning in particular may be inappropriate due to the risk of radiation damage to patients with inherited DNA repair defects. However, if total colonoscopy is not possible despite expert referral, low radiation-dose CT colonography with quality assurance111 is the preferred modality, as it has similar test performance to colonoscopy for CRC.110
Lynch syndrome
We recommend that for all people when first diagnosed with CRC, testing using immunohistochemistry (IHC) for MMR proteins or microsatellite instability is used to identify tumours with deficient DNA MMR, and to guide further sequential testing for LS.
(GRADE of evidence: strong; Strength of recommendation: strong)
Consensus reached: 100% agreement.
LS is a condition defined by the presence of pathogenic variants in the coding sequence or regulatory domains of the four MMR genes: MLH1, MSH2, MSH6 and PMS2. Patients with EPCAM mutations which embrace the regulatory domain of MSH2 should be managed as those with MSH2 pathogenic variants. Selection methods based on family history of cancer, or other clinical parameters, such as the Amsterdam or Bethesda criteria,112 113 were used historically to identify high-risk patients who may benefit from interventions and/or genetic testing; however, advances in constitutional testing in the past two decades have facilitated the accurate genetic diagnosis of LS. A comprehensive review performed by NICE of the clinical- and cost-effectiveness of universal diagnostic testing for LS was published in 2017.18 We recommend the use of colonoscopic biopsies as the preferred source material for tumour MMR testing.114
Colonoscopic surveillance and LS
We recommend that colonoscopic surveillance should be performed at a 2 yearly interval for all LS patients.
(GRADE of evidence: moderate; Strength of recommendation: strong)
Consensus reached: 85% agreement.
Surveillance colonoscopy in LS does not completely eradicate the risk of CRC, with the well-recognised phenomenon of interval cancers related to multiple factors including adherence and timeliness of colonoscopy. However, the optimal interval for surveillance colonoscopy is yet to be established.
The literature around colonoscopic surveillance is mixed with few studies reporting on recognised key performance indicators including adenoma detection rate, or caecal intubation rate or compliance with the screening interval. A study in the UK identified that hospital recall systems, clinician or patient related issues affect compliance with LS surveillance intervals.115 In this study variable colonoscopy quality indicators were highlighted with a caecal intubation rate was 92%, and approximately 10% had inadequate bowel preparation. In a retrospective, two centre Dutch study 31 interval CRCs were diagnosed in 29 patients with LS, within 2 years of previous colonoscopy, all of whom were MLH1 and MSH2 pathogenic variant carriers, and 84% were located in the proximal colon.85 In three of a total of five patients where colon examination was not achieved during the previous colonoscopy, the interval CRC was found in the unexamined proximal segment. In six of nine patients with a previous adenoma, the interval CRC was detected in the same colon segment, raising the possibility of incomplete endoscopic resection.
There is some evidence that earlier tumour stage may be observed in those with more frequent colonoscopy.116 117 More recent prospective data have described cancer incidence and survival in LS in patients undergoing surveillance.118–121 The incidence of CRC was influenced by the LS-associated gene; cumulative CRC incidence at 70 years by gene was greater in those with MLH1 (46%) or MSH2 (35%) constitutional pathogenic variants compared with those with MSH6 (20%) or PMS2 (0%) pathogenic variants (although with wide confidence intervals). The prognosis from interval CRC was good, with a 5 year survival of 94% (90–98%), and a 10 year survival of 91% (84–95%). This concords with an earlier Dutch study which reported a non-significantly increased risk of interval CRC in those with MLH1 or MSH2 constitutional pathogenic variants.122 In the prospective data published by Moller and colleagues, the interval between last surveillance colonoscopy and CRC was analysed; 100/145 (69%) CRCs were diagnosed >2 years after last colonoscopy (interval post colonoscopy range 0–125 months).121
An observational study by Engel et al 123 compared prospective colonoscopic data from three countries with different LS surveillance policies (Germany: annual surveillance; Netherlands: 1–2 yearly surveillance; Finland: 2–3 yearly surveillance) and found no significant difference in cumulative CRC incidence or stage at detection among the countries. The study included data from 16 327 colonoscopic examinations of 2747 LS patients (MLH1, MSH2, MSH6 pathogenic variant carriers) over 23 309 person-years of cumulative observation time. The 10 year cumulative CRC incidence ranged from 4.1% to 18.4% for patients with low- and high-risk profiles, respectively, and was influenced by age, gender, LS gene, and prior detection of CRC or adenoma. The authors conclude that a 2 year surveillance interval might be appropriate, and short surveillance intervals may only be beneficial to LS patients with high-risk factors. The findings of this study should be interpreted with caution, however, as there are some limitations including unavailable data regarding key performance indicators and non-compliance with country-specific surveillance protocols.
The largest study to date has recently reported cancer risk estimates for PMS2 pathogenic variant carriers,124 demonstrating a small increased risk of CRC (cumulative risk to age 80 years of 13% for males and 12% for females, compared with the general population risk of 6.6% and 4.7%, respectively). Based on this finding and data from the prospective LS database, the authors have suggested that extending the colonoscopic surveillance interval may be justified in PMS2 pathogenic variant carriers. However, most guidelines recommend surveillance colonoscopy between 1 and 2 yearly.35 117 125 126 The data regarding differences between the LS-associated genes are not sufficiently robust such that the surveillance interval can be stratified by LS gene. A lower penetrance of CRC in those with an MSH6 pathogenic variant is likely to account for the lower risk of interval CRC on surveillance. The surveillance interval is determined by tumour biology and the accelerated pathway of carcinogenesis in LS.127 There are no data to suggest that the speed of carcinogenesis in those with PMS2 or MSH6 constitutional pathogenic variants is different from those with MLH1 or MSH2 constitutional pathogenic variants; therefore until such data exist, the surveillance interval should be the same for all patients with LS, irrespective of the underlying LS-associated gene pathogenic variant.
Lynch syndrome: gene and gender specific guidelines
We recommend that age of onset of surveillance colonoscopy should be stratified according to the LS-associated gene. We recommend colonoscopy from the age of 25 years for MLH1 and MSH2 mutation carriers and 35 years for MSH6 and PMS2 mutation carriers. There are insufficient data to support stratifying age of onset of surveillance by gender.
(GRADE of evidence: moderate; Strength of recommendation: strong)
Consensus reached: 95% agreement.
Outcomes from the Dutch Hereditary Non-Polyposis Colorectal Cancer (HNPCC) registry showed that only 2/246 (0.8%) patients with LS developed CRC before the age of 20 years and another two between the age of 20 and 25 years.128 A number of other studies have confirmed that the risk of developing CRC before the age of 25 years is very low.129–132 It is largely based on these studies that most groups have recommended starting surveillance colonoscopy at the age of 20–25 years.35 117 126 However, a meta-analysis of the data for MLH1 and MSH2 pathogenic variant carriers has questioned whether surveillance colonoscopy was justified before the age of 30 years.133
The US multi-society task force guidelines recommend starting surveillance colonoscopy at 20–25 years (or 2–5 years younger than youngest affected relative if diagnosed <25 years) but to consider starting at 30 and 35 years for MSH6 and PMS2 pathogenic variant carriers, respectively.125
Ten Broeke et al described a series of 377 patients with constitutional PMS2 pathogenic variants.124 They observed that the median age at first CRC was 52 years (26–86 years), and noted gender differences in CRC risk. The cumulative risk (%) in men and women up to the age of 69 years was 18.8% and 10.5%, respectively. They recommended that commencement of colonoscopic surveillance could be deferred. A nationwide French study of patients with LS supports a genotype-phenotype correlation.134 There were no PMS2 pathogenic variant carriers in this cohort. Overall there was a cumulative CRC risk by 70 years of 38% in males and 31% in females. When analysed by genotype the cumulative CRC risks were 46% MLH1, 48% MSH2 and 12% MSH6. Furthermore, age (years) at CRC also varied by genotype: MLH1 45 (15–90), MSH2 44 (16–95), MSH6 54 (24–85). These data are supported by analyses of a prospective LS database.119 121 These reports observed that there was genotype specific penetrance. Furthermore, within each genotype the incidence of CRC was age dependent and CRC occurred as a stochastic event. In the report addressing first cancer incidence and survival in patients with LS receiving surveillance, Moller et al observed cumulative CRC cancer incidence to the age of 70 years was 46% MLH1, 35% MSH2, 20% MSH6 and 0% PMS2. Overall, there was no difference between gender. The most recent report from this prospective database120 describes cancer risk and survival up to age 75 years, analysing the data both by gene and gender. The cohort includes 3119 patients with 24 475 observation years. The previously described genotype dependent penetrance was confirmed; of note the number of PMS2 carriers in this cohort is small.
Further studies are required to further stratify risk for MSH6 and PMS2 pathogenic variant carriers. Until such data are available, based on the current sparse literature it appears reasonable to defer initiation of surveillance for these patients. The current literature is sufficiently robust to recommend that MSH6 and PMS2 pathogenic variant carriers should have different ages of onset for colorectal surveillance (figure 2).
Figure 2.
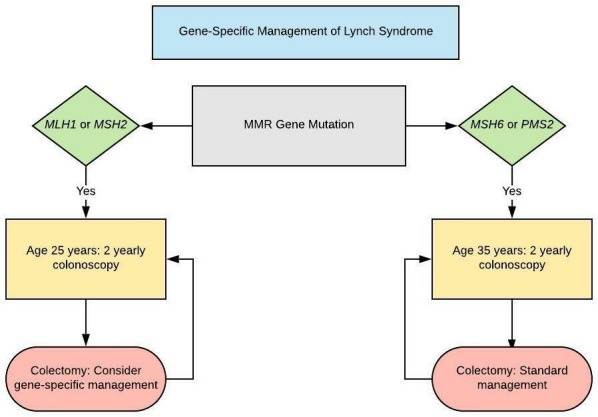
Gene-specific management of Lynch syndrome. MMR, mismatch repair.
Surgery in LS patients with CRC
We suggest that for LS patients with MLH1 or MSH2 mutations who develop colon cancer or colonic neoplasia not amenable to endoscopic control, the decision to perform segmental versus total/near total colectomy should balance the risks of metachronous cancer, the functional consequences of surgery, the patient ’s age and patient ’s wishes.
(GRADE of evidence: Moderate; Strength of recommendation: Strong)
Consensus reached: 100% agreement.We recommend that for LS patients with MSH6 or PMS2 mutations there is insufficient evidence for oncological benefit of extended colectomy over segmental resection.
(GRADE of evidence: low; Strength of recommendation: strong)
Consensus reached: 89% agreement.
When abdominal-perineal excision can be avoided, a standard low anterior resection is a reasonable option to treat rectal cancers in LS patients, even though the residual colon is at high risk of metachronous neoplasia.(GRADE of evidence: low; Strength of recommendation: weak)
Consensus reached: 88% agreement.
The decision to perform a total/subtotal colectomy or segmental colectomy involves consideration of:
The risk of metachronous CRC
Survival from metachronous CRC
Functional consequences and quality of life (QoL) following surgery
This decision regarding which operation is preferable should be made on the basis of individual patient factors and preferences, with special emphasis on the risk of metachronous CRC, age and the preparedness of the patient to continue colonoscopic surveillance. High-quality patient information and shared decision-making between patient and surgeon will facilitate this decision.
Risk of metachronous cancer
Parry et al found the cumulative risk of metachronous CRC to be 16% at 10 years, 41% at 20 years and 62% at 30 years after segmental colectomy. In contrast none of 50 subjects who had extensive colectomy developed metachronous CRC. They calculated that the risk of metachronous CRC was reduced by 31% for every 10 cm of large bowel removed. Kalady et al reported 296 patients (253 with segmental colectomy and 43 with total colectomy/ileorectal anastomosis). Of the 253 segmental colectomy patients, 55 patients (25%) developed a second CRC at a median of 69 months after index surgery. Stages of the metachronous cancers were I-16, II-18, III-12, and IV-2. By comparison, four of 38 patients (11%) who underwent total colectomy developed subsequent high-risk adenomas and only three (8%) developed metachronous cancer.135
A Finnish study reported the cumulative risk of subsequent CRC to be 20% within 10 years and 47% within 25 years after standard resection and 4% and 9% after extended surgery.136 A further study showed metachronous CRC in 6.3% of pathogenic variant carriers treated with total/subtotal colectomy compared with 27% treated by segmental colectomy.137 In meta-analysis much of the excess risk of metachronous CRC appears to be in carriers of pathogenic variants in the MLH1 and MSH2 pathogenic carriers, with insufficient data to suggest excess risk in MSH6 or PMS2 variant carriers.138
Survival from metachronous cancer
Moller et al in an international collaborative study found the cumulative incidence of metachronous CRC was 36% from 40 to 70 years. Five and 10 year crude survival after colectomy for metachronous CRC was 94% and 91%, respectively, with no significant difference between the pathogenic variants of the different genes.119
Natarajan et al found no significant difference in survival time between patients undergoing extended colectomy and limited resection in patients with LS.139 The potential health effects in terms of life expectancy for patients undergoing subtotal colectomy or hemicolectomy for CRC were analysed. The 10 year risk of CRC after subtotal colectomy was 4% and after hemicolectomy was 16% and stages of CRCs detected within a 2 year surveillance interval were 32% Dukes' A, 54% Dukes' B, and 14% Dukes' C (derived from two cohort studies). The overall LE gain of subtotal colectomy compared with hemicolectomy at ages 27, 47, and 67 was 2.3, 1, and 0.3 years, respectively.140 The authors concluded that unless surveillance results improve, subtotal colectomy was the preferred treatment for CRC in LS in view of the difference in life expectancy and that for older patients, hemicolectomy may be an option as there was no appreciable difference in life expectancy is this age group.
Functional consequences and QoL
In a Dutch study, no difference in global QoL was noted between 51 LS patients who underwent partial colectomy and 53 patients who underwent subtotal colectomy, although functional outcome (stool frequency and social impact) was worse after subtotal colectomy than after segmental colectomy.141
Maeda et al constructed a state-transition (Markov) model to compare segmental colectomy and total abdominal colectomy with ileorectal anastomosis.142 Quality-adjusted life years (QALYs) were calculated based on utility states for patients based on the colectomy they received. Multiple sensitivity analyses were planned to examine the impact of each assumption on model results. For young (30-year-old) patients with LS, mean survival was slightly better with total colectomy than with segmental resection (34.8 vs 35.5 years). When QALYs were considered, the two strategies were approximately equivalent, with QALYs per patient of 21.5 for segmental colectomy and 21.2 for total colectomy. They suggested that with advancing age, segmental colectomy becomes a more favourable strategy.
The decision regarding which operation is preferable should be made on the basis of individual patient factors and preferences, with special emphasis on the risk of metachronous CRC, age and the preparedness of the patient to continue colonoscopic surveillance.
Patients presenting with rectal cancer
A standard low anterior resection or abdominal perineal resection is a reasonable option to treat rectal cancers in LS patients, even though the residual colon is at high risk of metachronous neoplasia. A retrospective study of 79 LS patients with rectal cancer who had undergone proctectomy found a cumulative risk of metachronous colon cancer to be 19% at 10 years, 47% at 20 years, and 69% at 30 years after surgical resection.143 Kalady et al followed 50 HNPCC patients with a primary diagnosis of rectal cancer treated by proctectomy. Forty-eight high-risk adenomas developed in 13 patients (39.4%) and five patients (15.2%) developed metachronous adenocarcinoma at a median of 6 years (range 3.5–16) after proctectomy, including three at an advanced stage. Overall 17 of 33 patients (51.5%) developed high-risk adenoma or cancer after proctectomy.144
Upper GI, pancreatic and small bowel risk management in LS
We recommend that gastric, small bowel, or pancreatic surveillance in LS patients is only performed in the context of a clinical trial.
(GRADE of evidence: low; Strength of recommendation: strong)
Consensus reached: 100% agreement.
We recommend screening for H pylori in patients with LS and subsequent eradication therapy if indicated.
(GRADE of evidence: low; Strength of recommendation: strong)
Consensus reached: 100% agreement.
Gastric cancer
In a retrospective study, Capelle et al estimated lifetime risks of gastric cancer in 2014 patients with LS from the Dutch Hereditary Cancer Registry.145 Gastric cancer was diagnosed in 32 patients (1.6%); 22 of these patients (69%) had a negative family history of gastric cancer. The lifetime risk of gastric cancer was reported to be 4.8% and 9% for MLH1 and MSH2 pathogenic variant carriers, respectively. None of the 378 MSH6 pathogenic variant carriers developed gastric cancer. The median age of diagnosis was 55 years (range 27–82 years).
In a recent prospective study, Moller et al reported gene-specific prospective cumulative cancer risks (up to the age of 75 years) for gastric cancer in 3119 patients with LS.146 The risk of gastric carcinoma was reported to be the highest for MLH1 and MSH2 pathogenic variant carriers: 7.1% (95% CI 3.5% to 10.8%) for MLH1, 7.7% (95% CI 1.9% to 13.6%) for MSH2, and 5.3% (95% CI 0.0% to 13.1%) for MSH6. No gastric cancers were observed in PMS2 pathogenic variant carriers. The study also reported a 5 year overall survival rate of 61% (95% CI 33% to 81%) for LS-associated gastric cancer. This compares favourably with the prognosis in unselected patients with resectable gastric cancer, who have been reported to have an overall 5 year survival of 10–30%.147
A Finnish study reported the characteristics of 62 gastric cancers that occurred in 570 family members from the Finnish HNPCC registry. There was an overrepresentation of intestinal gastric cancers, which was found in 79% of cases, with only 13% of cases being diffuse gastric cancers. As the development of intestinal gastric cancers is thought to be closely associated with H.pylori-associated chronic gastritis, the authors investigated whether atrophic gastritis and H. p ylori infection could be markers of gastric cancer risk in patients with LS. Twenty percent of the LS-associated gastric cancers were H. pylori positive. The median age at diagnosis was 56 years.147
Renkonen-Sinisalo et al assessed the diagnostic yield of upper endoscopy in a series of 73 MMR pathogenic variants carriers (median age 47 years).148 The authors found no early gastric cancers or premalignant lesions (diagnostic yield 0). A single screened-detected small bowel cancer was identified: an advanced stage duodenal cancer. No additional studies evaluating the benefit of gastric surveillance in LS were identified from the literature.
The cumulative lifetime risk of gastric cancer is relatively low. Aspirin chemoprophylaxis may reduce the risk of all LS-associated cancers in patients with LS. In conclusion, there is no convincing evidence to support the utility of gastric surveillance in patients with LS.
Small bowel cancer
The cumulative lifetime risk of developing small bowel cancer has been estimated to be 4.2% in patients with constitutional MLH1 and MSH2 pathogenic variants.149 Small bowel cancers have rarely been reported in patients with constitutional MSH6 and PMS2 pathogenic variants. A recent study has reported gene-specific prospective cumulative cancer risks (up to the age of 75 years) for duodenal carcinoma in 3119 patients with LS. The risk of duodenal carcinoma was reported to be the highest for MLH1 pathogenic variant carriers (6.5%, 95% CI 1.7% to 10.2% for MLH1; 2.0%, 95% CI 0.1% to 4.0% for MSH2), and no small bowel cancers were observed in patients with constitutional MSH6 or PMS2 pathogenic variants.146
LS-related small bowel carcinomas have an earlier onset compared with sporadic tumours with a median age of onset of 52 years (range 23–69 years). LS-related small bowel carcinomas most commonly occur in the duodenum (49%), and decrease in frequency from the jejunum (29%) to the ileum (12%).150 Patients with LS who develop small bowel cancers have been demonstrated to have a better prognosis compared with patients who develop sporadic tumours. Moller et al, in a recent study, reported a 5 year survival rate of 67% (95% CI 28% to 88%) in LS patients with small bowel cancers diagnosed below the age of 65.146 This compares favourably with the 5 year survival of sporadic small bowel adenocarcinoma, which has been estimated to be 25–30%.151
Several studies have investigated the diagnostic yield of surveillance for small bowel cancers in patients with LS. In 2010 Saurin et al compared the use of CT enteroclysis and video-capsule endoscopy (VCE) in 35 asymptomatic patients with LS.152 Histologically confirmed small bowel tumours were identified in three patients (diagnostic yield of 8.6%): one jejunal adenocarcinoma (T3N0M0) and two adenomas with low-grade dysplasia. VCE identified all three tumours, but CT enteroclysis would have missed the two adenomas.152 In a Dutch study, Haanstra et al investigated the prevalence of small bowel tumours in 200 asymptomatic pathogenic variant carriers (aged 35–70 years) using VCE.153 Caecal visualisation was achieved in 95% of procedures. Histologically confirmed small bowel tumours were identified in two patients (diagnostic yield of 1%): one adenocarcinoma (TisN0Mx) and one adenoma, both located in the duodenum. In addition, another patient was diagnosed with a duodenal cancer (T2N0Mx) 7 months after a negative VCE (incidence of 1.5%). This suggests that VCE may miss some small bowel tumours.153 In a follow-up study, asymptomatic LS patients who underwent a VCE were invited to undergo a second VCE procedure 2 years later. A total of 155 (78%) of the initial 200 pathogenic variant carriers underwent a second VCE. Potentially significant lesions were identified in 17 patients (11%), which required further investigations: eight gastroduodenoscopies and nine balloon-assisted endoscopies were carried out, but no small bowel tumours were identified.154
In the CAPP2 randomised trial, Burn et al examined the effect of aspirin in 861 patients with LS who were randomly assigned in a two-by-two factorial design to 600 mg of aspirin or aspirin placebo or 30 g of resistant starch or starch placebo, for up to 4 years.47 The primary outcome was the incidence of new primary CRCs, but the incidence of all LS cancers was also examined. Following a re-analysis of the data at a mean follow-up of 55.7 months, for participants completing 2 years of intervention (258 aspirin; 250 aspirin placebo), per-protocol analysis yielded a hazard ratio (HR) of 0.45 (95% CI 0.26 to 0.79; p=0.005) for all LS-related cancers.
The absolute lifetime risk of small bowel cancer in patients with LS is 4.2%; this risk is likely to be most significant for MLH1 pathogenic variant carriers. LS-related small bowel cancers have been demonstrated to have a better prognosis than sporadic small bowel cancers. Aspirin chemoprophylaxis has been demonstrated to significantly reduce the risk of all LS-associated cancers in patients with LS. Video capsule endoscopy has been used to successfully identify small bowel tumours in patients with LS. However, there have been a limited number of studies and the diagnostic yield has been variable (1–8.6%). VCE may have missed some cancers. No small bowel tumours were detected on follow-up surveillance after an interval of 2 years. In addition, a high false positive rate (11%) has been reported, resulting in asymptomatic patients requiring time consuming and invasive tests.
Pancreatic cancer
In a retrospective study, Kastrinos et al estimated pancreatic cancer risks in individuals (n=6342) from 147 families with constitutional MLH1, MSH2 and MSH6 pathogenic variants.155 Forty-seven pancreatic cancer cases were reported in 31 families: 18 patients had a negative family history (38%). The cumulative risk for pancreatic cancer up to the age of 70 was reported to be 3.7% (95% CI 1.45% to 5.88%); this represented an 8.6-fold increased risk compared with the general population. Most of the pancreatic cases were observed in individuals from MSH2 families (31/47) and MLH1 families (13/47). Only three of the pancreatic cancers were diagnosed in the MSH6 families. The median age of pancreatic cancer diagnosis was 51.5 years (range 19–85).
Prospective lifetime risks stratified by the MMR gene have now also been reported for pancreatic cancer. Moller et al reported a cumulative risk of pancreatic cancer (up to the age of 75) for MLH1 of 6.2% (95% CI 2.6% to 9.8%), for MSH2 of 0.5% (95% CI 0.0% to 1.5%) and for MSH6 of 1.4% (95% CI 0.0% to 4.2%). No pancreatic cancers were observed in PMS2 pathogenic variant carriers. The authors also calculated 5 year overall survival for the LS patients with pancreatic cancer. The poor prognosis of pancreatic cancer patients is well established, and none of the affected carriers was alive at 5 years.119
Surveillance for pancreatic cancer has been recommended in high-risk groups (defined as >5%), including patients from familial pancreatic cancer pedigrees with an affected FDR, patients with PJS, and CDKN2A (P16), BRCA2 and MMR pathogenic variant carriers with at least one affected FDR.156 The goal of screening is to identify and treat early stage pancreatic cancers (T1N0M0) and high-risk precursor lesions, high-grade pancreatic intraepithelial neoplasia (PanIN-3) and intraductal papillary mucinous neoplasia (IPMN) with high-grade dysplasia. Signoretti et al conducted a systematic review and meta-analysis of 16 pancreatic surveillance studies in high-risk groups.157 A relatively low diagnostic yield of pancreatic cancers and relevant precursor lesions was reported (3.3%) using endoscopic ultrasound and MRI as first line screening tests. A significant proportion (25%) of the screen-detected cancers were also unresectable or metastatic. Some patients underwent surgery for precursor lesions and were not found to have high-risk precursor lesions. A significant morbidity (up to 40%) and mortality (0.5–6%) have been reported for the surgical treatment of suspicious pancreatic findings.154 Pancreatic surveillance has not been demonstrated to reduce pancreatic-cancer specific mortality in patients with LS.
The cumulative lifetime risk of pancreatic cancer is relatively low for MLH1 pathogenic variant carriers, and low (<5%) for MSH2, MSH6 and PMS2 pathogenic variant carriers. Aspirin chemoprophylaxis has also been demonstrated to significantly reduce the risk of all LS-associated cancers in patients with LS. The utility of pancreatic screening remains unproven, and there is a danger of overtreatment with patients undergoing surgery for benign or low-risk lesions. There is a significant morbidity and mortality associated with pancreatic surgery.
Lynch-like syndrome
We recommend that deficient MMR tumours without hypermethylation/BRAF pathogenic variant and no pathogenic constitutional pathogenic variant in MMR genes should undergo somatic tumour testing with a CRC gene panel.
(GRADE of evidence: low; Strength of recommendation: strong)
Consensus reached: 100% agreement.
We recommend that if double somatic MMR pathogenic variants are identified, manage proband and their FDRs based on the FHCC.
(GRADE of evidence: low; Strength of recommendation: strong )
Consensus reached: 95% agreement.
We suggest that if no or one somatic pathogenic variant is identified, the proband and their FDRs should be managed as per LS.
(GRADE of evidence: low; Strength of recommendation: weak)
Consensus reached: 100% agreement.
LLS describes a subgroup of patients with CRC or other LS-related tumours that manifest MMR deficiency (microsatellite instability (MSI) and/or loss of MMR protein expression) that is neither explained by somatic MLH1 promoter hypermethylation, BRAF pathogenic variant or a detectable pathogenic constitutional variant in an MMR gene or EPCAM LLS cases, and therefore cannot be readily assigned to either the sporadic or inherited MMR deficiency categories, respectively.158–160 Combining data from published studies, using current diagnostic approaches, an estimated 59% (95% CI 55% to 64%) of dMMR CRC cases are unexplained and categorised as LLS.23 129 161
Cancer risks
Three studies have investigated CRC risks for patients with LLS and their relatives. Overbeek et al compared the characteristics of 76 families with a constitutional variant in a DNA MMR gene with those of 18 families with unexplained dMMR tumours.162 Although the mean age of CRC onset of the index case was comparable at 44 years, a significantly higher proportion of the families with LS fulfilled the Amsterdam II criteria compared with the families with LLS (66% vs 11%; p<0.0001).
Rodríguez-Soler et al were the first to quantify the risk of CRC for FDRs of CRC cases with LLS.161 This population-based study examined the risk of CRC in the FDRs of CRC cases with LLS, LS and sporadic pMMR tumours. The authors found the highest risk of CRC for the FDRs of the LS cases (standardised incidence ratio (SIR) for LS 6.04, 95% CI 3.58 to 9.5), an intermediate risk for the FDRs of LLS cases (SIR for LLS 2.12, 95% CI 1.16 to 3.56), and the lowest risks for the FDRs of sporadic pMMR CRC cases (SIR for sporadic 0.48, 95% CI 0.27 to 0.79; p<0.001).
In a large family cohort study, Win et al subsequently confirmed that FDRs of LLS cases have a higher CRC risk compared FDRs of sporadic pMMR CRC cases, but a lower risk compared with FDRs of LS cases.23 Compared with FDRs of sporadic pMMR CRC cases, a higher risk of CRC was estimated for FDRs of LLS cases (HR 2.06, 95% CI 1.59 to 2.67), and an even higher risk for the FDRs of LS cases (HR 5.37, 95% CI 4.16 to 6.94).23 119 140 155–162
Potential aetiologies
Studies have shown that up 70% of patients with LLS have double (biallelic) somatic pathogenic variants in the MMR genes.163–165
In a study of 25 LLS cases with unexplained immunohistochemical absence of the MLH1 or MSH2 protein, Mesenkamp et al identified biallelic somatic pathogenic variants (one pathogenic sequence variant and loss of heterozygosity) in 13 cases (52%; 8/18 in MLH1 and 5/7 in MSH2).165 Other studies have subsequently confirmed the presence of a high proportion of biallelic somatic pathogenic variant LLS cases. In a study of 36 LLS cases, Geurts-Giele et al found double somatic pathogenic variants in 21 cases (58%; 16/21 in MLH1 and 5/12 in MSH2).166 In a study of 32 LLS cases, Haraldsdottir et al identified double somatic pathogenic variants in 22 cases (69%; seven for MLH1, 11 for MSH2, three for MSH6 and one for PMS2).164
Biallelic somatic pathogenic variants may be in the form of point pathogenic variants coupled with loss of heterozygosity, or another point pathogenic variant. Double variant somatic pathogenic variants are likely to be in trans (one on each allele), and therefore if identified provide a potential explanation for the tumour MMR deficiency in patients with LLS. A recent study has shown that a significant proportion of LLS cases may also be explained by false positive screening results, caused by an incorrect interpretation of MMR immunohistochemistry (IHC) results. The authors found discordant findings on IHC and MSI in six out 32 LLS cases (19%).164 However, there remains a possibility that some patients with LLS may actually have LS, due to constitutional pathogenic variants in the MMR genes that are not detected using current diagnostic methods, for example, pathogenic variants within regulatory/promoter regions and complex structural variants.158 Finally some patients with LLS may carry constitutional or somatic pathogenic variants in other genes, which may explain the deficient MMR tumour phenotype and/or occur in conjunction with somatic pathogenic variants in the MMR genes.167–169 Jasen et al investigated 62 LLS cases using gene panel sequencing including the POLE, POLD1 and MMR genes. The authors found somatic (n=7) or constitutional variants (n=2) in the POLE/POLD1 exonuclease domain in nine tumours (14.5%), which showed an ultramutated phenotype. Six of these cases also were found to carry somatic MMR variants.169 In a study by Morak et al, MUTYH diagnostic testing was carried out in 85 patients with LLS and biallelic constitutional pathogenic variants were found in one patient (1.18%).167
Consequently, LLS cases have been found to have heterogeneous aetiologies, ranging from biallelic somatic pathogenic variants to unidentifiable constitutional pathogenic variants in MMR genes. This is likely to explain the intermediate cancer risks, which have been reported for the FDRs of LLS cases, and has implications for the clinical management of LLS families.
Thus double somatic pathogenic variants in the MMR genes may explain up to 70% of LLS cases. Therefore, tumour sequencing of the DNA MMR genes should be undertaken in LLS cases, as this would help guide genetic counselling and the management recommendations for LLS cases and their FDRs (figure 3). If double somatic pathogenic variants are identified, we would recommend that these patients and their FDRs be managed based on the FHCC, and not as LS. However, if no somatic pathogenic variants or only one somatic pathogenic variant or loss of heterozygosity of one allele is identified we would recommend that these cases and the FDRs be managed as potential LS cases and follow colorectal surveillance guidelines for LS. This is based on the possibility that these cases could have LS due to an unidentifiable constitutional pathogenic variant in an MMR gene. These recommendations are consistent with the NCCN (National Comprehensive Cancer Network) guidelines170 and may help reduce variability in practice in the UK.171
Figure 3.
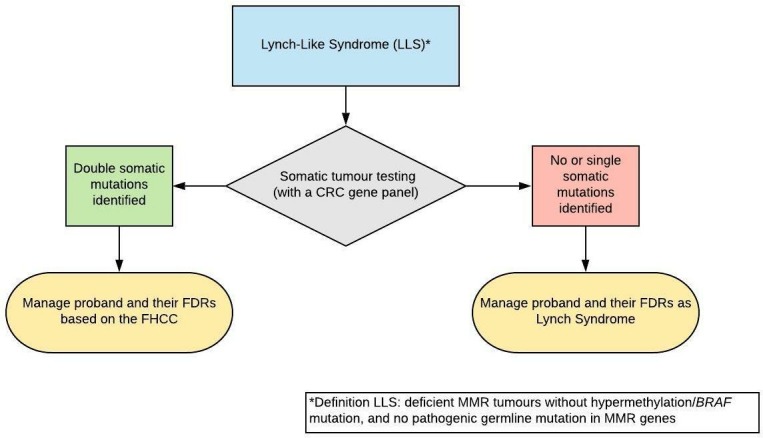
Management of Lynch-like syndrome. CRC, colorectal cancer; FDRs, first degree relatives; FHCC, family history of colorectal cancer, MMR, mismatch repair.
Early onset CRC
We recommend that in patients under 30 years of age with dMMR CRC, an LS constitutional panel test should be performed, followed by tumour testing for somatic testing if constitutional testing is negative.
(GRADE of evidence: low; Strength of recommendation: strong)
Consensus reached: 91% agreement.
We recommend that in patients under 30 years of age with pMMR CRC, a constitutional CRC multiple gene panel test should be performed.
(GRADE of evidence: low; Strength of recommendation: strong)
Consensus reached: 100% agreement.
Several studies have sought to define the prevalence and spectrum of constitutional cancer predisposition gene pathogenic variants among patients diagnosed with early-onset CRC (EOCRC), defined most commonly as CRC onset below age 50 years.172–174
In a retrospective cohort of very young patients (age 35 or younger) with CRC referred for genetic evaluation, syndrome-specific genetic testing guided by patient phenotype and family history, identified highly penetrant CRC syndromes in 35% of patients (67 of 193; 23 LS, 22 pathogenic variant-negative LS, 16 FAP, two constitutional MMR deficiency (CMMRD), two MAP, and one Li-Fraumeni syndrome). LS (23.3%; including patients with unexplained MMR deficiency, variants of uncertain significance, and incomplete LS investigations) and FAP (8.3%) were the most commonly identified CRC syndromes173
In a prospective cohort, panel testing of 25 cancer predisposition genes (APC, ATM, BARD1, BMPR1A, BRCA1, BRCA2, BRIP1, CDH1, CDK4, CDKN2A, CHEK2, EPCAM, MLH1, MSH2, MSH6, MUTYH, NBN, PALB2, PMS2, PTEN, RAD51C, RAD51D, SMAD4, STK11, TP53) was performed in 450 patients diagnosed with early-onset CRC (aged <50). Forty-eight patients (10.7%) had dMMR tumours and 402 (89.3%) had pMMR tumours. Sixteen percent of patients (72 of 450) were found to have a pathogenic variant in at least one cancer predisposition gene.172
Of the 48 patients with dMMR tumours, 40 patients (83.3%) had at least one constitutional pathogenic variant: 37 patient had LS (13 MLH1, 16 MSH2, one MSH2/monoallelic MUTYH, two MSH6, five PMS2), one patient had the low penetrant APC c.3920T>A, p.I1307K variant and a PMS2 variant of uncertain significance, and two patients had biallelic MUTYH pathogenic variants. Of the 402 patients with pMMR tumours, 32 (8%) had at least one constitutional pathogenic variant in a cancer predisposition gene: nine (2.2%) patients had pathogenic variants in high penetrant CRC predisposition genes (five APC, one APC/PMS2, two biallelic MUTYH pathogenic variants), 13 patients had pathogenic variants in genes not traditionally linked with CRC susceptibility (three ATM, one TM/CHEK2, two BRCA1, four BRCA2, one CDKN2A and two PALB2), three patients had the low penetrant APC c.3920T>A, p.I1307K variant, and seven patients had monoallelic MUTYH pathogenic variants.172
A constitutional pathogenic variant in a high penetrance CRC gene was identified in 22% of patients with CRC diagnosed between 20–29 years (4/18): two MSH2 pathogenic variants in patients with dMMR tumours and two APC pathogenic variants in patients with pMMR tumours.172 Of note, in those patients with a pathogenic variant identified in a high penetrance gene over the age of 30 years, all either had a significant family history, multiple primaries or exhibited dMMR on tumour testing.
A recent retrospective evaluation of patients with early-onset CRC (aged <50) reported comparable findings. In this study, 430 patients with early-onset CRC were referred for genetic assessment at a single tertiary care cancer centre from 1998 until 2015. Of the 430 patients, 41 had an dMMR tumour (9.5%), 161 had an Microsatellite stable (MSS) tumour (37.3%), and 228 had tumours with an unknown MMR status (53%). Clinical constitutional sequencing in 315 patients, based on their personal and family history, identified cancer predisposition gene pathogenic variants in 79 patients (18% of entire cohort, 25% of patients who underwent clinical sequencing), although five were in genes not associated with CRC syndromes. Fifty-six patients had a pathogenic variant in a DNA MMR gene (25 MSH2, 24 MLH1, five MSH6 and two PMS2), 10 in APC, eight in MUTYH (seven biallelic pathogenic variants) and two in SMAD4. Three patients were found to carry pathogenic variants in other cancer predisposition genes (one TP53, one BRCA1 and one CHEK2).174
In addition, 117 patients who had uninformative clinical evaluations underwent research-based next generation sequencing (NGS) using a multigene panel. This identified constitutional cancer predisposition pathogenic variants in six other patients (one PMS2, one MSH6, one MUTYH, one POLE, one APC and one TP53). In total, 85 patients (20%; 85/430) with early onset-CRC were found to carry a pathogenic variant in a constitutional cancer predisposition gene. However, only 79 were in genes associated with bowel cancer. LS was the most common genetic diagnosis (58/85; 68%). Seventeen patients with a constitutional MMR pathogenic variant had a dMMR tumour, 37 patients had a tumour with unknown MMR status and four (7%) patients had a pMMR tumour.174
In conclusion
The prevalence of cancer predisposition gene pathogenic variants in patients with early onset CRC (age <50) has been reported to range from approximately 15% to 20%. LS is the most common genetic diagnosis, accounts for up to 68% of cancer predisposition pathogenic variants in patients with early onset CRC (age <50 years), and the vast majority will have dMMR tumours.
In patients with pMMR tumours, the most common genetic diagnosis is an adenomatous polyposis syndrome (FAP and MAP), and the prevalence of associated constitutional pathogenic variants has been reported to reach the 10% threshold for patients diagnosed with CRC below the age of 30.
In patients with pMMR tumours between the age of 30 and 50 years at diagnosis of CRC, a genetic diagnosis may be suspected on the basis of FHCC or other clinical parameters. Altering the threshold for genetic testing in this patient cohort also needs to be balanced between clinical need and the ethical and logistical considerations of population testing.
Colonoscopic surveillance in EOCRC
We suggest that people diagnosed with CRC under the age of 50 years, where hereditary CRC syndromes have been excluded, undergo standard post-CRC surveillance for 3 years, then continue 5 yearly colonoscopic surveillance until the age they are eligible for national screening.
(GRADE of evidence: low; Strength of recommendation: weak)
Consensus reached: 80% agreement.
The risk of metachronous CRC is highest during the 36 months after surgery; subsequently this risk decreases.175 The most plausible explanation is that many early, apparently metachronous cancers are actually due to prevalent cancers or advanced adenomas missed at the time of the primary CRC diagnosis. As a consequence, new guidelines (in development) for sporadic CRC may recommend that standard post-CRC colonoscopic surveillance cease after 3 years.
Recently published, large, population-based cancer registry studies, including ones that specifically excluded patients with LS,176 177 recommend closer surveillance in high-risk populations, however.
There have been no studies specifically assessing metachronous CRC risk or the role of colonoscopic surveillance in EOCRC patients. However, when patients with hereditary CRC syndromes are excluded from CRC cohorts there is no evidence that metachronous CRC risk is significantly different from the sporadic population. Nevertheless, it may not be appropriate to discharge EOCRC cases from surveillance after 3 years in the absence of published surveillance outcome data. Thus, it may be considered prudent to offer 5 yearly colonoscopic surveillance until they are eligible for national screening in patients where LS and polyposis have been excluded.
Serrated polyposis syndrome
Definition
2019 Updated WHO clinical criteria for the diagnosis of serrated polyposis178
Criterion 1
At least 5 serrated lesions/polyps proximal to the rectum all being ≥ 5 mm in size, with 2 or more ≥ 10 mm in size
Criterion 2
More than 20 serrated lesions/polyps of any size distributed throughout the large bowel, with at least 5 proximal to the rectum
Any histological subtype of serrated lesion/polyp (hyperplastic polyp, sessile serrated lesion without or with dysplasia, traditional serrated adenoma, and unclassified serrated adenoma) is included in the final polyp count. The polyp count is cumulative over multiple colonoscopies
Diagnosis
We recommend a diagnosis of SPS should be made in accordance with the new WHO 2019 criteria for SPS. Since causative gene pathogenic variants for SPS have not been identified, a definitive diagnosis of SPS should be phenotype-driven.
(GRADE of evidence: moderate; Strength of recommendation: strong)
Consensus reached: 95% agreement.
Other intestinal polyposis syndromes may present with serrated lesions. If (i) the patient is under 50 or (ii) there are multiple affected individuals within a kindred or (iii) there is dysplasia within any of the polyps, then other polyposis syndromes should be excluded by gene panel testing before making a definitive diagnosis of SPS.
(GRADE of evidence: very low; Strength of recommendation: weak)
Consensus reached: 90% agreement.
We recommend the cumulative number of serrated polyps from all endoscopic examinations should be used when applying the WHO 2019 diagnostic criteria for SPS.
We recommend the cumulative number of serrated polyps from all endoscopic examinations should be used when applying the WHO 2019 diagnostic criteria for SPS.
(GRADE of evidence: moderate; Strength of recommendation: strong)
Consensus reached: 94% agreement.
SPS most probably comprises a phenotypically and genetically heterogeneous group of diseases. The phenotypic criteria for a diagnosis of SPS have recently been revised by the WHO in 2019 to include the following: (1) at least five serrated polyps proximal to the rectum, all >5 mm in size with at least two >10 mm in size; (2) at least 20 serrated polyps (of any size) with at least five located proximal to the rectum.178 Fulfilment of either criterion is sufficient for a diagnosis of SPS. Importantly, the previous WHO 2010 criterion 2, which required the presence of just one serrated lesion in a patient who has an FDR with SPS, has been removed. Given that the prevalence of serrated lesions in the Western population may be up to 39%, most of which will be sporadic, the chances of a false positive diagnosis using this criterion were high.
The prevalence of SPS in the West is generally considered to be around 1:3000 in screening populations. This may change as awareness of the condition among clinicians increases. Egoavil et al 179 found that patients with multiple serrated polyps but failing to fulfil the WHO criteria had a similar risk of CRC as those who did fulfil the criteria. Dovetailing with this to some degree (and possibly explaining this), Crowder et al 180 concluded that SPS was underdiagnosed due to failure to consider cumulative polyp numbers (rather than at a single episode). By evaluating “cumulative pathology” in a cohort of 927 consecutive patients undergoing colonoscopy, they found that up to 1.8% of patients with a serrated lesion at first colonoscopy eventually fulfilled the criteria for SPS.
Patients with SPS have an overall lifetime risk of CRC of approximately 7–70%181 and there is an increased risk in FDRs of patients with SPS. The risk of CRC thus may be reduced with appropriate management.181 182 There is an increased risk of CRC in FDRs of patients with SPS, but it does not follow a classical Mendelian pattern of inheritance. A single causative gene has not been identified and there is phenotypic overlap with some well characterised intestinal polyposis syndromes (MUTYH-associated polyposis, hereditary mixed polyposis syndrome, attenuated FAP). Most, but not all, of the tumours arising in SPS follow the serrated neoplasia pathway characterised by microsatellite instability (due to MLH1 promoter methylation) and BRAF pathogenic variant.183 This raises the possibility of accelerated tumorigenesis as well as phenotypic overlap with LS. Recently, constitutional truncating pathogenic variant in RNF43 pathogenic variant has been identified and shown to segregate with phenotype in one kindred with SPS.184 185 The same pathogenic variant was also identified in two patients in a separate study.
In the absence of a single causative gene (and the possibility that some forms of SPS may be polygenic in nature), the diagnosis of SPS should be phenotype-driven. Given the overlap with other well-defined syndromes, these should be excluded in all patients in whom a diagnosis of SPS is made. A specific gene panel for SPS may include testing for RNF43 and GREM1; however the frequency of these pathogenic variants is too low to recommend routinely.186
Colonoscopic surveillance in SPS
We recommend that patients with SPS should have colonoscopic surveillance yearly once the colon has been cleared of ALL lesions > 5 mm in size. If no polyps ≥ 10 mm in size are identified at subsequent surveillance examinations the interval can be extended to 2 year ly
(GRADE of evidence: moderate; Strength of recommendation: strong)
Consensus reached: 94% agreement.
Colonoscopic surveillance is currently recommended following detection and resection for adenomas in the colorectum due to an increased risk of advanced adenomas and CRC in such patients; however, there is another major pathway to CRC “serrated pathway” which accounts for 15–30% of CRC and has serrated lesions as cancer precursors. SPS is common in bowel cancer screening programmes which use guaiac faecal occult blood test (gFOBT) or FIT as a screening test, with estimates of SPS prevalence ranging from 1:150 to 1:300.187 188 A recent Spanish FIT based cohort followed up all their patients with proximal serrated polyps, tripling the number of additional cases of SPS, for a final prevalence of 1:100.189 Therefore, especially when using FIT in bowel cancer screening, colonoscopists should be alert to a diagnosis of SPS.
The BSG position statement on serrated polyps in the colon and rectum recommended 1–2 yearly surveillance for patients meeting the WHO criteria for serrated polyposis syndrome.190 This recommendation was on the basis that in early cohorts future risk of CRC was elevated as much as 7% at 5 years191 192; however in larger cohorts with rigorous surveillance performed every 1–2 years, with all lesions larger than 5 mm in size resected, at academic centres, the risk appeared much lower with CRC only diagnosed at 1.9 cases per 1000 years of patient follow-up.181 182 In a US study following up SPS patients which extended surveillance intervals for SPS patients to 2 years after colon clearance with no lesion ≥10 mm found at surveillance, no cancer developed or surgery was needed.193 A similar multicentre European study that individualised surveillance after colonic clearance to 1 or 2 year follow-up dependent on lesion size (≥10 mm), number and pathology also showed no difference in advanced neoplasia detection with a 2 year surveillance interval once the colon was cleared.194
No new data directly relevant to this area have been published since the BSG position statement. However, a study of patients with multiple serrated polyps and adenomas, not fulfilling the criteria for SPS, also noted that their risk for CRC was equivalent to patients who met the WHO definition of SPS, and that their FDRs had a comparable risk of CRC.179
Colonoscopic surveillance in unaffected FDRs of patients with SPS
We recommend all FDRs of patients with SPS on the basis of the new WHO 2019 SPS criteria 1 or 2 should be offered an index colonoscopic screening examination at the age of 40 years or 10 years before the diagnosis of the index case.
(GRADE of evidence: moderate; Strength of recommendation: strong)
Consensus reached: 89% agreement.
We suggest all FDRs of serrated polyposis patients have a surveillance examination every 5 years unless polyp burden indicates an examination is required earlier according to postpolypectomy surveillance guidelines.
(GRADE of evidence: low; Strength of recommendation: strong)
Consensus reached: 84% agreement.
SPS may be a familial condition with between 1.3% and 7.7% of index cases having an FDR who meets the original WHO criteria 1 or 3 for 195 196SPS. A further 14.3%–24.4% of FDRs meet original WHO criterion two for SPS. Current guidelines recommend a one-off screening examination of all FDRs to detect these familial SPS cases; however, the majority of FDR will not meet the original WHO criteria for SPS after this initial examination. The risk of CRC for FDRs of SPS patients is estimated to be substantially elevated above that of the general population by three- to fivefold.179 197 198 This risk may also apply to those with multiple serrated polyps not meeting WHO criteria for SPS. It is unclear how this risk is distributed in the SPS FDR population.
In three series the mean or median age at diagnosis for CRC in FDRs of SPS patients ranged from 55 to 62. CRC was very rare in those aged less than 40 years with the youngest case being aged 25. Authors have recommended a screening examination starting at 35 or between 40 and 50 for FDRs of SPS cases, or starting 5–10 years before the index case179 195–197
In a follow-up study of 78 patients, those who did not meet any WHO criteria for SPS seem to have low risk for subsequent polyp or advanced neoplasia development; however, those who meet WHO criteria 2 do seem to develop polyps with five of 14 patients developing three or more adenomas and two others of the 14 developing a serrated lesion ≥10 mm in size.196 High rates of polyps in patients who met WHO 2 criteria were also seen by Hazewinkel et al.195 Therefore a significant proportion of WHO 2 SPS patients would meet criteria for 3 yearly surveillance according to current guidelines. No cancers developed. No data are available to look at the colonoscopic yield in FDRs of index patients who meet WHO criteria 2, that is, a second degree relative of a patient with SPS WHO 1 or 3, although the risk of CRC in second degree relatives of index cases of SPS is only slightly elevated (SIR 1.38, 95% CI 1.01 to 1.91).198
The WHO definition of SPS has recently changed with criteria 2 abandoned completely so that FDRs with serrated polyps are not considered to meet the definition for SPS. Therefore a blanket recommendation for colonoscopic surveillance for FDRs of SPS every 5 years based on the three- to fivefold increase in SIR of CRC is suggested from age 40 or 10 years before the diagnosis for the index case, with more frequent surveillance in line with sporadic recommendations if additional polyps are detected.
FDRs of patients with multiple serrated polyps (MSP, 10 or more polyps in total of which 50% are serrated) but who do not meet the 2019 WHO criteria for SPS might be considered for similar surveillance approaches for FDRs.
The risk of CRC in FDRs of SPS patients is sufficiently elevated that they should be offered colonoscopic screening and regular surveillance.
Multiple colorectal adenomas
We suggest an individualised approach to germline testing of patients with MCRA (defined as having 10 or more metachronous adenomas). Consider this testing for:
Patients under 60 years of age with lifetime total of ≥ 10 adenomas; or
-
Patients from 60 years of age with lifetime total of:
≥ 20 adenomas, or
≥ 10 adenomas and an FHCC or polyposis
(GRADE of evidence: low; Strength of recommendation: weak)
Consensus reached: 91% agreement.
We suggest that patients with a finding of 10 or more polyps (adenomas or serrated lesions) should, at their next colonoscopy, have a high-quality colonoscopic assessment with pancolonic dye spray in order to accurately define the multiple polyp phenotype.
(GRADE of evidence: very low; Strength of recommendation: weak)
Consensus reached: 89% agreement.
We suggest that the endoscopic management of patients with 10 or more metachronous adenomas, without MUTYH or APC gene mutations, should be individualised according to phenotype.
(GRADE of evidence: very low; Strength of recommendation: weak)
Consensus reached: 91% agreement.
We suggest annual colonoscopic surveillance for patients with 10 or more metachronous adenomas after the colon has been cleared of all lesions >5mm in size. If no polyps 10mm or greater in size are identified at subsequent surveillance examinations the interval can be extended to 2 yearly.
(GRADE of evidence: very low; Strength of recommendation: weak)
Consensus reached: 80% agreement.
Patients with multiple adenomas but without classical familial adenomatous polyposis are frequently encountered in clinical practice. Approximately 1.1% of patients undergoing colonoscopy in the English bowel cancer screening programme have 10 or more adenomas.199 There is an association of adenoma multiplicity with metachronous advanced adenoma and/or CRC risk, with the degree of risk correlating with increasing adenoma number and size. With less than three adenomas as reference, Cubiella et al (n=5401) reported a statistically increased risk for AN when three or four adenomas (14.8%), and also when five to nine adenomas (18.4%), were present at index colonoscopy.200
In the largest study to date of individuals with 10–19 adenomas,172 3789 patients with 10 or more colorectal polyps underwent constitutional testing of the prevalence of pathogenic variants with a 17-gene panel. The diagnostic yield of pathogenic variants remained above 5% in all ages and cohorts, despite a decrease with age. In the multiple adenoma cohort, the yield was higher in those patients with a personal or family history of cancer. In 1342 patients with 10–19 adenomas, 7.8% had a pathogenic variant in one of the panel genes, with 2.2% in “traditional”’ polyposis predisposition genes, and 2.8% in MMR genes. Thus an unbiased multi-gene panel test approach may be associated with a higher diagnostic yield.
In a study of 7225 individuals with MCRA and oligopolyposis, pathogenic variants in APC or common European founder pathogenic biallelic variants in MUTYH were identified in 87/970 (9%) individuals with 10–19 adenomas and 559/3253 (17%) individuals with 20–99 adenomas.201 There was an incremental increase in the odds of a pathogenic variant with an increasing number of adenomas and earlier age at adenoma diagnosis, particularly under the age of 50 years.
In addition to APC and MUTYH there is an evolving range of other multiple adenoma susceptibility genes including NTHL1, GREM1, POLE, POLD1 and MSH3. Spier et al in 2015 demonstrated POLE pathogenic variants in 1.5% of individuals with greater than 20 synchronous or 40 metachronous adenomas although this percentage increased with a family history.202 There is also some evidence of polygenic risk contributing to this attenuated polyposis phenotype.203
In summary, it is advisable to consider a diagnosis of a highly penetrant syndrome in patients with multiple adenomas. A nuanced approach to patient selection for constitutive testing should incorporate patient age and personal and family history.
With regard to colonoscopic surveillance in the multiple adenoma patient population, the GDG suggests constitutional gene testing to rule out known hereditary syndromes. Subsequently an individualised approach should be taken depending on adenoma size and number, with the goal of clearance of colorectal adenomas >5 mm in size. However, surgical resection should be considered where surveillance is not feasible.
In patients with 10–99 adenomas without APC or MUTYH pathogenic variants, the clinical phenotype is similar to that of attenuated polyposis, with both an upper GI (21% with duodenal adenomas) and colorectal phenotype.204 In a study of 83 patients also of 10–99 adenomas without APC or MUTYH pathogenic variants from the UK and Holland, the upper GI phenotype was reported in a minority of patients (9.6%), all of whom had Spigelman I or II disease (ie, early stage).205
For FDRs of MCRA patients, clinicians may consider a colonoscopic assessment of risk at the time of referral, and/or a repeat assessment at age 50 years. This is in order that patients who may be at risk of high penetrance cancer predisposition syndromes may be effectively identified (for example, the children of individuals with unidentified mosaic pathogenic variants of the APC gene206). However the GDG did not reach consensus on this recommendation, and recommend that multicentre outcome data in these patients be collected and published to help guide future recommendations.
Familial adenomatous polyposis
FAP is defined by the presence of pathogenic variants in the APC gene with a prevalence of about 1/8500. It is a dominantly inherited multisystem cancer predisposition syndrome with a characteristic phenotype characterised by colorectal and upper GI polyposis.
Colorectal surveillance in FAP
We recommend that colonic surveillance should normally commence at the age of 12–14 years in those confirmed to have FAP on predictive genetic testing.
(GRADE of evidence: low; Strength of recommendation: strong)
Consensus reached: 100% agreement.
We suggest that for those with FAP, intervals between surveillance colonoscopy may be individualised depending on colonic phenotype every 1–3 years.
(GRADE of evidence: low; Strength of recommendation: weak)
Consensus reached: 94% agreement.
We suggest that colonoscopy screening is performed for individuals who have an FDR with a clinical diagnosis of FAP (ie, “at-risk”) and in whom an APC mutation has not been identified, starting at the age of 12–14 years, and should continue on 5 yearly surveillance until either a clinical diagnosis is made and they are then managed as FAP, or they reach the age at which they can enrol in national screening.
(GRADE of evidence: very low; Strength of recommendation: weak)
Consensus reached: 95% agreement.
Current international guidelines207 208 recommend starting colonoscopy surveillance or screening at the age of 12–14 years, after diagnostic genetic testing has been performed in at-risk children. There are no new data to recommend any change to this recommendation of age at which to start. The risk of CRC under the age of 20 years is very small and under 15 years extremely rare.207 In patients with FAP-related symptoms such as rectal bleeding, diarrhoea or mucous discharge should lead to a colonoscopy at any age, particularly in those with a constitutional pathogenic variant at codon 1309, which is associated with a greater risk of a more severe colonic phenotype.
Colonoscopic surveillance has been shown to lead to a reduction in CRC and CRC-associated mortality. Data detailing the results of polyposis registries have shown that in symptomatic patients the incidence of CRC was 50–70% compared with 3–10% in those that were identified by registry initiated surveillance.209–211 Surveillance and prophylactic intervention has reduced CRC associated mortality.212 213
Colonoscopic surveillance enables assessment of adenoma burden and distribution, which can guide the timing of and type of prophylactic surgery required. Previous guidelines recommended the use of flexible sigmoidoscopy. However, most endoscopic procedures are performed under general anaesthesia in children and young teenagers; therefore a full colonoscopy is recommended in an affected patient, to better determine their phenotype, as polyp distribution is not uniform and the rectum and sigmoid may be normal despite the presence of more proximal adenomas. It is not recommended therefore for flexible sigmoidoscopy to be used routinely in screening/surveillance in FAP.
Current guidelines advocate annual endoscopic assessment. There is no evidence for accelerated carcinogenesis in FAP and the rather scant data that are available do not indicate a rapid increase in polyp number in teenagers/young adults.214 Therefore it does not seem logical to mandate an annual assessment for all affected patients, particularly given that there is significant variability in colonic phenotype. Those with an attenuated phenotype (<100 adenomas) may not require such frequent colonoscopic surveillance as those with a classical phenotype (>100 adenomas). In addition, if an individual only has adenomas of 1–2 mm their surveillance could perhaps be longer than those with larger polyps, for example, 8–9 mm. Personalising surveillance interval according to phenotype, 1–3 yearly would appear safe as long as families are not lost to follow-up and this would concord with current paediatric guidelines.208 Extending the interval, however, should be for those with an attenuated phenotype, in the setting of good quality colonoscopy with robust systems in place to ensure appropriate recall.
There are a few patients with a particularly attenuated phenotype. In this group, primary endoscopic management by surveillance and polypectomy may be considered either to defer surgery or possibly to avoid the need for surgery altogether. However, there are no robust data to support this approach.
At risk patients, where predictive genetic testing is not possible, should be screened by colonoscopy every 5 years from the age of 12–14 years. If adenomas are identified, the patient should undergo repeat colonoscopy at a frequency depending on the colonic phenotype as described above. If no phenotype has been observed by the age of 50 years, FAP is unlikely and so the patient could be discharged from routine colonoscopic screening to have continued screening under the auspices of the national bowel cancer screening programme.
Upper GI surveillance in patients affected with FAP
We recommend upper GI surveillance for FAP patients starting at the age of 25 years.
(GRADE of evidence: low; Strength of recommendation: strong)
Consensus reached: 90% agreement.
We suggest that for those considered at risk, where predictive genetic testing is not possible, screening with upper GI endoscopy is not routinely recommended but should be started if/when a clinical diagnosis of FAP is made based on colorectal phenotype.
(GRADE of evidence: very low; Strength of recommendation: weak)
Consensus reached: 89% agreement.
Lifetime risk of duodenal polyposis approaches 100% in FAP.215 The absolute lifetime risk of developing duodenal cancer in FAP is estimated to be around 5%.216 Because of this, surveillance has been recommended and survival benefit for those diagnosed with duodenal cancer by surveillance compared with those who presented symptomatically has been demonstrated.217
The Spigelman classification (table 3) is the system that is most widely used for staging non-ampullary duodenal disease2182; it has been shown to correlate with cancer risk219 and is recommended to determine surveillance intervals.207 There is debate about how to incorporate ampullary disease in this classification system. A staging system for ampullary disease has been proposed,220 which in one series correlated to the development of ampullary cancer.221 A surveillance interval determined by the combination of these staging systems may be the most helpful and reliably replicated clinical means of managing duodenal surveillance (see figure 1). A duodenoscope is required to reliably assess and/or biopsy the periampullary region and ampulla itself. Although there are reports of chromoendoscopy increasing duodenal adenoma detection,222 223 its utility in clinical practice is not established. It may increase the number of adenomas detected but there remains no evidence that dye spray increases the pick-up of larger, more clinically meaningful lesions.223 There are no data to suggest that it alters the need for endoscopic or surgical intervention. The concern is that it may increase the pick-up of small lesions and may artificially “upstage” the duodenal disease but without reflecting a higher cancer risk, compared with white light endoscopy. Recent data suggest that it is polyp size and the presence of high-grade dysplasia that are the most important predictors of cancer risk, polyp multiplicity.224 It should be noted that the Spigelman classification system was developed and validated using white light endoscopy, without high definition.
Table 3.
Staging the duodenum and ampulla and recommended OGD surveillance intervals
| Points allocated | |||
| 1 | 2 | 3 | |
| Number of polyps | 1–4 | 5–20 | >20 |
| Polyp size (mm) | 1–4 | 5–10 | >10 |
| Histological type | Tubular | Tubulovillous | Villous |
| Degree of dysplasia | Mild | Moderate | Severe |
OGD, oesophago-gastro-duodenoscopy.
| Total points | Spigelman stage | Recommended follow-up interval |
| 0 | 0 | 5 years |
| 1–4 | I | 5 years |
| 5–6 | II | 3 years |
| 7–8 | III | Annual and consider endoscopic therapy |
| 9–12 | IV | 6–12 months and consider endoscopic or surgical therapy |
| Normal ampulla | Minor polyposis | Major polyposis | |
| Ampulla size | n/a | Less than 1 cm | More than 1 cm |
| Villous histology | n/a | None | Present |
| Degree of dysplasia | n/a | Mild | Moderate or severe |
| Recommended surveillance | 5 yearly | 3 yearly | Annual |
The role of endoscopic therapy in the duodenum and ampullary is not well established. It has an acceptable safety profile225 226; however, there are no long-term data to demonstrate it reduces cancer risk or indeed prevents or delays the need for prophylactic surgery. Nevertheless, it is widely performed in centres dealing with a large cohorts of patients with FAP. It would be most prudent for patients being considered for endoscopic therapy for duodenal disease to be referred to their local specialist centre, so that assessment and work up to decide as to the appropriateness of endoscopic and surgical intervention can be performed.
The data regarding endoscopic management of ampullary disease are rather scant. However, it appears to be less safe with risk of haemorrhage, pancreatitis and perforation, morbidity rates up to 45%225 and with high rates of recurrence, up to 58%.227 It cannot be routinely recommended. Referral to a specialist hepato-pancreatico-biliary (HPB) centre should be made for those in whom endoscopic ampullectomy is being considered.
Gastric lesions are also common in adult patients with FAP. Fundic gland polyps (FGPs) are seen in up to 80% of patients with FAP228 and although there is some debate they are likely to be an entirely benign entity without malignant potential. Gastric adenomas are being seen more commonly, as indeed is gastric cancer. Although historically gastric cancer risk was not thought to be elevated in patients with FAP, there are reports that gastric adenomas and cancer are becoming an important clinical problem.229 230 There are no data published regarding outcomes of endoscopic therapy for gastric adenomas in FAP. Referral to a specialist centre for assessment and management seems prudent given the lack of evidence and absence of consensus guidelines.
Congenital hypertrophy retinal pigmentation epithelium
We suggest that patients with congenital hypertrophy retinal pigmentation epithelium ( CHRPE ) be referred for a specialist ophthalmic review. Patients with bilateral and multiple CHRPE lesions should be referred for screening for FAP and considered for genetic testing and colonoscopy.
(GRADE of evidence: low; Strength of recommendation: weak)
Consensus reached: 100% agreement.
Up to two thirds of patients with FAP have CHRPE identified at ophthalmoscopy,231 compared with a prevalence in the general population of 1–4%. CHRPE lesions associated with FAP are most often multiple, bilateral (in 86% of cases) and oval or pisciform in shape. Multiple retinal lesions have a very high specificity as a phenotypic marker for FAP.232 Referral for a specialist ophthalmic review will assist in characterising those with lesions which need to be considered for screening for FAP. Such patients should be referred to a specialist centre for consideration of screening for FAP by genetic testing and thereafter colonoscopy, age 12–14 years, or at time of diagnosis if diagnosed at an older age.
FAP and surgery
We recommend that for patients with FAP who are undergoing colonoscopic surveillance, relative indications for surgery are polyps > 10 mm in diameter, high grade dysplasia within polyps and a significant increase in polyp burden between screening examinations.
(GRADE of evidence: low; Strength of recommendation: strong)
Consensus reached: 82% agreement.
For most patients the choice of surgery will be between total colectomy with ileorectal anastomosis (IRA) and proctocolectomy and ileal pouch anal anastomosis (IPAA). The choice of surgery will depend on rectal polyp number, size and presence of high-grade dysplasia, genotype and the functional consequences of the surgical procedure. IPAA should also be considered for those patients who are likely to be poorly compliant with follow-up surveillance.
Total proctocolectomy with end ileostomy can be considered for patients with poor sphincter function, incontinence, distal rectal cancer, cancers requiring radiation, or for those who desire to avoid the functional consequences of an ileoanal pouch.
In patients with FAP, colon cancer will inevitably develop if the colon is not removed. Total colectomy will prevent colon cancer in FAP patients. Prophylactic surgery can usually be planned at a time which is suitable to the patient, based on the risk of cancer as assessed colonoscopically. The timing and choice of surgical procedure should take into account the educational, social, family planning and emotional development of the patient and their reliability for attending follow-up evaluations.
Indications for surgery include polyps >10 mm diameter, polyps with high-grade dysplasia, and marked increases in polyp number between examinations. Symptoms from polyps should be rare in those undergoing regular screening and surgery should be performed before symptoms from polyps develop.
Total colectomy with ileorectal anastomosis (TAC-IRA) can be offered to patients with relative rectal sparing (<20 polyps) if all rectal adenomas are <5 mm in diameter and any polyps >5 mm can be endoscopically removed. However, flexibility with regard to a threshold polyp number should be employed with the advent of high-definition or chromo-endoscopic techniques.
The decision to retain the rectum is made based on future rectal cancer risk, polyposis phenotype in the rectum, functional considerations and the genotype.
Bülow et al evaluated 776 patients who had IRA, including 576 before the ileoanal pouch era, and 200 after the ileoanal pouch became available in these centres.233 The cumulative risk of rectal cancer by Kaplan-Meier analysis was 10% in the pre-pouch era versus 2% in the pouch era.
A cohort study from Church et al of 213 patients with FAP included 165 patients who had rectal-sparing surgery, with 128 of these having <20 polyps and 37 having >20 polyps.234 The rectal cancer incidence was 1.6% in the patients with <20 polyps, compared with 10.8% in the patients with >20 polyps.
Predicting future rectal excision may also aid decision making about choice of surgery. A study from St Marks hospital of 427 patients who underwent IRA found that by the age of 60 years, half of the patients retained their rectum. Rectal polyp count exceeding 20, APC pathogenic variant codon 1250–1450, colonic polyp count ≥500 and age <25 years at the time of surgery were independent predictors of progressive rectal disease.235 Church et al also found rectal polyp count >20 to predict future rectal excision.236
Genotype can also be utilised specifically when discussing surgical options. A study of four national polyposis registries included 475 polyposis patients with a previous colectomy. Cumulative risks of secondary proctectomy 20 years after primary colectomy were 10%, 39% and 61% in the attenuated, intermediate and severe genotype groups, respectively (p<0.05, groups compared separately).237 Patients with a severe genotype have a high-risk of rectal excision after primary colectomy and may be better directed to proctocolectomy with IPAA. Influence of genotype on survival has also been reported by Newton et al.238
For patients undergoing IRA, 1–3 flexible sigmoidoscopies are required as per FAP patients with intact colons.
There have been no randomised trials comparing the functional outcome of IRA versus IPAA. The study by Aziz et al provides a fair summary of the literature.239 They performed a meta-analysis on 12 non-randomized studies published between 1991 and 2003 and containing over 1000 patients (47% IRA vs 53% IPAA). Bowel frequency, nocturnal defaecation and use of incontinence pads were significantly less in the ileorectal group, although faecal urgency was reduced with the ileal pouch. There was no significant difference between the techniques in terms of sexual dysfunction, dietary restriction or postoperative complications.
The fecundity of women with FAP before operation and after colectomy with ileorectal anastomosis has been reported to be similar to that of the general population. However, fecundity dropped to 54 per cent following proctocolectomy with ileal pouch-anal anastomosis. It is recommended that the significant reduction in female fecundity after IPAA should be communicated to young women with FAP before surgery.240 The risk of developing postoperative fertility problems is not associated significantly with the type of surgery, indication for surgery, complications or other comorbid conditions. Postoperative fertility problems appear to be more common among women who had their first surgical procedure at a younger age.241
Indications for rectal excision following IRA include: the development of rectal cancer, polyps >10 mm diameter, polyps with high-grade dysplasia, and marked increases in polyp number between examinations. When conversion of an IRA to IPAA is required, functional outcomes appear similar to primary IPAA procedures. Complication rates and pouch failure rates are reported to be similar, but conversion to IPAA will not be possible in a small percentage of patients.242
Proctocolectomy with IPAA is the treatment of choice in the presence of: rectal cancer, a large rectal polyp burden (>20 synchronous adenomas, adenoma with high-grade dysplasia, large (>10 mm) adenomas) or a severe phenotype (>1000 synchronous adenomas).
Risk of neoplasia and cancer in the pouch
There is a very small risk of adenocarcinoma after an IPAA. Most cancers develop in residual rectal or in the anal transitional zone (ATZ) mucosa. Cancer can also develop within the ileal component of the pouch. Currently nearly all pouches are constructed with the use of stapling devices which results in the ATZ mucosa being preserved. Von Roon reported on 206 patients with a median follow-up of 10.3 years.243 The risk of adenoma of the IPAA at 10 years was 51% after stapled IPAA; no patient developed cancer. van Duijvendijk et al reported the risk of developing a polyp of the IPAA after 7 years was 31% after stapled IPAA.244
Of 212 patients followed up in the Dutch polyposis registry, the cumulative risk of developing an adenoma in the pouch at 10 year follow-up was 45%. Twenty-five patients (11.8%) developed an adenoma with advanced pathology and four (1.9%) developed a carcinoma. The cumulative risk of developing a pouch carcinoma at 10 year follow-up was 1%.245 A 2013 review of 24 studies reporting 92 pouch-related cancers found that 23 of 92 cancers (25%) developed in the pouch mucosa and 69 (75%) in the ATZ.246 A Mayo clinic study on 117 patients showed a median time to development of dysplasia was 149 months. Adenocarcinoma developed in one patient after 284 months. Risk of dysplasia at 10, 20 and 25 years was 17%, 45% and 69%, respectively.247
Thus, although the risk of severe mucosal dysplasia and cancer is low, annual endoscopic surveillance of any remaining rectal mucosa, ATZ mucosa and ileal pouch are recommended for life.
FAP and desmoid disease
We suggest that FAP patients should be counselled about the risk of post-operative desmoid disease formation.
(GRADE of evidence: low; Strength of recommendation: weak)
Consensus reached: 95% agreement.
For FAP patients before colectomy, consider determining genotypes or family history of desmoid disease which may be predictive of desmoid formation.
(GRADE of evidence: low; Strength of recommendation: weak)
Consensus reached: 78% agreement.
We suggest that sulindac in combination with high-dose selective oestrogen receptor modulators may be effective in FAP patients with intra-abdominal desmoids and desmoids located at the abdominal wall.
(GRADE of evidence: very low; Strength of recommendation: weak)
Consensus reached: 100% agreement.
We recommend the role of elective surgery for intra-abdominal desmoids should be restricted to treating secondary effects of the desmoid disease, and this surgery should be performed in expert centres.
(GRADE of evidence: low; Strength of recommendation: strong)
Consensus reached: 95% agreement.
Desmoid tumours will develop in around 15% of patients with FAP. Risk factors include abdominal surgery, positive family history for desmoids and site of the pathogenic variant.248 249 In individuals with APC pathogenic variants in the desmoid region 3' to codon 1399, abdominal surgery was associated with a 65% risk of developing mesenteric desmoids.249 As desmoids can cause significant complications and pose a low risk of death, genotypes predictive of desmoid formation and family history of desmoids should be determined for all patients before colectomy. Identification of at-risk patients will enable appropriate counselling and consent before surgery and allow informed decision about the timing of surgery. This decision should sensibly balance the risk of malignancy with the risk of mesenteric desmoid disease.
Some desmoid tumours spontaneously stop growing, some regress and others remorselessly increase in size. In a small proportion, this increase may be rapid and uncontrollable. There are no proven predictors of growth pattern.
The distinction between intra- and extra-abdominal desmoids is important. Abdominal wall desmoids cause pain (variable) and mass effect. Intra-abdominal lesions, in addition, are at risk of causing secondary effects: ureteric and small bowel obstruction, fistula formation or small bowel ischaemia. The quality of data on which to make decisions about treatment of abdominal wall and intra-abdominal desmoid disease is limited and largely consists of small, non-controlled studies. As such, the treatment of patients with desmoid tumours remains controversial. For abdominal wall lesions, there is an expanding role of percutaneous ablative treatments and in some institutions ablation has become the primary treatment.250
Church et al 251 combined symptoms (pain, restriction, hospitalisation and sensation of a mass) with size and growth rate in a proposed staging system for intra-abdominal desmoid disease. Decisions about treatment are partly based on size and symptoms, but are more often determined by secondary effects of the desmoid on the urinary and GI tract. Following diagnosis, if no secondary effects are present, serial imaging with CT or MRI scanning every 6–12 months depending on growth rate is reasonable. Radiological screening at 12 months following surgery for those who are at high-risk of desmoid development might be appropriate.
If there is concern about the size or growth rate of desmoid disease, first line treatment should be with high-dose selective oestrogen receptor modulators and sulindac, according to the regime outlined by Quast et al.252 In this observational series all desmoid patients treated and followed at their institution had completed at least 1 year of treatment.252 Response was defined as stable size or regression of desmoid size between two CT or MRI scans. Of the 134 patients included, half had a confirmed diagnosis of FAP. Eighty-five per cent of patients showed regression or had stable desmoid size. The mean time to reach at least stable size was 14.9±9.1 months. After regression or stabilisation, medication was tapered in 60% of the treated patients with only one long-term recurrence after >10 years.
In patients with progressive intra-abdominal desmoid disease that does not respond to this treatment, chemotherapy (eg, doxorubicine and dacarbazine or methotrexate and vinblastine)253 254 or radiation therapy is indicated.
Surgery should be reserved for secondary effects of the desmoid disease.
MUTYH-associated polyposis
Colorectal surveillance in MAP
We recommend that colorectal surveillance is commenced in MAP starting at the age of 18–20 years. If surgery is not undertaken then annual surveillance is suggested.
(GRADE of evidence: low; Strength of recommendation: strong)
Consensus reached: 83% agreement.
We recommend that for monoallelic MUTYH pathogenic variant carriers, the risk of CRC is not sufficiently different to population risk to meet thresholds for screening, and routine colonoscopy is not recommended.
(GRADE of evidence: moderate; Strength of recommendation: strong)
Consensus reached: 94% agreement.
MAP is a recessively inherited cancer and polyposis predisposition syndrome caused by pathogenic variants in the MUTYH base-excision repair gene, with significant phenotypic overlap with FAP. It is classically said to be associated with a more attenuated phenotype than FAP but there is significant phenotypic variation. Past guidelines have suggested that colonoscopy should commence at the age of 18–20 years, on the basis that CRC in MAP is rare below the age of 30 years207 and be performed up to 2 yearly, as the colonic phenotype is usually more attenuated. This attenuated phenotype also lead the authors to comment that endoscopic management may suffice if there is an attenuated phenotype.
CRC in MAP is more likely to be right sided and synchronous.255 256 A cumulative CRC risk of 63% at 60 years has been reported.257 The median age of CRC seems dependent on the underlying genotype.256 MAP accounts for up to 29% of those patients with 10–100 adenomas but also up to 29% of those with a classical phenotype (100–1000 adenomas). Biallelic pathogenic variants have also been reported in patients with <10 adenomas and also in patients with CRC but no adenomas.258–260 There are further data to suggest that CRC risk in MAP may not correlate to adenoma number.257
Colonoscopy surveillance may not be effective in MAP and it has been postulated that there may be accelerated carcinogenesis. Nieuwenhuis and colleagues reported that of those who presented with a polyposis phenotype but without CRC, 9% developed CRC during 5 years of surveillance. If the presentation was with CRC, then they observed a 5 year metachronous cancer risk of 11%.257 In light of these data, annual colonoscopy would appear appropriate if colonoscopy surveillance is pursued, but it appears that surgery may be the more appropriate management strategy, balanced against age, co-morbidity and expected functional outcome.
Upper GI surveillance in patients affected with MAP
We suggest that upper GI surveillance should be considered in MAP, starting at the age of 35 years. We recommend that the surveillance interval is determined as outlined for FAP.
(GRADE of evidence: low; Strength of recommendation: weak)
Consensus reached: 94% agreement.
The data regarding the upper GI tract phenotype in MAP is far less extensive than in FAP. It is generally suggested that the two conditions should be managed in a similar manner.207 261 Duodenal adenomas have been reported in 17–34% of those with MAP.262 263 The median age at which duodenal adenomas are diagnosed has been reported as 50 years.262 The lifetime risk of developing duodenal cancer is estimated to be around 4%.263 Two patients were diagnosed with duodenal or ampullary cancer in one series, age 83 and 63 years, respectively; both were diagnosed at index upper GI endoscopy but no cancer arose in patients who were on a surveillance programme.262 Polyps appear to progress through the development of villous features and increasing size in this series, rather than progression in number or dysplasia.262 Certainly the phenotype does appear to be different and polyp multiplicity in MAP does not seem to be observed in the same manner as it is in FAP. There appears to be scientific evidence to suggest that MAP duodenal polyps may behave differently to those in FAP. A higher burden of somatic pathogenic variants in MAP duodenal adenomas is reported, compared with FAP, despite a lower Spigelman stage disease. It is postulated that this may reflect an increased cancer risk in the context of apparently less severe benign disease264; however, long-term data to support this hypothesis are lacking.
Because of the reduced frequency of duodenal adenomas, compared with FAP and the later onset of duodenal adenomas, it has been suggested that starting upper GI endoscopic surveillance could be deferred until the age of 35 years.262 In the absence of more robust data, determining surveillance intervals according to duodenal and ampullary staging, as has been recommended in FAP, appears to be a pragmatic approach.
There are few data regarding gastric lesions in MAP. FGPs have been reported in 6% of a cohort with MAP and gastric adenomas in 3%.263 This would suggest that gastric polyposis is seen less commonly than in FAP. In the same series gastric cancer was observed but the incidence was not statistically different to the risk for the general population. Further data regarding the gastric phenotype are required to determine risk and how best to manage such lesions.
Peutz-Jeghers syndrome
PJS is an autosomal dominant genetic disorder characterised by the development of benign hamartomatous polyps in the GI tract and hyperpigmented macules on the lips and oral mucosa (which fade with age). The prevalence of PJS is between 1/8300 and 1/29 000 and most patients have pathogenic variants in the STK11 (also called LKB1) gene.
Diagnostic criteria265: A diagnosis of PJS in an individual may be made when any one of the following is present:
Two or more histologically confirmed PJ polyps
Any number of PJ polyps detected in one individual who has a family history of PJS in a close relative
Characteristic mucocutaneous pigmentation in an individual who has a family history of PJS in a close relative
Any number of PJ polyps in an individual who also has characteristic mucocutaneous pigmentation
A pathogenic variant in STK11.
The risk of GI tract cancer in PJS
GI tract carcinogenesis in PJS is controversial. The malignant potential of the PJS polyp is unclear. A hamartoma–adenoma–carcinoma sequence has been proposed but robust supporting data are lacking. There are data suggesting that the PJS polyp is non-neoplastic, including descriptions of polyclonality and the rarity of dysplasia in PJS polyps.266
It is widely accepted that there is an increased risk of malignancy in PJS. However, the risk is difficult to quantify, with the majority of the literature being small cohort studies with inherent bias, potentially leading to overestimation of risk. A meta-analysis has been performed by Hearle et al creating a cohort of 419 patients with PJS.267 This offers the most comprehensive data for cancer risk with a luminal GI cancer risk of 1%, 9%, 15%, 33% and 57% at 30, 40, 50, 60 and 70 years, respectively. More recent data, although from a smaller cohort, support the proposition that GI cancers are less of a clinical problem and that pancreatic and breast cancers are the most commonly diagnosed malignancies in PJS.266
GI surveillance and PJS
We suggest that in an asymptomatic patient with PJS, GI surveillance by upper GI endoscopy, colonoscopy and VCE commence at the age of 8 years. We recommend that small bowel surveillance should continue 3 year ly. If baseline colonoscopy and OGD are normal, then they can be safely deferred until the age of 18 years ; however, if polyps are found at baseline examination, then they should be repeated 3 year ly. Earlier investigation of the GI tract should be performed in symptomatic patients.
(GRADE of evidence: low; Strength of recommendation: weak)
Consensus reached: 82% agreement.
Small bowel polyps resulting in intussusception is the major clinical problem affecting children with PJS. The cumulative intussusception risk is estimated at 50–68% during childhood,268 269 with 15–30% requiring surgery before the age of 10 years with a median age of the first intussusception of 10–16 years. These data include patients who preceded routine small bowel surveillance. They also combine patients with an established diagnosis of PJS undergoing surveillance, with those not under surveillance in whom the diagnosis of PJS was made at presentation with an intussusception. The recommendation to start small bowel surveillance at the age of 8 years, and earlier if symptomatic, is based on these data.270
Gastroscopy and colonoscopy are the preferred investigation to assess the upper GI tract and colon, respectively. Barium follow through has been replaced by VCE and MRI enterography, both of which are better tolerated by patients, have a similar accuracy in detecting clinically significant polyps (>1 cm) and avoid the need for repeated ionising radiation.271–276 VCE and MR enterography may be complimentary. While VCE may be better at detecting smaller polyps, MRI enterography is better at localisation and accurate sizing.
Double balloon enteroscopy (DBE) should not recommended as a surveillance tool as it is technically challenging, limited by size of the abdomen and requires both oral and rectal approaches; therefore a general anaesthetic is required to reliably visualise the whole length of the small bowel. The main role of DBE is therapeutic, for targeted polypectomy.
Role of endoscopic polypectomy in PJS
We suggest elective polypectomy to prevent polyp related complications. Small bowel polyps >1.5 – 2 cm in size (or smaller if symptomatic) should be considered for elective resection to prevent intussusception.
(GRADE of evidence: low; Strength of recommendation: weak)
Consensus reached: 88% agreement.
Polypectomy at surveillance is recommended in PJS. It is not clear whether this approach modifies cancer risk and indeed, given the lack of dysplasia in PJS polyps, it is unlikely to do so. The main role for polypectomy therefore is to prevent polyp-related complications. Prevention of anaemia and bleeding, however, is difficult to quantify and there are no data regarding this. Intussusception is the most important clinical problem in children with PJS, with an accumulative risk of 50–68% in childhood.268 269
Existing guidelines suggest intervening in small bowel polyps which are 1.5–2 cm, earlier if symptomatic.270 This is supported by data which report a median polyp size of 35 mm (15–60 mm) in cases of confirmed intussusception.269 In addition, in a cohort in whom routine surveillance was performed, with intervention planned as outlined above, no patient required emergency surgery for intussusception during 683 patient-years follow-up.266
Options to remove a PJS polyp include endoscopy, surgery or combined approaches. There are no data to state that one modality is superior to another and choice will be dependent on patient (eg, age, past abdominal surgery), polyp factors (size, location and multiplicity) and local availability of techniques. For those who undergo surgery for small bowel polyps, intraoperative enteroscopy and a “clean sweep” is recommended and has been demonstrated to reduce the need for subsequent laparotomy.277 Polyp clearance by intraoperative enteroscopy in the PJS may be effective in the long term.277 278
Juvenile polyposis syndrome
JPS is an autosomal dominant condition characterised by predisposition to hamartomatous polyps in the GI tract. The prevalence is between 1/100 000 and 1/160 000, with the following diagnostic criteria279:
>5 juvenile polyps in the colon or rectum
Juvenile polyps in other parts of the GI tract
Any number of juvenile polyps and a positive family history
A pathogenic variant in SMAD4 or BMPR1A.
About 40% of families have mutations in SMAD4 and a further 40% in BMPR1A. Some patients have large chromosomal deletions encompassing both PTEN and BMPR1A genes. There is a requirement to develop an evidence base for the personalised management of JPS-related cancers, which is beyond the scope of this guideline.
JPS and colorectal surveillance
We suggest colonoscopic surveillance should commence from the age of 15 years or earlier if symptomatic. The surveillance interval should be 1–3 yearly, personalised according to colorectal phenotype.
(GRADE of evidence: low; Strength of recommendation: weak)
Consensus reached: 85% agreement.
The polyps in JPS are thought to have the potential for malignant transformation with dysplasia present in 8% of resected polyps in one series.280 The risk of CRC is ill-defined, being based on small series, with inherent bias. Such cohorts have reported CRC in 14–22% of patients280 281 with a reported lifetime CRC risk of 39–68%.281 282 CRC in childhood is not a clinically significant problem. The mean age of CRC in two of the larger studies was 44 years.280 282
The risk of CRC is undoubtedly elevated, and the current consensus is that colonoscopic surveillance is required, with the aim of preventing or detecting early cancers. Colonoscopy and polypectomy also has the potential to prevent polyp-related morbidity (bleeding, anaemia or abdominal pain). There are few data regarding the outcomes of surveillance in JPS. In one of the largest cohorts, colonoscopy was safe. Two patients developed CRC on surveillance—one in the setting of carpeting of polyps, and the other arose following what was almost certainly an incomplete colonoscopy in the mid 1970s.280
For those with a confirmed genetic diagnosis of JPS, or individuals with a clinical diagnosis of JPS in whom molecular genetic test results were uninformative, colonoscopic surveillance can be delayed until the age of 15 years if asymptomatic.
There are no data to guide the most appropriate interval for colonoscopy surveillance in JPS. There are no data to support accelerated carcinogenesis in JPS. A personalised approach with the surveillance interval (1–3 yearly) based on individual colonic phenotype appears to be a pragmatic approach and is in line with recent paediatric polyposis guidelines.279 Colonic resection may be considered for those with a polyp burden not manageable endoscopically. For those unaffected individuals from families with a clinical but not molecular diagnosis, 5 yearly colonoscopy would seem adequate. If normal, the interval to be reviewed if a JPS phenotype is found or other sporadic polyps are identified.
Upper GI surveillance and JPS
We suggest that for those with a confirmed clinical or genetic diagnosis, upper GI endoscopic surveillance should start at the age of 18 years for SMAD4 pathogenic variant carriers and the age of 25 years for BMPR1A pathogenic variant carriers and those without an identified mutation. The surveillance interval should be 1 – 3 yearly, personalised according to upper GI tract phenotype.
(GRADE of evidence: low; Strength of recommendation: weak)
Consensus reached: 83% agreement.
We suggest that for those with an FDR with a clinical diagnosis of JPS and in whom a mutation has not been identified, screening of the upper GI tract is not required routinely but should be initiated if/when a clinical diagnosis is made on the basis of colonic phenotype. It may however be considered if there is a family history suggestive of hereditary haemorrhagic telangiectasia (HHT), even in the absence of colonic polyps.
(GRADE of evidence: low; Strength of recommendation: weak)
Consensus reached: 83% agreement.
Upper GI endoscopy is not required in those unaffected individuals in whom the diagnosis in the family is clinical without a confirmed molecular diagnosis. It is clear that the colon is the site most likely to be affected in JPS and therefore any upper GI tract assessment can be deferred until a colonic phenotype of JPS has been established in that individual.
There is a paucity of data evaluating the upper tract in JPS. Historical series report the incidence of gastric polyps between 65% and 83% and duodenal polyps between 14% and 33%.283 284 More recently, 28/41 patients had neither macroscopic nor microscopic features of upper GI tract polyps and 13/41 had confirmed histological upper GI tract involvement at a median age of 33 years.285 In this cohort five patients underwent OGD before the age of 15 years and three had changes consistent with JPS, but none had dysplasia. In another series, gastroduodenal polyps were seen in 37%; in this cohort none of the six patients who had upper GI endoscopy before the age of 18 years had gastroduodenal polyps.280
Due to limited data, it is difficult to determine the exact lifetime risk of gastric cancer in JPS. There were two gastric cancers in a cohort of 44 patients with JPS (median age 56 years)280 and a further two (age not specified) in a cohort of 56 patients who appear to have been mostly JPS but some of whom had phenotypic features overlapping with Cowden syndrome.286 In another series, 3/42 patients had either high grade dysplasia or cancer in the stomach285 and a further two patients had prophylactic gastrectomy for benign gastric polyp burden.
The risk of severe gastric polyposis and gastric cancer phenotype appears to be increased in patients with SMAD4 pathogenic variants.287 288 All patients with advanced gastric polyposis were SMAD4 pathogenic variant carriers in one cohort. Aretz et al reported a significantly higher risk of gastric polyposis in SMAD4 pathogenic variant carriers (73% vs 8%; p<0.001) and again all cases of gastric cancer in this cohort occurred in patients with SMAD4 pathogenic variants.288
Current published guidelines for the age at which to start upper GI surveillance ranges from 12 years261 to 25 years of age.35 Given the emerging genotype–phenotype association and the lack of significant pathology being found in childhood, it is recommended that those with a SMAD4 mutation undergo upper GI tract surveillance 1–3 yearly from the age of 18 years, and those with a BMPR1A mutation from 25 years of age.
Additional investigations required in patients with a SMAD4 mutation
We suggest thatpatientswith a SMAD4 pathogenic variant should be evaluated for haemorrhagic telangiectasia (HHT), and that those at risk of, or with a confirmed diagnosis of HHT are best managed in conjunction with a specialist HHT centre.
(GRADE of evidence: low; Strength of recommendation: weak)
Consensus reached: 90% agreement.
The coexistence of JPS and HHT was reported early in the 1980s.289 It is now recognised that this is due to SMAD4 mutations. Up to 76% of patients with JPS due to SMAD4 mutations have features of HHT.290 Thoracic aortic disease and mitral valve dysfunction have also been reported.280 290 Aortopathy has been described in 38% of patients with a SMAD4 mutation, irrespective of the JPS phenotype.291
Patients may lack overt clinical symptoms of HHT but are at risk of asymptomatic arteriovenous malformation (AVMs) which can result in life threatening complications. Patients with a SMAD4 pathogenic variant are probably best managed in conjunction with a specialist centre with experience in evaluating and managing HHT patients. The recent British Thoracic Society guidelines on pulmonary AVMs may act as a useful resource.292
PTEN-hamartoma syndrome overlap and additional investigations indicated in a patient with microdeletions involving BMPR1A
Patients with JPS and a microdeletion involving BMPR1A and PTEN are at risk of the clinical manifestations of both JPS and PTEN-hamartoma tumour syndrome (PHTS). We suggest that they should be referred to their local genetics centre for further advice and to coordinate their surveillance needs.
(GRADE of evidence: low; Strength of recommendation: weak)
Consensus reached: 89% agreement.
PHTS encompasses four major clinically distinct syndromes associated with constitutional pathogenic variants in the tumour suppressor PTEN. These are associated with macrocephaly, dysmorphism, developmental delay and an increased risk of extra-intestinal cancers and possibly intestinal cancers. BMPR1A is located in the same chromosomal region as PTEN and deletions involving both genes have been reported. Microdeletions involving both genes have important clinical implications. There are numerous case reports suggesting that if the pathogenic variant (microdeletion) is found in both genes, these patients may present with a severe form of JPS with onset in early childhood and may also have an increased risk of the extra-intestinal manifestations of PHTS.293–295
Footnotes
Twitter: @kevinjmonahan, @sdolwani
Collaborators: Hereditary CRC guidelines eDelphi consensus group (professional societies or other roles in brackets) Toni Seppälä (EHTG, INSIGHT), Sue Clark (EHTG, BSG, INSIGHT, ACPGBI, ESCP), Omar Faiz (ACPGBI, ESCP), Francesc Balaguer (AEG, EHTG), Monique van Leerdam (EHTG, ESGE), D Gareth Evans (UKCGG, EHTG), Rodrigo Jover (EHTG, ESGE), Marc Tischkowitz (UKCGG), Helen Hanson (UKCGG), Sarah Gibson (UKCGG), Amy Taylor (UKCGG), Gabreilla Moeslein (EHTG, ESCP), Anja Wagner (EHTG), JC Saurin (ESGE), Tracy Smith (PPI, Lynch Syndrome UK), Jane Ashford (PPI, Lynch Syndrome UK), Jennifer Martin (PPI, Lynch Syndrome UK), Jennifer Gunningham (PPI, Polyposis representative), Mark Cooper (PPI, Lynch Syndrome UK).
Contributors: The document was written by all members of the guideline development group (GDG) and edited by the lead author. The guideline development process was contributed by all members of the GDG as outlined in the methods section.
Funding: This study was funded by of British Society of Gastroenterology. Prof. James East was funded by the National Institute for Health Research (NIHR) Oxford Biomedical Research Centre (BRC). The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR or the Department of Health.
Competing interests: KJM: Medical advisory board of Bowel Cancer UK, Lynch Syndrome UK. JH and FL: FAP trial (now closed) with funding awarded to NHS trust research facility. JE: Advisory board Lumendi, Boston Scientific; Speaker fees Olympus, Falk. MDR: Speaker fees: SwissSCWeb, Pentax; Research Grant: Olympus; Consultancy: Norgine.
Patient consent for publication: Not required.
Provenance and peer review: Commissioned; externally peer reviewed.
Data availability statement: All data relevant to the study are included in the article.
Contributor Information
Hereditary CRC guidelines eDelphi consensus group:
Toni Seppälä, Sue Clark, Omar Faiz, Francesc Balaguer, Monique van Leerdam, D Gareth Evans, Rodrigo Jover, Marc Tischkowitz, Helen Hanson, Sarah Gibson, Amy Taylor, Gabreilla Moeslein, Anja Wagner, JC Saurin, Tracy Smith, Jane Ashford, Jennifer Martin, Jennifer Gunningham, and Mark Cooper
References
- 1. Lichtenstein P, Holm NV, Verkasalo PK, et al. Environmental and heritable factors in the causation of cancer — analyses of cohorts of twins from Sweden, Denmark, and Finland. N Engl J Med 2000;343:78–85. 10.1056/NEJM200007133430201 [DOI] [PubMed] [Google Scholar]
- 2. Mitchell RJ, Campbell H, Farrington SM, et al. Prevalence of family history of colorectal cancer in the general population. Br J Surg 2005;92:1161–4. 10.1002/bjs.5084 [DOI] [PubMed] [Google Scholar]
- 3. NICE NICE CG164. Familial breast cancer: classification, care and managing breast cancer and related risks in people with a family history of breast cancer | guidance | NICE, 2017. Available: https://www.nice.org.uk/guidance/cg164 [Accessed 27 May 2019]. [PubMed]
- 4. Taylor A, Brady AF, Frayling IM, et al. Consensus for genes to be included on cancer panel tests offered by UK genetics services: guidelines of the UK cancer genetics group. J Med Genet 2018;372:jmedgenet-2017-105188 10.1136/jmedgenet-2017-105188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Brouwers MC. Agree next steps Consortium membership. Available: www.agreetrust.org
- 6. Boulkedid R, Abdoul H, Loustau M, et al. Using and reporting the Delphi method for selecting healthcare quality indicators: a systematic review. PLoS One 2011;6:e20476 10.1371/journal.pone.0020476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Jaeschke R, Guyatt GH, Dellinger P, et al. Use of grade grid to reach decisions on clinical practice guidelines when consensus is elusive. BMJ 2008;337 10.1136/bmj.a744 [DOI] [PubMed] [Google Scholar]
- 8. Guyatt GH, Oxman AD, Kunz R, et al. What is "quality of evidence" and why is it important to clinicians? BMJ 2008;336:995–8. 10.1136/bmj.39490.551019.BE [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Monahan KJ, Alsina D, Bach S, et al. Urgent improvements needed to diagnose and manage Lynch syndrome. BMJ 2017;356 10.1136/bmj.j1388 [DOI] [PubMed] [Google Scholar]
- 10. Barrow P, Khan M, Lalloo F, et al. Systematic review of the impact of registration and screening on colorectal cancer incidence and mortality in familial adenomatous polyposis and Lynch syndrome. Br J Surg 2013;100:1719–31. 10.1002/bjs.9316 [DOI] [PubMed] [Google Scholar]
- 11. Monahan KJ, Clark SK, on behalf of the British Society of Gastroenterology (BSG) Cancer Group . A national survey of hereditary colorectal cancer services in the UK. Frontline Gastroenterol 2014;5:130–4. 10.1136/flgastro-2013-100362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Adelson M, Pannick S, East JE, et al. Uk colorectal cancer patients are inadequately assessed for Lynch syndrome. Frontline Gastroenterol 2014;5:31–5. 10.1136/flgastro-2013-100345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bowel Cancer UK Improving services for Lynch syndrome: who’s responsible?.
- 14. Seppen J, Bruzzone L. Lynch syndrome: the patients' perspective. Fam Cancer 2013;12:341–5. 10.1007/s10689-013-9630-2 [DOI] [PubMed] [Google Scholar]
- 15. NHS England 2018/2019 final draft National GenomicTest directory FAQ, 2018. Available: https://www.england.nhs.uk/wp-content/uploads/2018/08/national-genomic-test-directory-faqs.pdf
- 16. Burton AM, Hovick SR, Peterson SK. Health behaviors in patients and families with hereditary colorectal cancer. Clin Colon Rectal Surg 2012;25:111–7. 10.1055/s-0032-1313782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Riley BD, Culver JO, Skrzynia C, et al. Essential elements of genetic cancer risk assessment, counseling, and testing: updated recommendations of the National Society of Genetic Counselors. J Genet Couns 2012;21:151–61. 10.1007/s10897-011-9462-x [DOI] [PubMed] [Google Scholar]
- 18. Excellence TNI for H and C Molecular testing strategies for Lynch syndrome in people with colorectal cancer (NICE diagnostics guidance DG27), 2017. Available: https://www.nice.org.uk/guidance/dg27
- 19. Johns LE, Houlston RS. A systematic review and meta-analysis of familial colorectal cancer risk. Am J Gastroenterol 2001;96:2992–3003. 10.1111/j.1572-0241.2001.04677.x [DOI] [PubMed] [Google Scholar]
- 20. Edwards E, Lucassen A. The impact of cancer pathology confirmation on clinical management of a family history of cancer. Fam Cancer 2011;10:373–80. 10.1007/s10689-010-9407-9 [DOI] [PubMed] [Google Scholar]
- 21. Lubbe SJ, Webb EL, Chandler IP, et al. Implications of familial colorectal cancer risk profiles and microsatellite instability status. J Clin Oncol 2009;27:2238–44. 10.1200/JCO.2008.20.3364 [DOI] [PubMed] [Google Scholar]
- 22. Bapat B, Lindor NM, Baron J, et al. The association of tumor microsatellite instability phenotype with family history of colorectal cancer. Cancer Epidemiol Biomarkers Prev 2009;18:967–75. 10.1158/1055-9965.EPI-08-0878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Win AK, Buchanan DD, Rosty C, et al. Role of tumour molecular and pathology features to estimate colorectal cancer risk for first-degree relatives. Gut 2015;64:101–10. 10.1136/gutjnl-2013-306567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bonnington SN, Rutter MD. Surveillance of colonic polyps: are we getting it right? World J Gastroenterol 2016;22:1925–34. 10.3748/wjg.v22.i6.1925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Graff RE, Möller S, Passarelli MN, et al. Familial risk and heritability of colorectal cancer in the Nordic twin study of cancer. Clin Gastroenterol Hepatol 2017;15:1256–64. 10.1016/j.cgh.2016.12.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Newton KF, Green K, Walsh S, et al. Metachronous colorectal cancer risk in patients with a moderate family history. Colorectal Dis 2013;15:309–16. 10.1111/codi.12005 [DOI] [PubMed] [Google Scholar]
- 27. Gupta A, Samadder J, Elliott E, et al. Prevalence of adenomas and advanced adenomas in patients in the 40- to 49-year age group undergoing screening colonoscopy because of a family history of adenoma/polyp in a first-degree relative. Gastrointest Endosc 2012;75:705–11. 10.1016/j.gie.2012.01.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Brenner H, Hoffmeister M, Stegmaier C, et al. Risk of progression of advanced adenomas to colorectal cancer by age and sex: estimates based on 840,149 screening colonoscopies. Gut 2007;56:1585–9. 10.1136/gut.2007.122739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Brenner H, Altenhofen L, Kretschmann J, et al. Trends in adenoma detection rates during the first 10 years of the German screening colonoscopy program. Gastroenterology 2015;149:356-66.e1 10.1053/j.gastro.2015.04.012 [DOI] [PubMed] [Google Scholar]
- 30. Wilschut JA, Habbema JDF, Ramsey SD, et al. Increased risk of adenomas in individuals with a family history of colorectal cancer: results of a meta-analysis. Cancer Causes Control 2010;21:2287–93. 10.1007/s10552-010-9654-y [DOI] [PubMed] [Google Scholar]
- 31. Morois S, Cottet V, Racine A, et al. Colonoscopy reduced distal colorectal cancer risk and excess cancer risk associated with family history. Cancer Causes Control 2014;25:1329–36. 10.1007/s10552-014-0438-7 [DOI] [PubMed] [Google Scholar]
- 32. Fuchs CS, Giovannucci EL, Colditz GA, et al. A prospective study of family history and the risk of colorectal cancer. N Engl J Med 1994;331:1669–74. 10.1056/NEJM199412223312501 [DOI] [PubMed] [Google Scholar]
- 33. Menges M, Fischinger J, Gärtner B, et al. Screening colonoscopy in 40- to 50-year-old first-degree relatives of patients with colorectal cancer is efficient: a controlled multicentre study. Int J Colorectal Dis 2006;21:301–7. 10.1007/s00384-005-0032-2 [DOI] [PubMed] [Google Scholar]
- 34. van der Meulen-de Jong AE, Morreau H, Becx MCJM, et al. High detection rate of adenomas in familial colorectal cancer. Gut 2011;60:73–6. 10.1136/gut.2010.217091 [DOI] [PubMed] [Google Scholar]
- 35. Cairns SR, Scholefield JH, Steele RJ, et al. Guidelines for colorectal cancer screening and surveillance in moderate and high risk groups (update from 2002). Gut 2010;59:666–89. 10.1136/gut.2009.179804 [DOI] [PubMed] [Google Scholar]
- 36. Wark PA, Wu K, van 't Veer P, et al. Family history of colorectal cancer: a determinant of advanced adenoma stage or adenoma multiplicity? Int J Cancer 2009;125:413–20. 10.1002/ijc.24288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Mesher D, Dove-Edwin I, Sasieni P, et al. A pooled analysis of the outcome of prospective colonoscopic surveillance for familial colorectal cancer. Int J Cancer 2014;134:939–47. 10.1002/ijc.28397 [DOI] [PubMed] [Google Scholar]
- 38. Dowling DJ, St John DJ, Macrae FA, et al. Yield from colonoscopic screening in people with a strong family history of common colorectal cancer. J Gastroenterol Hepatol 2000;15:939–44. 10.1046/j.1440-1746.2000.02254.x [DOI] [PubMed] [Google Scholar]
- 39. Forsberg AM, Kjellström L, Agréus L, et al. Prevalence of colonic neoplasia and advanced lesions in the normal population: a prospective population-based colonoscopy study. Scand J Gastroenterol 2012;47:184–90. 10.3109/00365521.2011.647062 [DOI] [PubMed] [Google Scholar]
- 40. Bradshaw N, Holloway S, Penman I, et al. Colonoscopy surveillance of individuals at risk of familial colorectal cancer. Gut 2003;52:1748–51. 10.1136/gut.52.12.1748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Tsai FC, Strum WB. Impact of a family history of colorectal cancer on the prevalence of advanced neoplasia at colonoscopy in 4,967 asymptomatic patients. Dig Dis Sci 2012;57:3234–9. 10.1007/s10620-011-2015-1 [DOI] [PubMed] [Google Scholar]
- 42. Dove-Edwin I, Sasieni P, Adams J, et al. Prevention of colorectal cancer by colonoscopic surveillance in individuals with a family history of colorectal cancer: 16 year, prospective, follow-up study. BMJ 2005;331:1047–9. 10.1136/bmj.38606.794560.EB [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Forsberg AM, Hagel E, Jaramillo E, et al. Predicting outcome in colonoscopic high-risk surveillance. Anticancer Res 2015;35:4813–9. [PubMed] [Google Scholar]
- 44. Hennink SD, van der Meulen-de Jong AE, Wolterbeek R, et al. Randomized comparison of surveillance intervals in familial colorectal cancer. J Clin Oncol 2015;33:4188–93. 10.1200/JCO.2015.62.2035 [DOI] [PubMed] [Google Scholar]
- 45. Lindor NM, Rabe K, Petersen GM, et al. Lower cancer incidence in Amsterdam-I criteria families without mismatch repair deficiency: familial colorectal cancer type X. JAMA 2005;293:1979–85. 10.1001/jama.293.16.1979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Dove-Edwin I, de Jong AE, Adams J, et al. Prospective results of surveillance colonoscopy in dominant familial colorectal cancer with and without Lynch syndrome. Gastroenterology 2006;130:1995–2000. 10.1053/j.gastro.2006.03.018 [DOI] [PubMed] [Google Scholar]
- 47. Burn J, Gerdes A-M, Macrae F, et al. Long-term effect of aspirin on cancer risk in carriers of hereditary colorectal cancer: an analysis from the CAPP2 randomised controlled trial. Lancet 2011;378:2081–7. 10.1016/S0140-6736(11)61049-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ait Ouakrim D, Dashti SG, Chau R, et al. Aspirin, ibuprofen, and the risk of colorectal cancer in Lynch syndrome. J Natl Cancer Inst 2015;107:djv170 10.1093/jnci/djv170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Burn J. ISRCTN - ISRCTN16261285: Finding the best dose of aspirin to prevent Lynch Syndrome cancers, 2015. Available: http://www.isrctn.com/ISRCTN16261285?q=capp3&filters=&sort=&offset=1&totalResults=1&page=1&pageSize=10&searchType=basic-search [Accessed 11 May 2019].
- 50. Smith SG, Foy R, McGowan J, et al. General practitioner attitudes towards prescribing aspirin to carriers of Lynch syndrome: findings from a national survey. Fam Cancer 2017;16:509–16. 10.1007/s10689-017-9986-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Burn J, Bishop DT, Chapman PD, et al. A randomized placebo-controlled prevention trial of aspirin and/or resistant starch in young people with familial adenomatous polyposis. Cancer Prev Res 2011;4:655–65. 10.1158/1940-6207.CAPR-11-0106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ishikawa H, Wakabayashi K, Suzuki S, et al. Preventive effects of low-dose aspirin on colorectal adenoma growth in patients with familial adenomatous polyposis: double-blind, randomized clinical trial. Cancer Med 2013;2:50–6. 10.1002/cam4.46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Iwama T, Akasu T, Utsunomiya J, et al. Does a selective cyclooxygenase-2 inhibitor (tiracoxib) induce clinically sufficient suppression of adenomas in patients with familial adenomatous polyposis? A randomized double-blind placebo-controlled clinical trial. Int J Clin Oncol 2006;11:133–9. 10.1007/s10147-005-0548-z [DOI] [PubMed] [Google Scholar]
- 54. Labayle D, Fischer D, Vielh P, et al. Sulindac causes regression of rectal polyps in familial adenomatous polyposis. Gastroenterology 1991;101:635–9. 10.1016/0016-5085(91)90519-Q [DOI] [PubMed] [Google Scholar]
- 55. Steinbach G, Lynch PM, Phillips RKS, et al. The effect of celecoxib, a cyclooxygenase-2 inhibitor, in familial adenomatous polyposis. N Engl J Med 2000;342:1946–52. 10.1056/NEJM200006293422603 [DOI] [PubMed] [Google Scholar]
- 56. Nugent KP, Farmer KC, Spigelman AD, et al. Randomized controlled trial of the effect of sulindac on duodenal and rectal polyposis and cell proliferation in patients with familial adenomatous polyposis. Br J Surg 1993;80:1618–9. 10.1002/bjs.1800801244 [DOI] [PubMed] [Google Scholar]
- 57. Higuchi T, Iwama T, Yoshinaga K, et al. A randomized, double-blind, placebo-controlled trial of the effects of rofecoxib, a selective cyclooxygenase-2 inhibitor, on rectal polyps in familial adenomatous polyposis patients. Clin Cancer Res 2003;9:4756–60. [PubMed] [Google Scholar]
- 58. Hirota C, Iida M, Aoyagi K, et al. Effect of indomethacin suppositories on rectal polyposis in patients with familial adenomatous polyposis. Cancer 1996;78:1660–5. [DOI] [PubMed] [Google Scholar]
- 59. Tonelli F, Valanzano R, Messerini L, et al. Long-term treatment with sulindac in familial adenomatous polyposis: is there an actual efficacy in prevention of rectal cancer? J Surg Oncol 2000;74:15–20. [DOI] [PubMed] [Google Scholar]
- 60. Cruz-Correa M, Hylind LM, Romans KE, et al. Long-term treatment with sulindac in familial adenomatous polyposis: a prospective cohort study. Gastroenterology 2002;122:641–5. 10.1053/gast.2002.31890 [DOI] [PubMed] [Google Scholar]
- 61. International Society for Gastrointestinal Hereditary Tumours-InSiGHT. Fam Cancer 2015;14(Suppl 1):1–91. 10.1007/s10689-015-9808-x [DOI] [PubMed] [Google Scholar]
- 62. West NJ, Clark SK, Phillips RKS, et al. Eicosapentaenoic acid reduces rectal polyp number and size in familial adenomatous polyposis. Gut 2010;59:918–25. 10.1136/gut.2009.200642 [DOI] [PubMed] [Google Scholar]
- 63. Phillips RKS, Wallace MH, Lynch PM, et al. A randomised, double blind, placebo controlled study of celecoxib, a selective cyclooxygenase 2 inhibitor, on duodenal polyposis in familial adenomatous polyposis. Gut 2002;50:857–60. 10.1136/gut.50.6.857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Samadder NJ, Neklason DW, Boucher KM, et al. Effect of sulindac and erlotinib vs placebo on duodenal neoplasia in familial adenomatous polyposis: a randomized clinical trial. JAMA 2016;315:1266–75. 10.1001/jama.2016.2522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Calabrese C, Praticò C, Calafiore A, et al. Eviendep® reduces number and size of duodenal polyps in familial adenomatous polyposis patients with ileal pouch-anal anastomosis. World J Gastroenterol 2013;19:5671–7. 10.3748/wjg.v19.i34.5671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. van Hattem WA, Brosens LAA, Marks SY, et al. Increased cyclooxygenase-2 expression in juvenile polyposis syndrome. Clin Gastroenterol Hepatol 2009;7:93–7. 10.1016/j.cgh.2008.07.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Kurland JE, Beck SE, Solomon CJ, et al. Cyclooxygenase-2 expression in polyps from a patient with juvenile polyposis syndrome with mutant BMPR1A. J Pediatr Gastroenterol Nutr 2007;44:318–25. 10.1097/MPG.0b013e31802e98e5 [DOI] [PubMed] [Google Scholar]
- 68. McGarrity TJ, Peiffer LP, Amos CI, et al. Overexpression of cyclooxygenase 2 in hamartomatous polyps of Peutz-Jeghers syndrome. Am J Gastroenterol 2003;98:671–8. 10.1111/j.1572-0241.2003.07328.x [DOI] [PubMed] [Google Scholar]
- 69. Whiteman DC, Webb PM, Green AC, et al. Cancers in Australia in 2010 attributable to modifiable factors: summary and conclusions. Aust N Z J Public Health 2015;39:477–84. 10.1111/1753-6405.12471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Islami F, Goding Sauer A, Miller KD, et al. Proportion and number of cancer cases and deaths attributable to potentially modifiable risk factors in the United States. CA Cancer J Clin 2018;68:31–54. 10.3322/caac.21440 [DOI] [PubMed] [Google Scholar]
- 71. Fardet A, Druesne-Pecollo N, Touvier M, et al. Do alcoholic beverages, obesity and other nutritional factors modify the risk of familial colorectal cancer? A systematic review. Crit Rev Oncol Hematol 2017;119:94–112. 10.1016/j.critrevonc.2017.09.001 [DOI] [PubMed] [Google Scholar]
- 72. Watson P, Ashwathnarayan R, Lynch HT, et al. Tobacco use and increased colorectal cancer risk in patients with hereditary nonpolyposis colorectal cancer (Lynch syndrome). Arch Intern Med 2004;164:2429–31. 10.1001/archinte.164.22.2429 [DOI] [PubMed] [Google Scholar]
- 73. Pande M, Lynch PM, Hopper JL, et al. Smoking and colorectal cancer in Lynch syndrome: results from the colon cancer family registry and the University of Texas M.D. Anderson cancer center. Clin Cancer Res 2010;16:1331–9. 10.1158/1078-0432.CCR-09-1877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Diergaarde B, Braam H, Vasen HF, et al. Environmental factors and colorectal tumor risk in individuals with hereditary nonpolyposis colorectal cancer. Clin Gastroenterol Hepatol 2007;5:736–42. 10.1016/j.cgh.2007.02.019 [DOI] [PubMed] [Google Scholar]
- 75. Winkels RM, Botma A, Van Duijnhoven FJB, et al. Smoking increases the risk for colorectal adenomas in patients with Lynch syndrome. Gastroenterology 2012;142:241–7. 10.1053/j.gastro.2011.10.033 [DOI] [PubMed] [Google Scholar]
- 76. Burn J, Bishop DT, Mecklin J-P, et al. Effect of aspirin or resistant starch on colorectal neoplasia in the Lynch syndrome. N Engl J Med 2008;359:2567–78. 10.1056/NEJMoa0801297 [DOI] [PubMed] [Google Scholar]
- 77. Botma A, Nagengast FM, Braem MGM, et al. Body mass index increases risk of colorectal adenomas in men with Lynch syndrome: the GEOLynch cohort study. J Clin Oncol 2010;28:4346–53. 10.1200/JCO.2010.28.0453 [DOI] [PubMed] [Google Scholar]
- 78. Win AK, Dowty JG, English DR, et al. Body mass index in early adulthood and colorectal cancer risk for carriers and non-carriers of germline mutations in DNA mismatch repair genes. Br J Cancer 2011;105:162–9. 10.1038/bjc.2011.172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Movahedi M, Bishop DT, Macrae F, et al. Obesity, aspirin, and risk of colorectal cancer in carriers of hereditary colorectal cancer: a prospective investigation in the CAPP2 study. J Clin Oncol 2015;33:3591–7. 10.1200/JCO.2014.58.9952 [DOI] [PubMed] [Google Scholar]
- 80. Shaw E, Farris MS, Stone CR, et al. Effects of physical activity on colorectal cancer risk among family history and body mass index subgroups: a systematic review and meta-analysis. BMC Cancer 2018;18:71 10.1186/s12885-017-3970-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Prichard I, Lee A, Hutchinson AD, et al. Familial risk for lifestyle-related chronic diseases: can family health history be used as a motivational tool to promote health behaviour in young adults? Health Promot J Austr 2015;26:122–8. 10.1071/HE14104 [DOI] [PubMed] [Google Scholar]
- 82. Tarr GP, Crowley A, John R, et al. Do high risk patients alter their lifestyle to reduce risk of colorectal cancer? BMC Gastroenterol 2014;14:22 10.1186/1471-230X-14-22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Anderson AS, Dunlop J, Gallant S, et al. Feasibility study to assess the impact of a lifestyle intervention ('LivingWELL') in people having an assessment of their family history of colorectal or breast cancer. BMJ Open 2018;8:e019410 10.1136/bmjopen-2017-019410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Rees CJ, Thomas Gibson S, Rutter MD, et al. UK key performance indicators and quality assurance standards for colonoscopy. Gut 2016;65:1923–9. 10.1136/gutjnl-2016-312044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Haanstra JF, Vasen HFA, Sanduleanu S, et al. Quality colonoscopy and risk of interval cancer in Lynch syndrome. Int J Colorectal Dis 2013;28:1643–9. 10.1007/s00384-013-1745-2 [DOI] [PubMed] [Google Scholar]
- 86. Boonstra JJ, de Vos Tot Nederveen Cappel WH, Langers AMJ, et al. Colonoscopy in Lynch syndrome: the need for a new quality score. Fam Cancer 2017;16:239–41. 10.1007/s10689-016-9950-0 [DOI] [PubMed] [Google Scholar]
- 87. Kaminski MF, Thomas-Gibson S, Bugajski M, et al. Performance measures for lower gastrointestinal endoscopy: a European Society of Gastrointestinal Endoscopy (ESGE) quality improvement initiative. Endoscopy 2017;49:378–97. 10.1055/s-0043-103411 [DOI] [PubMed] [Google Scholar]
- 88. Clark BT, Rustagi T, Laine L. What level of bowel prep quality requires early repeat colonoscopy: systematic review and meta-analysis of the impact of preparation quality on adenoma detection rate. Am J Gastroenterol 2014;109:1714–23. 10.1038/ajg.2014.232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Connor A, Tolan D, Hughes S, et al. Consensus guidelines for the safe prescription and administration of oral bowel-cleansing agents. Gut 2012;61:1525–32. 10.1136/gutjnl-2011-300861 [DOI] [PubMed] [Google Scholar]
- 90. Forsberg AM, Hagel E, Jaramillo E, et al. Surveillance of colonic polyps: are we getting it right? Gut 2015;22:101–10. [Google Scholar]
- 91. Rondagh EJA, Gulikers S, Gómez-García EB, et al. Nonpolypoid colorectal neoplasms: a challenge in endoscopic surveillance of patients with Lynch syndrome. Endoscopy 2013;45:257–64. 10.1055/s-0032-1326195 [DOI] [PubMed] [Google Scholar]
- 92. Kamiński MF, Hassan C, Bisschops R, et al. Advanced imaging for detection and differentiation of colorectal neoplasia: European Society of Gastrointestinal Endoscopy (ESGE) guideline. Endoscopy 2014;46:435–57. 10.1055/s-0034-1365348 [DOI] [PubMed] [Google Scholar]
- 93. Lecomte T, Cellier C, Meatchi T, et al. Chromoendoscopic colonoscopy for detecting preneoplastic lesions in hereditary nonpolyposis colorectal cancer syndrome. Clin Gastroenterol Hepatol 2005;3:897–902. 10.1016/S1542-3565(05)00403-9 [DOI] [PubMed] [Google Scholar]
- 94. Stoffel EM, Turgeon DK, Stockwell DH, et al. Missed adenomas during colonoscopic surveillance in individuals with Lynch syndrome (hereditary nonpolyposis colorectal cancer). Cancer Prev Res 2008;1:470–5. 10.1158/1940-6207.CAPR-08-0098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. East JE, Suzuki N, Stavrinidis M, et al. Narrow band imaging for colonoscopic surveillance in hereditary non-polyposis colorectal cancer. Gut 2008;57:65–70. 10.1136/gut.2007.128926 [DOI] [PubMed] [Google Scholar]
- 96. Rahmi G, Lecomte T, Malka D, et al. Impact of chromoscopy on adenoma detection in patients with Lynch syndrome: a prospective, multicenter, blinded, tandem colonoscopy study. Am J Gastroenterol 2015;110:288–98. 10.1038/ajg.2014.423 [DOI] [PubMed] [Google Scholar]
- 97. Haanstra JF, Dekker E, Cats A, et al. Effect of chromoendoscopy in the proximal colon on colorectal neoplasia detection in Lynch syndrome: a multicenter randomized controlled trial. Gastrointest Endosc 2019;90:624–32. 10.1016/j.gie.2019.04.227 [DOI] [PubMed] [Google Scholar]
- 98. Sánchez LR. High definition white-light colonoscopy versus chromoendoscopy for surveillance of Lynch syndrome. A multicenter, randomized and controlled study (ENDOLYNCH study). United Eur Gastroenterol J 2018;6(Suppl 1). [Google Scholar]
- 99. Brown SR, Baraza W, Din S, et al. Chromoscopy versus conventional endoscopy for the detection of polyps in the colon and rectum. Cochrane Database Syst Rev 2016;4:CD006439 10.1002/14651858.CD006439.pub4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. McGowan LA, Hopkins LA, Monahan KJ. PWE-362 detection of neoplasia using dye spray chromoendoscopy in patients with a high risk of familial colorectal cancer: Abstract PWE-362 Table 1. Gut 2015;64:A369.2 10.1136/gutjnl-2015-309861.808 [DOI] [Google Scholar]
- 101. Ket SN, Bird-Lieberman E, East JE. Electronic imaging to enhance lesion detection at colonoscopy. Gastrointest Endosc Clin N Am 2015;25:227–42. 10.1016/j.giec.2014.11.011 [DOI] [PubMed] [Google Scholar]
- 102. Hüneburg R, Lammert F, Rabe C, et al. Chromocolonoscopy detects more adenomas than white light colonoscopy or narrow band imaging colonoscopy in hereditary nonpolyposis colorectal cancer screening. Endoscopy 2009;41:316–22. 10.1055/s-0028-1119628 [DOI] [PubMed] [Google Scholar]
- 103. Matsumoto T, Esaki M, Fujisawa R, et al. Chromoendoscopy, narrow-band imaging colonoscopy, and autofluorescence colonoscopy for detection of diminutive colorectal neoplasia in familial adenomatous polyposis. Dis Colon Rectum 2009;52:1160–5. 10.1007/DCR.0b013e31819ef6fe [DOI] [PubMed] [Google Scholar]
- 104. Wallace MH, Frayling IM, Clark SK, et al. Attenuated adenomatous polyposis coli: the role of ascertainment bias through failure to dye-spray at colonoscopy. Dis Colon Rectum 1999;42:1078–80. 10.1007/bf02236707 [DOI] [PubMed] [Google Scholar]
- 105. Hurt C, Ramaraj R, Farr A, et al. Feasibility and economic assessment of chromocolonoscopy for detection of proximal serrated neoplasia within a population-based colorectal cancer screening programme (CONSCOP): an open-label, randomised controlled non-inferiority trial. Lancet Gastroenterol Hepatol 2019;4:364–75. 10.1016/S2468-1253(19)30035-4 [DOI] [PubMed] [Google Scholar]
- 106. Katsoula A, Paschos P, Haidich A-B, et al. Diagnostic accuracy of fecal immunochemical test in patients at increased risk for colorectal cancer: a meta-analysis. JAMA Intern Med 2017;177:1110–8. 10.1001/jamainternmed.2017.2309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Goede SL, Rabeneck L, Lansdorp-Vogelaar I, et al. The impact of stratifying by family history in colorectal cancer screening programs. Int J Cancer 2015;137:1119–27. 10.1002/ijc.29473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Adrián-de-Ganzo Z, Alarcón-Fernández O, Ramos L, et al. Uptake of colon capsule endoscopy vs colonoscopy for screening relatives of patients with colorectal cancer. Clin Gastroenterol Hepatol 2015;13:2293–301. 10.1016/j.cgh.2015.06.032 [DOI] [PubMed] [Google Scholar]
- 109. Florie J, Jensch S, Nievelstein RAJ, et al. MR colonography with limited bowel preparation compared with optical colonoscopy in patients at increased risk for colorectal cancer. Radiology 2007;243:122–31. 10.1148/radiol.2431052088 [DOI] [PubMed] [Google Scholar]
- 111. Pickhardt PJ, Mbah I, Pooler BD, et al. Ct colonographic screening of patients with a family history of colorectal cancer: comparison with adults at average risk and implications for guidelines. AJR Am J Roentgenol 2017;208:794–800. 10.2214/AJR.16.16724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Plumb AA, Halligan S, Nickerson C, et al. Use of CT colonography in the English bowel cancer screening programme. Gut 2014;63:964–73. 10.1136/gutjnl-2013-304697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Vasen HF, Watson P, Mecklin JP, et al. New clinical criteria for hereditary nonpolyposis colorectal cancer (HNPCC, Lynch syndrome) proposed by the International Collaborative Group on HNPCC. Gastroenterology 1999;116:1453–6. 10.1016/S0016-5085(99)70510-X [DOI] [PubMed] [Google Scholar]
- 113. Umar A, Boland CR, Terdiman JP, et al. Revised Bethesda guidelines for hereditary nonpolyposis colorectal cancer (Lynch syndrome) and microsatellite instability. J Natl Cancer Inst 2004;96:261–8. 10.1093/jnci/djh034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Cavazza A, Radia C, Harlow C, et al. Experience of the implementation and outcomes of universal testing for Lynch syndrome in the United Kingdom. Colorectal Dis 2019;21:760–6. 10.1111/codi.14597 [DOI] [PubMed] [Google Scholar]
- 115. Newton K, Green K, Lalloo F, et al. Colonoscopy screening compliance and outcomes in patients with Lynch syndrome. Colorectal Dis 2015;17:38–46. 10.1111/codi.12778 [DOI] [PubMed] [Google Scholar]
- 116. de Vos tot Nederveen Cappel WH, Nagengast FM, Griffioen G, et al. Surveillance for hereditary nonpolyposis colorectal cancer: a long-term study on 114 families. Dis Colon Rectum 2002;45:1588–94. 10.1007/s10350-004-7244-3 [DOI] [PubMed] [Google Scholar]
- 117. Vasen HFA, Blanco I, Aktan-Collan K, et al. Revised guidelines for the clinical management of Lynch syndrome (HNPCC): recommendations by a group of European experts. Gut 2013;62:812–23. 10.1136/gutjnl-2012-304356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Seppälä T, Pylvänäinen K, Evans DG, et al. Colorectal cancer incidence in path_MLH1 carriers subjected to different follow-up protocols: a prospective Lynch syndrome database report. Hered Cancer Clin Pract 2017;15:18 10.1186/s13053-017-0078-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Møller P, Seppälä T, Bernstein I, et al. Incidence of and survival after subsequent cancers in carriers of pathogenic MMR variants with previous cancer: a report from the prospective Lynch syndrome database. Gut 2017;66:1657–64. 10.1136/gutjnl-2016-311403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Møller P, Seppälä TT, Bernstein I, et al. Cancer risk and survival in path_MMR carriers by gene and gender up to 75 years of age: a report from the prospective Lynch syndrome database. Gut 2018;67:1306–16. 10.1136/gutjnl-2017-314057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Møller P, Seppälä T, Bernstein I, et al. Cancer incidence and survival in Lynch syndrome patients receiving colonoscopic and gynaecological surveillance: first report from the prospective Lynch syndrome database. Gut 2017;66:464–72. 10.1136/gutjnl-2015-309675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Vasen HFA, Abdirahman M, Brohet R, et al. One to 2-year surveillance intervals reduce risk of colorectal cancer in families with Lynch syndrome. Gastroenterology 2010;138:2300–6. 10.1053/j.gastro.2010.02.053 [DOI] [PubMed] [Google Scholar]
- 123. Engel C, Vasen HF, Seppälä T, et al. No difference in colorectal cancer incidence or stage at detection by colonoscopy among 3 countries with different Lynch syndrome surveillance policies. Gastroenterology 2018;155:1400–9. 10.1053/j.gastro.2018.07.030 [DOI] [PubMed] [Google Scholar]
- 124. Ten Broeke SW, van der Klift HM, Tops CMJ, et al. Cancer risks for PMS2-associated Lynch syndrome. J Clin Oncol 2018;36:2961–8. 10.1200/JCO.2018.78.4777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Giardiello FM, Allen JI, Axilbund JE, et al. Guidelines on genetic evaluation and management of Lynch syndrome: a consensus statement by the US Multi-society Task force on colorectal cancer. Am J Gastroenterol 2014;109:1159–79. 10.1038/ajg.2014.186 [DOI] [PubMed] [Google Scholar]
- 126. Rubenstein JH, Enns R, Heidelbaugh J, et al. American Gastroenterological Association Institute guideline on the diagnosis and management of Lynch syndrome. Gastroenterology 2015;149:777–82. 10.1053/j.gastro.2015.07.036 [DOI] [PubMed] [Google Scholar]
- 127. Rijcken FEM, Hollema H, Kleibeuker JH. Proximal adenomas in hereditary non-polyposis colorectal cancer are prone to rapid malignant transformation. Gut 2002;50:382–6. 10.1136/gut.50.3.382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. de Jong AE, Nagengast FM, Kleibeuker JH, et al. What is the appropriate screening protocol in Lynch syndrome? Fam Cancer 2006;5:373–8. 10.1007/s10689-006-0008-6 [DOI] [PubMed] [Google Scholar]
- 129. Hampel H, Frankel WL, Martin E, et al. Screening for the Lynch syndrome (hereditary nonpolyposis colorectal cancer). N Engl J Med 2005;352:1851–60. 10.1056/NEJMoa043146 [DOI] [PubMed] [Google Scholar]
- 130. Hendriks YMC, Wagner A, Morreau H, et al. Cancer risk in hereditary nonpolyposis colorectal cancer due to MSH6 mutations: impact on counseling and surveillance. Gastroenterology 2004;127:17–25. 10.1053/j.gastro.2004.03.068 [DOI] [PubMed] [Google Scholar]
- 131. Jenkins MA, Baglietto L, Dowty JG, et al. Cancer risks for mismatch repair gene mutation carriers: a population-based early onset case-family study. Clin Gastroenterol Hepatol 2006;4:489–98. 10.1016/j.cgh.2006.01.002 [DOI] [PubMed] [Google Scholar]
- 132. Quehenberger F, Vasen HFA, van Houwelingen HC. Risk of colorectal and endometrial cancer for carriers of mutations of the hMLH1 and hMSH2 gene: correction for ascertainment. J Med Genet 2005;42:491–6. 10.1136/jmg.2004.024299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Jenkins MA, Dowty JG, Ait Ouakrim D, et al. Short-term risk of colorectal cancer in individuals with Lynch syndrome: a meta-analysis. J Clin Oncol 2015;33:326–31. 10.1200/JCO.2014.55.8536 [DOI] [PubMed] [Google Scholar]
- 134. Bonadona V, Bonaïti B, Olschwang S, et al. Cancer risks associated with germline mutations in MLH1, MSH2, and MSH6 genes in Lynch syndrome. JAMA 2011;305:2304–10. 10.1001/jama.2011.743 [DOI] [PubMed] [Google Scholar]
- 135. Kalady MF, McGannon E, Vogel JD, et al. Risk of colorectal adenoma and carcinoma after colectomy for colorectal cancer in patients meeting Amsterdam criteria. Ann Surg 2010;252:105–12. discussion 511-3 10.1097/SLA.0b013e3181f20bd2 [DOI] [PubMed] [Google Scholar]
- 136. Renkonen-Sinisalo L, Seppälä TT, Järvinen HJ, et al. Subtotal colectomy for colon cancer reduces the need for subsequent surgery in Lynch syndrome. Dis Colon Rectum 2017;60:792–9. 10.1097/DCR.0000000000000802 [DOI] [PubMed] [Google Scholar]
- 137. Hiatt MJ, Casey MJ, Lynch HT, et al. Efficacy of proximal colectomy for surgical management of right-sided first colorectal cancer in Lynch syndrome mutation carriers. Am J Surgery 2018;216:99–105. 10.1016/j.amjsurg.2017.11.003 [DOI] [PubMed] [Google Scholar]
- 138. Malik SS, Lythgoe MP, McPhail M, et al. Metachronous colorectal cancer following segmental or extended colectomy in Lynch syndrome: a systematic review and meta-analysis. Fam Cancer 2018;17:557–64. 10.1007/s10689-017-0062-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Natarajan N, Watson P, Silva-Lopez E, et al. Comparison of extended colectomy and limited resection in patients with Lynch syndrome. Dis Colon Rectum 2010;53:77–82. 10.1007/DCR.0b013e3181c702de [DOI] [PubMed] [Google Scholar]
- 140. de Vos tot Nederveen Cappel WH, Buskens E, van Duijvendijk P, et al. Decision analysis in the surgical treatment of colorectal cancer due to a mismatch repair gene defect. Gut 2003;52:1752–5. 10.1136/gut.52.12.1752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Haanstra JF, de Vos Tot Nederveen Cappel WH, Gopie JP, et al. Quality of life after surgery for colon cancer in patients with Lynch syndrome: partial versus subtotal colectomy. Dis Colon Rectum 2012;55:653–9. 10.1097/DCR.0b013e31824f5392 [DOI] [PubMed] [Google Scholar]
- 142. Maeda T, Cannom RR, Beart RW, et al. Decision model of segmental compared with total abdominal colectomy for colon cancer in hereditary nonpolyposis colorectal cancer. J Clin Oncol 2010;28:1175–80. 10.1200/JCO.2009.25.9812 [DOI] [PubMed] [Google Scholar]
- 143. Win AK, Parry S, Parry B, et al. Risk of metachronous colon cancer following surgery for rectal cancer in mismatch repair gene mutation carriers. Ann Surg Oncol 2013;20:1829–36. 10.1245/s10434-012-2858-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Kalady MF, Lipman J, McGannon E, et al. Risk of colonic neoplasia after proctectomy for rectal cancer in hereditary nonpolyposis colorectal cancer. Ann Surg 2012;255:1121–5. 10.1097/SLA.0b013e3182565c0b [DOI] [PubMed] [Google Scholar]
- 145. Capelle LG, Van Grieken NCT, Lingsma HF, et al. Risk and epidemiological time trends of gastric cancer in Lynch syndrome carriers in the Netherlands. Gastroenterology 2010;138:487–92. 10.1053/j.gastro.2009.10.051 [DOI] [PubMed] [Google Scholar]
- 146. Møller P, Seppälä TT, Bernstein I, et al. Cancer risk and survival in path_MMR carriers by gene and gender up to 75 years of age: a report from the Prospective Lynch Syndrome Database. Gut 2018;67:1306–16. 10.1136/gutjnl-2017-314057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Dicken BJ, Bigam DL, Cass C, et al. Gastric adenocarcinoma: review and considerations for future directions. Ann Surg 2005;24:27–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Renkonen-Sinisalo L, Sipponen P, Aarnio M, et al. No support for endoscopic surveillance for gastric cancer in hereditary non-polyposis colorectal cancer. Scand J Gastroenterol 2002;37:574–7. 10.1080/00365520252903134 [DOI] [PubMed] [Google Scholar]
- 149. ten Kate GL, Kleibeuker JH, Nagengast FM, et al. Is surveillance of the small bowel indicated for Lynch syndrome families? Gut 2007;56:1198–201. 10.1136/gut.2006.118299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Schulmann K, Brasch FE, Kunstmann E, et al. HNPCC-associated small bowel cancer: clinical and molecular characteristics. Gastroenterology 2005;128:590–9. 10.1053/j.gastro.2004.12.051 [DOI] [PubMed] [Google Scholar]
- 151. Cheung DY, Choi M-G. Current advance in small bowel tumors. Clin Endosc 2011;44:13–21. 10.5946/ce.2011.44.1.13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Saurin J-C, Pilleul F, Soussan EB, et al. Small-bowel capsule endoscopy diagnoses early and advanced neoplasms in asymptomatic patients with Lynch syndrome. Endoscopy 2010;42:1057–62. 10.1055/s-0030-1255742 [DOI] [PubMed] [Google Scholar]
- 153. Haanstra JF, Al-Toma A, Dekker E, et al. Prevalence of small-bowel neoplasia in Lynch syndrome assessed by video capsule endoscopy. Gut 2015;64:1578–83. 10.1136/gutjnl-2014-307348 [DOI] [PubMed] [Google Scholar]
- 154. Haanstra JF, Al-Toma A, Dekker E, et al. Incidence of small bowel neoplasia in Lynch syndrome assessed by video capsule endoscopy. Endosc Int Open 2017;5:E622–6. 10.1055/s-0043-111723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Kastrinos F, Mukherjee B, Tayob N, et al. Risk of pancreatic cancer in families with Lynch syndrome. JAMA 2009;302 10.1001/jama.2009.1529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Canto MI, Harinck F, Hruban RH, et al. International Cancer of the Pancreas Screening (CAPS) Consortium Summit on the management of patients with increased risk for familial pancreatic cancer. Gut 2013;62:339–47. 10.1136/gutjnl-2012-303108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Signoretti M, Bruno MJ, Zerboni G, et al. Results of surveillance in individuals at high-risk of pancreatic cancer: a systematic review and meta-analysis. United Eur Gastroenterol J 2018;6:489–99. 10.1177/2050640617752182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Carethers JM. Differentiating Lynch-like from Lynch syndrome. Gastroenterology 2014;146:602–4. 10.1053/j.gastro.2014.01.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Pai RK, Pai RK. A practical approach to the evaluation of gastrointestinal tract carcinomas for Lynch syndrome. Am J Surg Pathol 2016;40:e17–34. 10.1097/PAS.0000000000000620 [DOI] [PubMed] [Google Scholar]
- 160. Buchanan DD, Tan YY, Walsh MD, et al. Tumor mismatch repair immunohistochemistry and DNA MLH1 methylation testing of patients with endometrial cancer diagnosed at age younger than 60 years optimizes triage for population-level germline mismatch repair gene mutation testing. J Clin Oncol 2014;32:90–100. 10.1200/JCO.2013.51.2129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Rodríguez-Soler M, Pérez-Carbonell L, Guarinos C, et al. Risk of cancer in cases of suspected Lynch syndrome without germline mutation. Gastroenterology 2013;144:926–32. 10.1053/j.gastro.2013.01.044 [DOI] [PubMed] [Google Scholar]
- 162. Overbeek LIH, Kets CM, Hebeda KM, et al. Patients with an unexplained microsatellite instable tumour have a low risk of familial cancer. Br J Cancer 2007;96:1605–12. 10.1038/sj.bjc.6603754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Sourrouille I, Coulet F, Lefevre JH, et al. Somatic mosaicism and double somatic hits can lead to MSI colorectal tumors. Fam Cancer 2013;12:27–33. 10.1007/s10689-012-9568-9 [DOI] [PubMed] [Google Scholar]
- 164. Haraldsdottir S, Hampel H, Tomsic J, et al. Colon and endometrial cancers with mismatch repair deficiency can arise from somatic, rather than germline, mutations. Gastroenterology 2014;147:1308–16. 10.1053/j.gastro.2014.08.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Mensenkamp AR, Vogelaar IP, van Zelst–Stams WAG, et al. Somatic mutations in MLH1 and MSH2 are a frequent cause of mismatch-repair deficiency in Lynch syndrome-like tumors. Gastroenterology 2014;146:643–6. 10.1053/j.gastro.2013.12.002 [DOI] [PubMed] [Google Scholar]
- 166. Geurts-Giele WRR, Leenen CHM, Dubbink HJ, et al. Somatic aberrations of mismatch repair genes as a cause of microsatellite-unstable cancers. J Pathol 2014;234:548–59. 10.1002/path.4419 [DOI] [PubMed] [Google Scholar]
- 167. Morak M, Heidenreich B, Keller G, et al. Biallelic MUTYH mutations can mimic Lynch syndrome. Eur J Hum Genet 2014;22:1334–7. 10.1038/ejhg.2014.15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Elsayed FA, Kets CM, Ruano D, et al. Germline variants in POLE are associated with early onset mismatch repair deficient colorectal cancer. Eur J Hum Genet 2015;23:1080–4. 10.1038/ejhg.2014.242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Jansen AM, van Wezel T, van den Akker BE, et al. Combined mismatch repair and POLE/POLD1 defects explain unresolved suspected Lynch syndrome cancers. Eur J Hum Genet 2016;24:1089–92. 10.1038/ejhg.2015.252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Gupta S, Provenzale D, Regenbogen SE, et al. NCCN guidelines insights: genetic/familial high-risk assessment: colorectal, version 3.2017. J Natl Compr Canc Netw 2017;15:1465–75. 10.6004/jnccn.2017.0176 [DOI] [PubMed] [Google Scholar]
- 171. Georgiou D, Kiesel V, Brady AF, et al. Response to "Histology of colorectal adenocarcinoma with double somatic mismatch-repair mutations is indistinguishable from those caused by Lynch syndrome". Hum Pathol 2019;89:115–6. 10.1016/j.humpath.2019.04.015 [DOI] [PubMed] [Google Scholar]
- 172. Pearlman R, Frankel WL, Swanson B, et al. Prevalence and spectrum of germline cancer susceptibility gene mutations among patients with early-onset colorectal cancer. JAMA Oncol 2017;3 10.1001/jamaoncol.2016.5194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Mork ME, You YN, Ying J, et al. High prevalence of hereditary cancer syndromes in adolescents and young adults with colorectal cancer. J Clin Oncol 2015;33:3544–9. 10.1200/JCO.2015.61.4503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Stoffel EM, Koeppe E, Everett J, et al. Germline genetic features of young individuals with colorectal cancer. Gastroenterology 2018;154:897–905. 10.1053/j.gastro.2017.11.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Fuccio L, Rex D, Ponchon T, et al. New and recurrent colorectal cancers after resection: a systematic review and meta-analysis of endoscopic surveillance studies. Gastroenterology 2019;156:1309–23. 10.1053/j.gastro.2018.12.006 [DOI] [PubMed] [Google Scholar]
- 176. Samadder NJ, Curtin K, Wong J, et al. Epidemiology and familial risk of synchronous and metachronous colorectal cancer: a population-based study in Utah. Clin Gastroenterol Hepatol 2014;12:2078–84. 10.1016/j.cgh.2014.04.017 [DOI] [PubMed] [Google Scholar]
- 177. le Clercq CMC, Winkens B, Bakker CM, et al. Metachronous colorectal cancers result from missed lesions and non-compliance with surveillance. Gastrointest Endosc 2015;82:333.e2 10.1016/j.gie.2014.12.052 [DOI] [PubMed] [Google Scholar]
- 183. Rosty C, Brosens LAA, Dekker E, et al. WHO Classification of Tumours Editorial Board : Digestive system tumours. 1 5th edn Lyon (France: International Agency for Research on Cancer: Serrated Polyposis, 2019. http://publications.iarc.fr/579 [Google Scholar]
- 179. Egoavil C, Juárez M, Guarinos C, et al. Increased risk of colorectal cancer in patients with multiple serrated polyps and their first-degree relatives. Gastroenterology 2017;153:106–12. 10.1053/j.gastro.2017.04.003 [DOI] [PubMed] [Google Scholar]
- 180. Crowder CD, Sweet K, Lehman A, et al. Serrated polyposis is an underdiagnosed and unclear syndrome: the surgical pathologist has a role in improving detection. Am J Surg Pathol 2012;36:1178–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. IJspeert JEG, Rana SAQ, Atkinson NSS, et al. Clinical risk factors of colorectal cancer in patients with serrated polyposis syndrome: a multicentre cohort analysis. Gut 2017;66:278–84. 10.1136/gutjnl-2015-310630 [DOI] [PubMed] [Google Scholar]
- 182. Carballal S, Rodríguez-Alcalde D, Moreira L, et al. Colorectal cancer risk factors in patients with serrated polyposis syndrome: a large multicentre study. Gut 2016;65:1829–37. 10.1136/gutjnl-2015-309647 [DOI] [PubMed] [Google Scholar]
- 183. He EY, Wyld L, Sloane MA, et al. The molecular characteristics of colonic neoplasms in serrated polyposis: a systematic review and meta-analysis. J Pathol Clin Res 2016;2:127–37. 10.1002/cjp2.44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Yan HHN, Lai JCW, Ho SL, et al. RNF43 germline and somatic mutation in serrated neoplasia pathway and its association with BRAF mutation. Gut 2017;66:1645–56. 10.1136/gutjnl-2016-311849 [DOI] [PubMed] [Google Scholar]
- 185. Gala MK, Mizukami Y, Le LP, et al. Germline mutations in oncogene-induced senescence pathways are associated with multiple sessile serrated adenomas. Gastroenterology 2014;146:520–9. 10.1053/j.gastro.2013.10.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Buchanan DD, Clendenning M, Zhuoer L, et al. Lack of evidence for germline RNF43 mutations in patients with serrated polyposis syndrome from a large multinational study. Gut 2017;66:1170–2. 10.1136/gutjnl-2016-312773 [DOI] [PubMed] [Google Scholar]
- 187. Biswas S, Ellis AJ, Guy R, et al. High prevalence of hyperplastic polyposis syndrome (serrated polyposis) in the NHS bowel cancer screening programme. Gut 2013;62:475. 10.1136/gutjnl-2012-303233 [DOI] [PubMed] [Google Scholar]
- 188. Moreira L, Pellisé M, Carballal S, et al. High prevalence of serrated polyposis syndrome in FIT-based colorectal cancer screening programmes. Gut 2013;62:476–7. 10.1136/gutjnl-2012-303496 [DOI] [PubMed] [Google Scholar]
- 189. Rivero-Sanchez L, Lopez-Ceron M, Carballal S, et al. Reassessment colonoscopy to diagnose serrated polyposis syndrome in a colorectal cancer screening population. Endoscopy 2017;49:44–53. 10.1055/s-0042-115640 [DOI] [PubMed] [Google Scholar]
- 190. East JE, Atkin WS, Bateman AC, et al. British Society of Gastroenterology position statement on serrated polyps in the colon and rectum. Gut 2017;66:1181–96. 10.1136/gutjnl-2017-314005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Boparai KS, Mathus-Vliegen EMH, Koornstra JJ, et al. Increased colorectal cancer risk during follow-up in patients with hyperplastic polyposis syndrome: a multicentre cohort study. Gut 2010;59:1094–100. 10.1136/gut.2009.185884 [DOI] [PubMed] [Google Scholar]
- 192. Edelstein DL, Axilbund JE, Hylind LM, et al. Serrated polyposis: rapid and relentless development of colorectal neoplasia. Gut 2013;62:404–8. 10.1136/gutjnl-2011-300514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193. MacPhail ME, Thygesen SB, Patel N, et al. Endoscopic control of polyp burden and expansion of surveillance intervals in serrated polyposis syndrome. Gastrointest Endosc 2019;90:96–100. 10.1016/j.gie.2018.11.016 [DOI] [PubMed] [Google Scholar]
- 194. Bleijenberg AG, IJspeert JE, van Herwaarden YJ, et al. Personalised surveillance for serrated polyposis syndrome: results from a prospective 5-year international cohort study. Gut 2020;69:112–21. 10.1136/gutjnl-2018-318134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Hazewinkel Y, Koornstra J-J, Boparai KS, et al. Yield of screening colonoscopy in first-degree relatives of patients with serrated polyposis syndrome. J Clin Gastroenterol 2015;49:407–12. 10.1097/MCG.0000000000000103 [DOI] [PubMed] [Google Scholar]
- 196. Oquiñena S, Guerra A, Pueyo A, et al. Serrated polyposis: prospective study of first-degree relatives. Eur J Gastroenterol Hepatol 2013;25:28–32. 10.1097/MEG.0b013e3283598506 [DOI] [PubMed] [Google Scholar]
- 197. Boparai KS, Reitsma JB, Lemmens V, et al. Increased colorectal cancer risk in first-degree relatives of patients with hyperplastic polyposis syndrome. Gut 2010;59:1222–5. 10.1136/gut.2009.200741 [DOI] [PubMed] [Google Scholar]
- 198. Win AK, Walters RJ, Buchanan DD, et al. Cancer risks for relatives of patients with serrated polyposis. Am J Gastroenterol 2012;107:770–8. 10.1038/ajg.2012.52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199. Alexander J, Johnston B, Smith T, et al. OWE-25 patients with multiple adenomas in bowel cancer screening program are not referred for genetic testing. Gut 2019;68:A188. [Google Scholar]
- 200. Cubiella J, Carballo F, Portillo I, et al. Incidence of advanced neoplasia during surveillance in high- and intermediate-risk groups of the European colorectal cancer screening guidelines. Endoscopy 2016;48:995–1002. 10.1055/s-0042-112571 [DOI] [PubMed] [Google Scholar]
- 201. Grover S, Kastrinos F, Steyerberg EW, et al. Prevalence and phenotypes of APC and MUTYH mutations in patients with multiple colorectal adenomas. JAMA 2012;308:485–92. 10.1001/jama.2012.8780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202. Spier I, Holzapfel S, Altmüller J, et al. Frequency and phenotypic spectrum of germline mutations in POLE and seven other polymerase genes in 266 patients with colorectal adenomas and carcinomas. Int. J. Cancer 2015;137:320–31. 10.1002/ijc.29396 [DOI] [PubMed] [Google Scholar]
- 203. Cheng THT, Gorman M, Martin L, et al. Common colorectal cancer risk alleles contribute to the multiple colorectal adenoma phenotype, but do not influence colonic polyposis in FAP. Eur J Hum Genet 2015;23:260–3. 10.1038/ejhg.2014.74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204. Tieu AH, Edelstein D, Axilbund J, et al. Clinical characteristics of multiple colorectal adenoma patients without germline APC or MYH mutations. J Clin Gastroenterol 2016;50:584–8. 10.1097/MCG.0000000000000416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205. Kallenberg FGJ, Latchford A, Lips NC, et al. Duodenal adenomas in patients with multiple colorectal adenomas without germline APC or MUTYH mutations. Dis Colon Rectum 2018;61:58–66. 10.1097/DCR.0000000000000868 [DOI] [PubMed] [Google Scholar]
- 206. Spier I, Drichel D, Kerick M, et al. Low-level APC mutational mosaicism is the underlying cause in a substantial fraction of unexplained colorectal adenomatous polyposis cases. J Med Genet 2016;53:172–9. 10.1136/jmedgenet-2015-103468 [DOI] [PubMed] [Google Scholar]
- 207. Vasen HFA, Möslein G, Alonso A, et al. Guidelines for the clinical management of familial adenomatous polyposis (FAP). Gut 2008;57:704–13. 10.1136/gut.2007.136127 [DOI] [PubMed] [Google Scholar]
- 208. Hyer W, Cohen S, Attard T, et al. Management of familial adenomatous polyposis in children and adolescents: position paper from the ESPGHAN Polyposis Working Group. J Pediatr Gastroenterol Nutr 2019;68:428–41. [DOI] [PubMed] [Google Scholar]
- 209. Bülow S. Clinical features in familial polyposis coli. Results of the Danish polyposis register. Dis Colon Rectum 1986;29:102–7. 10.1007/bf02555389 [DOI] [PubMed] [Google Scholar]
- 210. Järvinen HJ, Husa A, Aukee S, et al. Finnish Registry for familial adenomatosis coli. Scand J Gastroenterol 1984;19:941–6. 10.1080/00365521.1984.12005837 [DOI] [PubMed] [Google Scholar]
- 211. Vasen HF, Griffioen G, Offerhaus GJ, et al. The value of screening and central registration of families with familial adenomatous polyposis. A study of 82 families in the Netherlands. Dis Colon Rectum 1990;33:227–30. 10.1007/bf02134185 [DOI] [PubMed] [Google Scholar]
- 212. Nugent KP, Spigelman AD, Phillips RK. Life expectancy after colectomy and ileorectal anastomosis for familial adenomatous polyposis. Dis Colon Rectum 1993;36:1059–62. 10.1007/BF02047300 [DOI] [PubMed] [Google Scholar]
- 213. Belchetz LA, Berk T, Bapat BV, et al. Changing causes of mortality in patients with familial adenomatous polyposis. Dis Colon Rectum 1996;39:384–7. 10.1007/BF02054051 [DOI] [PubMed] [Google Scholar]
- 214. Sarvepalli S, Burke CA, Monachese M, et al. Natural history of colonic polyposis in young patients with familial adenomatous polyposis. Gastrointest Endosc 2018;88:726–33. 10.1016/j.gie.2018.05.021 [DOI] [PubMed] [Google Scholar]
- 215. Bulow S, et al. Duodenal adenomatosis in familial adenomatous polyposis. Gut 2004;53:381–6. 10.1136/gut.2003.027771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216. Vasen HF, Bülow S, Myrhøj T, et al. Decision analysis in the management of duodenal adenomatosis in familial adenomatous polyposis. Gut 1997;40:716–9. 10.1136/gut.40.6.716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217. Bülow S, Christensen IJ, Højen H, et al. Duodenal surveillance improves the prognosis after duodenal cancer in familial adenomatous polyposis. Colorectal Dis 2012;14:947–52. 10.1111/j.1463-1318.2011.02844.x [DOI] [PubMed] [Google Scholar]
- 218. Spigelman AD, Williams CB, Talbot IC, et al. Upper gastrointestinal cancer in patients with familial adenomatous polyposis. Lancet 1989;2:783–5. 10.1016/S0140-6736(89)90840-4 [DOI] [PubMed] [Google Scholar]
- 219. Groves CJ, Saunders BP, Spigelman AD, et al. Duodenal cancer in patients with familial adenomatous polyposis (FAP): results of a 10 year prospective study. Gut 2002;50:636–41. 10.1136/gut.50.5.636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220. Kashiwagi H, Spigelman AD, Debinski HS, et al. Surveillance of ampullary adenomas in familial adenomatous polyposis. Lancet 1994;344:1582 10.1016/S0140-6736(94)90395-6 [DOI] [PubMed] [Google Scholar]
- 221. Latchford AR, Neale KF, Spigelman AD, et al. Features of duodenal cancer in patients with familial adenomatous polyposis. Clin Gastroenterol Hepatol 2009;7:659–63. 10.1016/j.cgh.2009.02.028 [DOI] [PubMed] [Google Scholar]
- 222. Dekker E, Boparai KS, Poley JW, et al. High resolution endoscopy and the additional value of chromoendoscopy in the evaluation of duodenal adenomatosis in patients with familial adenomatous polyposis. Endoscopy 2009;41:666–9. 10.1055/s-0029-1214980 [DOI] [PubMed] [Google Scholar]
- 223. Hurley JJ, Thomas LE, Walton S-J, et al. The impact of chromoendoscopy for surveillance of the duodenum in patients with MUTYH-associated polyposis and familial adenomatous polyposis. Gastrointest Endosc 2018;88:665–73. 10.1016/j.gie.2018.04.2347 [DOI] [PubMed] [Google Scholar]
- 224. Thiruvengadam SS, Lopez R, O’Malley M, et al. Spigelman stage IV duodenal polyposis does not precede most duodenal cancer cases in patients with familial adenomatous polyposis. Gastrointest Endosc 2019;89:345–54. 10.1016/j.gie.2018.07.033 [DOI] [PubMed] [Google Scholar]
- 225. Moussata D, Napoleon B, Lepilliez V, et al. Endoscopic treatment of severe duodenal polyposis as an alternative to surgery for patients with familial adenomatous polyposis. Gastrointest Endosc 2014;80:817–25. 10.1016/j.gie.2014.03.012 [DOI] [PubMed] [Google Scholar]
- 226. Jaganmohan S, Lynch PM, Raju RP, et al. Endoscopic management of duodenal adenomas in familial adenomatous polyposis–a single-center experience. Dig Dis Sci 2012;57:732–7. 10.1007/s10620-011-1917-2 [DOI] [PubMed] [Google Scholar]
- 227. Ma T, Jang EJ, Zukerberg LR, et al. Recurrences are common after endoscopic ampullectomy for adenoma in the familial adenomatous polyposis (FAP) syndrome. Surg Endosc 2014;28:2349–56. 10.1007/s00464-014-3467-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228. Bianchi LK, Burke CA, Bennett AE, et al. Fundic gland polyp dysplasia is common in familial adenomatous polyposis. Clin Gastroenterol Hepatol 2008;6:180–5. 10.1016/j.cgh.2007.11.018 [DOI] [PubMed] [Google Scholar]
- 229. Walton S-J, Frayling IM, Clark SK, et al. Gastric tumours in FAP. Fam Cancer 2017;16:363–9. 10.1007/s10689-017-9966-0 [DOI] [PubMed] [Google Scholar]
- 230. Mankaney G, Leone P, Cruise M, et al. Gastric cancer in FAP: a concerning rise in incidence. Fam Cancer 2017;16:371–6. 10.1007/s10689-017-9971-3 [DOI] [PubMed] [Google Scholar]
- 231. Wallis YL, Macdonald F, Hultén M, et al. Genotype-phenotype correlation between position of constitutional APC gene mutation and CHRPE expression in familial adenomatous polyposis. Hum Genet 1994;94:543–8. 10.1007/BF00211023 [DOI] [PubMed] [Google Scholar]
- 232. Traboulsi EI, Krush AJ, Gardner EJ, et al. Prevalence and importance of pigmented ocular fundus lesions in Gardner's syndrome. N Engl J Med 1987;316:661–7. 10.1056/NEJM198703123161104 [DOI] [PubMed] [Google Scholar]
- 233. Bülow S, Bülow C, Vasen H, et al. Colectomy and ileorectal anastomosis is still an option for selected patients with familial adenomatous polyposis. Dis Colon Rectum 2008;51:1318–23. 10.1007/s10350-008-9307-3 [DOI] [PubMed] [Google Scholar]
- 234. Church J, Burke C, McGannon E, et al. Risk of rectal cancer in patients after colectomy and ileorectal anastomosis for familial adenomatous polyposis: a function of available surgical options. Dis Colon Rectum 2003;46:1175–81. 10.1007/s10350-004-6710-2 [DOI] [PubMed] [Google Scholar]
- 235. Sinha A, Tekkis PP, Rashid S, et al. Risk factors for secondary proctectomy in patients with familial adenomatous polyposis. Br J Surg 2010;97:1710–5. 10.1002/bjs.7202 [DOI] [PubMed] [Google Scholar]
- 236. Church J, Burke C, McGannon E, et al. Predicting polyposis severity by proctoscopy: how reliable is it? Dis Colon Rectum 2001;44:1249–54. 10.1007/bf02234779 [DOI] [PubMed] [Google Scholar]
- 237. Nieuwenhuis MH, Bülow S, Björk J, et al. Genotype predicting phenotype in familial adenomatous polyposis: a practical application to the choice of surgery. Dis Colon Rectum 2009;52:1259–63. 10.1007/DCR.0b013e3181a0d33b [DOI] [PubMed] [Google Scholar]
- 238. Newton KF, Mallinson EKL, Bowen J, et al. Genotype-phenotype correlation in colorectal polyposis. Clin Genet 2012;81:521–31. 10.1111/j.1399-0004.2011.01740.x [DOI] [PubMed] [Google Scholar]
- 239. Aziz O, Athanasiou T, Fazio VW, et al. Meta-analysis of observational studies of ileorectal versus ileal pouch-anal anastomosis for familial adenomatous polyposis. Br J Surg 2006;93:407–17. 10.1002/bjs.5276 [DOI] [PubMed] [Google Scholar]
- 240. KØ O, Juul S, Bülow S, et al. Female fecundity before and after operation for familial adenomatous polyposis. Br J Surg 2003;90:227–31. [DOI] [PubMed] [Google Scholar]
- 241. Nieuwenhuis MH, Douma KF, Bleiker EM, et al. Female fertility after colorectal surgery for familial adenomatous polyposis: a nationwide cross-sectional study. Ann Surg 2010;252:341–4. [DOI] [PubMed] [Google Scholar]
- 242. von Roon AC, Tekkis PP, Lovegrove RE, et al. Comparison of outcomes of ileal pouch-anal anastomosis for familial adenomatous polyposis with and without previous ileorectal anastomosis. Br J Surg 2008;95:494–8. 10.1002/bjs.6005 [DOI] [PubMed] [Google Scholar]
- 243. von Roon AC, Will OCC, Man RF, et al. Mucosectomy with handsewn anastomosis reduces the risk of adenoma formation in the anorectal segment after restorative proctocolectomy for familial adenomatous polyposis. Ann Surg 2011;253:314–7. 10.1097/SLA.0b013e318f3f498 10.1097/SLA.0b013e318f3f498 [DOI] [PubMed] [Google Scholar]
- 244. van Duijvendijk P, Vasen HF, Bertario L, et al. Cumulative risk of developing polyps or malignancy at the ileal pouch-anal anastomosis in patients with familial adenomatous polyposis. J Gastrointest Surg 1999;3:325–30. 10.1016/S1091-255X(99)80075-4 [DOI] [PubMed] [Google Scholar]
- 245. Friederich P, de Jong AE, Mathus-Vliegen LM, et al. Risk of developing adenomas and carcinomas in the ileal pouch in patients with familial adenomatous polyposis. Clin Gastroenterol Hepatol 2008;6:1237–42. 10.1016/j.cgh.2008.06.011 [DOI] [PubMed] [Google Scholar]
- 246. Smith JC, Schäffer MW, Ballard BR, et al. Adenocarcinomas after prophylactic surgery for familial adenomatous polyposis. J Cancer Ther 2013;04:260–70. 10.4236/jct.2013.41033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247. Boostrom SY, Mathis KL, Pendlimari R, et al. Risk of neoplastic change in ileal pouches in familial adenomatous polyposis. J Gastrointest Surg 2013;17:1804–8. 10.1007/s11605-013-2319-x [DOI] [PubMed] [Google Scholar]
- 248. Nieuwenhuis MH, Lefevre JH, Bülow S, et al. Family history, surgery, and APC mutation are risk factors for desmoid tumors in familial adenomatous polyposis: an international cohort study. Dis Colon Rectum 2011;54:1229–34. 10.1097/DCR.0b013e318227e4e8 [DOI] [PubMed] [Google Scholar]
- 249. Speake D, Evans DG, Lalloo F, et al. Desmoid tumours in patients with familial adenomatous polyposis and desmoid region adenomatous polyposis coli mutations. Br J Surg 2007;94:1009–13. 10.1002/bjs.5633 [DOI] [PubMed] [Google Scholar]
- 250. Kurup AN, Callstrom MR. Expanding role of percutaneous ablative and consolidative treatments for musculoskeletal tumours. Clin Radiol 2017;72:645–56. 10.1016/j.crad.2017.02.019 [DOI] [PubMed] [Google Scholar]
- 251. Church J, Berk T, Boman BM, et al. Staging intra-abdominal desmoid tumors in familial adenomatous polyposis: a search for a uniform approach to a troubling disease. Diseases of the Colon & Rectum 2005;48:1528–34. 10.1007/s10350-005-0018-8 [DOI] [PubMed] [Google Scholar]
- 252. Quast DR, Schneider R, Burdzik E, et al. Long-term outcome of sporadic and FAP-associated desmoid tumors treated with high-dose selective estrogen receptor modulators and sulindac: a single-center long-term observational study in 134 patients. Fam Cancer 2016;15:31–40. 10.1007/s10689-015-9830-z [DOI] [PubMed] [Google Scholar]
- 253. Gega M, Yanagi H, Yoshikawa R, et al. Successful chemotherapeutic modality of doxorubicin plus dacarbazine for the treatment of desmoid tumors in association with familial adenomatous polyposis. JCO 2006;24:102–5. 10.1200/JCO.2005.02.1923 [DOI] [PubMed] [Google Scholar]
- 254. Skapek SX, Ferguson WS, Granowetter L, et al. Vinblastine and methotrexate for desmoid fibromatosis in children: results of a pediatric Oncology Group phase II trial. JCO 2007;25:501–6. 10.1200/JCO.2006.08.2966 [DOI] [PubMed] [Google Scholar]
- 255. Cleary SP, Cotterchio M, Jenkins MA, et al. Germline MutY human homologue mutations and colorectal cancer: a multisite case-control study. Gastroenterology 2009;136:1251–60. 10.1053/j.gastro.2008.12.050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256. Lubbe SJ, Di Bernardo MC, Chandler IP, et al. Clinical Implications of the colorectal cancer risk associated with MUTYH mutation. JCO 2009;27:3975–80. 10.1200/JCO.2008.21.6853 [DOI] [PubMed] [Google Scholar]
- 257. Nieuwenhuis MH, Vogt S, Jones N, et al. Evidence for accelerated colorectal adenoma–carcinoma progression in MUTYH-associated polyposis? Gut 2012;61:734–8. 10.1136/gut.2010.229104 [DOI] [PubMed] [Google Scholar]
- 258. Croitoru ME, Cleary SP, Di Nicola N, et al. Association between biallelic and monoallelic germline MYH gene mutations and colorectal cancer risk. J Natl Cancer Inst 2004;96:1631–4. 10.1093/jnci/djh288 [DOI] [PubMed] [Google Scholar]
- 259. Farrington SM, Tenesa A, Barnetson R, et al. Germline susceptibility to colorectal cancer due to base-excision repair gene defects. The American Journal of Human Genetics 2005;77:112–9. 10.1086/431213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260. Knopperts AP, Nielsen M, Niessen RC, et al. Contribution of bi-allelic germline MUTYH mutations to early-onset and familial colorectal cancer and to low number of adenomatous polyps: case-series and literature review. Fam Cancer 2013;12:43–50. 10.1007/s10689-012-9570-2 [DOI] [PubMed] [Google Scholar]
- 261. Syngal S, Brand RE, Church JM, et al. ACG clinical guideline: genetic testing and management of hereditary gastrointestinal cancer syndromes. Am J Gastroenterol 2015;110:223–62. quiz 263 10.1038/ajg.2014.435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262. Walton S-J, Kallenberg FGJ, Clark SK, et al. Frequency and features of duodenal adenomas in patients with MUTYH-associated polyposis. Clin Gastroenterol Hepatol 2016;14:986–92. 10.1016/j.cgh.2016.02.020 [DOI] [PubMed] [Google Scholar]
- 263. Vogt S, Jones N, Christian D, et al. Expanded extracolonic tumor spectrum in MUTYH-associated polyposis. Gastroenterology 2009;137:1976–85. 10.1053/j.gastro.2009.08.052 [DOI] [PubMed] [Google Scholar]
- 264. Thomas LE, Hurley JJ, Meuser E, et al. Burden and profile of somatic mutation in duodenal adenomas from patients with familial adenomatous- and MUTYH-associated polyposis. Clin Cancer Res 2017;23:6721–32. 10.1158/1078-0432.CCR-17-1269 [DOI] [PubMed] [Google Scholar]
- 265. Latchford A, Cohen S, Auth M, et al. Management of Peutz-Jeghers syndrome in children and adolescents: a position paper from the ESPGHAN Polyposis Working Group. J Pediatr Gastroenterol Nutr 2019;68:442–52. [DOI] [PubMed] [Google Scholar]
- 266. Latchford AR, Neale K, Phillips RKS, et al. Peutz-Jeghers syndrome: intriguing suggestion of gastrointestinal cancer prevention from surveillance. Dis Colon Rectum 2011;54:1547–51. 10.1097/DCR.0b013e318233a11f [DOI] [PubMed] [Google Scholar]
- 267. Hearle N, Schumacher V, Menko FH, et al. Frequency and spectrum of cancers in the Peutz-Jeghers syndrome. Clin Cancer Res 2006;12:3209–15. 10.1158/1078-0432.CCR-06-0083 [DOI] [PubMed] [Google Scholar]
- 268. Hinds R, Philp C, Hyer W, et al. Complications of childhood Peutz-Jeghers syndrome: implications for pediatric screening. J Pediatr Gastroenterol Nutr 2004;39:219–20. 10.1097/00005176-200408000-00027 [DOI] [PubMed] [Google Scholar]
- 269. van Lier MGF, Mathus-Vliegen EMH, Wagner A, et al. High cumulative risk of intussusception in patients with Peutz-Jeghers syndrome: time to update surveillance guidelines? Am J Gastroenterol 2011;106:940–5. 10.1038/ajg.2010.473 [DOI] [PubMed] [Google Scholar]
- 270. Beggs AD, Latchford AR, Vasen HFA, et al. Peutz-Jeghers syndrome: a systematic review and recommendations for management. Gut 2010;59:975–86. 10.1136/gut.2009.198499 [DOI] [PubMed] [Google Scholar]
- 271. Mata A, Llach J, Castells A, et al. A prospective trial comparing wireless capsule endoscopy and barium contrast series for small-bowel surveillance in hereditary Gi polyposis syndromes. Gastrointest Endosc 2005;61:721–5. 10.1016/S0016-5107(05)00289-0 [DOI] [PubMed] [Google Scholar]
- 272. Kurugoglu S, Aksoy H, Kantarci F, et al. Radiological work-up in Peutz-Jeghers syndrome. Pediatr Radiol 2003;33:766–71. [DOI] [PubMed] [Google Scholar]
- 273. Brown G, Fraser C, Schofield G, et al. Video capsule endoscopy in Peutz-Jeghers syndrome: a blinded comparison with barium follow-through for detection of small-bowel polyps. Endoscopy 2006;38:385–90. 10.1055/s-2006-925028 [DOI] [PubMed] [Google Scholar]
- 274. Caspari R, von Falkenhausen M, Krautmacher C, et al. Comparison of capsule endoscopy and magnetic resonance imaging for the detection of polyps of the small intestine in patients with familial adenomatous polyposis or with Peutz-Jeghers’ syndrome. Endoscopy 2004;36:1054–9. 10.1055/s-2004-826041 [DOI] [PubMed] [Google Scholar]
- 275. Maccioni F, Al Ansari N, Mazzamurro F, et al. Surveillance of patients affected by Peutz-Jeghers syndrome: diagnostic value of MR enterography in prone and supine position. Abdom Imaging 2012;37:279–87. 10.1007/s00261-011-9739-4 [DOI] [PubMed] [Google Scholar]
- 276. Gupta A, Postgate AJ, Burling D, et al. A prospective study of MR enterography versus capsule endoscopy for the surveillance of adult patients with Peutz-Jeghers syndrome. AJR Am J Roentgenol 2010;195:108–16. 10.2214/AJR.09.3174 [DOI] [PubMed] [Google Scholar]
- 277. Oncel M, Remzi FH, Church JM, et al. Benefits of 'clean sweep' in Peutz-Jeghers patients. Colorect Dis 2004;6:332–5. 10.1111/j.1463-1318.2004.00623.x [DOI] [PubMed] [Google Scholar]
- 278. Edwards DP, Khosraviani K, Stafferton R, et al. Long-term results of polyp clearance by intraoperative enteroscopy in the Peutz-Jeghers syndrome. Dis Colon Rectum 2003;46:48–50. 10.1007/s10350-004-6495-3 [DOI] [PubMed] [Google Scholar]
- 279. Cohen S, Hyer W, Mas E, et al. Management of juvenile polyposis syndrome in children and adolescents. J Pediatr Gastroenterol Nutr 2019;68:453–62. 10.1097/MPG.0000000000002246 [DOI] [PubMed] [Google Scholar]
- 280. Latchford AR, Neale K, Phillips RKS, et al. Juvenile polyposis syndrome: a study of genotype, phenotype, and long-term outcome. Dis Colon Rectum 2012;55:1038–43. [DOI] [PubMed] [Google Scholar]
- 281. Jass JR, Williams CB, Bussey HJ, et al. Juvenile polyposis--a precancerous condition. Histopathology 1988;13:619–30. 10.1111/j.1365-2559.1988.tb02093.x [DOI] [PubMed] [Google Scholar]
- 282. Brosens LAA, van Hattem A, Hylind LM, et al. Risk of colorectal cancer in juvenile polyposis. Gut 2007;56:965–7. 10.1136/gut.2006.116913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283. Järvinen HJ, Sipponen P. Gastroduodenal polyps in familial adenomatous and juvenile polyposis. Endoscopy 1986;18:230–4. 10.1055/s-2007-1018386 [DOI] [PubMed] [Google Scholar]
- 284. Howe JR, Mitros FA, Summers RW. The risk of gastrointestinal carcinoma in familial juvenile polyposis. Ann Surg Oncol 1998;5:751–6. 10.1007/BF02303487 [DOI] [PubMed] [Google Scholar]
- 285. Ma C, Giardiello FM, Montgomery EA. Upper tract juvenile polyps in juvenile polyposis patients: dysplasia and malignancy are associated with foveolar, intestinal, and pyloric differentiation. Am J Surg Pathol 2014;38:1618–26. 10.1097/PAS.0000000000000283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286. Woodford-Richens K, Bevan S, Churchman M, et al. Analysis of genetic and phenotypic heterogeneity in juvenile polyposis. Gut 2000;46:656–60. 10.1136/gut.46.5.656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287. Friedl W, Uhlhaas S, Schulmann K, et al. Juvenile polyposis: massive gastric polyposis is more common in MADH4 mutation carriers than in BMPR1A mutation carriers. Hum Genet 2002;111:108–11. 10.1007/s00439-002-0748-9 [DOI] [PubMed] [Google Scholar]
- 288. Aretz S, Stienen D, Uhlhaas S, et al. High proportion of large genomic deletions and a genotype phenotype update in 80 unrelated families with juvenile polyposis syndrome. J Med Genet 2007;44:702–9. 10.1136/jmg.2007.052506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289. Baert AL, Casteels-Van Daele M, Broeckx J, et al. Generalized juvenile polyposis with pulmonary arteriovenous malformations and hypertrophic osteoarthropathy. AJR Am J Roentgenol 1983;141:661–2. 10.2214/ajr.141.4.661 [DOI] [PubMed] [Google Scholar]
- 290. Wain KE, Ellingson MS, McDonald J, et al. Appreciating the broad clinical features of Smad4 mutation carriers: a multicenter chart review. Genet Med 2014;16:588–93. 10.1038/gim.2014.5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291. Heald B, Rigelsky C, Moran R, et al. Prevalence of thoracic aortopathy in patients with juvenile polyposis syndrome-hereditary hemorrhagic telangiectasia due to Smad4. Am J Med Genet A 2015;167A:1758–62. 10.1002/ajmg.a.37093 [DOI] [PubMed] [Google Scholar]
- 292. Shovlin CL, Condliffe R, Donaldson JW, et al. British Thoracic Society clinical statement on pulmonary arteriovenous malformations. Thorax 2017;72:1154–63. 10.1136/thoraxjnl-2017-210764 [DOI] [PubMed] [Google Scholar]
- 293. Menko FH, Kneepkens CMF, de Leeuw N, et al. Variable phenotypes associated with 10q23 microdeletions involving the PTEN and BMPR1A genes. Clin Genet 2008;74:145–54. 10.1111/j.1399-0004.2008.01026.x [DOI] [PubMed] [Google Scholar]
- 294. Delnatte C, Sanlaville D, Mougenot J-F, et al. Contiguous gene deletion within chromosome arm 10q is associated with juvenile polyposis of infancy, reflecting cooperation between the BMPR1A and PTEN tumor-suppressor genes. Am J Hum Genet 2006;78:1066–74. 10.1086/504301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295. Dahdaleh FS, Carr JC, Calva D, et al. Juvenile polyposis and other intestinal polyposis syndromes with microdeletions of chromosome 10q22-23. Clin Genet 2012;81:110–6. 10.1111/j.1399-0004.2011.01763.x [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
gutjnl-2019-319915supp001.pdf (78.1KB, pdf)
gutjnl-2019-319915supp002.pdf (220.8KB, pdf)