

Abstract
A craniotomy involves the removal of a skull fragment to access the brain, such as during tumor or epilepsy surgery, which is immediately replaced intra-operatively. The infection incidence after craniotomy ranges from 0.8–3%, with approximately half caused by Staphylococcus aureus (S. aureus). To mitigate infectious complications following craniotomy, we engineered a 3D bioprinted bone scaffold to harness the potent antibacterial activity of macrophages (MΦs) together with antibiotics using a mouse S. aureus craniotomy-associated biofilm model that establishes persistent infection on the bone flap, subcutaneous galea, and brain. The 3D scaffold contained rifampin and daptomycin printed in a composite slurry, with viable MΦs incorporated into a hydrogel-based bioink, which was assessed for both the treatment and prevention of craniotomy-associated infections in the mouse model. For the treatment paradigm, the bone flap was removed at day 7 post-infection after a mature biofilm had formed, and replaced with a 3D printed antibiotic scaffold, with or without MΦ incorporation. Bacterial burdens in the galea and brain were reduced by at least 100-fold at early time points, which was potentiated by bioprinting viable MΦs into the 3D antibiotic scaffold. We also examined a prevention paradigm, where scaffolds were placed at the time of surgery and challenged with S. aureus one day later at the surgical site. Interestingly, unlike the treatment paradigm, the incorporation of viable MΦs into the 3D antibiotic scaffold did not enhance bacterial clearance compared to antibiotic alone. With further refinement, our 3D bioprinted scaffold represents a potential treatment modality, since it delivers therapeutic antibiotic levels more rapidly than systemic administration, based on its proximity to the infection site. In addition, the incorporation of viable MΦs into the 3D scaffold is an important advance, which demonstrated improved therapeutic benefit for the treatment of established biofilms that represent the most clinically challenging scenario.
Keywords: Craniotomy, Staphylococcus aureus, biofilm infection, 3D bioprinting, macrophage
BACKGROUND
Decompressive craniectomy refers to the surgical removal of a portion of the skull following traumatic brain injury (TBI) or other head-related injuries, such as stroke or cranial hemorrhage, to control subsequent brain edema and prevent death1. Upon removal, the bone fragment (or bone flap) is cryopreserved or implanted subcutaneously in the abdominal wall of the patient for future replacement once brain edema has subsided. A craniotomy refers to the temporary removal of a skull fragment to access the brain, such as during tumor resection or epilepsy treatment, which is replaced intra-operatively. The incidence of infection after craniotomy/craniectomy ranges from 0.8–3% in the modern surgical era, with approximately half attributed to Staphylococcus aureus (S. aureus), which forms a biofilm on the native bone2–3. Several factors have been identified that increase the risk for infectious complications after craniotomy, including the presence of another infection, number of operations, cerebrospinal fluid (CSF) leakage, extent of CSF drainage, and venous sinus entry. It has been proposed that CSF leakage promotes the retrograde movement of bacteria, resulting in intracranial infection, which increases intracranial pressure and further CSF leakage, perpetuating a vicious cycle4.
Historically, the standard-of-care for managing bone flap infection following craniotomy has been intra-operative debridement of the affected tissue and bone flap removal, whereupon after a prolonged course of antibiotic treatment, a cranioplasty is performed. However, some studies have reported successful regimens for salvaging infected bone flaps, which include aggressive debridement of the surgical site, scrubbing and soaking the bone flap in povidone-iodine or other antiseptics, and in some instances indwelling antibiotic irrigations systems have been used to bathe the bone flap in antibiotics upon reinsertion5–8. In all of these studies, patients received i.v. antibiotics on average for one week followed by a longer duration of oral antibiotics (i.e. 2–3 months)5–8. Although these studies have shown good efficacy in preventing infection recurrence, they report small sample sizes (i.e. ≤ 14 subjects) and have not yet been adapted into mainstream clinical practice. In instances where the infected bone flap cannot be salvaged, patients are typically subjected to at least two additional surgeries. The first is to remove the infected bone, and after a variable period of antibiotic treatment that can last for weeks-months, a second surgery occurs to seal the cranial cavity with an alloplastic prosthesis or autologous bone graft9. Prolonged absence of the skull flap during antibiotic treatment can lead to “syndrome of the trephined” in approximately 13% of patients, which can include headache, seizures, mood imbalances, and behavioral disturbances10. Treatment of trephine syndrome is replacement of the original bone flap or synthetic device11–12; however, this cannot occur until convincing evidence exists that any residual infection has been eliminated, and some patients experience lingering cognitive impairment. Although less common, patients with craniotomy/craniectomy infections can experience chronic seizures and focal neurological deficits13. Despite the extensive steps taken both pre- and post-operatively to prevent infectious complications following craniotomy, including surgical scrub techniques, peri-operative antibiotics, post-operative wound care, and discontinuation of wound healing retardant medications, infections still occur and some require the bone flap to be discarded. Here we sought to evaluate the utility of a 3D bioprinted bone scaffold as a proof-of-concept for sustained local antibiotic delivery in combination with the incorporation of immune cells to augment bacterial clearance.
This study utilized a mouse model of S. aureus craniotomy-associated biofilm, which mimics aspects of craniotomy/craniectomy-related infections in humans, based on ultrastructural features of the biofilm, magnetic resonance imaging (MRI) presentation, and persistence14. In the current report, we developed a 3D printed bone scaffold to prevent or treat these infections by incorporating sustained-release antibiotics into the scaffold in combination with macrophages (MΦ) that possess potent anti-microbial activity. Prior work from our laboratory demonstrated that the introduction of exogenous MΦs into sites of S. aureus biofilm infection were capable of partially reducing bacterial burdens by promoting biofilm dispersal15. Biofilms are bacterial communities encased in a self-produced matrix composed of extracellular DNA, polysaccharides, and proteins that are difficult to treat with antibiotics based on their metabolic dormancy16. Once MΦ-mediated biofilm dispersal is initiated, bacteria may become antibiotic-sensitive, since most currently available antibiotics rely on active cell division and protein synthesis, both of which are limited in intact biofilms. Nevertheless, additional factors can influence the metabolic state of bacteria (i.e. oxygen and nutrient availability)17 and their potential to maintain antibiotic recalcitrance following biofilm dispersal must also be considered.
Another complication of craniotomy-associated infection is recurrence, since residual biofilm remains after the infected bone flap is removed. Our 3D bioprinted antibiotic scaffold features sustained antibiotic release directed to the site of infection, which was magnified by the presence of bioprinted MΦs that presumably facilitated biofilm dispersal, effectively transforming dormant biofilm-associated bacteria into metabolically active cells that are sensitive to antibiotic action.
MATERIALS AND METHODS
Generation of 3D bioprinted scaffolds containing antibiotics and viable macrophages.
3D printed scaffolds were engineered using a high-resolution 3D bioprinter (3D-Bioplotter® Manufacturer Series, EnvisionTEC; Dearborn, MI). A polycaprolactone (PCL, Mw 80,000, Sigma-Aldrich, St. Louis, MO)/hydroxyapatite (HAp nanocrystals, avg. 100 nm, Berkeley Advanced Biomaterials, Inc.; Berkeley, CA) slurry was used to generate multiple PCL/HAp frames in each layer throughout the construct. Another printing head deposited methacrylated hyaluronic acid (Me-HA, ~1,200 kDa, NovaMatrix; Sandvika, Norway) and methacrylated gelatin (Me-Gel, from type B gelatin, Sigma)-based hydrogels between the PCL/HAp frames18–19. An antibiotic cocktail containing both hydrophobic and hydrophilic antibiotics (rifampin and daptomycin, respectively) was incorporated into the scaffold, where rifampin was included in the PCL/HAp slurry and daptomycin was added to the HA/Gel hydrogel. The drug components and doses are controllable and in vitro and in vivo pilot studies identified an optimal concentration of 16 μg rifampin and 18 μg daptomycin per mg scaffold (i.e. ~20 mg/ml in the bioinks). The antibiotic scaffold was dried after printing and UV sterilized for 2 h. For MΦ incorporation, a total of 107 viable bone marrow-derived MΦs were printed on the surface in a HA/Gel hydrogel matrix in L929 supernatant as a source of macrophage colony-stimulating factor (M-CSF) to maximize cell viability. Bone marrow-derived MΦs were prepared as previously described20, and approximately 104 MΦs were printed per 3 mm2 scaffold that was inserted into the cranial cavity. For the treatment paradigm, scaffolds were trimmed to size and placed in the space of the voided infected bone flap following tissue debridement, whereupon the skin was sutured closed. In the case of the prevention paradigm, scaffolds were inserted at the time the craniotomy was performed, as described above. The 3D bioprinted implant was approximately 1 mm thick and not malleable following hydration. It was printed in its final shape and trimmed to fill the voided space following removal of the infected bone flap. Structurally, the 3D printed implant is distinct from the attributes of the explanted bone flap in that it is not a solid structure but instead has a lattice configuration.
Antibiotic release from 3D bioprinted scaffolds.
3D printed scaffolds (~8 mm in diameter and ~1 mm in thickness) with low (~3.2 μg rifampin and ~3.6 μg daptomycin per mg scaffold) and standard (~16 μg rifampin and ~18 μg daptomycin per mg scaffold) antibiotic doses were immersed in 1 mL sterile PBS and incubated at 37 °C. At predetermined time points, the PBS release solution was removed and stored at −80 °C until testing, whereupon fresh PBS was added to scaffolds. The concentration of released rifampin was calculated from the absorbance measured with an UV spectrophotometer at 474 nm21. The concentration of released daptomycin was determined using a high performance liquid chromatography (HPLC) system (Agilent Technologies; Santa Clara, CA) with a Poroshell 120 (EC-C18, 2.7 μm) column, and the specific chromatographic conditions were as follows: 0.7 mL/min flow; injection volume of 20 mL; mobile phase of 35% acetonitrile and 65% PBS (pH 7.4) and detection at 230 nm.
In vitro toxicity testing.
Primary human osteoblasts (hOBs) were purchased from Promocell (Heidelberg, Germany) and cultured in DMEM/F12 medium supplemented with 10% FBS and 1% penicillin/streptomycin (all from Invitrogen; Carlsbad, CA) in 5% CO2 at 37 °C. Medium was replaced every 2 days and cells were used between passages 4–6.
To assess whether the 3D bioprinted scaffolds exhibited any cytotoxicity, hOBs were seeded at a density of 105 cells per UV sterilized scaffold. Cell viability was examined using a Live/Dead assay (Invitrogen) after 14 days of culture by confocal microscopy (LSM 710, Carl Zeiss)22. Quantitative assessments of hOB viability on various scaffold formulations were performed using an MTT assay after 7 and 14 days in culture23. To determine whether sustained antibiotic release would negatively affect the brain, the survival of mouse primary mixed neuron-glial cultures was examined. Mixed neuron-glial cultures were prepared from neonatal C57BL/6 mice as previously described24. For toxicity assessments, 3D printed antibiotic scaffolds (8 mm diameter) were incubated in media to release antibiotics for up to 14 days. At days 1, 7, and 14, conditioned media was collected and mixed neuron-glia cultures were treated with 10-fold dilutions of supernatants for 48 h, whereupon cell viability was assessed using a lactate dehydrogenase (LDH) assay (Promega; Madison, WI). Both MTT and LDH release assays are widely used as reliable assessments of cell viability and provide complementary information.
Mouse model of S. aureus craniotomy-associated biofilm infection.
Male and female C57BL/6 mice were purchased from Charles River Laboratories (Wilmington, MA) and were utilized between 8–10 weeks of age. Animals were housed in an AAALAC-accredited facility at the University of Nebraska Medical Center, provided with food and water ad libitum, and housed under 12 h light/dark cycles. As described previously, mice were anesthetized using ketamine/xylazine, and an incision was made in the skin opposite to the side the bone flap14. A high-speed pneumatic drill was used to create a bone flap (approximately 3 mm in diameter) that was incubated in a broth culture of S. aureus (USA300 LAC13C25) for 5 min to allow for bacterial adherence, rinsed, and immediately reinserted into the skull, whereupon the skin incision was closed by suturing. The procedure results in S. aureus colonization of the bone flap, which ultimately leads to infection persistence in the subcutaneous galea and brain14. Of note, bone flap infections in humans with an intact dura do not always present with subdural infection, which differs from our mouse model, and neurosurgeons avoid penetrating the dura when treating these infections unless it is unavoidable. Although great care is taken to preserve the integrity of the dura during the craniotomy procedure in the mouse, damage to the dura may occur. Bacteria are present on both surfaces of the bone flap in our mouse model, which was also observed at the ultrastructural level in our previous study of a bone flap from a patient with confirmed MRSA infection14, making the possibility of bacterial translocation into the brain a possibility. For the prevention paradigm, a bone flap was created as described above, but was immediately reinserted, with a 3D scaffold placed on top. The following day, 105-108 CFU of S. aureus was injected subcutaneously in the area of the scaffold/flap. For the treatment paradigm, infected bone flaps were removed and discarded at day 7, and replaced with 3D bioprinted scaffolds under ketamine/xylazine anesthesia. For systemic antibiotic (Abx) administration, mice received once daily i.p. injections of daptomycin and rifampin (5 and 25 μg/ml, respectively). The animal use protocol was approved by the University of Nebraska Medical Center Animal Care and Use Committee (approval #16–123-10).
Tissue processing and bacterial quantification.
At the indicated time points post-infection, mice were sacrificed using an overdose of inhaled isoflurane and transcardially perfused with PBS. The bone flap and scaffold were removed first, followed by the galea, which represents the subcutaneous tissue and associated purulent exudate. Next, the ipsilateral brain hemisphere associated with the infected bone flap was removed and placed in PBS. The bone flap and scaffolds were vortexed in PBS for 30 sec followed by a 5 min sonication to dislodge biofilm-associated bacteria. The galea was dissociated in PBS using the blunt end of a plunger from a 3 cc syringe and the brain was homogenized by being pressed through a 70 μm cell strainer using the blunt end of a syringe plunger and rinsed with PBS. Once all of the tissues were processed, an aliquot was removed to quantify bacterial titers. Titers were determined by serial dilutions on TSA plates supplemented with 5% sheep blood (Remel, Lenexa, KS) and are expressed as Log10 colony forming units (CFUs). The contralateral hemisphere was examined during initial experiments; however, the degree of bacterial dissemination was low, which precluded an accurate assessment of 3D scaffold efficacy and therefore, was not continued for the remainder of the study.
Flow Cytometry.
After aliquots were removed for quantifying S. aureus titers, the galea and brain were further processed for flow cytometry to determine how the 3D bioprinted scaffolds affected leukocyte infiltrates. Briefly, the brain homogenate was incubated in a dissociation buffer containing HBSS, collagenase IV, and DNaseI (both from Sigma-Aldrich) for 20 min at 37oC, whereupon 20% FBS was added to stop enzymatic activity followed by centrifugation at 300xg for 10 min. The pellet was layered over a 25% Percoll (GE Healthcare; Marlborough, MA) gradient containing 3% FBS and centrifuged at 520xg for 20 min with no brake26. The upper myelin layer down to the pellet was discarded, and the pellet was resuspended in PBS and filtered to remove remaining particulate material. The galea sample was also centrifuged and filtered, and cells from both the galea and brain were incubated with TruStain fcX (BioLegend; San Diego, CA) to block nonspecific antibody binding to Fc receptors. Cells were then stained with CD11b-FITC, CD45-APC, Ly6G-PE, Ly6C-PerCP-Cy5.5, and F4/80-PE-Cy7 (BioLegend and BD Biosciences; San Diego, CA). Dead cells were excluded using a Live/Dead Fixable Cell Stain Kit (Invitrogen) according to the manufacturer’s instructions. Analysis was performed using BD FACSDiva software and results are presented as the percentage of live, CD45+ leukocytes. Myeloid-derived suppressor cells (MDSCs) were classified as CD11bhighLy6C+Ly6G+F4/80-, neutrophils as CD11blowLy6C+Ly6G+F4/80-, and monocytes Ly6C+Ly6G-CD11b+F4/80-27.
Determination of blood-brain barrier (BBB) integrity.
Mice were anesthetized with isoflurane and subsequently administered 100 μl of a solution of 2.0% Evans blue dye in PBS via the retro-orbital vein. Animals were euthanized 60 min following Evans blue injection and perfused transcardially with 20 ml of PBS using a peristaltic pump to remove residual dye from the circulation. The brain was immediately removed and imaged to depict the extent of Evan’s blue accumulation in the brain parenchyma, reflective of BBB permeability.
Statistical methods.
Significant differences between groups were determined using an unpaired Student’s t-test with Welch’s correction or a One-way ANOVA with Tukey’s multiple comparison test using GraphPad Prism version 6 (La Jolla, CA) where a p-value < 0.05 was considered statistically significant.
RESULTS
Persistence of S. aureus craniotomy-associated biofilm infection and recalcitrance to systemic antibiotics.
Our laboratory has developed a mouse model of S. aureus craniotomy-associated infection where bacteria colonize both surfaces of the bone flap to establish infection in the subcutaneous galea and brain14. Both chronicity and recalcitrance to systemic antibiotics are characteristics of biofilms, which was demonstrated in the craniotomy model by the persistence of infection out to 9 months (Fig 1A) and resistant to systemic rifampin and daptomycin (Fig 1B), an antibiotic regimen used to treat staphylococcal infections28–29. However, antibiotic accumulation in the CNS following systemic administration is slow due to restriction by the blood-brain barrier (BBB)30; therefore, we developed a 3D printed scaffold to deliver antibiotics directly at the site of infection using a slow release formulation, in either treatment or prevention paradigms. A second approach was to determine if harnessing the antibacterial activity of MΦs, by incorporation into the 3D printed scaffold, would augment antibiotic action to facilitate S. aureus clearance.
Figure 1. Craniotomy-associated infections are persistent and recalcitrant to systemic antibiotics.
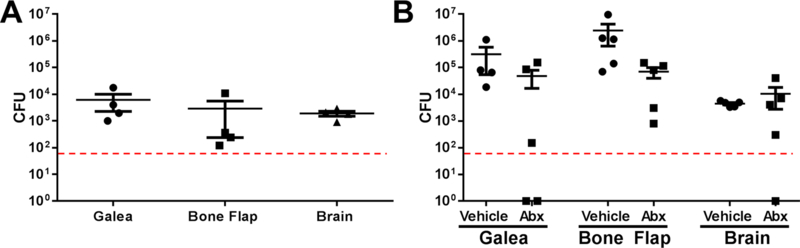
S. aureus craniotomy infection was established in C57BL/6 mice (n=5 per group) and animals were (A) sacrificed 9 months later to quantify bacterial persistence in the galea, brain, and bone flap. (B) Mice received systemic antibiotics (Abx; daptomycin and rifampin, once/daily) or vehicle (PBS) beginning at day 7 post-infection and continuing for one week, whereupon mice were sacrificed to determine S. aureus titers. Results are expressed as the number of colony forming units (CFU) per region.
Development of a 3D bioprinted scaffold containing antibiotics and viable macrophages.
We utilized a high-resolution 3D bioprinter that can support different materials for printing synthetic bone constructs. Previous studies from our group have optimized the 3D printing parameters, material compositions, and material usage to guarantee controllability, reproducibility, and quality of the printed scaffolds18–19. We first utilized a PCL/HAp slurry containing rifampin to generate a PCL/HAp frame in each layer (Fig. 2A). Next, another printing head was used to deposit the Me-HA/Me-Gel hydrogel-based bioink containing daptomycin between the PCL/HAp frame on the same layer. Me-HA/Me-Gel bioinks were further photocrosslinked after 3D printing18. Incorporation of antibiotics into each printing formulation was based on their solubility properties. Once the antibiotic scaffold was dried and sterilized, viable MΦs were printed on the surface in a HA/Gel hydrogel, and the scaffolds were trimmed to the size of bone flaps for implantation at the craniotomy site (Fig 2A). The drug components and doses are controllable; therefore, we initiated our studies by testing scaffolds with low (~3.2 μg rifampin/mg scaffold and ~3.6 μg daptomycin/mg scaffold, respectively) and high (~16 μg rifampin/mg scaffold and ~18 μg daptomycin/mg scaffold, respectively) antibiotic concentrations. Both 3D antibiotic scaffolds demonstrated robust killing of the community-acquired S. aureus clinical isolate, USA300 LAC13C25, whereas empty scaffolds had no activity (Fig. 2B). To evaluate the longevity of antibiotic action following release from the bioprinted scaffolds in vitro, scaffolds were incubated in medium for either 7 or 14 days, whereupon they were placed on a lawn of S. aureus to determine the extent of bactericidal activity. While both the high and low antibiotic scaffolds inhibited growth following 7 days of release in a dose-dependent manner, the lower concentration was not as effective after 14 days, whereas the high dose scaffold retained antimicrobial activity (Fig. 2B). In vitro kinetic release studies demonstrated that both antibiotics had significant burst release at day 1, with sustained release for at least 14 days, with both antibiotics demonstrating similar release rates (Fig. 2C). Since maximal scaffold efficacy in vivo was envisioned to be achieved by sustained release of high antibiotic concentrations, the 3D bioprinted scaffold harboring high dose antibiotics was used for all subsequent experiments.
Figure 2. Release profile and bactericidal effect of 3D printed scaffolds containing daptomycin and rifampin.
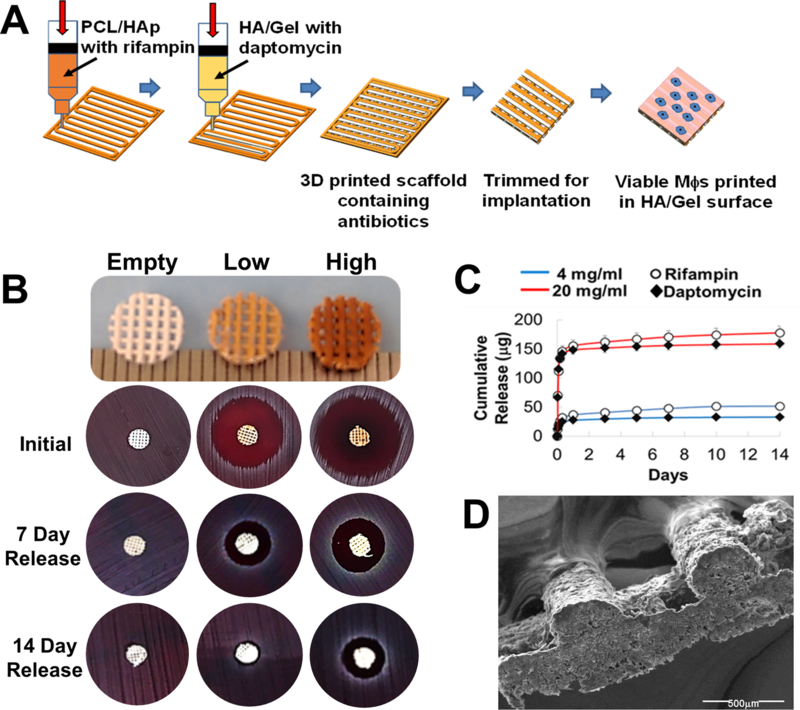
(A) A polycaprolactone (PCL) (polyester) /hydroxyapatite (HAp) slurry was used to generate multiple PCL/HAp frames in each layer throughout the construct. Another printing head deposited hyaluronic acid (HA) and gelatin-based hydrogels between the PCL/HAp frames. Rifampin was incorporated in the PCL/HAp slurry and daptomycin was added to the HA/Gel hydrogel. Both low (~0.6 μg and 1.6 μg rifampin and daptomycin per mg scaffold, respectively) and high (~16 μg and 18 μg rifampin and daptomycin per mg scaffold, respectively) were examined. Once the antibiotic scaffold was dried, viable MΦs were printed on the surface in a HA/Gel hydrogel matrix and immediately implanted into mice. (B) Efficacy of 3D bioprinted scaffolds to kill a lawn of S. aureus when used fresh or after incubation in PBS for 7 or 14 days to simulate antibiotic release in vivo. (C) Quantitation of antibiotic release profile from 3D printed scaffolds over the course of 14 days in vitro. (D) Scanning electron microscopy image of a 3D printed antibiotic scaffold depicting the lattice patterning (80x magnification).
To assess the potential toxicity of 3D bioprinted scaffolds, effects on primary hOBs and mixed neuron-glial cultures were examined. Culture of hOBs on scaffolds for 14 days revealed no evidence of cell death, as determined by a Live/Dead assay (Fig. 3A) or MTT assay, where hOB proliferation over time was evident (Fig. 3B). To evaluate potential CNS toxicity, mouse neuron-glial cultures were exposed to medium where antibiotics had been released from the 3D printed scaffold over a 1, 7, or 14 day period. No adverse effects of antibiotic exposure on brain cell viability were observed at any of the concentrations examined (Fig. 3C). Collectively, these results suggest that the 3D bioprinted scaffolds should be well tolerated and safe for in vivo testing.
Figure 3. 3D antibiotic scaffolds are not cytotoxic.
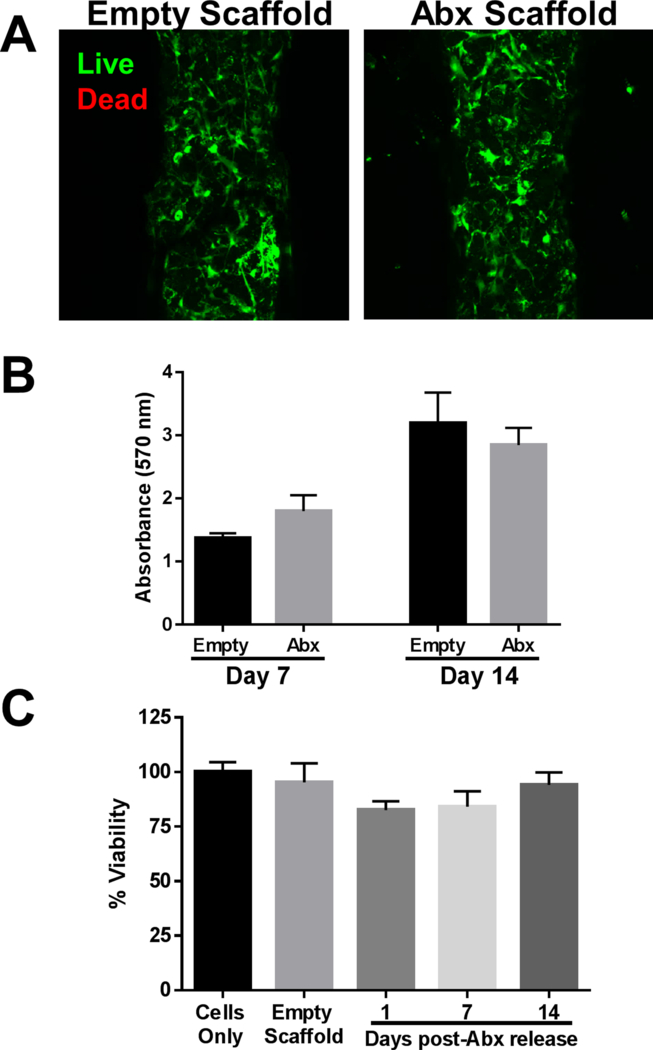
Primary human osteoblasts were cultured on 3D antibiotic (Abx) scaffolds in vitro for 7 or 14 days, whereupon (A) osteoblast viability was examined by confocal microscopy and (B) cell survival was demonstrated by MTT assay. (C) Survival of primary mouse neuron-glial cultures after exposure to conditioned medium from 3D antibiotic scaffolds incubated for 1–14 days in medium as assessed by LDH release assays. Results are reported as the mean ± SD of three biological replicates.
3D bioprinted antibiotic scaffolds containing macrophages promote S. aureus clearance during craniotomy-associated biofilm infection.
The treatment of established biofilm infections represents a challenging clinical scenario due to the large bacterial biomass and microdomains of metabolic dormancy17, 31. To evaluate the efficacy of 3D bioprinted scaffolds against an existing biofilm, infected bone flaps were replaced at day 7 post-infection with 3D bioprinted antibiotic scaffolds or empty scaffolds as a control (Fig. 4A). Treatment was initiated at day 7 post-infection, since our earlier studies demonstrated that bona fide biofilm formation has occurred based on recalcitrance to systemic antibiotics (Fig. 1B). We first evaluated whether the 3D antibiotic scaffold modulated BBB integrity during infection using Evan’s blue. The 3D antibiotic scaffold led to a dramatic reduction in BBB permeability (Fig. 4B), suggesting improved outcomes in terms of decreased bacterial burdens and inflammation.
Figure 4. 3D antibiotic scaffolds reduce blood-brain barrier (BBB) permeability associated with S. aureus craniotomy infection.
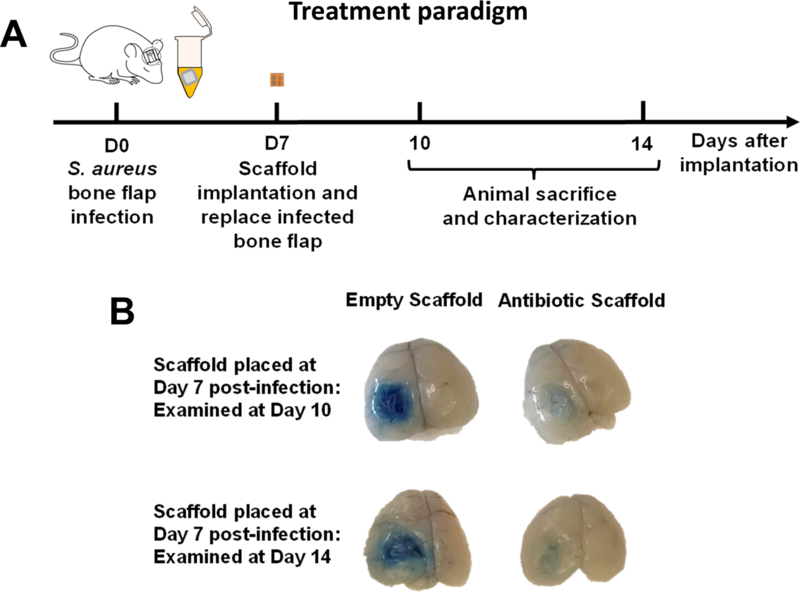
(A) Schematic depicting the paradigm used to assess the treatment efficacy of 3D bioprinted scaffolds. (B) 3D antibiotic scaffolds (daptomycin + rifampin) were inserted at day 7 after S. aureus craniotomy infection, whereupon BBB permeability was assessed 3 or 7 days later using Evan’s blue. Results are representative of 5 mice per group.
To examine this further, biofilm titers and leukocyte infiltrates were quantified in animals receiving 3D bioprinted antibiotic or empty scaffolds at day 7 post-infection, whereupon assessments were performed 3 or 7 days later (corresponding to days 10 and 14 after infection, respectively). While the 3D antibiotic scaffold reduced S. aureus burden by 2–3 log in the galea and brain (Fig. 5A), residual infection was still evident. To augment the bactericidal activity of the 3D antibiotic scaffold, viable MΦs were also incorporated based on their potent antimicrobial activity. The addition of MΦs further reduced S. aureus titers compared to the 3D antibiotic scaffold alone, which was most pronounced in the brain at day 3 after placement (Fig 5A). The beneficial effect of MΦ addition was transient, in that no additional decrease in titers was observed 7 days after treatment (data not shown). This is likely explained by the limited half-life of MΦs when exposed to the large number of bacteria contained in the biofilm that produce lytic toxins32. The combined action of antibiotics and MΦs was required for maximal bacterial clearance, since scaffolds containing macrophages only had no impact on biofilm burdens (Fig. 5A).
Figure 5. 3D antibiotic scaffolds reduce bacterial burdens during S. aureus craniotomy infection, which is augmented by viable macrophages.
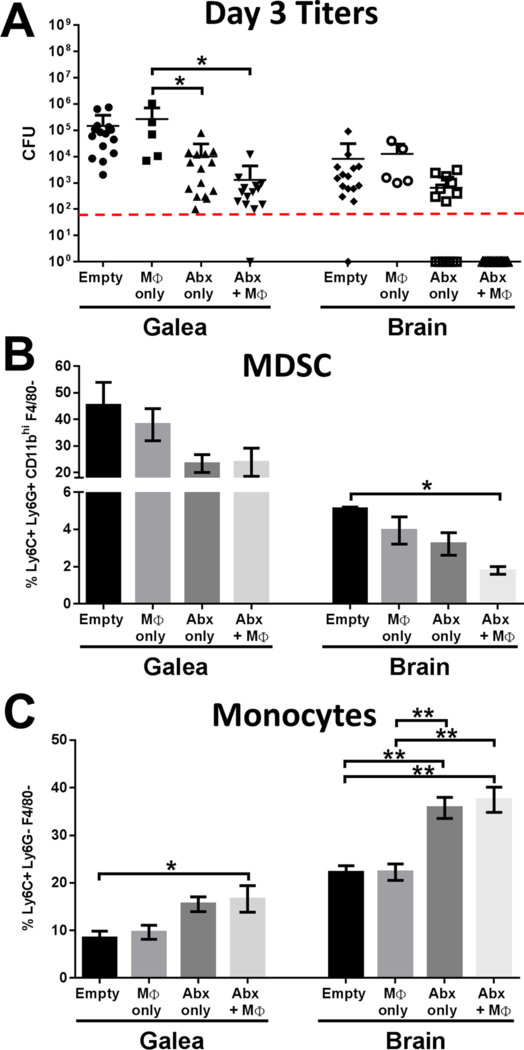
3D bioprinted scaffolds containing antibiotics (Abx) only (daptomycin + rifampin), macrophages (MΦs) only, antibiotics + MΦs, or empty scaffolds were placed at day 7 after S. aureus craniotomy infection, whereupon (A) bacterial burdens, (B) MDSC, and (C) monocyte infiltrates in the galea and brain were determined 3 days later (i.e. day 10 post-infection; n=5–15 mice per group). Flow cytometry results are presented from one experiment (n=5 mice) that was representative of three independent studies. Results were analyzed by One-way ANOVA with Tukey’s multiple comparison test (*, p < 0.05; **, p < 0.01).
The 3D bioprinted scaffolds also influenced leukocyte recruitment into the galea and brain, as revealed by flow cytometry (Fig. 5B and C). For this study we focused on two leukocyte populations that are prominent during biofilm-associated infection, namely MDSCs and monocytes27. MDSCs are immature myeloid cells that we have previously shown inhibit monocyte proinflammatory activity during S. aureus biofilm infection and contribute, in part, to infection persistence33–35. In situations where MDSC recruitment is reduced this typically corresponds with an increase in effector cell recruitment (i.e. monocytes) and reduced biofilm burdens33–34. MDSCs are the most abundant leukocyte population in the galea (Fig 5B), which represents the region with highest bacterial titers (Fig. 5A). In contrast, MDSC infiltrates are less numerous in the brain (Fig. 5B). 3D antibiotic scaffolds decreased MDSC influx in the galea by approximately 50% compared to empty scaffolds (Fig. 5B), which coincided with increased monocyte recruitment (Fig. 5C). Similar trends with the 3D antibiotic scaffold were observed with MDSC and monocyte recruitment in the brain (Fig. 5B and C). MΦ incorporation into the Abx scaffold led to significant changes in leukocyte recruitment in the galea and brain compared to empty scaffolds (Fig. 5B and C), and correlated with a reduction in bacterial burdens (Fig. 5A).
Prevention of craniotomy-associated infection with 3D bioprinted antibiotic scaffolds.
We next evaluated a prophylactic approach to determine whether 3D bioprinted scaffolds could protect against infectious complications following craniotomy. A craniotomy was performed and a 3D scaffold was placed directly above the sterile bone flap upon reinsertion in the skull. Mice were challenged with various doses of live S. aureus (105 to 107 CFU) 1 day later by injecting bacteria subcutaneously along the suture site (Fig. 6A). Various inocula were tested to determine the maximal challenge dose that the 3D bioprinted antibiotic scaffold could eradicate to produce a sterile site and facilitate wound healing and recovery. The 3D antibiotic scaffolds were effective at preventing infection in response to challenge with 105 and 106 S. aureus and bacterial burdens were near the lower limit of detection with the 107 inoculum (Fig. 6B).
Figure 6. Efficacy of 3D bioprinted antibiotic scaffolds to prevent S. aureus craniotomy-associated infection.
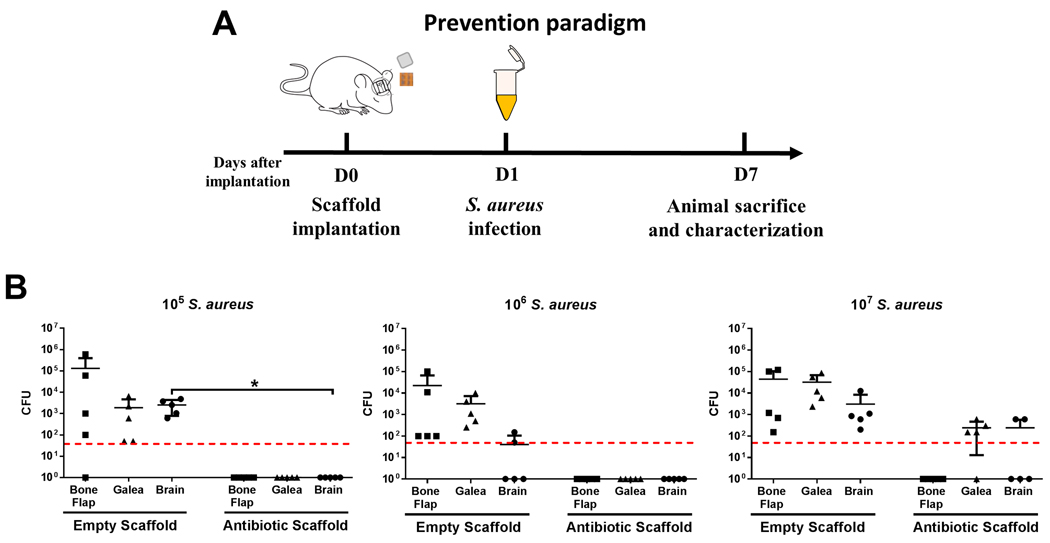
(A) Schematic depicting the experimental paradigm to assess the ability of 3D bioprinted scaffolds to prevent S. aureus craniotomy infection. (B) 3D bioprinted scaffolds ± antibiotics (daptomycin + rifampin) were placed at the time of craniotomy, whereupon mice (n=5 per group) where challenged 1 day later with 105−107 CFU live S. aureus at the surgical site. Bacterial burdens were determined 7 days later. Results were analyzed by an unpaired Student’s t-test with Welch’s correction (*, p < 0.05).
To evaluate whether MΦs would enhance the efficacy of 3D antibiotic scaffolds, a challenge dose of 108 CFU was used to ensure that sufficient bacteria would remain to detect a potential effect of MΦ addition. Unlike the treatment paradigm, the incorporation of MΦs into the 3D scaffold did not potentiate bacterial clearance compared to antibiotics alone (Fig. 7). Nevertheless, the 3D antibiotic scaffold reduced S. aureus titers compared to empty scaffolds and demonstrated excellent efficacy at doses that well exceed what would be encountered during a native infection in patients.
Figure 7. Viable macrophages do not augment antibiotic efficacy in a prevention paradigm.
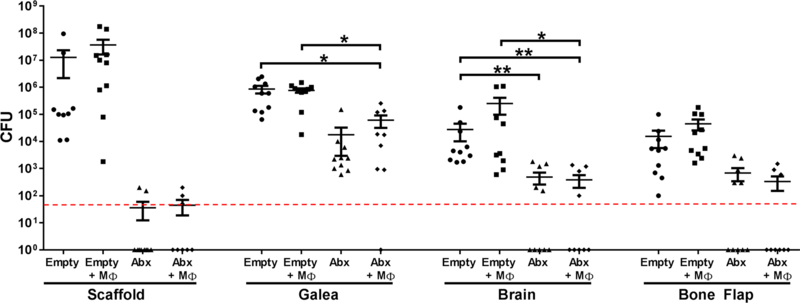
3D bioprinted scaffolds containing antibiotics only (Abx; daptomycin + rifampin), macrophages (MΦs) only, antibiotics + MΦs, or empty scaffolds were placed at the time of craniotomy, whereupon mice (n=8–12 per group) where challenged 1 day later with 108 CFU live S. aureus at the surgical site. Bacterial burdens were determined 7 days later. Results were analyzed by One-way ANOVA with Tukey’s multiple comparison test (*, p < 0.05; **, p < 0.01).
Long-term effectiveness of local 3D antibiotic scaffolds and combination with systemic antibiotic therapy.
In terms of translational potential, it was important to determine whether the 3D antibiotic scaffold sterilized the infection site or whether bacterial outgrowth would resume after the antibiotic depot had become exhausted (estimated at occur at day 14 based on our in vitro studies; Fig. 2C). Macrophages were not examined in these experiments, since they only exhibited beneficial effects at acute time points (i.e. 3 days; Fig. 5). Using our standard antibiotic dose (~16 μg rifampin /mg scaffold and ~18 μg daptomycin /mg scaffold), bacterial burdens began to remerge at day 14 on the scaffold and in the galea, which were further increased at day 28 (Fig. 8). To determine whether outgrowth could be prevented by increasing the antibiotic dose, we repeated these experiments with 3D antibiotic scaffolds that contained 5-fold more antibiotic cocktail. This formulary completely prevented S. aureus recurrence at day 14 following scaffold placement, but by day 28 bacterial outgrowth was again evident (Fig. 8).
Figure 8. Assessment of the longevity of 3D printed antibiotic scaffolds to treat S. aureus craniotomy infection.
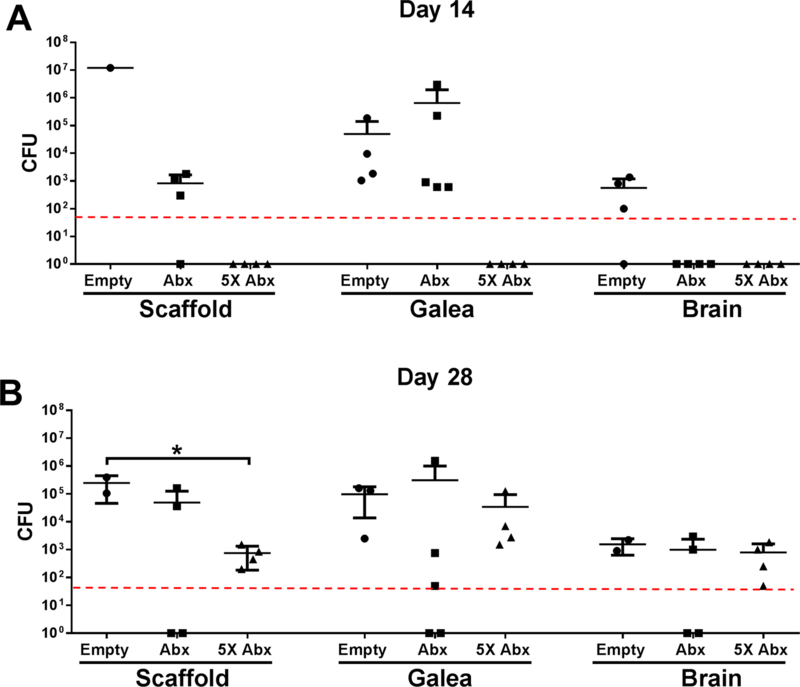
3D bioprinted scaffolds that were empty, containing 20 μg/ml antibiotics (Abx; daptomycin + rifampin), or 5X higher antibiotic concentrations (5X) were placed at day 7 after S. aureus craniotomy infection (n=3–5 mice/group), whereupon bacterial burdens were determined at (A) 14 days (i.e. day 21 post-infection) or (B) 28 days (i.e. day 35 post-infection) following scaffold insertion. Results were analyzed by One-way ANOVA with Tukey’s multiple comparison test (*, p < 0.05).
Although we showed that systemic antibiotics had a limited effect on craniotomy-associated burdens (Fig. 1B), from a clinical perspective patients with these infections receive systemic antibiotics as a standard-of-care. Therefore, we assessed whether the combination of systemic antibiotics and the 3D antibiotic scaffold would exert an additive effect. In our earlier studies (Figs. 6 and 8), higher bacterial inoculums (108 CFU) were used to prevent complete clearance of the infection, such that effects of MΦs could be assessed. For these experiments, the bacterial inoculum was reduced to 5×105 CFU to recapitulate the lower inoculum that likely occurs in human craniotomy-associated infection. 3D antibiotic scaffolds were placed at day 7 following craniotomy infection coincident with the initiation of daily antibiotic treatment (daptomycin/rifampin) for a period of 7 days. This approach was effective at reducing bacterial burdens near or below the level of detection, indicating progression towards an effective clinical outcome (Fig. 9).
Figure 9. 3D antibiotic scaffolds synergize with systemic antibiotics to clear established S. aureus craniotomy-associated infection.
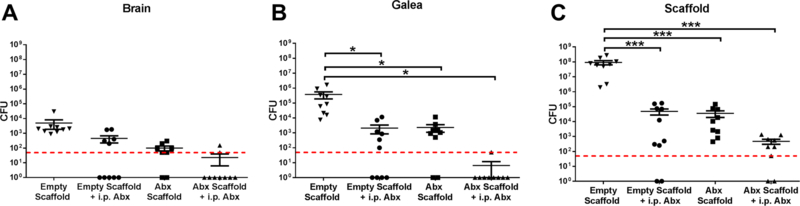
3D bioprinted scaffolds that were empty or containing antibiotics only (Abx; daptomycin + rifampin) were placed at day 7 after S. aureus craniotomy infection (n=10 mice/group), whereupon mice received daily i.p. injections of daptomycin and rifampin until sacrifice 7 days later (i.e. day 14 post-infection) and bacterial burdens in the (A) brain, (B) galea, and (C) scaffold were quantified. Results were analyzed by One-way ANOVA with Tukey’s multiple comparison test (*, p < 0.05; ***, p < 0.001).
DISCUSSION
Biofilm infections are difficult to treat because a subpopulation of bacteria are metabolically dormant17, 31. In terms of craniotomy-associated infection this is magnified by the protracted period required for therapeutic antibiotic concentrations to be achieved in the brain due to BBB exclusion30. This study examined the efficacy of a first-generation device designed to demonstrate proof-of-concept at reducing bacterial burdens following craniotomy-associated infection. We considered that a localized delivery approach with a 3D bioprinted bone scaffold would facilitate more rapid therapeutic benefit based on proximity to the infection site coupled with consistent slow-release to ensure steady therapeutic concentrations. In general, our results support these findings, where systemic antibiotics alone were less effective at clearing craniotomy-associated infection, but local delivery with 3D printed antibiotic scaffolds reduced S. aureus burdens. Furthermore, the combination of the 3D antibiotic scaffold and systemic antibiotic delivery demonstrated superior efficacy at clearing infection. This is likely because the 3D scaffold reduced biofilm burdens, which would be expected to alter biofilm architecture, resulting in the transition to a planktonic growth state that is amenable to antibiotic action.
We also examined whether an immune-based approach incorporated into the 3D scaffold would synergize with antibiotics to promote biofilm clearance. This was realized by bioprinting viable MΦs into the antibiotic scaffold. MΦs are professional phagocytes and were envisioned to promote biofilm dispersal, which would transform dormant biofilm-associated bacteria into a metabolically active planktonic state, sensitizing them to antibiotic clearance17. Interestingly, a beneficial effect of MΦs to potentiate bacterial clearance was most evident in the brain, whereas a trend towards reduced titers was also detected in the galea, but was less robust. This might be explained by the lower bacterial burdens in the brain compared to galea, which were more amenable to immune-mediated clearance. Currently, the mechanism whereby MΦs facilitate S. aureus clearance is not known. One possibility is that MΦs migrate out of the porous gel scaffold into the brain parenchyma to exert direct antibacterial activity. Alternatively, activated MΦs could secrete cytokines/chemokines that augment the antimicrobial activity of other glia/leukocytes associated with the infection. However, neither of these possibilities are mutually exclusive and both could be operative. Nevertheless, our results demonstrate that the beneficial effects of MΦs are short-lived, since their impact was only observed 3 days following 3D scaffold treatment, which was lost at day 7. One unexpected finding was that MΦs were only effective in facilitating S. aureus clearance from established biofilms (i.e. treatment initiated at day 7 post-infection), whereas they did not improve bacterial killing in the prevention paradigm. One possibility to explain this observation is that a robust milieu of cytokines and growth factors is present in an established biofilm14, 33–34, which may deliver pro-survival signals to MΦs within the 3D scaffold and promote their antibacterial activity. In contrast, when 3D antibiotic/MΦ scaffolds are placed at the time of surgery and infection is delayed until the following day, MΦs are not bathed in a growth factor-enriched environment and conceivably to do not receive signals to promote their viability or prime their pro-inflammatory activity in response to a subsequent bacterial challenge. However, these possibilities currently remain speculative.
One limitation of the current study is the finding that empty scaffolds behaved as a foreign body, which raises concerns about the material serving as a potential nidus for infection after the antibiotic has dissipated from the scaffold. Indeed, this was observed when 3D antibiotic scaffolds remained in the skull for longer periods in vivo, where bacterial titers were undetectable out to day 14 but began to remerge at 28 days later. This was partially mitigated by increasing the antibiotic concentration printed into the 3D scaffold; however, additional studies are needed to optimize the 3D antibiotic scaffold formulation to prevent bacterial growth once antibiotics are no longer present. This may be achieved by modifying matrix components to utilize materials that are more inert or possess inherent antimicrobial properties. Such second-generation 3D scaffolds could also be engineered to contain immune-modifying molecules to augment the pro-inflammatory properties of biofilm-associated leukocytes and/or glia, rather than incorporating live immune cells as was done for the current report. Another limitation is that since the treatment model involved removal of the bone flap, the antibiotic scaffolds were only tested for clearance of bacteria from the galea and brain and the effects on bone-associated, established biofilm were not assessed. We are currently working to design a second-generation implant by the continued refinement of 3D printing materials with the long-term goal of integrating the implant into the cranial cavity.
Finally, the 3D antibiotic scaffold prevented infection establishment with a challenge dose of 105 S. aureus, which is expected to exceed the number of organisms that would seed a site during native infection in humans. When considering the potential translation of this 3D scaffold to protect against possible infectious complications following craniotomy, one question that remains is whether the device would need to be removed, since it could conceivably act as a foreign body. Alternatively, a second-generation formulation could be engineered to be biodegradable, which would provide temporary protection against infection during the critical post-surgical period. Nevertheless, these studies demonstrate the feasibility of a 3D bioprinting approach to mitigate bacterial burdens when scaffolds are placed at the site of craniotomy infection in combination with systemic antibiotics. In addition, efficacy can be potentiated by incorporating an immune-mediated strategy to promote biofilm dispersal.
ACKNOWLEDGEMENTS
This work was supported by the National Institutes of Health/National Institute of Neurological Disorders and Stroke R01 NS107369 (to TK). The authors thank Drs. Cortney Heim, Megan Bosch, and Kelsey Yamada for assistance with mouse craniotomy surgeries, Rachel Fallet for managing the mouse colony, Shaohua Wu for performing the antibiotic release test, and Tom Barger in the UNMC Electron Microscopy Core Facility. The authors wish to acknowledge the support of the Fred & Pamela Buffett Cancer Center Flow Cytometry Research Facility Shared Resource, supported by the National Cancer Institute under award number P30 CA036727.
Footnotes
CONFLICT OF INTEREST
A provisional patent has been filed with the US Patent and Trademark Office covering the application of 3D bioprinted scaffolds for the treatment of craniotomy-associated infections (EFS ID 29315578; TK, BD, AA, and MK).
REFERENCES
- (1).Stiver SI Complications of Decompressive Craniectomy for Traumatic Brain Injury. Neurosurg Focus 2009, 26 (6), E7. [DOI] [PubMed] [Google Scholar]
- (2).McClelland S 3rd; Hall WA Postoperative Central Nervous System Infection: Incidence and Associated Factors in 2111 Neurosurgical Procedures. Clin Infect Dis 2007, 45 (1), 55–59. [DOI] [PubMed] [Google Scholar]
- (3).Chiang HY; Steelman VM; Pottinger JM; Schlueter AJ; Diekema DJ; Greenlee JD; Howard MA 3rd; Herwaldt LA Clinical Significance of Positive Cranial Bone Flap Cultures and Associated Risk of Surgical Site Infection After Craniotomies or Craniectomies. J Neurosurg 2011, 114 (6), 1746–1754. [DOI] [PubMed] [Google Scholar]
- (4).Fang C; Zhu T; Zhang P; Xia L; Sun C. Risk Factors of Neurosurgical Site Infection After Craniotomy: A Systematic Review and Meta-analysis. Am J Infect Control 2017, 45 (11), e123–e134. [DOI] [PubMed] [Google Scholar]
- (5).Widdel L; Winston KR Pus and Free Bone Flaps. J Neurosurg Pediatr 2009, 4 (4), 378–382. [DOI] [PubMed] [Google Scholar]
- (6).Auguste KI; McDermott MW Salvage of Infected Craniotomy Bone Flaps with the Wash-in, Wash-out Indwelling Antibiotic Irrigation System. Technical Note and Case Series of 12 Patients. J Neurosurg 2006, 105 (4), 640–644. [DOI] [PubMed] [Google Scholar]
- (7).Bruce JN; Bruce SS Preservation of Bone Flaps in Patients with Postcraniotomy Infections. J Neurosurg 2003, 98 (6), 1203–1207. [DOI] [PubMed] [Google Scholar]
- (8).Wallace DJ; McGinity MJ; Floyd JR 2nd, Bone Flap Salvage in Acute Surgical Site Infection After Craniotomy for Tumor Resection. Neurosurg Rev 2018, 41 (4), 1071–1077. [DOI] [PubMed] [Google Scholar]
- (9).Baumeister S; Peek A; Friedman A; Levin LS; Marcus JR Management of Postneurosurgical Bone Flap Loss Caused by Infection. Plast Reconstr Surg 2008, 122 (6), 195e–208e. [DOI] [PubMed] [Google Scholar]
- (10).Honeybul S; Ho KM Long-term Complications of Decompressive Craniectomy for Head Injury. J Neurotrauma 2011, 28 (6), 929–935. [DOI] [PubMed] [Google Scholar]
- (11).Sinclair AG; Scoffings DJ Imaging of the Post-operative Cranium. Radiographics 2010, 30 (2), 461–482. [DOI] [PubMed] [Google Scholar]
- (12).Yang XF; Wen L; Shen F; Li G; Lou R; Liu WG; Zhan RY Surgical Complications Secondary to Decompressive Craniectomy in Patients with a Head Injury: A Series of 108 Consecutive Cases. Acta Neurochir (Wien) 2008, 150 (12), 1241–1248. [DOI] [PubMed] [Google Scholar]
- (13).Bhaskar IP; Inglis TJ; Lee GY Clinical, Radiological, and Microbiological Profile of Patients with Autogenous Cranioplasty Infections. World Neurosurg 2014, 82 (3–4), e531–e534. [DOI] [PubMed] [Google Scholar]
- (14).Cheatle J; Aldrich A; Thorell WE; Boska MD; Kielian T. Compartmentalization of Immune Responses During Staphylococcus aureus Cranial Bone Flap Infection. Am J Pathol 2013, 183 (2), 450–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Hanke ML; Heim CE; Angle A; Sanderson SD; Kielian T. Targeting Macrophage Activation for the Prevention and Treatment of Staphylococcus aureus Biofilm Infections. J Immunol 2013, 190 (5), 2159–2168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Otto M. Staphylococcal Biofilms. Curr Top Microbiol Immunol 2008, 322, 207–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Lewis K. Persister Cells, Dormancy and Infectious Disease. Nat Rev Microbiol 2007, 5 (1), 48–56. [DOI] [PubMed] [Google Scholar]
- (18).Kuss MA; Wu S; Wang Y; Untrauer JB; Li W; Lim JY; Duan B. Prevascularization of 3D Printed Bone Scaffolds by Bioactive Hydrogels and Cell Co-culture. J Biomed Mater Res B Appl Biomater 2018, 106 (5), 1788–1798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Wang Y; Wu S; Kuss M; Streubel P; Duan B. Effects of Hydroxyapatite and Hypoxia on Chondrogenesis and Hypertrophy in 3D Bioprinted ADMSC Laden Constructs. ACS Biomaterials Science & Engineering 2017, 3, 826–835. [DOI] [PubMed] [Google Scholar]
- (20).Yamada KJ; Heim CE; Aldrich AL; Gries CM; Staudacher AG; Kielian T. Arginase-1 Expression in Myeloid Cells Regulates Staphylococcus aureus Planktonic but Not Biofilm Infection. Infect Immun 2018, 86 (7). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Mohyeldin SM; Mehanna MM; Elgindy NA The Relevancy of Controlled Nanocrystallization on Rifampicin Characteristics and Cytotoxicity. Int J Nanomedicine 2016, 11, 2209–2222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Kuss M; Kim J; Qi D; Wu S; Lei Y; Chung S; Duan B. Effects of Tunable, 3D-bioprinted Hydrogels on Human Brown Adipocyte Behavior and Metabolic Function. Acta Biomater 2018, 71, 486–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Wu S; Peng H; Li X; Streubel PN; Liu Y; Duan B. Effect of Scaffold Morphology and Cell Co-culture on Tenogenic Differentiation of HADMSC on Centrifugal Melt Electrospun poly (Llactic acid) Fibrous Meshes. Biofabrication 2017, 9 (4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Esen N; Kielian T. Effects of Low Dose GM-CSF on Microglial Inflammatory Profiles to Diverse Pathogen-associated Molecular Patterns (PAMPs). J Neuroinflammation 2007, 4, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Thurlow LR; Hanke ML; Fritz T; Angle A; Aldrich A; Williams SH; Engebretsen IL; Bayles KW; Horswill AR; Kielian T. Staphylococcus aureus Biofilms Prevent Macrophage Phagocytosis and Attenuate Inflammation In Vivo. J Immunol 2011, 186 (11), 6585–6596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).LaFrance-Corey RG; Howe CL Isolation of Brain-infiltrating Leukocytes. J Vis Exp 2011, (52). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Heim CE; West SC; Ali H; Kielian T. Heterogeneity of Ly6G(+) Ly6C(+) Myeloid-Derived Suppressor Cell Infiltrates during Staphylococcus aureus Biofilm Infection. Infect Immun 2018, 86 (12). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Saleh-Mghir A; Muller-Serieys C; Dinh A; Massias L; Cremieux AC Adjunctive Rifampin is Crucial to Optimizing Daptomycin Efficacy Against Rabbit Prosthetic Joint Infection Due to Methicillin-resistant Staphylococcus aureus. Antimicrob Agents Chemother 2011, 55 (10), 4589–4593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Lefebvre M; Jacqueline C; Amador G; Le Mabecque V; Miegeville A; Potel G; Caillon J; Asseray N. Efficacy of Daptomycin Combined with Rifampicin for the Treatment of Experimental Methicillin-resistant Staphylococcus aureus (MRSA) Acute Osteomyelitis. Int J Antimicrob Agents 2010, 36 (6), 542–544. [DOI] [PubMed] [Google Scholar]
- (30).Nau R; Sorgel F; Eiffert H. Penetration of Drugs Through the Blood-cerebrospinal fluid/Blood-brain Barrier for Treatment of Central Nervous System Infections. Clin Microbiol Rev 2010, 23 (4), 858–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Wood TK; Knabel SJ; Kwan BW Bacterial Persister Cell Formation and Dormancy. Appl Environ Microbiol 2013, 79 (23), 7116–7121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Scherr TD; Hanke ML; Huang O; James DB; Horswill AR; Bayles KW; Fey PD; Torres VJ; Kielian T. Staphylococcus aureus Biofilms Induce Macrophage Dysfunction Through Leukocidin AB and Alpha-Toxin. MBio 2015, 6 (4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Heim CE; Vidlak D; Kielian T. Interleukin-10 Production by Myeloid-derived Suppressor Cells Contributes to Bacterial Persistence During Staphylococcus aureus Orthopedic Biofilm Infection. J Leukoc Biol 2015, 98 (6), 1003–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Heim CE; Vidlak D; Scherr TD; Hartman CW; Garvin KL; Kielian T. IL-12 Promotes Myeloid-derived Suppressor Cell Recruitment and Bacterial Persistence During Staphylococcus aureus Orthopedic Implant Infection. J Immunol 2015, 194 (8), 3861–3872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Heim CE; Vidlak D; Scherr TD; Kozel JA; Holzapfel M; Muirhead DE; Kielian T. Myeloid-derived Suppressor Cells Contribute to Staphylococcus aureus Orthopedic Biofilm Infection. J Immunol 2014, 192 (8), 3778–3792. [DOI] [PMC free article] [PubMed] [Google Scholar]