

Abstract
High serum concentrations of kidney-derived protein uromodulin (Tamm-Horsfall protein or THP) have recently been shown to be independently associated with low mortality in both older adults and cardiac patients, but the underlying mechanism remains unclear. Here, we show that THP inhibits the generation of reactive oxygen species (ROS) both in the kidney and systemically. Consistent with this experimental data, the concentration of circulating THP in patients with surgery-induced acute kidney injury (AKI) correlated with systemic oxidative damage. THP in the serum dropped after AKI, and was associated with an increase in systemic ROS. The increase in oxidant injury correlated with post-surgical mortality and need for dialysis. Mechanistically, THP inhibited the activation of the transient receptor potential cation channel, subfamily M, member 2 (TRPM2) channel. Furthermore, inhibition of TRPM2 in vivo in a mouse model, mitigated the systemic increase in ROS during AKI and THP deficiency. Our results suggest that THP is a key regulator of systemic oxidative stress by suppressing TRPM2 activity and our findings might help to explain how circulating THP deficiency is linked with poor outcomes and increased mortality.
One Sentence Summary:
Uromodulin inhibits systemic oxidative stress via TRPM2.
Editor’s Summary
The oxidative hypothesis
Elevated plasma concentration of the kidney-derived protein uromodulin (THP) has been associated with better kidney function and decreased mortality in patients with and without kidney diseases. However, causal relationship and underlying mechanisms remain unclear. Now, La Favers et al. used transgenic mice and showed that THP deletion resulted in systemic oxidative damage induced by activation of the transient receptor potential cation channel, subfamily M, member 2 (TRPM2) channel. In plasma samples from patients with kidney injury, THP was negatively correlated with ROS expression and oxidative damage was associated with increased mortality. TRPM2 inhibition in a mouse model of kidney injury reduced the injury-mediated ROS increase.
Introduction
Uromodulin (also known as Tamm-Horsfall protein or THP) is a glycoprotein uniquely expressed in the kidney by cells of the thick ascending limb (TAL) of the loop of Henle (1–3). THP is predominantly targeted to the apical surface of cells through a glycophosphatidylinositol anchor and secreted in the urine upon cleavage by a serine protease recently identified as hepsin (4). However, a smaller, but substantial, amount of THP is targeted towards the basolateral domain, making its way into the interstitium and the circulation (1, 5, 6). The mechanism by which THP is directed toward the basolateral side of the TAL is still unclear. This secretory pathway appears to be favored during stress states, such as during recovery from AKI(6, 7). Several studies show that the concentration of circulating THP correlates with kidney function and may serve as an inverse biomarker to identify early stages of chronic kidney disease (8, 9).
More recently, THP has emerged as an independent predictor of survival in patients with and without kidney disease, whereby elevated concentrations of both urinary and serum THP were associated with decreased mortality (10–13). This association, particularly for circulating THP, remained valid even after controlling for kidney function (estimated glomerular filtration rate or eGFR), suggesting that THP may play an independent physiological role, rather than simply acting as a marker of nephron mass and renal activity. Although the mechanism underlying this relationship remains unclear, serum THP was inversely correlated with inflammatory markers (C-reactive protein) and markers of cardiovascular risk (N-terminal pro-B-type natriuretic peptide) (13). It is possible that these systemic effects observed with low concentrations of THP are due to activation of inflammatory pathways within the kidney that extend systemically. In fact, we previously showed that THP deficiency activates the IL-23/IL-17 axis by the kidney, resulting in stimulation of granulopoiesis and systemic neutrophilia (14). However, it is also possible that THP has direct effects outside the kidney, which could underlie the heretofore unexplained systemic outcomes associated with low serum THP.
We previously demonstrated that, within the kidney, interstitial THP is an important regulator of the sensitivity of S3 proximal tubules to injury by inhibiting pro-inflammatory signaling within these tubular epithelial cells (5, 6, 14–16). We showed that basolaterally released THP can bind to the S3 segments, where its presence is essential to inhibit the production of cytokines and chemokines such as CXCL2 and IL-23 (5, 6). We believe that the crosstalk between THP and proximal tubule segments provides an ideal model for elucidating the cellular and molecular mechanisms triggered by THP. By uncovering this signaling within kidney cells, we could also identify an underlying mechanism for the protective effect of THP systemically.
In this manuscript, we utilized an unbiased combined “omics” methodology to identify the molecular signals regulated by THP by taking advantage of our ability to study signaling within specific kidney cells/structures, in vivo in mice, using fluorescence-based laser microdissection and tissue cytometry. Using this approach, we discovered that RAC1 and c-JUN N-terminal kinase (JNK) are activated in proximal tubules from THP−/− kidneys. In addition, we determined that THP−/− mice have increased oxidative stress not only within the kidney, but also systemically in the serum and in the lung, which does not produce THP. The translatability of our findings to human disease and the correlation between THP and systemic ROS burden was demonstrated in a cohort of patients who developed AKI after liver transplant surgery. In this setting, AKI resulted in a drop in serum THP, which was associated with elevated post-surgery oxidative damage. High concentrations of oxidative damage were associated with systemic outcomes such as 60-day mortality. Lastly, we also identified the transient receptor potential cation channel, subfamily M, member 2 (TRPM2) as a key target for THP in inhibiting oxidative stress. Findings from this work might contribute to the understanding of the modulatory role of THP in the kidney and systemically during health and disease.
Results
Concurrent transcriptomic and proteomic profiling uncovers THP-dependent signaling in kidney proximal tubules.
To perform transcriptomic analysis with high precision on isolated S3 (pars recta) proximal epithelial tubular cells from THP−/− and THP+/+ mouse kidneys, we performed fluorescence-based laser microdissection as described recently (17) (fig. S1A), followed by gene microarrays on the extracted RNA (Fig. 1A). Analysis of the data uncovered 956 genes that were up-regulated and 848 genes that were downregulated in S3 cells from THP−/− vs. THP+/+ kidneys, respectively. Bioinformatic analysis using Ingenuity (Qiagen) identified several canonical pathways predicted to be differentially regulated in THP−/− vs. THP+/+ S3 proximal epithelial cells, shown in Fig. 1B (p <0.05). Among these identified pathways, c-JUN-N-terminal kinase (JNK) signaling had the highest z-score for activation (z-score 3.3, fig. 1B, fig. S1B, C). Disease annotation analysis showed that the differential gene expression pattern caused by THP loss in S3 cells was associated with patterns observed in renal damage and injury pathways (fig. S1D).
Fig. 1. THP inhibits activation of the RAC1/p-JNK signaling pathway.
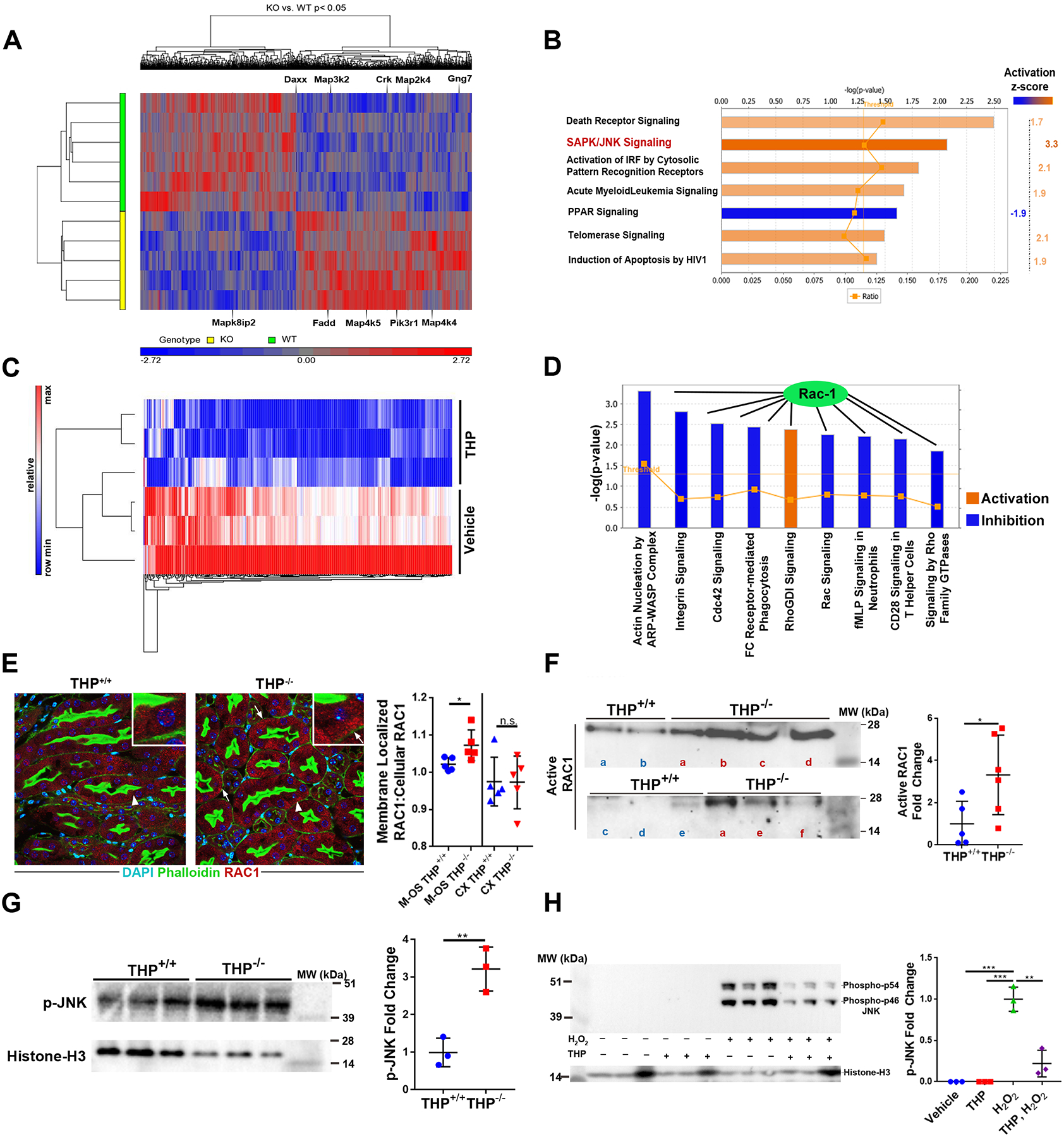
(A) Transcriptomic analysis of laser micro-dissected S3 proximal tubules from THP−/− and THP+/+ mouse kidney.
(B) Canonical pathway analysis (Ingenuity) of differentially expressed transcripts and pathways with Z score >1.5 or <−1.5 are shown with their predicated activation state in the S3 tubules of THP−/− mice.(C) Triplicate groups of HK-2 cells were treated with THP (1μg/ml) or vehicle for 6 hours. Protein extraction was followed by quantitative label-free proteomics. Differentially expressed proteins were identified (shown in heat map).
(D) Canonical pathway analysis of the proteomic data from C was performed and pathways with Z score >2 or <−2 are shown with their predicated activation state in the THP-stimulated HK-2 cells.RAC1
(E) In the left panel, immunofluorescence confocal microscopy shows shifting of RAC1 (red) in THP−/− mice to the basolateral domain of S3 segments (right) compared to a cytoplasmic localization in THP+/+ (left). Insets show a higher magnification of the areas indicated by arrowheads (Arrow points to basolateral RAC1). RAC1 shift is a surrogate for its activation. In the right panel, THP−/− mouse kidneys (n=5 per group) are shown.
(F) Left panel: active RAC1 was immunoprecipated from total kidney lysates of THP+/+ and THP−/− kidneys (n=5–6 per group, sample “a” of the THP−/− group bridges the two immunoblots).Right panel: quantification of the immunoprecipitation experiment.
(G) Phosphorylated JNK was assayed from nuclear extracts of total kidney lysates by immunoblot (left) and quantitated (right) by band densitometry.
(H) Immunoblot of H2O2-dependent JNK activation following treatment with 1 μg/mL of THP in the left panel, quantitated in the right panel.
Scatter plots are mean ± standard deviation. * denotes statistical significance (p<0.05), ** denotes statistical significance (p<0.01), *** denotes statistical significance (p<0.001). MW = molecular weight, kDa = kiloDaltons.
Next, we adopted an orthogonal approach to discover the protein expression changes elicited by treatment of proximal tubular epithelial cells with THP using label-free quantitative mass spectrometry-based proteomics (LFQMS) (18, 19). We incubated HK-2 human proximal tubular cells with purified human THP or vehicle, after which we measured the changes in expression of 1881 proteins identified by LFQMS (Fig. 1C). At 6 hours, THP caused changes in the expression of 406 proteins. Using bioinformatic analysis on the differentially expressed proteins, most of the top canonical pathways affected by THP treatment were linked to the inhibition of RAC1 signaling (Fig. 1D). Since RAC1 activation occurs at the plasma membrane and can lead to downstream activation of JNK signaling (20), RAC1 itself could link THP to JNK activation (fig. S2). Therefore, together with the transcriptomics data, our findings suggest that THP might inhibit the RAC1/JNK signaling pathway in proximal tubules.
THP deficiency activates the RAC1/JNK/c-JUN signaling pathway in proximal epithelial cells.
To validate and further link the transcriptomic and proteomic data, we next surveyed RAC1 activation in kidneys from THP−/− and THP+/+ mice. Confocal microscopy showed that RAC1 shifted from a cytoplasmic localization to the basolateral domain of S3 tubular segments in the absence of THP in the outer stripe, but not in the cortical S1/S2 proximal tubules (Fig. 1E, fig. S3). Because RAC1 is activated at the basolateral domain, its shift is a reliable surrogate for activation (21). Immunoprecipitation of activated RAC1 was performed on kidney lysates. RAC1 activation was significantly increased in the setting of THP deficiency (Fig. 1F, p<0.05). Phosphorylated (activated) JNK in nuclear extracts from total kidney lysates was significantly increased (p<0.05) in THP−/− kidneys compared to THP+/+ kidneys (Fig. 1G). Consistent with the changes in JNK activity observed in the THP−/− kidney, we also observed that THP inhibits JNK activation in HK-2 proximal tubular cells (Fig. 1H).
Furthermore, active JNK leads to phosphorylation of c-JUN (p-c-JUN), which is a member of the AP-1 transcription factor and has been implicated in the transcription of a myriad of inflammatory cytokines, including IL-23 (22). We utilized 3D quantitative imaging and analysis to quantitate the number of p-c-JUN+ cells in THP−/− and THP+/+ in sections from the outer medulla and cortex of the kidney (Fig. 2A, B) using volumetric tissue exploration and analysis software (VTEA, (23)). Since these kidney sections originated from naïve mice, expression of p-c-JUN in THP+/+ was negligible (0.03 ± 0.01% in the outer medulla, 0.08 ± 0.08 % in the cortex, Fig. 2B). In THP−/− outer medulla and cortex sections, p-c-JUN expression was significantly increased compared to THP+/+ (0.98 ± 0.31% and 1.0 ± 0.74%, respectively, p<0.05, Fig. 2B). A three dimensional rendering overlaying phospho-c-JUN staining of nuclei with phospho-c-JUN positive cells gated by VTEA demonstrated the accuracy of the gating approach and confirmed that c-JUN activation was limited to proximal tubules (Fig. 2C). Cumulatively, these data show increased activation of RAC1/JNK/c-JUN in THP−/− kidneys. For RAC1, this appears to be predominantly evident in S3 proximal tubules, whereas increased c-JUN activation occurs in most proximal tubules within the kidney. Taken together with the transcriptomic and proteomic analyses shown so far, we concluded that THP deficiency stimulates the activation of RAC1/JNK/c-JUN in the kidney in mice.
Fig. 2. THP inhibits activation of p-C-JUN.

(A-C) Sections from the outer medulla of kidneys from THP+/+ and THP−/− mice were stained for phosphorylated (active) c-JUN (p-C-JUN, A). Volumetric tissue exploration and analysis (VTEA) was used to quantify the percentage of cells positive for p-c-JUN staining (B). In C, 3D rendering of a representative p-c-JUN stained image is overlaid with the VTEA gating.
Scatter plot error bars represent mean ± standard deviation, * denotes (p< 0.05), ** denotes (p<0.01). M-OS = Medulla, outer stripe. CX = Cortex.
Proteomic analysis of S3 proximal cells reveals dysregulation of the free radical scavenging network in THP−/− mice.
To further characterize the changes elicited in S3 proximal epithelial tubular segments in the setting of THP deficiency, we performed targeted proteomic analysis on S3 proximal epithelial cells in mice. This was achieved by isolating S3 segments by laser microdissection from THP+/+ and THP−/− kidneys, followed by protein extraction and two-dimensional differential gel electrophoresis (2D-DIGE), as described in a recent publication (fig. S4A–C) (17). Differentially expressed proteins were identified by mass spectrometry (fig. S4D). Bioinformatics analysis of the differentially expressed proteins identified the free radical scavenging network as the top network of proteins that was differentially regulated in THP−/−mice (fig. S4E). This analysis predicted increased generation of reactive oxygen species (ROS) in S3 segments from THP−/− compared to THP+/+ mice. (fig. S4F).
The predicted redox imbalance within the THP−/− S3 tubules was consistent with the observed activation of the RAC1/JNK/c-JUN signaling pathway in these mice. This connection between ROS and RAC1/JNK signaling was highlighted in the combined signaling/oxidative stress bioinformatics model (fig. S5).
THP deficiency leads to increased oxidative stress and damage within the kidney.
Given the predicted redox imbalance of S3 segments within THP−/− kidneys, we performed live imaging of kidneys from mice injected with a redox sensitive dye, H2DCFDA,. We found that THP−/− mice have a higher burden of oxidative stress throughout the kidney cortex (Fig. 3A, p<0.05). We extended these findings to the kidney medulla using another redox sensitive dye (CellROX Deep Red) which helped to localize a ROS signal to the S3 segments (fig. S6A, B, p<0.05). To further quantitate the differences in oxidative stress found in THP−/− mice, we measured hydrogen peroxide (H2O2) directly using a colorimetric assay and found that its concentration was significantly increased in total kidney lysates from THP−/− kidneys compared to THP+/+ (Fig. 3B, p<0.05). Taken together, these results suggest that kidneys from THP−/− mice have an increased ROS burden throughout the kidney, particularly within the S3 tubules.
Fig. 3. THP−/− mice have increased oxidative stress and damage within the kidneys and systemically.

(A) Live imaging with the ROS-sensitive dye H2DCFDA in kidneys of THP+/+ and THP−/− mice (inset: background fluorescence) in the left panel is quantitated in the right panel (n=3–4 per group)
(B) H2O2 concentrations in kidney lysates from THP+/+ and THP−/− mice (n=5 per group). Values were normalized to the amount of protein present in each lysate.
(C) Heat map of oxidized phosphatidylcholine (PC) and phosphatidylethanolamine (PE) concentrations in the kidneys of THP+/+ and THP−/− mice (n=3 per group).
(D-F) Concentration of 8OHdG, a marker of oxidative DNA damage, in the kidney (D, n = 7–15 per group), serum (E, n = 15–17 per group) and lungs (F, n = 8 per group) of THP−/− mice compared to THP+/+ mice. Scatter plots errors bars represent mean ± standard deviation.
* denotes statistical significance between the two groups (p< 0.05). *** denotes statistical significance between the two groups (p< 0.001).
To determine what types of damage might be occurring as a result of the increased oxidative stress burden within the THP−/− kidneys, we first quantitated the oxidized forms of the major classes of phospholipids in kidney lysates using targeted lipidomics. A heat map showing the relative quantity of oxidized phospholipids in THP+/+ and THP−/− is shown in Fig. 3C. Specifically, we found that THP−/− kidneys have increased amounts of oxidized phosphatidylcholine (Fisher’s exact test, p<0.0001) and phophatidylethanolamine (Fisher’s exact test, p<0.0001) lipids as compared to THP+/+ kidneys (Fig. 3C, Data File S3). Since oxidative stress is known to cause organelle dysfunction, we investigated mitochondrial morphology in S3 proximal tubules using toluidine blue staining and electron microscopy studies. We observed increased mitochondrial swelling and deformation in S3 cells of THP−/− compared to THP+/+ mouse kidneys (fig. S7A–C, two-way ANOVA, p<0.0001). Collectively, these data further establish that THP deficiency increases oxidative stress in the kidney, particularly in proximal tubular segments. Given our previous data that THP−/− kidneys have increased expression of IL-23 within the S3 tubules, we hypothesized that increased oxidative stress within the S3 tubular cells leads to increased IL-23 expression. In fact, we found that treatment of HK-2 cells with H2O2 leads to an increase in IL-23 mRNA expression (fig. S8), providing a potential link between these two observations.
THP Deficiency Leads to Systemic Oxidative Damage.
Given the role that THP deficiency plays in increasing oxidative stress and damage within the kidney, along with our previous data showing that the loss of THP can lead to systemic effects on inflammation (24) and neutrophil homeostasis (14), we hypothesized that the basolateral release of THP into the interstitium and circulation could mitigate the buildup of oxidative damage systemically. We measured oxidative DNA damage in the kidneys, serum and lungs in THP+/+ and THP−/− mice using an assay for 8-hydroxy-2’-deoxyguanosine(8OHdG) (25), which provides a robust method to quantify oxidative damage that could be used in both tissue lysates and serum. As expected, THP−/− mice had higher DNA damage in the kidneys (Fig. 3D, p<0.05), but we also observed higher concentrations in the serum (Fig. 3E, p<0.001) and the lungs (Fig. 3F, p<0.05). From this data, we conclude that the loss of THP leads to increased oxidative damage not only in the kidneys, but also in the serum and in the lung, an organ that is not known to produce THP. These findings suggest that circulating THP, through an effect distant from the kidney, might mitigate systemic oxidative damage.
To test the relevance of these findings in disease, we subjected THP+/+ mice to AKI using the ischemia reperfusion injury (IRI) model. We found that serum THP decreased twenty-four hours post-surgery (Fig. 4A), while oxidative DNA damage and kidney function/injury markers increased (Fig. 4B, C). Therefore, these results extend the findings from the THP−/− mice to a disease model in THP+/+ mice, where a decrease in systemic THP was observed in early AKI, concomitant with increased systemic oxidative stress.
Fig. 4. Serum THP decreases and oxidative DNA damage increases following ischemic AKI in THP+/+ mice.

(A) Serum THP concentrations of THP+/+ mice before and 24 hours following IRI surgery (n=15 mice)
(B) Serum DNA damage concentrations of THP+/+ mice before and 24 hours following IRI surgery (n=15 mice)
(C) Serum concentrations of kidney injury markers Cystatin C, NGAL and Osteopontin in THP+/+ mice before and 24 hours following IRI surgery (n=15 mice)
Scatter plots are mean ± standard deviation. ** denotes statistical significance (p<0.01). *** denotes statistical significance (p<0.001). **** denotes statistical significance (p<0.0001).
Plasma THP concentrations drop during human AKI, and are associated with increased systemic oxidative damage and outcomes
To test the translatability and relevance of these findings to human disease and validate the relationship of THP with oxidative stress, we measured changes in plasma THP and oxidative stress among 41 patients with liver diseases before and 18 hours after liver transplantation. Demographics and clinical variables for this cohort are shown in Tables 1 and S1. Baseline measurements were not different compared to a small number of control subjects who did not undergo liver transplantation (Table S1), supporting that the liver disease burden in this cohort did not affect the baseline measurements. In patients who did not develop post-surgery AKI, pre-surgery baseline eGFR was higher (Table S1), consistent with previous reports that pre-surgical kidney function is an important risk factor of post-surgery AKI (26, 27).
Table 1. Characteristics of the AKI cohort.
Demographic and clinical data for a cohort of liver transplant recipients who developed acute kidney injury following transplant surgery.
| Sex | 56% |
| Male | 44% |
| Female | |
| Age (years) | 53.5 ± 11.3 |
| Race | |
| Caucasian | 85% |
| African | 3% |
| Hispanic | 9% |
| Asian | 3% |
| Body Mass Index (kg/m2) | 27.5 ± 5.03 |
| Pre-surgery Plasma 8OHdG (ng/mL) | 11.8 ± 8.14 |
| Post-surgery Plasma 8OHdG (ng/mL) | 16.3 ± 11.8 |
| Pre-surgery Plasma THP (ng/mL) | 291 ± 258 |
| Post-surgery Plasma THP (ng/mL) | 102 ± 93.6 |
| Pre-Surgery eGFR (mL/min/1.73 m2) | 67.8 ± 33.0 |
| Pre-surgery Plasma Creatinine (mg/dL) | 1.32 ± 0.807 |
| Post-surgery Plasma Creatinine (mg/dL) | 1.53 ± 0.689 |
| Acute Kidney Injury Stage | |
| Stage 1 | 15% |
| Stage 2 | 29% |
| Stage 3 | 56% |
| Received Dialysis | 47% |
| 60 Day Survival | 77% |
In 34 patients who developed AKI based on the Kidney Disease Improving Global Outcomes (KDIGO) serum creatinine criterion (28), THP concentrations were inversely correlated with systemic DNA damage (Fig. 5A, R2= 0.06, p<0.05), which is consistent with the mouse data above. When compared to pre-surgery concentrations, plasma THP decreased significantly at 18 hours following surgery in patients with AKI (Fig. 5B, Table 1, 291 ±258 ng/ml vs. 102 ± 93.6 ng/ml, p<0.001). These findings confirm, in humans, that AKI results in a state of acute systemic THP deficiency. This decrease in THP was concomitant with a significant increase in oxidative DNA damage (Fig. 5C, Table 1, 11.8 ± 8.14 ng/ml vs. 16.3 ± 11.8, p<0.05). Pre-surgery concentrations of THP in patients with AKI were inversely correlated with post-surgery DNA damage (Fig. 5D, R2 = 0.23, p<0.05), demonstrating that higher baseline THP correlated with lower oxidative damage incurred post-surgery. In patients who did not develop post-surgery AKI, there was no statistical difference between pre and post-surgery THP, although concentrations were lower, on average, post-surgery (fig. S9).
Fig. 5. Plasma THP decreases and oxidative DNA damage increases in AKI patients following transplant surgery.
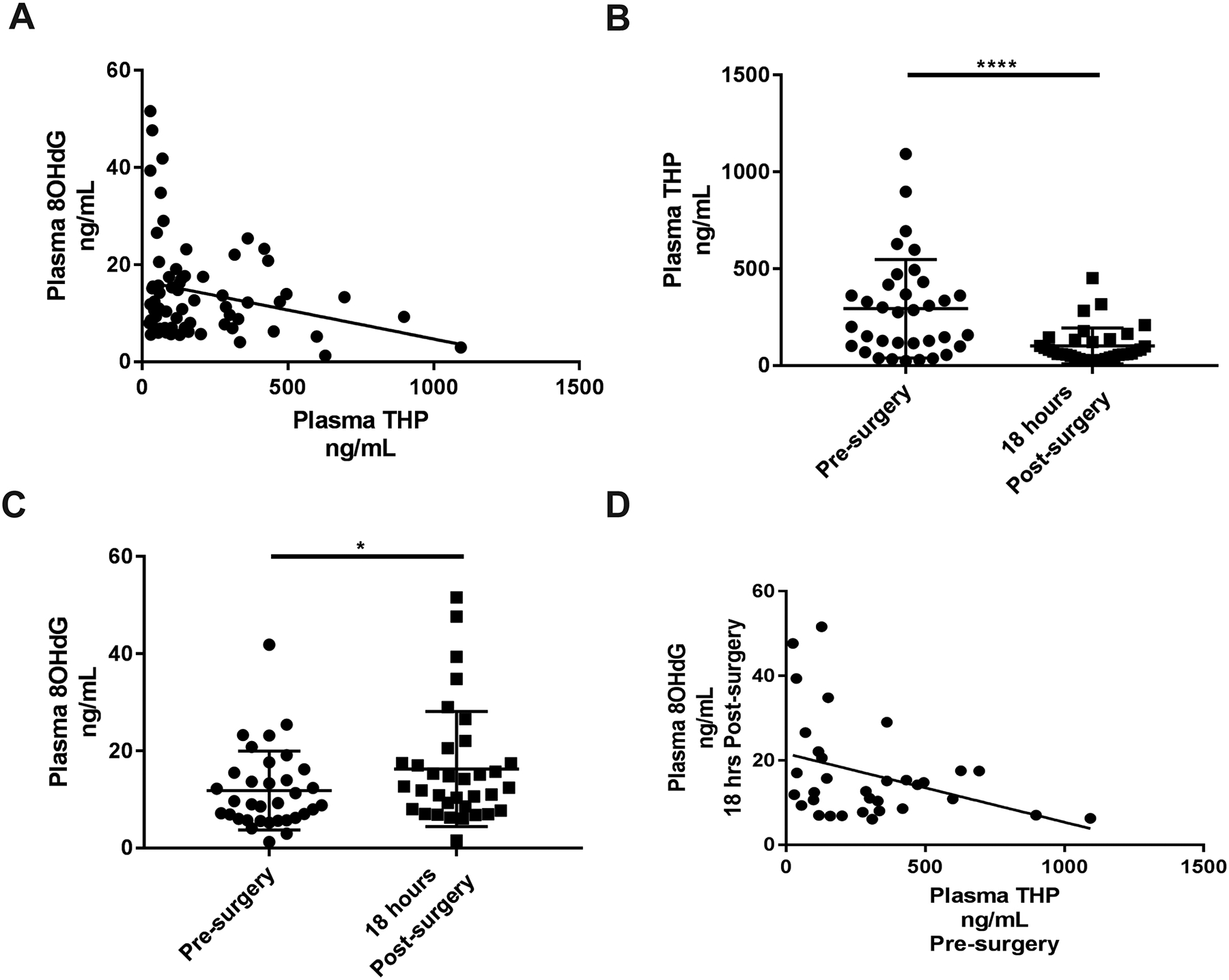
(A) Correlation of THP and 8OHdG concentrations from all time-points for AKI cohort and control participants (n=43)
(B) Pre-surgery plasma THP concentrations compared to 18 hours post-surgery concentrations (paired analysis, n=34)
(C) Pre-surgery plasma 8OHdG concentrations compared to 18 hours post-surgery concentrations (paired analysis, n=34)
(D) Correlation of pre-surgery plasma concentrations of THP with plasma 8OHdG levels collected 18 hours post-surgery (n=34, R2 = 0.23, p < 0.05)
Scatter plot error bars are mean ± standard deviation. * denotes statistical significance (p<0.05). **** denotes statistical significance (p<0.0001).
Although this human study was not designed, nor powered, to detect a relationship between THP and/or oxidative stress concentrations with other endpoints such as mortality or need for dialysis, we did find that the post-surgery oxidative DNA damage plasma concentrations were increased in non-survivors (Fig. 6B, 14.0 ± 9.94 ng/ml in survivors vs. 23.6 ± 14.7 ng/ml in non-survivors, p<0.05). Pre-surgery concentrations of plasma oxidative DNA damage did not differ between survivors and non-survivors (Fig. 6A) suggesting that elevated oxidative damage in the plasma as early as 18 hours post-surgery may be a predictor of mortality. However, plasma THP both before (Fig. 6C) and after (Fig. 6D) development of AKI was not different in non-survivors compared to survivors. Oxidative DNA damage pre and post-surgery was higher in patients who subsequently required dialysis (p<0.05, Fig. 6E, F). Together with the animal data presented above, these results suggest that high concentrations of circulating THP may be protective from increased oxidative stress.
Fig. 6. Oxidative DNA damage is associated with mortality and need for dialysis in AKI patients following transplant surgery.

(A-B) Pre-surgery (A) and post-surgery (B) plasma concentrations of 8OHdG in patients who did or did not survive following transplant surgery and development of AKI. (n=34)
(C-D) Pre-surgery (C) and post-surgery (D) plasma concentrations of THP in patients who did or did not survive following transplant surgery and development of AKI. (n=34)
(E-F) Pre-surgery (E) and post-surgery (F) plasma concentrations of 8OHdG in patients who did or did not receive dialysis following transplant surgery and development of AKI. (n=34)
** denotes statistical significance between the two groups (p< 0.01).
*** denotes statistical significance between the two groups (p< 0.001).
THP inhibits systemic oxidative stress through TRPM2.
Having established the signaling pathways activated during THP deficiency and the resultant oxidative damage, we set out to determine the molecular target linking THP with increased oxidative stress and activated RAC1/JNK signaling. We hypothesized that THP could targets a RAC1 modulator. One of the candidate molecules is the transient receptor potential cation channel, subfamily M, member 2 (TRPM2). TRPM2 is a non-voltage activated, Ca2+-permeable, non-selective cation channel that plays an important role in ROS-coupled diseases (41, 42) Therefore, we hypothesized that THP inhibits TRPM2 and the downstream RAC1/oxidative stress/JNK signaling pathway. THP was recently shown to regulate the activity of a related channel, TRPM6 (29).We first investigated whether THP can directly alter the function of TRPM2 using a HEK 293 recombinant cell line expressing an inducible copy of TRPM2 in which channel specific activation can be monitored by measuring the kinetics of calcium influx after stimulation with H2O2, which increases the open state of the channel (30). Using this approach, we showed that THP inhibited TRPM2 activity (Fig. 7A), leading to complete inhibition of TRPM2-mediated calcium influx. This effect was specific (Fig. 7B) to THP, since similar inhibition was not shown with human serum albumin (HSA), and dose dependent (Fig. 7C). The potency of this inhibition was also equivalent to N-(p-amylcinnamoyl) anthranilic acid (ACA), a known TRP channel blocker (31) (Fig. 7D). Therefore, our data demonstrate that THP specifically inhibits the activation of TRPM2. Consistent with this finding, inhibition of TRPM2 in HK-2 cells with ACA blocked JNK activation, as was previously demonstrated with THP (fig. S4, Fig. 8), suggesting that the inhibitory effect of THP on JNK activation is at least partially dependent on TRPM2.
Fig. 7. THP inhibits TRPM2 calcium influx in vitro and TRPM2 inhibition in vivo reduces oxidative damage.

(A-D) HEK293 cells expressing a tetracycline-inducible TRPM2 channel were treated with doxycycline or vehicle control (vehicle control, no TRPM2) for 24 hours before loading with the calcium indicator Fluo-4. Cells were pre-treated with THP, human serum albumin (HSA) or N-(p-amylcinnamoyl) anthranilic acid (ACA), respectively, prior to activation of the TRPM2 channel with H2O2. Calcium influx was measured following addition of extracellular calcium (CaCl2). In (A) pre-treatment with 1 μg/ml THP is shown. In (B) pre-treatment with 1 μg/ml THP is compared to 1 μg/ml HSA. In (C), pre-treatment with 0.01 μg/ml, 0.1 μg/ml and 1 μg/ml THP are compared. In (D), treatment with 1 μg/ml THP is compared to 25 μM ACA, a small molecule inhibitor of TRPM2. Representative results are shown from a single experiment (n=3 wells per treatment condition).
(E) Concentration of oxidative DNA damage (8OHdG) in THP−/− mice treated with the TRPM2 inhibitor 2-aminoethyoxydiphenyl borate (2-APB, 16 mg/kg) or vehicle (10% DMSO/90% sterile saline) for three days (n=7–8 mice/group).
(F) Concentration of oxidative DNA damage (8OHdG) in THP−/− and THP+/+ mice treated with TRPM2 inhibitor 2-APB (16 mg/kg) or vehicle (10% DMSO/90% sterile saline) for three days before performing a 22-minute bilateral clamping of the renal artery followed by six hour recovery. (n=7–8 mice/group).
Scatter plots are mean ± standard deviation. * denotes statistical significance (p<0.05). **** denotes statistical significance (p<0.0001)
Fig. 8. Inhibition of TRPM2 blocks JNK activation in HK-2 cells.

(A) Immunoblot for TRPM2 in HK-2 cell lysate.
(B) Immunoblot of H2O2-dependent JNK activation following treatment with 25 μM ACA in the left panel, quantitated in the right panel.
Scatter plots are mean ± standard deviation. * denotes statistical significance (p<0.05) *** denotes statistical significance (p<0.001). **** denotes statistical significance (p<0.0001). kDa = kiloDaltons.
TRPM2 is expressed at high concentrations in the brain and immune system, and to a varying degree in other organs such as heart, spleen, liver, pancreas and kidney (32). Due to its broad expression profile in immune cells and the demonstrated inhibition by THP in vitro, we hypothesized that TRPM2 mediates the systemic oxidative damage observed in THP−/− mice. To test this hypothesis, we treated THP−/− mice with the TRPM2 inhibitor 2-Aminoethoxydiphenyl borate (2-APB), previously used for in vivo studies, (33) and found that 2-APB significantly lowered systemic oxidative DNA damage in these mice (Fig. 7E, p<0.05). To extend these findings to AKI, where TRPM2 was shown to promote ischemia reperfusion injury (IRI) (34), we subjected THP+/+ and THP−/− mice to IRI following TRPM2 inhibition with 2-APB and measured oxidative damage in the serum 6 hours later. The vehicle-treated THP−/− mice showed increased oxidative damage as compared to vehicle-treated THP+/+ mice, mirroring the results in uninjured animals (Fig. 7F, p<0.05). Furthermore, pre-treatment of both the THP+/+ and THP−/− mice with 2-APB significantly reduced oxidative damage post-IRI (Fig. 7F, p<0.05 and p<0.01, respectively). The difference in oxidative damage seen between the THP+/+ and THP−/− mice was lost upon treatment with 2-APB, implying that TRPM2 is a main target for the inhibitory effect of THP on systemic oxidative stress.
Discussion
In this study, using a combination of experimental approaches and a small human cohort, we show a role for THP in maintaining redox balance not just in the kidney, but also systemically. This protective effect of THP against systemic oxidative stress occurs by inhibiting TRPM2 channel activation. Since TRMP2 is expressed in many organs throughout the body, particularly in the immune system (32), uncovering the THP-TRPM2 link provides a potential mechanism by which the kidney impacts other organs following the release of THP into the circulation. It is important to note that non-specific targeting of TRPM2 itself is likely to be risky, because of its pleotropic constitutive expression and tissue specific effects (35). However, targeted interventions that modulate the concentration of systemic THP may hold a future therapeutic potential, especially in view of recent experimental data that support such interventions (36).
We found that there is increased oxidative stress in proximal tubules, both within the cortex (predominantly S1/S2 proximal tubules) and the outer medulla (predominantly S3 proximal tubules) in kidneys from THP−/− mice. Oxidative stress results from an imbalance between generation of ROS such as superoxide and hydrogen peroxide, and the reductive buffer system that quenches the excess of these toxic molecules (37). Within the cells, ROS at low levels are essential byproducts of oxidative phosphorylation and needed for signal transduction (37–40). However, excess ROS will damage the cell through multiple pathways, such as direct damage to organelles, macromolecules, and impairment of normal functioning systems, leading eventually to cell death. Interestingly, ROS can directly stimulate inflammatory signaling, such as NFkB and AP-1-dependent pathways, leading to the production of a myriad of pro-inflammatory cytokines (37, 38). Therefore, by inhibiting oxidative stress, THP can potentially regulate both cell fate of S3 segments and the production of inflammatory mediators, such as IL-23.
We also found that the RAC1/JNK/c-JUN pathway is activated in these tubules. Oxidative stress stimulates RAC1/JNK/c-JUN signaling (41) and links the proteomics analysis of S3 tubules to the transcriptomic analysis of S3 tubules. Many of the proteins dysregulated in the S3 tubules of THP−/− mice, including ALBU, ALDH3, GPX3, and SODC, are also linked to RAC1/JNK signaling, either as upstream regulators or targets (42–53). Our data suggested that the burden of oxidative stress may be accentuated in S3 segments, which is also supported by the differential shift of RAC1 to the basolateral membrane in outer medulla cells. This is not surprising, given the fact that the absence of THP may be more impactful in the outer medulla than the cortex, due to a relatively higher frequency of TALs in this region, which are likely releasing higher amounts of THP into the interstitium surrounding S3 tubules (6). JNK activation, as measured by phosphorylation of c-JUN, is present in both the cortex and the medulla, which may reflect the generalized presence of oxidative stress and the high sensitivity of the 3D imaging assay used.
Circulating THP has been shown to correlate with kidney function (8, 9), is inversely associated with CKD and predicts the risk of developing CKD (54). Of note, risk variants in the UMOD promoter region may be independent modifiers of CKD risk and further studies are needed to investigate their interaction (55), especially how they may be associated with the concentrations of circulating THP. Our study does not specifically address how circulating THP is linked to the risk of CKD. It is possible that the decrease in THP production and systemic release observed in CKD (54) can lead to increased ROS within the kidney, thereby predisposing it to ongoing tubular injury and fibrosis (56). This hypothesis will need to be tested in models of chronic kidney disease.
TRPM2 is a multifunctional non-selective calcium ion channel. It is expressed in high levels in the brain, but also in other organs such as heart, spleen, liver, bone marrow, pancreas and kidney (32). Within organs, its expression is uneven with specific cells exhibiting a high level of expression (32). In the kidney specifically, the link between TRPM2, RAC1-dependent signaling and oxidative stress in epithelial cells has been recently demonstrated in the context of AKI (34). In our study, we extended these findings by showing that THP could be an important regulator of TRPM2, RAC1 and oxidative stress. We also showed that THP deficiency, whether genetic or due to AKI, was associated with elevated systemic oxidative damage. The increased systemic ROS burden was mitigated by treatment with a TRPM2 inhibitor, suggesting that it is at least partially TRPM2 dependent. The relationship between ROS and TRPM2 is complex because TRPM2 is both activated by ROS and can also amplify downstream oxidant damage (57). Our work may help clarify this relationship by identifying THP as an upstream mediator of TRPM2 and ROS.
Findings from this study provide a mechanism that could help to explain how serum THP is linked with systemic outcomes such as all-cause mortality and may have a high clinical impact. When interpreted with other clinical data linking reduced serum THP to mortality (11, 13), our current findings suggest that regulation of systemic oxidative stress is a mechanism that helps explain this link. The increased susceptibility to oxidative stress has particular implications in diseases leading to acute THP deficiency, such as AKI, which is supported by our data exploring the relationship between THP and oxidative damage in human AKI. Our study also provides a rationale to explore the sensitivity of serum THP as a biomarker for early AKI in large studies, especially with a trend in decreased THP even observed in patients who did not develop AKI by standard clinical criteria. Finally, our findings also raise the possibility of using THP and oxidative stress measurements to prognosticate and guide overall clinical management post-AKI. These prospects will require validation in a large prospective study before they can be utilized in the clinic.
A major strength of our experimental design was the use of a multi-disciplinary approach, combining complementary “omics” and imaging techniques that converged on a specific finding, which we were able to translate from experimental models to patients. Though this strategy could help to overcome experimental limitations, it is important to note these limitations to facilitate interpretation. Although the exact sub-segmental origin (S1–S2 or S3) of the HK-2 cells utilized for the proteomic studies of THP-treated cells is not well established, there is currently no suitable alternative, because there are no available S3 specific pure human cells (58). In addition, HK-2 cells are among the most characterized and commonly used human tubular epithelial cells, which can facilitate the reproducibility of this work. The use of these human cells also allows the extension of the findings to human pathology. For the THP treatment cell studies, the monomeric THP used to mimic basolaterally released THP is cleaved differently than serum THP, which is a potential limitation of this work (36). Additionally, the interpretation of these findings needs to be balanced by the fact that the dose of THP used in the HK-2 cell experiments is likely in the pharmacological range, compared to circulating concentrations. However, in the TRPM2 inhibition experiments even physiological concentrations inhibit TRPM2-mediated calcium influx. For the human studies, the statistical power to establish relationships between THP, oxidative stress and clinical variables is limited by the sample size, which also restricted our ability to make comparisons between patients who developed more or less severe kidney injury. Finally, although the use of a transgenic cell line for the TRPM2 studies allowed us to show specific inhibition of the TRPM2 channel, we are unable to determine which TRPM2-expressing cells are the primary targets of THP inhibition in vivo, which will require additional studies to establish.
In conclusion, our study suggests that circulating THP is an important regulator of renal and systemic oxidative stress. Findings from this work could advance our understanding of the modulatory role of THP in the kidney and systemically during health and disease.
Materials and Methods
Study Design
In this study, we studied the S3 proximal tubules in THP−/− mice to generate, and subsequently test, hypotheses about THP’s function. We utilized laser microdissection to isolate S3 proximal tubules from THP+/+ and THP−/− mice and performed transcriptomic and proteomic studies on the samples. In parallel, we treated a proximal tubular cell line with THP prior to proteomic analysis. Additional experiments were performed to validate, explore and expand on these results, as described in the following methods sections. Sample sizes were established for different analyses as follows. For in vitro experiments, all experiments were performed with three technical replicates and were repeated a minimum of 1–2 additional times to ensure reproducibility. For animal experiments in uninjured animals, a pilot experiment with 3–5 animals per group was first conducted to measure effect size and conduct power calculations to scale up the experiment accordingly. For animal experiments using the IRI model of AKI, experiments were conducted with 8–10 mice per group, based on the number of mice required for significance in uninjured animals along with feasibility considerations. The subjects were not randomly assigned to experimental groups due to animal housing considerations. The study was not blinded. All data acquired is presented in the main figures and supplementary data.. These criteria were established prospectively. Statistical outliers were defined using the Tukey’s fences methods, unless otherwise described in the specific methods sections.
Mice
Animal experiments and protocols were approved by the Indianapolis VA Animal Care and Use Committee. Age matched 8–12 week-old Tamm-Horsfall protein (THP) knockout male mice (129/SvEv THP−/−) and wild type background strain were used as described previously (6, 7). For 2-APB dosing experiments, mice were dosed as described previously (34), with either 16 mg/kg 2-APB in 90% saline/10% DMSO or vehicle control (90% saline/10% DMSO), once daily for three days before collecting organ and serum samples. Ischemia reperfusion injury was performed as described previously (16), following dosing with 2-APB or vehicle control for three days as indicated for each experiment. The final dose of 2-APB or vehicle control was given prior to the beginning of ischemia reperfusion injury surgery. Mice were allowed to recover for six hours before collecting organ and serum samples.
Immuno-fluorescence confocal microscopy and 3D tissue cytometry
Immuno-fluorescence staining for RAC1 (Millipore, Cat #05–389), DAPI (Sigma Aldrich, Cat #D9542), p-c-JUNC-JUN (Cell Signaling Technology, Cat #3270S) and FITC-phalloidin (Molecular Probes, Cat. #D1306 and #F432) was performed on 50 μm vibratome sections of kidneys fixed with 4% para-formaldehyde as described previously (6). Briefly, immunostaining was done at room temperature, overnight, with primary antibody in PBS + 2% BSA + 0.1 % Triton X-100 and secondary antibody + FITC-phalloidin + DAPI in the same buffer. After mounting on glass slides, stained sections were viewed under Olympus Fluoview laser-scanning confocal microscope. Images were collected under X20 and X60 magnification. Quantitation of the cellular distribution of RAC1 fluorescence in proximal tubules (identified by Oregon 488-green phalloidin staining of the brush borders) was performed on high power fields (2 fields per section, 1 section per mouse, 5 mice per each experimental group), using ImageJ. Basolateral membrane associated RAC1 average intensity was measured along the basolateral border (outlined by F-actin staining) in a 4 pixel (1.56 μm) spanning region of interest using a brush tool (Fig. S3). Cellular RAC1 average intensity for each corresponding tubule was measured in the remaining area. The ratio of membrane associated RAC1: cellular RAC1 was calculated for each tubule. 3D tissue cytometry was performed with Volumetric Tissues Exploration and Analysis (v0.5.2) (23). Briefly, cells were segmented by their nuclei. Identifying cell-types or cell characteristics was accomplished by measuring the intensity of markers either within the putative nuclei or a small surrounding volume generated with a morphological dilation. These measurements are collated and plotted on an interactive scatter plot, which can be interrogated with gating approaches. Here, p-c-JUN intensity was assessed in the nuclei of segmented cells and positive cells were gated and counted.
Electron Microscopy
Upon harvesting, kidneys were perfused-fixed with 3% glutaraldehyde buffered with 0.15M sodium cacodylate, and subsequently embedded in Poly Bed 812 embedding mixture resin. For light microscopy, 0.5μm-thick sections were cut using an ultramicrotome, and stained with toluidine blue. For transmission electron microscopy, 70–90 nm sections were cut, collected on 200 mesh copper grids, viewed and photographed on a Tecnai electron microscope. We quantified mitochondrial swelling by measuring the maximum length of the short axis for all mitochondria within 5 cells from the representative images shown for THP+/+ and THP−/− mice.
Laser Micro-dissection
Kidneys were extracted after euthanasia. Sections from each kidney were snap frozen in OCT compound (Tissue-Tek, Cat. #4583) on dry ice and kept at −80° C until use. They were subsequently cut using a cryotome at 10 μm sections and mounted on Leica PPS membrane slides (Leica, Cat. #11505268). The protocol for staining S3 proximal tubules, described previously in detail (17), identifies these tubules by the intense brush border stain in the outer medulla. Following rapid dehydration/re-hydration steps the samples were stained with FITC-phalloidin (1:20) + DAPI (Molecular Probes, 1:300), washed with PBS and the membranes were allowed to air dry. Following staining, sections were immediately taken to a Leica LMD6000 Laser micro-dissection microscope. Dissection was performed at 40x magnification under fluorescence. There were 200–250 segments dissected in each 90-minute session (average dissected area varies on type of tubules, for S3 segments this ranges from 350,000 – 500,000 μm2). RNA extraction was performed using Arcturus PicoPure RNA isolation kit (Applied Biosystems).
Micro-arrays
Total RNA was analyzed using the Agilent Bioanalyzer (Agilent Technologies) to assess the quality. Samples were processed in two batches with THP−/− and THP+/+ samples in each batch. Two ng of total RNA for each sample was labeled using the standard protocol for the Affymetrix WT Pico kit (Affymetrix). Individual labeled samples were hybridized to the Mouse Gene 2.0 ST GeneChipsfor 17 hours then washed, stained and scanned with the standard protocol using Affymetrix GeneChip Command Console Software (AGCC) to generate data (CEL files). Arrays were visually scanned for abnormalities or defects; none were found.
CEL files were imported into Partek Genomics Suite 6.6 (Partek, Inc.) for analysis. RMA (Robust multichip average (59)) expression values were generated for all probe sets using the RMA background correction, Quantile normalization and summarization by Median Polish. Summarized signals for each probe set were log2 transformed. These log transformed signals were used for Principal Components Analysis, hierarchical clustering and signal histograms to determine if there were any outlier arrays. No outliers were found. Data were analyzed using a 2-way ANOVA with factors for genotype (THP−/−/THP+/+) and labeling batch. The contrast for THP−/−/ vs. THP+/+ was performed, and fold changes were calculated using the untransformed RMA signals. Probe sets with log2 expression values < 3.0 were below background. Probe sets (transcripts) with average expression values < 3.0 for all experimental groups (60) and Affymetrix control probe sets were removed.
Hierarchical clustering of differentially expressed genes was done in Partek, using Euclidean distance for arrays and Pearson’s dissimilarity for genes with average linkage.
Protein extraction, 2-dimensional differential gel electrophoresis (2D-DIGE) and Mass spectrometry
This experiment has been described in detail in an initial publication (17). Briefly, 2D-DIGE was performed on S3 segments isolated from wild type (THP+/+) and Tamm-Horsfall Protein knockout (THP−/−) mice using IF-LMD (performed by Applied Biomics). In brief, an initial analytical gel was done by using 50 μg of protein extract from each S3 sample (THP−/− and THP+/+). Each sample was labeled with Cy2 or Cy3, mixed and separated on a 2D gel. In-gel data analyses for protein spots and comparison of the integrated volumetric ratios were done using the DeCyder software. Spots chosen by the DeCyder software were identified using mass spectrometry (MALDI/TOF then MALDI/TOF/TOF) on the basis of peptide fingerprint mass mapping and peptide fragmentation mapping. The MASCOT search engine was used to identify proteins from primary sequence databases.
Measurement of oxidative stress in vivo
A hydrogen peroxide assay (Abcam, Cat. #102500) was used to measure the concentration of H2O2 in kidney lysates from THP−/− and THP+/+ mice, following the manufacturer’s instructions. Concentrations were normalized to the amount of protein present in each sample.
Using intravital microscopy, oxidative stress was measured in the live animals with H2DCFDA (Life Technologies). H2DCFDA was administered intravenously as a 7 mg/kg bolus from a stock dissolved in ethanol and rediluted in PBS. Images were collected before injection and 20 minutes after H2DCFDA administration, as we described previously (61). Fluorescence intensity was measured using ImageJ in 3–4 fields (20X objective) per animal; the values for each animal were expressed as a fold change over background intensity (image taken just prior to injection).
To localize the site of oxidative stress, we used an in vivo redox dye CellROX Deep Red (Life Technologies, Cat. #C10422). THP+/+ and THP−/− mice were injected with Hoechst 33342 (50 μg/mouse, Molecular Probes, Cat. #H3570) and CellROX (50 μl/mouse of 2.5 mM solution) intraperitoneally, and mice were euthanized 2 hours later and immediately sectioned for imaging using confocal microscopy. Another set of kidneys were fixed with 4% paraformaldehyde and imaged following co-staining with FITC-phalloidin. Fluorescence intensity was measured using ImageJ in 2–4 fields (20X objective) per animal and the values for each animal were expressed as a fold change over the negative control (THP+/+ mouse injected with Hoechst 33342 only).
Tamm-Horsfall protein purification
Tamm-Horsfall protein was purified from normal human urine according to the method described by Tamm and Horsfall (62, 63). THP was subsequently treated with urea, and resolved using gel filtration to recover a non-aggregated form in the range of 60–120 kiloDaltons (kDa), as we described recently (36). The purified THP was kept in a 5% Dextrose water solution. In all in vitro experiments, the concentration of endotoxin was < 0.02 EU/ml, measured using Limulus Amebocyte Lysate (LAL) assay.
HK-2 cells
HK-2 cells were obtained from ATCC (Cat. # CRL-2190) and grown in KSFM medium (Gibco Cat. #1074–019) according to manufacturer’s instructions. THP at a concentration of 1 μg/ml or vehicle (5% Dextrose) were added in the experimental wells. For RNA studies, cells were incubated for 1 and 4 hours, and RNA was extracted as described below. For proteomic studies, cells were harvested at 6 hours and processed for protein extraction as previously described (36). For the indicated experiments, hydrogen peroxide (1mM) was added to HK-2 cells for 1 hour, and cells were allowed to recover in media for 1 hour before RNA extraction. All experiments were performed in triplicate. For the JNK activation experiments, cells were incubated with THP (1μg/ml), H2O2 (1 mM) or ACA (25 μM) as indicated for 1 hour. In pre-treatment experiments, cells were incubated with THP or ACA alone as indicated prior to adding H2O2 (1mM) for an additional half an hour. All cells were allowed to recover in media for 1 hour before processing for protein extraction.
Real time PCR
RNA extraction from whole kidneys and from HK-2 cells was done using TriReagent (Ambion, Cat. #AM9738) according to manufacturer’s protocol, and reverse transcribed as previously described (6). Real-time PCR was performed in an Applied Biosystems (AB) ViiA 7 system using TaqMan Gene Expression Assays also from AB. The following primers were used for human cell studies: IL-23: Hs 00900828_g1; 18s rRNA: Hs 99999901_s1. Cycling parameters were: 50°C for 2 min, then 95°C for 10 min followed by 40 cycles of 95°C for 15 sec and 60°C for 1 min. Expression values were normalized to endogenous controls and reported as fold change compared to control using the delta-delta CT method, according to the manufacturer’s instructions.
RAC1 activation assay
RAC1 activation assay was performed using a kit from Millipore (Cat. #17–283). Band densitometry was done using ImageJ (NIH).
Label-free quantitative mass spectrometry (LFQMS) proteomic studies
Triplicate protein extracts from HK-2 cells, treated with THP or vehicle, were analyzed using LFQMS as previously described (18, 19). In brief, HK-2 proteins were digested with trypsin and peptides analyzed using a Thermo-Finnigan linear ion-trap (LTQ) mass spectrometer coupled with a Surveyor autosampler and MS HPLC system (Thermo-Finnigan). The data were collected in the “Data dependent MS/MS” mode with the ESI interface and searched against the International Protein Index (IPI) HUMAN database using SEQUEST (v. 28 rev. 12) algorithms in Bioworks (v. 3.3). The searched peptides and proteins were validated by PeptideProphet (64) and ProteinProphet (65) in the Trans-Proteomic Pipeline (TPP, v. 3.3.0) (http://tools.proteomecenter.org/software.php). Only proteins with probability ≥ 0.9000 and peptides with probability ≥ 0.8000 were used for quantitation.
Lipidomics and multiple reaction monitoring profiling
Lipid extraction from kidney tissue was performed according to the Bligh & Dyer protocol (66). The multiple reaction monitoring (MRM) profiling approach was applied to investigate the lipid profile of oxidized phospholipids. MRM uses direct sample injection and functional group profiling by using class-specific product ion (67). For the analysis of phospholipids (PL), the Lipid Maps database was used to pull the parent ions and the specific lipid class-related product ion was added to obtain a list of transitions to be monitored by MRM scan. For the oxidized PL profile methods, we added the expected m/z value shift for one or two oxidation events to the parent mass of each lipid.
The lipid extracts were diluted in ACN+MeOH+300mM NH4Ac 3:6.65:0.35 (v/v). Mass spectrometry was performed as described in detail in de Lima et al (67). Briefly, for the MS analysis we used a capillary pump (G1376A, Agilent Technologies), operated with a flow of 20μL/min and a pressure of 200 bar, connected to a autosampler (1100 series, Agilent Technologies) to deliver 8μL of sample to the ESI source of an Agilent 6410 QQQ mass spectrometer (Agilent Technologies), operated at the source with capillary voltage of 3.5–5 kVand gas flow of 5.1 L/min at 300°C. Xcalibur data files (Agilent Technologies) were processed using an in-house script and lists containing MRM transitions and the respective ion intensity values were exported to Microsoft Excel. Absolute ion intensity of all monitored transitions were compared to blank samples. Transitions that yielded a signal above the value seen in blank samples were used for comparative analysis. Subsequent statistical analyses were performed using MetaboAnalyst software (68). Samples were normalized by kidney weight. The Fisher’s exact test was used to compare the relative fold changes of statistically different lipids by grouping increased or decreased fold changes together as a categorical variable.
Oxidative DNA Damage Assay
Oxidative DNA damage was measured using an ELISA kit against 8-hydroxy-2’-deoxyguanosine (8OHdG) from Enzo Life Sciences (Cat #ADI-EKS-350) according to the manufacturer protocol. Samples from the lung and kidney were lysed in RIPA lysis and extraction buffer (ThermoFisher Scientific, Cat. #89900) supplemented with Halt Protease and Phospatase single use inhibitor cocktail (ThermoFisher Scientific, Cat. #78442) in the Precellys tissue homogenizer (Bertin). Total protein concentration was measured using a modified Lowry assay from Bio-Rad (Cat. #5000111). The concentration of 8OHdG in tissue lysates was normalized to total protein concentration. Oxidative DNA damage for the serum samples from IRI-injured mice was measured using an ELISA kit against 8-hydroxy-2’-deoxyguanosine (8OHdG) from Cell Biolabs (Cat. #STA-320) according to the manufacturer’s protocol.
Measurement of Tamm Horsfall Protein (THP)
THP concentrations in human plasma samples were measured using an ELISA kit from Sigma Aldrich (Cat. #RAB0751) according to the manufacturer protocol.
Clinical Studies
During a 24-month period, from June 2013 through June 2015, all planned liver transplant recipients (LT) at University of Sao Paulo, Brazil, were screened prior to surgery for enrollment. Exclusion criteria were: age less than 18 years, need for dialysis preoperatively, combined liver and kidney transplant, chronic kidney disease stage 5, and previous kidney or liver transplant. One hundred eligible patients were enrolled after voluntary informed consent was obtained as per the guidelines of the Institution Ethics Committee. The University of Sao Paulo Ethics Committee approved the study under the protocol number:CAAE:06636513.4.0000.0068., following the Principles of the Declaration of Istanbul and the Declaration of Helsinki. Conforming to national guidelines, the protocol is registered in https://clinicaltrials.gov by the identifier NCT 02095431.
Of this cohort, we selected 41 patients, 7 with no AKI and 34 patients who developed at least stage 1 AKI, to include in our analysis. Baseline characteristics of these 34 patients were compared to 9 control subjects enrolled in a study evaluating Normal Ranges of Biomarkers of Kidney Injury in a Healthy Population. The control cohort included subjects between the ages of 18–90 with no known medical conditions enrolled following informed consent.
Baseline kidney function and pertinent clinical variables were extracted from electronic medical records (EMR). Blood samples were collected simultaneously in the perioperative period of LT, before induction of anesthesia and at various points after surgery. Vital signs, process of care and lab results were recorded daily for 7 days after LT. We recorded past medical history and baseline information and collected blood (~10mL), and urine (~250mL) samples. Samples from these cohorts were stored at −70 °C in the O’Brien Center Core A Laboratory at University of California San Diego.
Outcomes including the development of AKI, severity stage using the KDIGO criteria (28), need for dialysis and mortality were assessed in the intensive care unit (ICU), at hospital discharge, and 60 days after enrollment.
HEK293-TRPM2 Calcium Influx Assay
TRPM2-HEK293 recombinant cell line was purchased from BPS Bioscience (Cat. # 90331). The cells were propagated in culture according to the manufacturer’s instructions, and TRPM2 channel expression was induced with 0.2 μg/ml Doxycycline (Fisher Scientific, Cat. # ICN 19504401) 24 hours prior to Ca2+ influx experiment. Ca2+ influx was measured in a 96-well format in triplicates, using reagents provided in the Fluo-4 NW Calcium Assay Kit (Molecular Probes, Cat. # F36206). Cells in Ca2+-free assay buffer, were loaded with the reporter dye Fluo-4 and stimulated with 1.7 mM H2O2. The fluorescence was monitored for ~10 min, prior to addition of 10 mM CaCl2, and the readings continued for another 10 min, at which point the plateau was reached. In the inhibition experiments, TRP channel blocker ACA was used at 10 μM, while THP was used at 1 μg/ml, unless otherwise indicated. Cells were incubated with the THP or ACA in assay buffer for 20 min prior to stimulation with H2O2. In addition to the triplicate format, most experiments were repeated a minimum of two additional times with consistent results.
Bioinformatic analysis
Bioinformatic analysis of differentially expressed proteins was performed using Ingenuity Pathway Analysis (IPA, Quiagen). Gene E (Broad Institute: https://software.broadinstitute.org/GENE-E/) and MetaboAnalyst 4.0 (68) were also used to perform statistical clustering and generate heat maps.
Statistical Analysis
Values of each experimental group are reported as mean ± standard deviation unless otherwise indicated. All statistical tests were performed using GraphPad Prism software unless otherwise noted. A two tailed t-test was used to examine the difference in means for continuous data. A paired t-test was used for samples from the same patient collected at different times. The Fisher’s exact test was used to determine differences between categorical variables. Simple linear regressions were used to determine relationships between two continuous variables. Statistical significance was determined at p< 0.05.
Supplementary Material
Fig. S1. Fluorescence-based laser micro-dissection (LMD) and downstream transcriptomic and bioinformatic analyses
Fig. S2. THP treatment of HK-2 cells results in inhibition of multiple RAC1-dependent pathways
Fig. S3. Quantitation of membrane associated and cellular RAC1 in kidney sections from THP+/+ and THP−/− mice
Fig. S4. Free radical scavenging proteins are downregulated in the S3 tubules of THP−/− mice
Fig. S5. Molecules of the RAC1/JNK/c-JUN signaling network involved in free radical synthesis
Fig. S6. Reactive oxygen species concentrations are increased within the S3 proximal tubules of THP−/− mice
Fig. S7. Electron microscopy studies of mitochondrial morphology in S3 segments
Fig. S8. H2O2 activates IL-23 expression in HK-2 cells
Fig. S9. THP plasma concentrations in liver transplant recipients who did not develop AKI
Table S1. Demographic and baseline clinical data for liver transplant surgery group and healthy controls.
Data file S1. Gene Arrays S3.xlsx
Data file S2. Proteomics HK-2 Cells THP Stimulation.xlsx
Data File S3. Oxidized Phospholipidomics from Kidneys.xlsx
Acknowledgments:
We acknowledge the Susan G. Komen Tissue bank for assistance with Laser Microdissection (www.komentissuebank.iu.edu) and Dr. Christina Ferreira of the Bindley Bioscience Center (Purdue University) for assistance in lipidomic analyses.
Funding: This work was supported by The National Institute of Diabetes Digestive and Kidney Disease (NIDDK: 1R01DK111651 and P30DK079312 to TME, K23DK114556 to PG), a Veterans Affairs Merit Award to TEA, a American Society of Nephrology Ben J. Lipps award to KL and the São Paulo Research Foundation (FAPESP) (process number 2013/12710-2 to CL). We acknowledge support from the UAB-UCSD O’Brien Core Center for Acute Kidney Injury Research (supported by NIH P30-DK079337) for this project.
Footnotes
Publisher's Disclaimer: “This manuscript has been accepted for publication in Science Translational Medicine. This version has not undergone final editing. Please refer to the complete version of record at www.sciencetranslationalmedicine.org/. The manuscript may not be reproduced or used in any manner that does not fall within the fair use provisions of the Copyright Act without the prior written permission of AAAS.”
Competing interests: TEA and RM have applied for a patent related to this work titled “Modified Tamm-Horsfall Protein and related compositions and methods of use”, publication number US 2018/0305420 A1.
Data and materials availability: All data associated with this study are available in the main text or the supplementary materials.
References and Notes:
- 1.El-Achkar TM, Wu XR, Uromodulin in kidney injury: an instigator, bystander, or protector? Am J Kidney Dis 59, 452–461 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rampoldi L, Scolari F, Amoroso A, Ghiggeri G, Devuyst O, The rediscovery of uromodulin (Tamm-Horsfall protein): from tubulointerstitial nephropathy to chronic kidney disease. Kidney Int 80, 338–347 (2011). [DOI] [PubMed] [Google Scholar]
- 3.Serafini-Cessi F, Malagolini N, Cavallone D, Tamm-Horsfall glycoprotein: biology and clinical relevance. Am J Kidney Dis 42, 658–676 (2003). [DOI] [PubMed] [Google Scholar]
- 4.Brunati M et al. , The serine protease hepsin mediates urinary secretion and polymerisation of Zona Pellucida domain protein uromodulin. Elife 4, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.El-Achkar TM, Dagher PC, Tubular cross talk in acute kidney injury: a story of sense and sensibility. Am J Physiol Renal Physiol 308, F1317–1323 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.El-Achkar TM et al. , Tamm-Horsfall protein translocates to the basolateral domain of thick ascending limbs, interstitium, and circulation during recovery from acute kidney injury. Am J Physiol Renal Physiol 304, F1066–1075 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Balasubramanian G et al. , Early nephrologist involvement in hospital-acquired acute kidney injury: a pilot study. Am J Kidney Dis 57, 228–234 (2011). [DOI] [PubMed] [Google Scholar]
- 8.Steubl D et al. , Plasma Uromodulin Correlates With Kidney Function and Identifies Early Stages in Chronic Kidney Disease Patients. Medicine (Baltimore) 95, e3011 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Thornley C, Dawnay A, Cattell WR, Human Tamm-Horsfall glycoprotein: urinary and plasma levels in normal subjects and patients with renal disease determined by a fully validated radioimmunoassay. Clin Sci (Lond) 68, 529–535 (1985). [DOI] [PubMed] [Google Scholar]
- 10.Garimella PS et al. , Urinary uromodulin, kidney function, and cardiovascular disease in elderly adults. Kidney Int 88, 1126–1134 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Delgado GE et al. , Serum Uromodulin and Mortality Risk in Patients Undergoing Coronary Angiography. J Am Soc Nephrol 28, 2201–2210 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Garimella PS et al. , Association of urinary uromodulin with kidney function decline and mortality: the health ABC study. Clin Nephrol 87, 278–286 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Leiherer A et al. , Serum uromodulin is a predictive biomarker for cardiovascular events and overall mortality in coronary patients. Int J Cardiol 231, 6–12 (2017). [DOI] [PubMed] [Google Scholar]
- 14.Micanovic R et al. , Tamm-Horsfall Protein Regulates Granulopoiesis and Systemic Neutrophil Homeostasis. J Am Soc Nephrol 26, 2172–2182 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.El-Achkar TM et al. , Tamm-Horsfall protein-deficient thick ascending limbs promote injury to neighboring S3 segments in an MIP-2-dependent mechanism. Am J Physiol Renal Physiol 300, F999–1007 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.El-Achkar TM et al. , Tamm-Horsfall protein protects the kidney from ischemic injury by decreasing inflammation and altering TLR4 expression. Am J Physiol Renal Physiol 295, F534–544 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Micanovic R, Khan S, El-Achkar TM, Immunofluorescence laser micro-dissection of specific nephron segments in the mouse kidney allows targeted downstream proteomic analysis. Physiol Rep 3, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lai X, Wang L, Tang H, Witzmann FA, A novel alignment method and multiple filters for exclusion of unqualified peptides to enhance label-free quantification using peptide intensity in LC-MS/MS. J Proteome Res 10, 4799–4812 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lai X, Wang L, Witzmann FA, Issues and applications in label-free quantitative mass spectrometry. Int J Proteomics 2013, 756039 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Minden A, Lin A, Claret FX, Abo A, Karin M, Selective activation of the JNK signaling cascade and c-Jun transcriptional activity by the small GTPases Rac and Cdc42Hs. Cell 81, 1147–1157 (1995). [DOI] [PubMed] [Google Scholar]
- 21.Prieto-Sanchez RM, Bustelo XR, Structural basis for the signaling specificity of RhoG and Rac1 GTPases. J Biol Chem 278, 37916–37925 (2003). [DOI] [PubMed] [Google Scholar]
- 22.Liu W et al. , AP-1 activated by toll-like receptors regulates expression of IL-23 p19. J Biol Chem 284, 24006–24016 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Winfree S et al. , Quantitative Three-Dimensional Tissue Cytometry to Study Kidney Tissue and Resident Immune Cells. J Am Soc Nephrol 28, 2108–2118 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu Y, El-Achkar TM, Wu XR, Tamm-Horsfall protein regulates circulating and renal cytokines by affecting glomerular filtration rate and acting as a urinary cytokine trap. J Biol Chem 287, 16365–16378 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kroese LJ, Scheffer PG, 8-hydroxy-2’-deoxyguanosine and cardiovascular disease: a systematic review. Curr Atheroscler Rep 16, 452 (2014). [DOI] [PubMed] [Google Scholar]
- 26.Grynberg K et al. , Early serum creatinine accurately predicts acute kidney injury post cardiac surgery. BMC Nephrol 18, 93 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Saratzis A, Melas N, Mahmood A, Sarafidis P, Incidence of Acute Kidney Injury (AKI) after Endovascular Abdominal Aortic Aneurysm Repair (EVAR) and Impact on Outcome. Eur J Vasc Endovasc Surg 49, 534–540 (2015). [DOI] [PubMed] [Google Scholar]
- 28.Kellum JA, Lameire N, K. A. G. W. Group, Diagnosis, evaluation, and management of acute kidney injury: a KDIGO summary (Part 1). Crit Care 17, 204 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nie M et al. , Uromodulin regulates renal magnesium homeostasis through the ion channel transient receptor potential melastatin 6 (TRPM6). J Biol Chem 293, 16488–16502 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fonfria E et al. , TRPM2 channel opening in response to oxidative stress is dependent on activation of poly(ADP-ribose) polymerase. Br J Pharmacol 143, 186–192 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kraft R, Grimm C, Frenzel H, Harteneck C, Inhibition of TRPM2 cation channels by N-(p-amylcinnamoyl)anthranilic acid. Br J Pharmacol 148, 264–273 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Uhlen M et al. , Proteomics. Tissue-based map of the human proteome. Science 347, 1260419 (2015). [DOI] [PubMed] [Google Scholar]
- 33.Togashi K, Inada H, Tominaga M, Inhibition of the transient receptor potential cation channel TRPM2 by 2-aminoethoxydiphenyl borate (2-APB). Br J Pharmacol 153, 1324–1330 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gao G et al. , TRPM2 mediates ischemic kidney injury and oxidant stress through RAC1. J Clin Invest 124, 4989–5001 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cheung JY, Miller BA, Transient Receptor Potential-Melastatin Channel Family Member 2: Friend or Foe. Trans Am Clin Climatol Assoc 128, 308–329 (2017). [PMC free article] [PubMed] [Google Scholar]
- 36.Micanovic R et al. , Tamm-Horsfall Protein Regulates Mononuclear Phagocytes in the Kidney. J Am Soc Nephrol 29, 841–856 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Marinho HS, Real C, Cyrne L, Soares H, Antunes F, Hydrogen peroxide sensing, signaling and regulation of transcription factors. Redox Biol 2, 535–562 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sies H, Role of metabolic H2O2 generation: redox signaling and oxidative stress. J Biol Chem 289, 8735–8741 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gough DR, Cotter TG, Hydrogen peroxide: a Jekyll and Hyde signalling molecule. Cell Death Dis 2, e213 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schrader M, Fahimi HD, Peroxisomes and oxidative stress. Biochim Biophys Acta 1763, 1755–1766 (2006). [DOI] [PubMed] [Google Scholar]
- 41.Kaneto H et al. , Oxidative stress, ER stress, and the JNK pathway in type 2 diabetes. J Mol Med (Berl) 83, 429–439 (2005). [DOI] [PubMed] [Google Scholar]
- 42.Zhang Y et al. , miRNA-192–5p impacts the sensitivity of breast cancer cells to doxorubicin via targeting peptidylprolyl isomerase A. Kaohsiung J Med Sci 35, 17–23 (2019). [DOI] [PubMed] [Google Scholar]
- 43.De Bessa TC et al. , Subverted regulation of Nox1 NADPH oxidase-dependent oxidant generation by protein disulfide isomerase A1 in colon carcinoma cells with overactivated KRas. Cell Death Dis 10, 143 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang Y et al. , Pokeweed antiviral protein attenuates liver fibrosis in mice through regulating Wnt/Jnk mediated glucose metabolism. Saudi J Gastroenterol 24, 157–164 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li Q, Zhang M, Xuan L, Liu Y, Chen C, Anagliptin inhibits neointimal hyperplasia after balloon injury via endothelial cell-specific modulation of SOD-1/RhoA/JNK signaling in the arterial wall. Free Radic Biol Med 121, 105–116 (2018). [DOI] [PubMed] [Google Scholar]
- 46.Kobayashi T et al. , Chloride intracellular channel 1 as a switch among tumor behaviors in human esophageal squamous cell carcinoma. Oncotarget 9, 23237–23252 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Qi X et al. , The Clinical Significance and Potential Therapeutic Role of GPx3 in Tumor Recurrence after Liver Transplantation. Theranostics 6, 1934–1946 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jang S et al. , Critical role of c-jun N-terminal protein kinase in promoting mitochondrial dysfunction and acute liver injury. Redox Biol 6, 552–564 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yu Y et al. , [Effects of activation of ALDH2 by ethanol on the expression of JNK in kidney of diabetic rats]. Zhongguo Ying Yong Sheng Li Xue Za Zhi 30, 270–273 (2014). [PubMed] [Google Scholar]
- 50.Prakasam A et al. , JNK1/2 regulate Bid by direct phosphorylation at Thr59 in response to ALDH1L1. Cell Death Dis 5, e1358 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Xin C et al. , Study of the insulin signaling pathways in the regulation of ACAT1 expression in cultured macrophages. Cell Biol Int 33, 602–606 (2009). [DOI] [PubMed] [Google Scholar]
- 52.Whaley-Connell AT et al. , Albumin activation of NAD(P)H oxidase activity is mediated via Rac1 in proximal tubule cells. Am J Nephrol 27, 15–23 (2007). [DOI] [PubMed] [Google Scholar]
- 53.Lee M, Choi I, Park K, Activation of stress signaling molecules in bat brain during arousal from hibernation. J Neurochem 82, 867–873 (2002). [DOI] [PubMed] [Google Scholar]
- 54.Leiherer A et al. , The value of uromodulin as a new serum marker to predict decline in renal function. J Hypertens 36, 110–118 (2018). [DOI] [PubMed] [Google Scholar]
- 55.Trudu M et al. , Common noncoding UMOD gene variants induce salt-sensitive hypertension and kidney damage by increasing uromodulin expression. Nat Med 19, 1655–1660 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jha JC, Banal C, Chow BS, Cooper ME, Jandeleit-Dahm K, Diabetes and Kidney Disease: Role of Oxidative Stress. Antioxid Redox Signal 25, 657–684 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yamamoto S, Shimizu S, Targeting TRPM2 in ROS-Coupled Diseases. Pharmaceuticals (Basel) 9, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ryan MJ et al. , HK-2: an immortalized proximal tubule epithelial cell line from normal adult human kidney. Kidney Int 45, 48–57 (1994). [DOI] [PubMed] [Google Scholar]
- 59.Bolstad BM, Irizarry RA, Astrand M, Speed TP, A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics 19, 185–193 (2003). [DOI] [PubMed] [Google Scholar]
- 60.McClintick JN, Edenberg HJ, Effects of filtering by Present call on analysis of microarray experiments. BMC Bioinformatics 7, 49 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hato T et al. , The macrophage mediates the renoprotective effects of endotoxin preconditioning. J Am Soc Nephrol 26, 1347–1362 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tamm I, Horsfall FL Jr., Characterization and separation of an inhibitor of viral hemagglutination present in urine. Proc Soc Exp Biol Med 74, 106–108 (1950). [PubMed] [Google Scholar]
- 63.Tamm I, Horsfall FL Jr., A mucoprotein derived from human urine which reacts with influenza, mumps, and Newcastle disease viruses. J Exp Med 95, 71–97 (1952). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Keller A, Nesvizhskii AI, Kolker E, Aebersold R, Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Analytical chemistry 74, 5383–5392 (2002). [DOI] [PubMed] [Google Scholar]
- 65.Nesvizhskii AI, Keller A, Kolker E, Aebersold R, A statistical model for identifying proteins by tandem mass spectrometry. Analytical chemistry 75, 4646–4658 (2003). [DOI] [PubMed] [Google Scholar]
- 66.Bligh EG, Dyer WJ, A rapid method of total lipid extraction and purification. Can J Biochem Physiol 37, 911–917 (1959). [DOI] [PubMed] [Google Scholar]
- 67.de Lima CB et al. , Comprehensive Lipid Profiling of Early Stage Oocytes and Embryos by MRM Profiling. J Mass Spectrom, (2018). [DOI] [PubMed] [Google Scholar]
- 68.Chong J et al. , MetaboAnalyst 4.0: towards more transparent and integrative metabolomics analysis. Nucleic Acids Res 46, W486–W494 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Related Resources
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Fluorescence-based laser micro-dissection (LMD) and downstream transcriptomic and bioinformatic analyses
Fig. S2. THP treatment of HK-2 cells results in inhibition of multiple RAC1-dependent pathways
Fig. S3. Quantitation of membrane associated and cellular RAC1 in kidney sections from THP+/+ and THP−/− mice
Fig. S4. Free radical scavenging proteins are downregulated in the S3 tubules of THP−/− mice
Fig. S5. Molecules of the RAC1/JNK/c-JUN signaling network involved in free radical synthesis
Fig. S6. Reactive oxygen species concentrations are increased within the S3 proximal tubules of THP−/− mice
Fig. S7. Electron microscopy studies of mitochondrial morphology in S3 segments
Fig. S8. H2O2 activates IL-23 expression in HK-2 cells
Fig. S9. THP plasma concentrations in liver transplant recipients who did not develop AKI
Table S1. Demographic and baseline clinical data for liver transplant surgery group and healthy controls.
Data file S1. Gene Arrays S3.xlsx
Data file S2. Proteomics HK-2 Cells THP Stimulation.xlsx
Data File S3. Oxidized Phospholipidomics from Kidneys.xlsx