

ABSTRACT
Group 2 innate lymphoid cells (ILC2s) are recently reported to play a more critical role in allergic diseases. We previously identified that mesenchymal stromal cells (MSCs) elicited therapeutic effects on allergic airway inflammation. Small extracellular vesicles (sEV) derived from MSCs possess striking advantages including low immunogenicity and high biosafety, and is extremely promising cell-free therapeutic agents. However, the effects of MSC-sEV on ILC2s are still unclear. Additionally, scalable isolation protocols are required for the mass production of homogenous MSC-sEV especially in clinical application. We previously reported that induced pluripotent stem cells-derived MSCs were the ideal cellular source for the large preparation of MSC-sEV. Here we developed a standardized scalable protocol of anion-exchange chromatography for isolation of MSC-sEV, and investigated the effects of MSC-sEV on ILC2 function from patients with allergic rhinitis and in a mouse ILC2-dominant asthma model. The characterization of MSC-sEV was successfully demonstrated in terms of size, morphology and specific markers. Using flow cytometry and human Cytokine Antibody Array, MSC-sEV but not fibroblasts-sEV (Fb-sEV) were found to significantly inhibit the function of human ILC2s. Similarly, systemic administration of MSC-sEV but not Fb-sEV exhibited an inhibition of ILC2 levels, inflammatory cell infiltration and mucus production in the lung, a reduction in levels of T helper 2 cytokines, and alleviation of airway hyperresponsiveness in a mouse model of asthma. Using RNA sequencing, miR-146a-5p was selected as the candidate to mediate the above effects of MSC-sEV. We next revealed the uptake of ILC2s to MSC-sEV, and that transfer of miR-146a-5p in MSC-sEV to ILC2s in part contributed to the effects of MSC-sEV on ILC2s in vitro and in a mouse model. In conclusion, we demonstrated that MSC-sEV were able to prevent ILC2-dominant allergic airway inflammation at least partially through miR-146a-5p, suggesting that MSC-sEV could be a novel cell-free strategy for the treatment of allergic diseases.
KEYWORDS: Small extracellular vesicles, exosomes, mesenchymal stromal cells, anion-exchange chromatography, scalable protocol, group 2 innate lymphoid cells, allergic airway inflammation, microRNA
Introduction
Asthma and allergic rhinitis are characterized by airway hyperresponsiveness (AHR), inflammation and remodelling, and have become a significant global public health concern [1]. The damage to the airway epithelium, the imbalance of T cells and dendritic cells (DCs) are involved in the pathogenesis of airway inflammation [2]. Traditionally, T helper 2 (Th2)-cells are thought to be dominant to drive allergic inflammatory disease. Recently, group 2 innate lymphoid cells (ILC2s) are reported to greatly contribute to the initiation and maintenance of type 2 allergic airway inflammation [3,4]. ILC2s are critical innate immunocytes that produce dramatic increases of IL-5, IL-9 and IL-13 in response to the epithelial cell-derived cytokines like IL-25, IL-33 and thymic stromal lymphopoietin [3–5]. We and others have previously found that the frequency of peripheral ILC2s was significantly increased in patients with asthma and allergic rhinitis [6,7]. Furthermore, ILC2 frequency correlated with persistent pulmonary eosinophilia in cases of severe asthma [8,9]. Therefore, ILC2s are the new and promising targets to inhibit allergic airway inflammation.
We previously reported that mesenchymal stromal cells (MSCs), multipotent cells that are capable of differentiating into three types of mesenchymal cells, were effective in the modulation of T-cell phenotypes [10], as well as differentiation and maturation of DCs [11], and contributed to amelioration of allergic airway inflammation, especially neutrophil-predominant asthma which is insensitive to glucocorticoid [12,13]. MSCs were reported to exert their immunomodulation via cell-cell contacts, some soluble factors and exosomes [14]. Exosomes are extracellular vesicles released by fusion of multivesicular bodies with the cellular membrane. They are approximately 30–150 nm in diameter and contain abundant of functional components such as microRNAs (miRNAs) and proteins [15]. It has been previously reported that exosomes derived from MSCs elicited similar therapeutic effects to their parent MSCs [16]. More importantly, exosomes released by MSCs possess striking advantages over whole-cell therapy, such as low immunogenicity, convenient storage and high biosafety. Thus, MSC exosomes have been identified as extremely promising cell-free therapeutic agents for some diseases.
To date, only two studies have investigated the therapeutic effects of MSC-sEV on allergic airway inflammation, in which MSC-sEV significantly reduced inflammation infiltration, airway remodelling and AHR in mice [17,18]. While these studies established a role for MSC-sEV in allergic airway inflammation, the mechanisms of MSC-sEV-mediated amelioration of allergic airway inflammation remain largely unknown. Particularly, the effects of MSC-sEV on ILC2s and the mechanisms are still unclear. In light of the crucial role of ILC2s in allergic airway inflammation, we hypothesized that the activation of ILC2s is a potential novel therapeutic target for MSC-sEV in allergic airway inflammation. In addition, we hypothesized that the survival, expansion and maintenance of ILC2s in IL-33 induced allergic inflammation could be regulated by miRNAs [19–22], one of the most important components of sEV. Therefore, we further postulated that the therapeutic effects of MSC-sEV in allergic airway inflammation could be attributed to sEV miRNAs.
Nevertheless, scalable isolation protocols are required for the mass production of homogenous MSC exosomes for clinical application. Though differential ultracentrifugation remains the most common method for isolation of exosomes [23], it is largely limited by the disadvantages of low efficiency, high heterogeneity and non-scalability. Alternatively, it has been reported that anion-exchange chromatography achieves scalable separation of biologically active exosomes released by bone marrow-derived MSCs (BM-MSCs). However, the limitation of using BM-MSCs for producing therapeutic exosomes is that the cells will quickly become senescent after extensive expansion in vitro, making it impossible to isolate homogeneous MSC exosomes for clinical therapies. We have previously demonstrated that MSCs derived from induced pluripotent stem cells (iPSCs) were phenotypically and functionally analogous to adult MSCs [24], and exhibited higher proliferation potential, longer life time, stronger immune privilege and lower heterogeneity [11,25], making them an ideal cellular source for large-scale preparation of exosomes. Therefore, in our current study, we first optimized an anion-exchange chromatography method for purification of exosomes derived from iPSC-MSCs, which are termed as small extracellular vesicles (sEV) according to MISEV 2018 [26].
In this study, we aimed to optimize a scalable protocol for isolation of MSC-sEV. We also sought to explore the therapeutic effects of MSC-sEV on ILC2-dominant allergic airway inflammation in mice, and identify the potentially functional miRNAs that may contribute to the therapeutic effects of MSC-sEV in allergic airway inflammation.
Materials and methods
Collection of supernatants of MSCs
Human iPSC-MSCs used in this study were prepared as described in our previous study [11]. The cells were frozen at passage 10 to create a working cell bank for the isolation of MSC-sEV. Fibroblasts were purchased from ATCC (CAT# PCS201012™; Manassas, VA, USA) and served as the negative control cells in our study. For the preparation of sEV-enriched medium, one vial of frozen cells (2 × 106/vial) was recovered into two 150 cm2 cell culture plates at day 0 with complete culture medium (CCM, Supplementary Table 1). The MSCs reached 70%-80% confluency after 3 days of culture (about 1 × 107 cells in total) and were further passaged and seeded in twenty 150 cm2 cell culture plates. After the cells reached 70–80% confluency (about 1 × 108 cells in total), usually in 3 to 5 days, the CCM was replaced with chemically defined and protein-free (CDPF, Supplementary Table 1) medium after three washes with 30 mL phosphate-buffered saline (PBS). Because MSCs incubated in CDPF medium were previously found to express high level of pro-inflammation cytokines and low level of anti-inflammation cytokines during the early stage [27], CDPF medium was discarded after 6 h of incubation and replaced with fresh CDPF medium, which was harvested for the isolation of sEV after another 42 h of incubation (Figure 1(a)). The harvested sEV-enriched supernatant was centrifuged immediately at 2000 g for 20 min to remove cell debris. Similarly, fibroblasts were cultured with the CCM as suggested by the manufacturer (Supplementary Table 1) and incubated with CDPF for isolation of fibroblasts-derived small extracellular vesicles (Fb-sEV).
Figure 1.
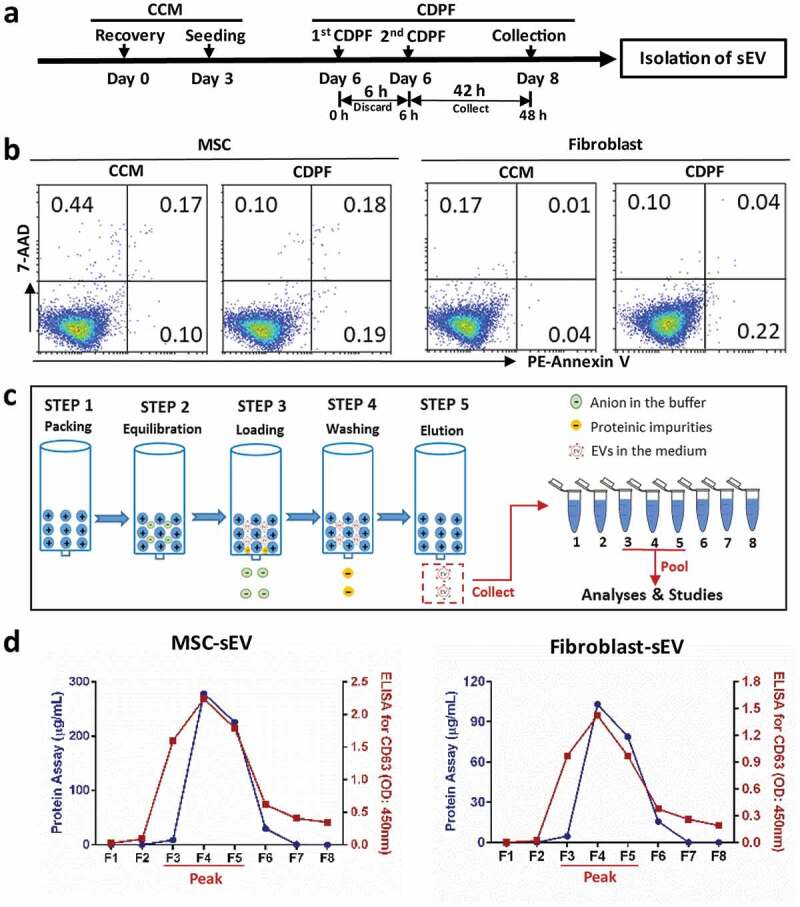
Isolation of MSC-sEV by anion chromatography. (a) Schematic diagram for the preparation of EV-enriched medium. (b) Apoptosis detection for the cells incubated in CCM or CDPF for 48 h. (c) Schematic diagram for the anion-exchange chromatographic isolation of MSC-sEV. (d) Levels of total protein and CD63 in the eight elution fractions collected from anion-exchange chromatographic isolation of sEV from MSCs and fibroblasts. CCM, Complete Culture Medium; CDPF, Chemically Defined Protein-Free Medium; MSC, mesenchymal stromal cell; sEV, small extracellular vesicles.
Isolation of sEV by anion-exchange chromatography
Isolation sEV was done as previously reported with some modifications (Figure 1(c)) [27]. An Econo-Pac column (Bio-Rad Laboratories, CD63 Hercules, CA, USA) was packed with 4 mL anion-exchange resin (Q Sepharose Fast Flow; GE Health Care Life Science, Pittsburgh, PA, USA) and equilibrated with 8 mL Equilibration Buffer (Supplementary Table 2). Then, the column was loaded with 150 mL supernatant, washed with 40 mL Wash Buffer to remove the proteomic impurities and eluted continuously with 1 mL Elution Buffer (Supplementary Table 2) for eight times. The concentrations of sEV in the eight fractions were estimated by Bradford protein assay and enzyme-linked immunosorbent assay (ELISA) for CD63. The fractions with peak concentration of sEV were pooled for PBS dialysis overnight at 4°C. Then, we further concentrated the sEV using Pierce™ Protein Concentrator (30 kDa, Thermo Fisher Scientific, Rockford, IL, USA), which were used for further analyses and studies. To prevent endotoxin contamination in sEV samples, all buffers and materials used for anion-exchange chromatography were endotoxin-free. The final protein and particle concentrations of sEV preparations were quantified by Bradford protein assay and nanoparticle tracking analysis (NanoSight NS300; Malvern, UK).
Differential ultracentrifugation
In this study, we made side by side comparison of ultracentrifugation and anion-exchange chromatography such as yield rate and time costing. Differential ultracentrifugation for isolation of MSC-sEV was performed as previously reported with minor modification [23]. Briefly, the supernatants were differentially centrifuged at 300 g for 5 min, 2000 g for 20 min, and 12,000 g for 30 min at 4°C. After the final centrifugation, the supernatants were further ultracentrifuged at 110,000 g for 2 h at 4°C using a Beckman Coulter Optima L-100 XP ultracentrifuge (SW 32 rotor). The pellets were then washed with ice-cold PBS and ultracentrifuged at 110,000 g for another 70 min at 4°C. The pellets were finally suspended with 100 μL PBS and used for further analysis.
Generation of sEV-depleted MSC supernatant
In order to exclude the possible role of soluble factors in MSC supernatant on ILC2 function, sEV-depleted MSC supernatant was prepared. As Figure 1(c), the column was loaded with 150 mL supernatant, and the fluid without sEV was collected as sEV-depleted biofluid after the sEV were bound to the resin at Step 3. In order to fully compare the activities of sEVs and sEV-depleted biofluid, sEV-depleted biofluid was concentrated by protein concentrator (Pierce) under the same volume of CDPF culture medium as sEV preparation.
Generation of RNA-depleted sEV
Purified MSC-sEV were treated with 1% Triton X-100 at 37°C for 30 min, then incubated at 37°C for 30 min with 2 mg/ml of DNase and protease-free RNase A (Thermo Fisher Scientific, Vilnius, LT, USA). The effect of RNase A was then stopped by the administration of RNase inhibitor (Thermo Fisher Scientific, Vilnius, LT, USA). Next, the RNA-depleted MSC-sEV were washed with PBS, and spun down using ultracentrifugation at 100,000 g for 70 min at 4°C.
Generation of mCherry-labelled MSC-sEV
Human MSCs were seeded at 2 × 105 cells/well in a 6-well plate and transfected with 2 × 107 lentivirus vectors with mCherry-CD63 fusion gene driven by EF1A promoter (pLV [Exp]-Puro-EF1A>mCherry (ns):hCD63) for 18 h. We observed that over 90% of MSCs were successfully transduced (Supplementary figure 1). The cells were further passaged and used for isolation of MSC-sEV as described above. Flow cytometry analysis revealed that more than 90% of MSC-sEV were mCherry-positive (Supplementary figure 1).
Loading sEV with miRNA mimics and inhibitors
Human MSCs were incubated with antibiotics-free CCM in twenty 150 cm2 cell culture plates and transfected directly with miR-146a-5p inhibitor or miRNA scramble control using riboFECTTM CP Transfection Kit for 48 h after the cell density reached about 50% confluency. The transfected MSCs were further incubated in CCM to 80% confluency, and the CCM was replaced with CDPF for isolation of MSC-sEV containing the inhibitor (MSC-sEVinhibitor) or a scrambled miRNA (MSC-sEVscramble). Similarly, human fibroblasts were transfected with 100 nM hsa-miR-146a mimics or miRNA scramble control to produce miR-146a-enriched Fb-sEV (Fb-sEVmimics) and miRNA scrambled control-enriched Fb-sEV (Fb-sEVscramble). Fibroblasts transfected with hsa-miR-146a-5p mimics but not scramble increased expression of has-miR-146-5p in fibroblasts (Supplementary figure 2). The miRNA inhibitors, miRNA mimics and transfection kit used were all purchased from RiboBio Co., Ltd (Guangzhou, Guangdong, CN). Level of miR-146-5p in MSCs or Fb was determined by quantitative real-time PCR.
Apoptosis detection for human MSCs and fibroblasts
Cells (5 × 104/well) were seeded in 6-well plates with CCM. Medium was replaced with fresh CCM or CDPF for 3 days. The cells were further incubated for 48 h and collected for flow cytometry analysis using the Apoptosis Detection Kit (BD Bioscience, San Jose, CA, USA) according to the manufacturer’s instructions.
Transmission electron microscopy for sEV
The sEV preparations were fixed with 2% glutaraldehyde (Sigma, Saint Louis, MO, USA). After 30 min, 10 µL of fixed samples was pipetted onto copper grids with carbon-coated formvar film and incubated for 10 min. Grids were washed three times with ddH2O and the excess liquid was removed by blotting. Next, 30 µL of 3% uranyl acetate (Sigma, Saint Louis, MO, USA, pH = 7.0) was pipetted onto the grids and incubated for 5 min. Micrographs of sEV were obtained with a Transmission electron microscopy (H7650; HITACHI, Tokyo, Japan). Transmission electron microscopy was technically supported by Guangdong Institute of Microbiology.
Quantitative real-time PCR for miR-146-5p
Total RNA was extracted from MSCs and fibroblasts with miRNeasy Kit (Qiagen, Hilden, Germany). Isolated RNA was used for synthetization of cDNA templates by using miScript® II RT Kit (Qiagen, Hilden, Germany), in which mature miRNAs were polyadenylated by poly(A) polymerase and converted into cDNA by reverse transcriptase with oligo-dT priming. Quantitative real-time PCR for miR-146a-5p was performed using SYBR Premix ExTaq (Takara Bio Inc., Japan) on Applied Biosystems™ 7500 Real-Time PCR System. The Ct values were normalized to U6 and miRNA levels were calculated by 2−ΔCt method.
sEV RNA sequencing
RNA from MSC-sEV and Fb-sEV was extracted using miRNeasy Kit (Qiagen, Hilden, Germany). RNA yield was assessed by using the Qubit® 2.0 (Life Technologies, USA). In total, 20 ng RNA was used to prepare small RNA libraries by NEBNext® Multiplex Small RNA Library Prep Set for Illumina (NEB, USA) according to the manufacturer’s instructions. The RNA libraries were sequenced by HiSeq 2500 (Illumina, USA) with single-end 50 bp reads at Ribobio Co. Ltd, Guangzhou, China. Differential expression was calculated with the edgeR algorithm between two sets of samples based on the significant criteria of |log2 (Fold Change) |≥1 and P-value <0.05. The target genes of the differentially expressed miRNAs were predicted by using TargetScan, miRDB, miRTarBase and miRWalkGO (Supplementary figure 3(a)). Gene Ontology (GO) analysis of the predicted target genes was performed using DAVID Bioinformatics Resources 6.8 (https://david.ncifcrf.gov/tools.jsp).
Subjects
This study included 40 patients with allergic rhinitis (17 males/23 females, aged 20–61 years). The diagnosis of allergic rhinitis was in accordance with the criteria of the initiative from Allergic Rhinitis and its Impact on Asthma (ARIA) [1]. All patients were positive for Der p/Der f on the basis of the positive skin prick test (SPT) result and specific IgE tests (sIgE) to Der p/Der f (>0.35 IU/mL, Pharmacia CAP system, Pharmacia Diagnostics, Uppsala, Sweden). The patients had not received antihistamines or intranasal steroid treatments for 1 month, or oral steroids for 3 months, prior to this study. This study was approved by The Ethics Committee of The First Affiliated Hospital, Sun Yat-sen University, and informed consents were obtained from all participants. Human blood buffy coats from “anonymous donors” were provided from Guangzhou Blood Centre; exemption of written informed consent was approved.
Isolation and treatment of PBMCs, Lineage− cells and ILC2s
Human peripheral blood mononuclear cells (PBMCs) from patients of allergic rhinitis were isolated by Ficoll-Paque PREMIUM density gradient centrifugation (GE Healthcare Life Sciences, England, UK) for the evaluation of ILC2 function using flow cytometry. Lineage− (Lin-) cells and ILC2s from the PBMCs of human buffy coats of volunteers (n = 12) were purified using the Lineage Cell Depletion Kit and the ILC2 Isolation Kit, respectively, following the manufacturer’s instructions. Both microbeads isolation kits were purchased from Miltenyi Biotec GmbH, Bergisch Gladbach, Germany. To assay the immunoregulatory effects of sEV on ILC2s, PBMCs and Lin− cells were seeded in 24-well plates at 1 × 106 cells per well in 1 mL RPMI 1640 plus 10% foetal bovine serum, and ILC2s were cultured with 200 µL medium in 96-well plates at a density of 5 × 105 cells per well. All cells were stimulated with recombinant human (rh)IL-2 (50 ng/mL), rhIL-25 (10 ng/mL) and rhIL-33 (10 ng/mL) (R&D Systems, Minneapolis, MN) in the presence or absence of 5 µg/mL or 40 µg/mL sEV for 2 days, in which each ILC2 was exposed to sEV isolated from 1.32 to 6.60 MSCs, respectively. Supernatants were collected for analyses of IL-5, IL-9 and IL-13, and PBMCs were used for flow cytometry analyses of IL-9+ILC2s and IL-13+ILC2s.
PBMCs from patients with AR were also treated with EV-depleted MSC supernatant or RNA-depleted sEV, and the levels of IL-13+ ILC2s were evaluated using flow cytometry.
Animals
Female C57BL/6 mice aged 4–6 weeks (n = 102) were purchased from Beijing Vital River Laboratory Animal Technology Co., Ltd., and the numbers about the animals for each experiment are indicated in supplementary Table 3. The animals were maintained in a specific pathogen-free environment in the Animal Experimental Centre of North Campus, Sun Yat-sen University. All procedures about animals performed in the study were approved by the Ethics Committee of Sun Yat-sen University.
Mouse model of ILC2-dominant eosinophilic airway inflammation
The ILC2-dominant eosinophilic airway inflammation mouse model was developed as previously reported with minor modifications [28,29]. As shown in Figure 4(a), the mice were administered intratracheally with 1 µg murine IL-33 (Peprotech, Rocky Hill, NJ, USA) in 20 µL PBS on days 1, 3, and 5. To study the therapeutic effects of iPSC-MSC-sEV, the mice were pretreated intravenously with 2 × 1010 (about 100 µg protein) Fb-sEV (IL-33/Fb-sEV) or iPSC-MSC-sEV (IL-33/MSC-sEV) per mouse on day 0. To study the role of miR-146a-5p in the therapeutic effects of MSC-sEV, mice were administered intravenously with iPSC-MSC-sEVscramble (IL-33/MSC-sEVscramble), iPSC-MSC-sEVinhibitor (IL-33/MSC-sEVinhibitor), Fb-sEVscramble (IL-33/Fb-sEVscramble) or Fb-sEVmimics (IL-33/Fb-sEVmimics). The negative control mice were pretreated with an equivalent volume of PBS and administered intratracheally with 20 µL PBS (PBS/PBS). All mice were sacrificed on day 6.
Figure 4.
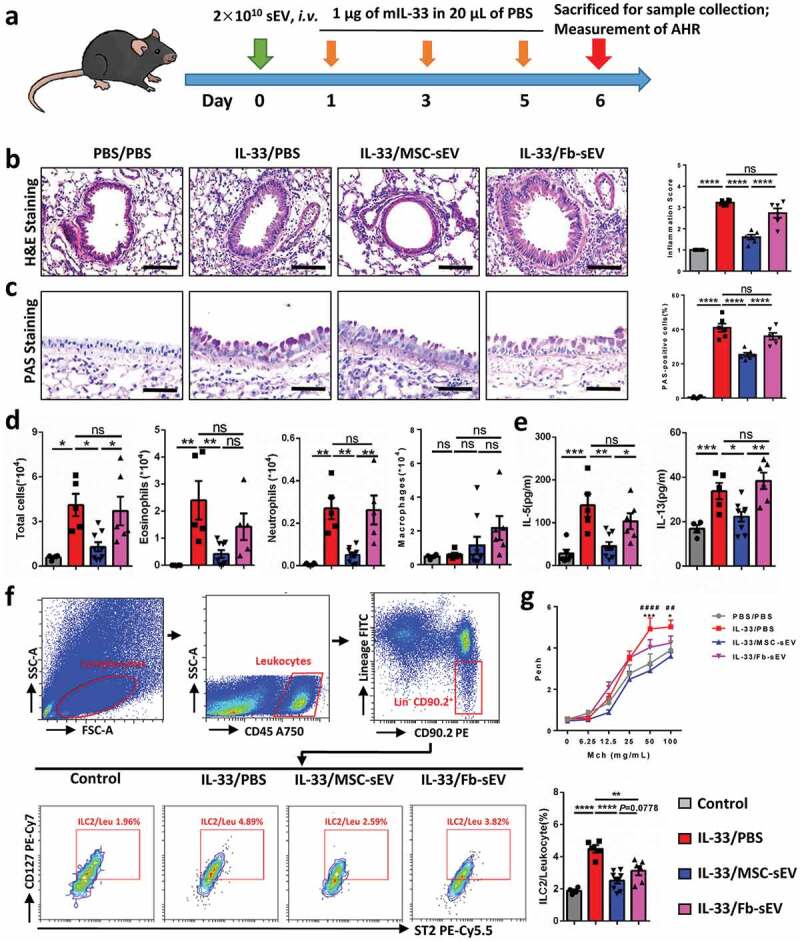
MSC-sEV significantly reduced ILC2-dominant allergic airway inflammation in mice. (a) Schematic diagram for the development of ILC2-dominant allergic airway inflammation in mice. (b-c) MSC-sEV but not Fb-sEV reduced infiltration of inflammatory cells in peritracheal area as determined by H&E staining (b), and numbers of epithelial goblet cells as determined by PAS staining (c) for mouse lung tissues, respectively. (d) Numbers of total inflammatory cells, eosinophils and neutrophils in BALF were decreased by MSC-sEV but not Fb-sEV as determined by flow cytometry. (e) Levels of IL-5 and IL-13 in BALF were decreased by MSC-sEV but not Fb-sEV. (f) Flow cytometry analyses of the ILC2s in lung tissues showed that MSC-sEV elicited more obvious effects on the reduction of ILC2s than Fb-sEV. (g) MSC-sEV but not Fb-sEV significantly reduced the airway hyperresponsiveness in ILC2-dominant allergic airway inflammation (*PBS/PBS versus IL-33/PBS; # IL-33/MSC-sEV versus IL-33/PBS). BALF, bronchoalveolar lavage fluids; Fb, Fibroblast; ILC2, Group 2 innate lymphoid cells; Leu, leukocytes; mIL-33, murine IL-33; MSC, mesenchymal stromal cell; ns, not significant; PBS, phosphate saline solution; sEV, small extracellular vesicles. *P< 0.05, ** or ##P< 0.01, ***P< 0.001, **** or ####P< 0.0001 analysed by one-way ANOVA. Scale bar, 100 μm for H&E Staining and 200 μm for PAS Staining.
Bronchoalveolar lavage fluid (BALF) was collected by performing lung lavage with 1 mL of pre-cooled PBS three times and centrifuged at 400 g for 5 min. After centrifugation, supernatants were collected for detection of Th2-related cytokines (R&D Systems, Minneapolis, MN, USA) and pellets were used for flow cytometry analyses of inflammatory cells. The left lobe of lung tissues was collected for histopathologic evaluation. The other lung tissues were minced and digested for the isolation of lung cells following the procedures described in our previous reports [12,13] and used for flow cytometry analyses of ILC2s.
Measurement of airway hyperresponsiveness
AHR to methacholine was measured using a four-chamber, whole-body plethysmograph (WBP system, Buxco Electronics, USA) on day 6. Mice were first placed in the chambers for 1 min for acclimatization, then were given nebulized PBS only and nebulized PBS with increasing methacholine concentrations (6.25, 12.5, 25, 50, 100 mg/mL) (Sigma-Aldrich, St. Louis, MO, USA). The time for nebulization was set to 1 min for each methacholine dosage and the reading interval was set to 5 min after nebulization. Enhanced pause (Penh) values were continuously collected, and mean values were selected to express changes of lung function.
The uptake effects of ILC2s on sEV in vitro and in vivo
To determine the uptake effects of ILC2s on sEV, PBMCs from patients with allergic rhinitis were co-cultured with 40 µg/mL mCherry-labelled sEV for the 12 h or 24 h, and MFI of mCherry in ILC2s was analysed using flow cytometry. Sorted human ILC2s from buffy coats of volunteers were co-cultured with 40 µg/mL mCherry-labelled sEV for 12 h or 24 h and the nuclei were counterstained using 4ʹ, 6-diamidino-2-phenylindole (DAPI, Invitrogen, Eugene, OR, USA). The ILC2s were then smeared on the slides, and were taken pictures using Leica Microsystems (DM6B, Wetzlar, Germany).
To study the in vivo uptake effects of ILC2s on MSC-sEV, mice were established with allergic airway inflammation as above, administered with mCherry-labelled MSC-sEV on day 6, and sacrificed at 4 h, 18 h and 24 h post-administration. MFI of mCherry in lung ILC2s was analysed using flow cytometry.
Flow cytometry analysis
For analysis of surface markers or mCherry of sEV, 20 µL of human anti-CD63-coated magnetic beads (Life Technologies, AS, Norway) were transferred to a 1.5 mL tube, washed with 200 µL of Isolation Buffer (PBS containing 0.1% BSA) and incubated with 100 µL of sEV samples overnight at 4°C with rotating. The bead-bound sEV were washed with 500 µL of Isolation Buffer three times and then incubated with antibodies for EV markers of PE-CD9, FITC-CD63 and APC-CD81, or antibodies for MSC surface markers of PC5.5-CD44, PC5.5-CD146, PE-CD73, APC-CD90, PE-CD105 at RT for 30 min. All of the antibodies were purchased from eBioscience, San Diego, CA, USA.
For flow cytometry analysis of ILC2s in human PBMCs [6], the cells were stained with Lineage markers (CD2, CD3, CD14, CD16, CD19, CD56, CD235a, FceR1)-FITC, CRTH2-PE and CD127-PE-Cy7. Gating strategies for flow cytometry analyses of ILC2s in PBMCs are shown as Figure 5(a). In analyses of IL-9 and IL-13 positive ILC2s, PBMCs were first stimulated with phorbol myristate acetate (50 ng/mL, Sigma, St. Louis, MO, USA), ionomycin (1000 ng/mL, St. Louis, Sigma, MO, USA) and Monensin (1:1000, Thermo Fisher Scientific, Carlsbad, CA, USA) for 5 h and then stained with anti-IL-9-APC and anti-IL-13-PB450 besides the above antibodies for gating ILC2s.
Figure 5.
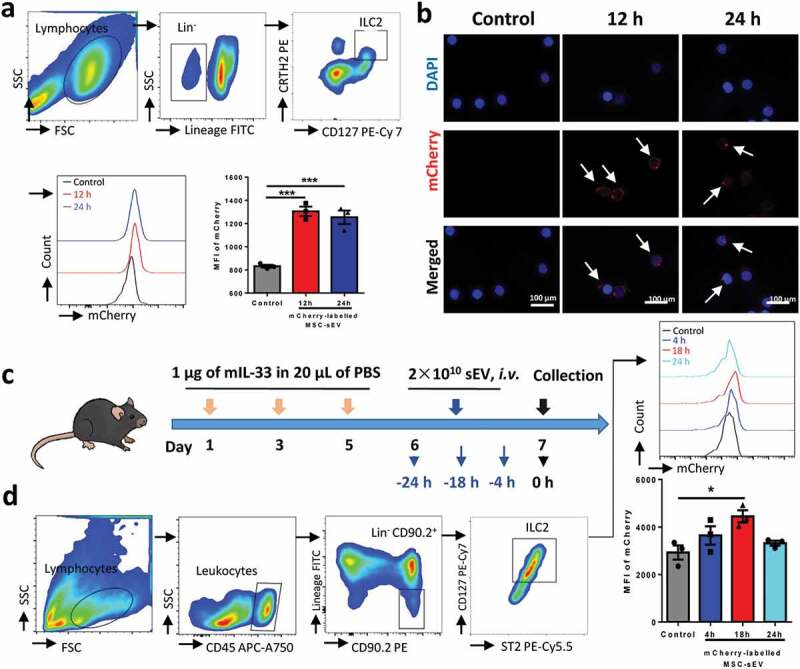
MSC-sEV were able to be taken in by ILC2s both in vitro and in vivo. PBMCs from patients with allergic rhinitis (a, n = 3) or sorted ILC2s from human buffy coats (b, n = 3) were incubated with mCherry-labelled MSC-sEV. (a) Gating strategy for human ILC2s and MFI of mCherry in ILC2s were finally analysed. MFI of mCherry in ILC2s were significantly increased at 12 h and 24 h. (b) Detection of mCherry (red) in purified human ILC2s by means of fluorescent microscopy. (c) Schematic diagram for studying the uptake effects of lung ILC2s on mCherry-labelled MSC-sEV. (d) Flow cytometry analysis suggested that MFI of mCherry in lung ILC2s in mice with allergic airway inflammation were significantly increased after systemic administration of mCherry-labelled MSC-sEV. Abbreviations: ILC2 Group 2 innate lymphoid cells, Lin− Lineage-negative, MFI mean fluorescent index, MSC mesenchymal stromal cell, sEV small extracellular vesicles. *P< 0.05 and ***P< 0.001 analysed by one-way ANOVA.
For analyses of eosinophils, neutrophils and macrophages in mouse BALF, the pellets were stained with antibodies to CD45-FITC, CD11b-APC-Cy7, CD64-PE, Ly-6G-Alexa 700 and Siglec-F-Alexa 647 and analysed based on gating strategies as previously reported (Supplementary figure 4 (a)) [30]. For analysis of ILC2s in mouse lung tissues, isolated lung cells were stained with antibodies to CD45-APC-Cy7, Lin+ markers (CD3, CD45R, CD11b, TER-119 and Ly-G6)-FITC, CD90.2-PE, ST2-PerCP-eFluor 710 and CD127-PE-Cy7 according to the gating strategies as previously reported [28,29].
After staining, samples were analysed on a CytoFLEX Flow Cytometer (Beckman Coulter, Hercules, CA, USA). The antibody to Lin+ markers was purchased from Thermo Fisher Scientific, San Diego, CA, USA. All the other antibodies were purchased from BD Biosciences, San Jose, CA, USA.
The detailed information about the antibodies used for flow cytometry analyses of mice and human ILC2s is presented in supplementary Table 4.
Human inflammatory cytokine array
To investigate the effects of sEV on ILC2 function, levels of 40 inflammatory cytokines in the supernatants of ILC2s with or without treatment with MSC-sEV were assayed using the Quantibody® Human Inflammation Array 3 according to the manufacturer’s instructions (RayBiotech Inc., Norcross, GA, USA).
Western blot analysis
The protein levels of sEV and cell lysates were quantified using the Pierce™ BCA protein assay (Thermo Fisher Scientific, Rockford, IL, USA), and 50 µg of protein was denatured in SDS loading buffer at 95°C for 5 min. Then, 10 µg of the samples were loaded, separated by SDS-PAGE and transferred to polyvinylidene difluoride membranes (Roche Diagnostics, Mannheim, Germany). The membranes were blocked with 5% skimmed milk at RT for 1 h and further incubated with rabbit antibodies against CD9 (1:2000), CD63 (1:2000), CD81 (1:2000), Alix (1:5000), TSG101 (1:1000) and Calnexin (1:1000) at 4°C overnight. After three washes, the membranes were incubated with horseradish peroxidase-conjugated anti-rabbit antibodies for 1 h at RT. The membranes were washed, incubated with Enhanced Chemiluminescence Plus (Millipore Corporation, Billerica, MA, USA), and photographed using an imaging analysis system (ImageQuant LAS 4000, GE Health Care Life Science, Uppsala, Sweden). Primary antibodies were purchased from Abcam, Cambridge, UK and the secondary antibody was purchased from Jackon ImmunoResearch, West Grove, PA, USA.
ELISA for levels of cytokines
Levels of Th2-related cytokines in human cell-culture supernatants were determined by Ready-SET-Go ELISA Kits (Thermo Fisher Scientific, San Diego, CA, USA). Mouse BALF samples were assessed for levels of IL-5 and IL-13 using ELISA kits purchased from R&D System Inc., Minneapolis, MN, USA. All procedures were performed according to the manufacturer’s instructions.
Statistical analyses
Statistical analyses were performed using Prism software (GraphPad Software, La Jolla, CA, USA). All values were expressed as mean ± SEM for each group. Statistical significance was assessed by using independent two-tailed Student’s t-test for single comparisons or one-way ANOVA with Tukey’s correction for multiple comparisons as indicated. A P value less than 0.05 was considered statistically significant.
Results
Isolation of sEV from supernatants
Human MSCs and fibroblasts were cultured in the indicated CCM for six days and then switched to CDPF medium that was specially optimized for production of sEV (Figure 1(a)). We observed that MSCs and fibroblasts did not exhibit the proliferation in the CDPF medium. The morphologies of cells incubated in CDPF medium for 48 h became more elongated compared with those cultured in CCM, suggesting a response by MSCs and fibroblasts to stress induced by the protein-free environment in the CDPF medium. Importantly, no difference was found in the percentages of apoptotic cells in MSCs and fibroblasts incubated in CDPF and CCM (Figure 1(b)), which were all much lower than the 5% specified in the 2013 ISEV position paper [31]. It suggests that CDPF medium can be a good candidate for sEV preparation from MSCs. We then separated sEV from harvested supernatants by a more efficient, stable and scalable anion-exchange chromatography method, as previously reported but with some modification [27]. sEV were eluted from the column after the sequential steps of resin packing, column equilibration, sample loading, column washing and elution of sEV (Figure 1(c)). Protein assay and CD63 ELISA of elution fractions 1 to 8 (F1 to F8) suggested that the concentration of sEV stably peaked at F3 to F5 (Figure 1(d)). Consistently, EV markers of CD63 and Alix were also peaked at F3 to F5 by Western blot analysis (Supplementary figure 5). F3 to F5 were then pooled together for dialysis and further analysis. Because endotoxin in the supernatant and buffer would also be captured by anion-exchange chromatography and eluted together with the sEV, we thus used endotoxin-free cell culture medium, buffers and materials in all of the progress for sEV isolation. The levels of endotoxin in the final sEV samples were about 1.2 EU/mL as determined by LAL endotoxin determination kit (Bioeddo Technologies Co LTD., Xiamen, CN). The total particle number of MSC-sEV was about 9.43 × 1010 ± 5.40 × 109 per mL (n = 3) as determined by Nanosight, and the concentration of protein for MSC-sEV was about 435.0 ± 13.08 µg per mL (n = 3) as determined by Bradford Assay.
We also made a side by side comparison of anion-exchange chromatography and differential ultracentrifugation for isolation of MSC-sEV (Table 1). We evaluated the yield rates and the time costing for these two types of sEV isolation. Finally, we obtained 1.74 × 109 ± 1.01 × 108 sEV (n = 3) per 106 MSCs using anion-exchange chromatography, while 9.58 × 108 ± 7.28 × 107 sEV (n = 3) by differential ultracentrifugation. Additionally, it only took about 3 h to separate sEV from 600 mL supernatant using anion-exchange chromatography, while it took about 15 h to process with 600 mL supernatant using differential ultracentrifugation. Moreover, compared with differential ultracentrifugation, anion-exchange chromatography has more advantages in MSC-sEV isolation, including low cost, high reproducibility, scalable possibility, no specialized equipment required, and without the damage for sEV.
Table 1.
Comparison of differential ultracentrifugation and anion-exchange chromatography for MSC-sEV isolation.
| Features | Differential ultracentrifugation | Anion-exchange chromatography |
|---|---|---|
| Yield rate (/106 cells) # | 9.58 × 108 ± 7.28 × 107 sEV | 1.74 × 109 ± 1.01 × 108 sEV |
| Efficiency | 15 h per 600 mL supernatant | 3 h per 600 mL supernatant |
| Cost | High | Low |
| Reproducibility | Low | High |
| Scalability | No | Yes |
| Damage of sEV | Yes | No |
| Equipment | Yes | No |
#Calculated based on three independent preparations of MSC-sEV.
Characterization of sEV isolated by anion-exchange chromatography
sEV derived from MSCs and fibroblasts had a mean diameter of approximately 100 nm as determined by Nanosight (Figure 2(a)). sEV were confirmed by TEM as diameters of 150 nm or less and revealed the characteristic lipid-bilayer (Figure 2(b)). Flow cytometric analysis of sEV showed the surface expression of tetraspanins CD9/CD63/CD81 (Figure 2(c)) on MSC-sEV and Fb-sEV. Using Western blot, we further confirmed that both MSC-sEV and Fb-sEV were positive for conventional sEV-specific markers of CD9, CD63, CD81, Alix and TSG101, and that the levels of these markers were higher in sEV than in their parent cells (Figure 2(d)). Additionally, both MSC-sEV and Fb-sEV lacked detectable Calnexin, an endoplasmic reticulum membrane marker expressed in cells but less in sEV. We also found that MSC-sEV did not express the general surface markers of MSCs as our previous study [11] including CD44, CD146, CD73, CD90 and CD105 (Supplementary figure 5), at least partly suggesting no much proportion of microvesicles originated by budding of the plasma membrane under the procedures used in this study. Taken together, the characterization of sEV in terms of size, morphology and specific makers suggested that our sEV preparations included canonical exosomes.
Figure 2.
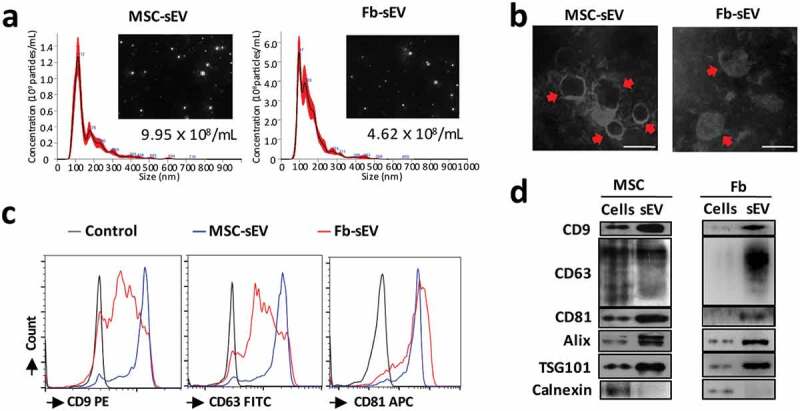
Characterization of sEV. (a) Representative results of nanoparticle tracking analyses of the sEV (1:100 dilution with particle-free PBS). (b) Representative results of the nano-size vesicles photographed by transmission electron microscope. (c) sEV markers of CD9, CD63 and CD81 were determined by bead-based flow cytometry. (d) sEV markers of CD9, CD63, CD81, Alix and TSG101 were positive in the sEV derived from MSCs or Fb as determined by Western Blot. Fb, Fibroblast; MSC, mesenchymal stromal cells; sEV, small extracellular vesicles. Scale bar, 100 nm.
MSC-sEV inhibited human ILC2 function from patients with allergic rhinitis in vitro
To evaluate the effects of MSC-sEV on the function of ILC2s, human PBMCs from patients with allergic rhinitis were treated with IL-2/25/33 in the presence or absence of MSC-sEV. We found that high levels of IL-5 and IL-13 in response to IL-2/25/33 were significantly reversed by MSC-sEV in a dose-dependent manner (Supplementary figure 6(a)). However, no effect was observed after treated with Fb-sEV (Supplementary figure 6(b)). Similar results were found with Lin− cells from the buffy coat of human volunteers, in which other IL-5- or IL-13-releasing immunocytes such as T cells were excluded (Supplementary figure 6(c)). Consistently, flow cytometry analyses demonstrated that levels of IL-9+ILC2s and IL-13+ILC2s were decreased by MSC-sEV but not Fb-sEV (Figure 3(a&b)). More importantly, MSC-sEV were found to inhibit the production of IL-9 and IL-13 in purified ILC2s under the stimulation of IL-2/25/33 (Figure 3(c)), showing that MSC-sEV were able to directly mitigate the activation of ILC2s.
Figure 3.
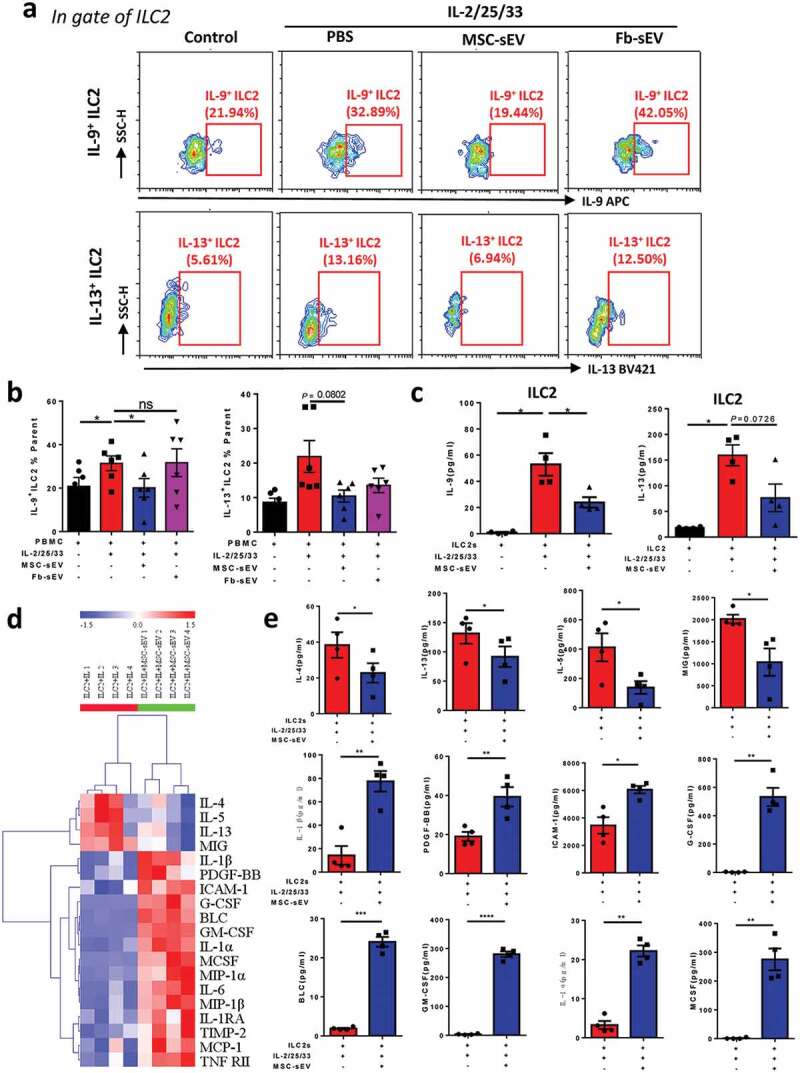
MSC-sEV significantly inhibited the function of human ILC2s in vitro. (a-b) Flow cytometry analyses showed that high levels of IL-9+ILC2s and IL-13+ILC2s were simultaneously decreased by MSC-sEV but not Fb-sEV in PBMCs from patients with allergic rhinitis in response to IL-2/25/33 (n = 6). (c) MSC-sEV significantly reversed the high levels of IL-9 and IL-13 in the supernatants of sorted ILC2s from human buffy coat in response to IL-2/25/33 (n = 4). (d-e) The effects of MSC-sEV on the levels of inflammatory cytokines in stimulated ILC2s were analysed by Human Inflammation Antibody Arrays (n = 4). Fb, Fibroblast; ILC2, Group 2 innate lymphoid cells; MSC, mesenchymal stromal cell; ns, not significant; PBS, phosphate saline solution; sEV, small extracellular vesicles. * P < 0.05, ** P < 0.01, *** P < 0.001, **** P < 0.0001 analysed by two-tailed Student’s t test for single comparison or one-way ANOVA for multiple comparisons.
To better understand the functional changes of ILC2s after MSC-sEV exposure, we assessed the levels of a broader range of cytokines using a Quantibody® Human Inflammation Array. Because we focused on the effects of MSC-sEV’s immunomodulation in this study, and we have already confirmed that Fb-sEV exhibited no effect on ILC2 function, here we only performed the Human Inflammation Assay with the treatment of MSC-sEV. Consistently, we found that the levels of ILC2 cytokines (e.g. IL-13 and IL-5) were significantly decreased, in contrast, the cytokines about group 3 ILC (ILC3) function such as granulocyte-macrophage colony stimulating factor (GM-CSF) were increased after sorted ILC2s were treated with MSC-sEV (Figure 3(d & e)).
Previous studies have reported that MSCs exerted their immunomodulation via some soluble factors [14]. To evaluate the possible effects of soluble factors involved in, we compared the effects of MSC-sEV and condensed EV-depleted supernatant on ILC2 function. Similarly, MSC-sEV significantly reduced the levels of IL-13+ILC2s after PBMCs from patients with AR were stimulated with IL-2/25/33 (Supplementary figure 6(d)). We observed that the sEV-depleted supernatant had some inhibition for ILC2 function but without significant difference (P = 0.1078). It suggests that sEV are the more important players than soluble molecules on ILC2 function by MSCs at least in our study.
Taken together, our findings suggested that MSC-sEV were able to inhibit the function of ILC2s.
Systemic administration of MSC-sEV ameliorated ILC2-dominant allergic airway inflammation in mice
To further investigate the effect of MSC-sEV on ILC2-dominant allergic airway inflammation, we developed an ILC2-dominant asthma mouse model via intratracheal administration of rmIL-33 (Figure 4(a)). Pathological analyses of lung tissues showed that both inflammatory infiltrations in the peritracheal area (Figure 4(b)) and numbers of epithelial goblet cells (Figure 4(c)) were significantly decreased in the mice treated with MSC-sEV. Total inflammatory cells, eosinophils and neutrophils in BALF were all significantly reduced by administration of MSC-sEV (Figure 4(d) & Supplementary figure 4(b)). Similarly, we observed that levels of type 2 cytokines, such as IL-5 and IL-13 in BALF, were significantly decreased by MSC-sEV (Figure 4(e)). More importantly, we found that the high levels of ILC2s in murine lung tissues were significantly decreased by MSC-sEV (Figure 4(f)). Additionally, we found that MSC-sEV were able to significantly reduce the AHR of the mice with asthmatic inflammation (Figure 4(g)), which further provided the strong evidence of the therapeutic effects of MSC-sEV. However, Fb-sEV failed to elicit any significant effects on allergic airway inflammation, including the pathological changes (Figure 4(b & c)), levels of inflammatory cells and cytokines in BALF (Figure 4(d & e)), and AHR (Figure 4(g)). Of course, we observed that Fb-sEV were able to reduce the levels of ILC2s in lung tissues, but the effect was lower than that of MSC-sEV (Figure 4(f)). It suggests that Fb-sEV might have some but very mild immunoregulatory function on ILC2s, and not strong enough to exhibit therapeutic effects on allergic airway inflammation. In total, our findings demonstrated the therapeutic effects of MSC-sEV on ILC2-dominant asthma inflammation in mice.
ILC2s were able to take MSC-sEV in vitro and in vivo
To further understand the mechanisms of the effects of MSC-sEV on ILC2 function, mCherry-labelled MSC-sEV were successfully prepared (Supplementary figure 1), and the uptake effects of ILC2s on MSC-sEV in vitro and in vivo were evaluated. We found that MSC-sEV were significantly taken in by human ILC2s after both 12 h and 24 h treatment of MSC-sEV by means of flow cytometry (Figure 5(a)). Furthermore, the in vitro uptake effects of sorted human ILC2s on MSC-sEV (red) were confirmed by using fluorescent microscopy (Figure 5(b)). Consistently, the MFI of mCherry in lung ILC2s was significantly increased at 18 h after administration of mCherry-labelled of MSC-sEV to mice with ILC2-dominant allergic airway inflammation (Figure 5(c & d)). Overall, these results indicated that ILC2s were able to take in MSC-sEV in the setting of ILC2-dominant allergic airway inflammation.
Small RNA sequencing for differential signature of miRNAs between MSC-sEV and Fb-sEV
It was reported that miRNAs are the important functional components in sEV [32]. To identify the miRNAs that contributed to the therapeutic effects of MSC-sEV on allergic airway inflammation, we performed total small RNA sequencing to detect the RNA profiles of MSC-sEV and Fb-sEV. In total, we identified approximately 400 miRNAs in both MSC-sEV and Fb-sEV. Total of 142 miRNAs were differentially expressed between MSC-sEV and Fb-sEV (Figure 6(a)), among which 74 of miRNAs were higher in MSC-sEV and 68 of miRNAs were higher in Fb-sEV (Figure 6(b), Supplementary Table 5). Go enrichment analysis revealed that the genes targeted by these differentially expressed miRNAs were significantly involved in the regulation of some biological processes, cellular components and molecular functions (Supplementary figure 3(b&c)). The miRNAs that obviously enriched in MSC-sEV included miR-146a-5p, hsa-miR-206, hsa-miR-184, hsa-miR-1290, hsa-miR-133a-3p and hsa-miR-122-5p, and hsa-miR-320a-5p was significantly enriched in Fb-sEV (Figure 6(b)). Among the miRNAs that were highly enriched in MSC-sEV, miR-146a-5p was previously reported to inhibit the function of activated ILC2s and ameliorate allergic airway inflammation [22,33]. We also found that miR-146a-5p was significantly highly expressed in MSCs compared with fibroblasts (Figure 6(c)). The difference of miR-146a-5p expression in MSC-sEV vs. Fb-sEV with a fold change (log2) of 5.85 (Supplementary Table 5) was much greater than that of MSCs v.s. Fb, which indicated the highly selective transfer of miRNA cargo into sEV in MSCs. Thus, miR-146a-5p was selected as a potential candidate mediating the therapeutic effects of MSC-sEV in our allergic airway inflammation.
Figure 6.
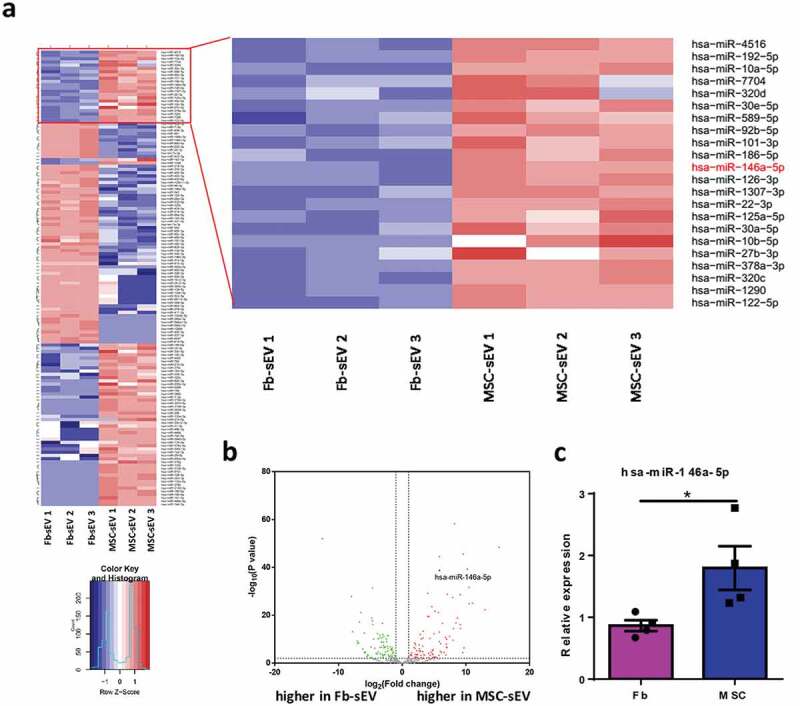
Small RNA sequencing for analysis of miRNAs in MSC-sEV and Fb-sEV. (a) Heatmap showing the differential expression of miRNAs between MSC-sEV and Fb-sEV. (b) Volcano plot comparing relative expression of the miRNAs between MSC-sEV and Fb-sEV, in which miR-146a-5p (black dot) was identified as one of the most significant upregulated miRNAs in MSC-sEV. (c) MSCs expressed higher level of hsa-miR-146a-5p than Fibroblasts as determined by qPCR. Fb, Fibroblast; MSC, mesenchymal stromal cell; sEV, small extracellular vesicles; *P< 0.05 analysed by two-tailed Student’s t test.
We hypothesized that miRNAs are critical for the therapeutic effect of MSC-sEV on ILC2 function. We next investigated the functional effects of MSC-sEV on ILC2s after depleting the RNAs using RNase A in MSC-sEV. We found that RNA-depleted MSC-sEV did not inhibit the high levels of IL-13+ILC2s in response to IL-2/25/33, further suggesting the important role of miRNAs in the effects of MSC-sEV on the inhibition of ILC2 function (as supplementary figure 6(e)).
hsa-miR-146a-5p mediated therapeutic effects of MSC-sEV on allergic airway inflammation
To further determine whether miR-146a-5p was involved in the effects of MSC-sEV on ILC2-dominant airway inflammation, we decreased its expression with miR-146a-5p inhibitor in MSCs, and increased its expression with miR-146a-5p mimics in fibroblasts, and checked their effects using human PBMCs or mouse model. We first examined the effects of miR-146a-5p on the function of ILC2s using IL-2/25/33 treated PBMCs from patients with allergic rhinitis. Similar to our above findings (Figure 3(a & b)), MSC-sEVscramble significantly decreased the high levels of IL-9+ILC2s and IL-13+ILC2s at 48 h after the treatment (Figure 7(a-c)). However, MSC-sEV from cells transfected with a miR-146a-5p inhibitor (MSC-sEVinhibitor) had no any effects on their high levels. In contrast, Fb-sEV from cells transfected with hsa-miR-146a-5p mimics (Fb-sEVmimics) got the ability to decrease ILC2 function as same as MSC-sEV (Figure 7(a-c)).
Figure 7.
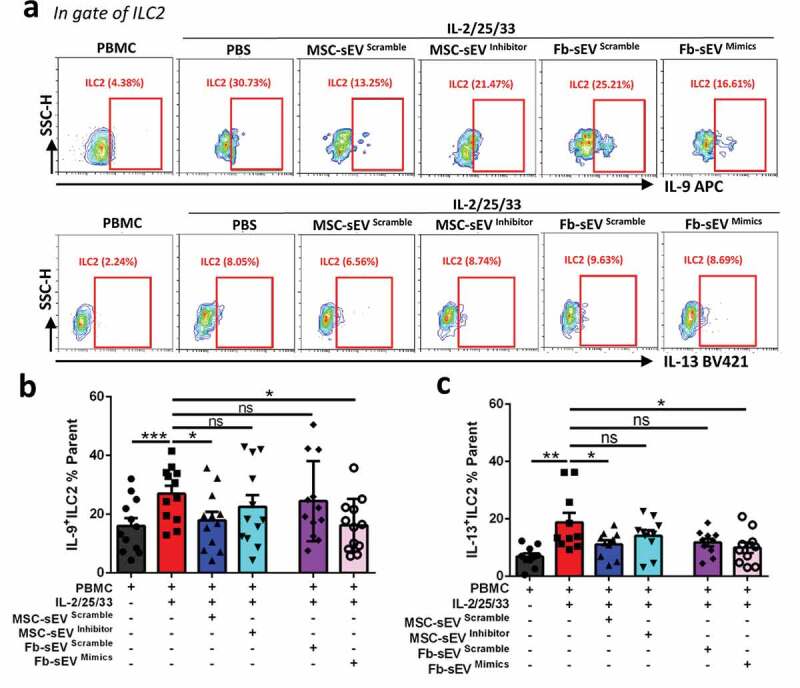
Immunoregulatory effect of MSC-sEV on human ILC2s was mediated by miR-146a-5p in vitro. (a) Represent contour plots for the percentages IL-9+ILC2s and IL-13+ILC2s in IL-2/25/33-stimulated PBMCs from patients with allergic rhinitis with the treatments of different sEV. (b-c) Effects of MSC-sEV on the percentages of IL-9+ILC2s and IL-13+ILC2s were both abrogated by antagonization of miR-146a-5p, while Fb-sEV overexpressed with miR-146a-5p acquired the function to inhibit the percentages of IL-9+ILC2s and IL-13+ILC2s (n = 12). Fb, Fibroblast; ILC2, Group 2 innate lymphoid cells; MSC, mesenchymal stromal cell; ns, not significant; PBMC, peripheral blood mononuclear cells; PBS, phosphate saline solution; sEV, small extracellular vesicles. *P< 0.05, **P< 0.01, ***P< 0.001 analysed by one-way ANOVA.
Furthermore, we evaluated the effects of MSC-sEVinhibitor and Fb-sEVmimics in a mouse model of allergic airway inflammation. Similar to the findings in vitro, we found that MSC-sEVinhibitor had weaker effects on the reduction of inflammatory infiltration and epithelial goblet cell hyperplasia compared with MSC-sEVscramble (Figure 8(a & b)). Similar results were found on the levels of type 2 cytokines (Figure 8(c)) and inflammatory cells in BALF (Figure 8(d) and Supplementary figure 4(c)). More importantly, we observed that the effects of MSC-sEVinhibitor on levels of ILC2s in lung tissues were mitigated compared to MSC-sEVscramble (Figure 8(e)). On the contrary, Fb-sEVmimics acquired the above effects on ILC2-dominant allergic airway inflammation.
Figure 8.
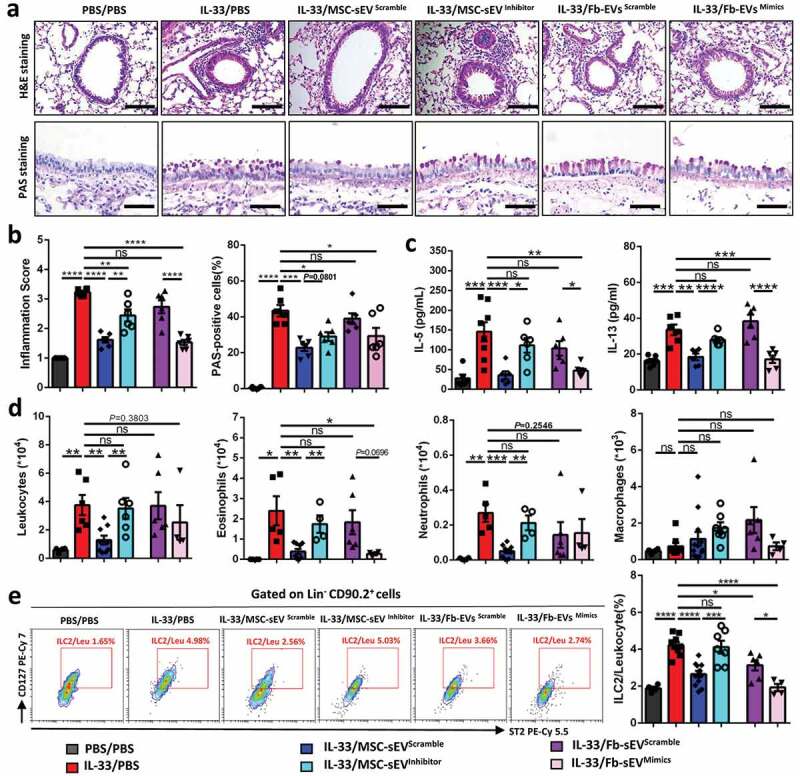
MSC-sEV ameliorated ILC2-dominant allergic airway inflammation through miR-146a. (a-b) H&E and PAS staining for lung tissues. MSC-sEVinhibitor elicited lower effects on infiltration of inflammatory cells and epithelial goblet cells compared with MSC-sEVScramble, while Fb-sEVmimics showed higher effects than Fb-sEVScramble. (c-d) Effects of MSC-sEV on levels of inflammatory cytokines (c) and cells (d) in BALF were abrogated after miR-146a-5p was antagonized, while Fb-sEV acquired the effects after overexpression of miR-146a-5p. (e) Effects of MSC-sEV on levels of ILC2s in lung tissues were abrogated after miR-146a-5p was antagonized, while the effect of Fb-sEV was enhanced after overexpression of miR-146a-5p. Fb, Fibroblast; ILC2, Group 2 innate lymphoid cells; MSC, mesenchymal stromal cell; ns, not significant; PBS, phosphate saline solution; sEV, small extracellular vesicles. *P< 0.05, **P< 0.01, ***P< 0.001 and ****P< 0.0001 analysed by one-way ANOVA. Scale bar, 100 μm for H&E Staining and 200 μm for PAS Staining.
Collectively, these data indicated that MSC-sEV ameliorated ILC2-dominant allergic airway inflammation in mice, which was partially attributed to the delivery of miR-146a-5p in MSC-sEV to ILC2s.
Discussion
In the current study, we successfully developed a standardized scalable protocol of anion-exchange chromatography for isolation of MSC-sEV. For the first time, we demonstrated that MSC-sEV were able to inhibit the function of human ILC2s and attenuate ILC2-dominant allergic airway inflammation in mice. Moreover, we revealed that miR-146a-5p in MSC-sEV at least partly contributed to the therapeutic effects of MSC-sEV on human ILC2 function and in mouse ILC2-dominant allergic airway inflammation.
MSCs have been intensively reported to display therapeutic effects in many human diseases through different mechanisms, including local differentiation to replace injured cells, secretion of soluble factors or transfer of functional molecules via exosomes [34]. In our previous studies, we have comprehensively demonstrated that MSCs derived from iPSCs elicited therapeutic effects on allergic airway inflammation through regulation on immunocytes [10–13] or mitochondrial transfer [35]. MSC-sEV have become extremely promising because they possess similar therapeutic effects to their parent MSCs in many diseases. However, the greatest challenges for large preparation of MSC-sEV are the huge number of homogeneous MSCs and the scalable isolation protocol. The standardized protocols for sEV isolation in our study would overcome these limitations. To minimize the variations among different preparations of MSCs, we first selected the highly proliferative iPSC-MSCs as the cell sources, which were expanded substantially to create a cell bank for production of iPSC-MSC-sEV. Moreover, we obtained sEV-enriched medium by incubating iPSC-MSCs with the chemically defined and protein-free medium, which was specifically optimized to minimize the cell death and impose stress on cells to increase the release of MSC-sEV. Additionally, we isolated iPSC-MSC-sEV from supernatants by anion-exchange chromatography, which was more efficient and could be readily scaled up to industrial-scale for clinical application in the future. The characterization of iPSC-MSC-sEV further suggested that our protocol was suitable to purify iPSC-MSC-sEV. Compared with differential ultracentrifugation, our protocol for MSC-sEV production has the advantages of high efficiency, homogeneity and scalability.
Asthma and allergic rhinitis are both characterized by airway inflammation, mucus hyperproduction, and airway hyperreactivity, which are mainly driven by Th2 cells and the cytokines they release. In recent years, increasing evidences have also demonstrated that ILC2s, activated by epithelial cell cytokines such as IL-25, IL-33 and thymic stromal lymphopoietin, are able to orchestrate the pathological features of asthma, and sometimes play a more important role than Th2 cells. Effects of MSC-sEV on allergic asthma have been reported, in which sEV released by bone marrow or adipose-derived MSCs ameliorated Th2/Th17-dominant allergic airway inflammation [17,18]. However, some drawbacks could be noted in these two studies. MSCs derived from human adult tissues would easily become senescent after in vitro expansion, challenging the preparation of sufficient homogenous MSCs for future mass production of MSC-sEV in clinical application. Moreover, both studies used differential ultracentrifugation for isolation of MSC-sEV; since ultracentrifugation is relatively unscalable, this method could not easily be applied to industrial production. More importantly, these studies merely evaluated the therapeutic potential of MSC-sEV on Th2/Th17-dominant allergic airway inflammation. In contrast, we innovatively investigated the effects of iPSC-MSC-sEV isolated by scalable anion-exchange chromatography in ILC2-dominant allergic airway inflammation. Furthermore, we further confirmed the immunomodulation of MSC-sEV on ILC2 function using human PBMCs from patients of allergic rhinitis, which has higher morbidities than asthma in clinical and are more easily available for us. Since ILC2s play a similar role in the pathogenesis of allergic rhinitis and asthma, our study would to some extent reveal the effects of MSC-sEV on ILC2s of patients with asthma. More importantly, using ILC2-dominant mouse model, we identified that MSC-sEV were able to effectively inhibit the activation of ILC2s, attenuate eosinophilic airway inflammation and decrease AHR. Fb-sEV, as the negative control, did not moderate ILC2 functions or ameliorate allergic airway inflammation, further reinforcing the immunoregulatory effects of iPSC-MSC-sEV. More importantly, we established mCherry-labelled MSC-sEV to investigate the uptake effects of ILC2s on MSC-sEV in vitro and in vivo. We found that MSC-sEV were significantly taken in by isolated human ILC2s and lung ILC2s in mice of asthma. It further suggests that ILC2s were able to take in MSC-sEV. Therefore, our data further extend current knowledge of the therapeutic effects of MSC-sEV on allergic airway inflammation. However, we fully acknowledge that sEV are merely one mode of action for MSCs. The effects of MSC-sEV are likely disease/model dependent, thus other models may require the other modes of MSCs.
It is been well acknowledged that miRNAs are a class of noncoding single-stranded RNAs that exert biological functions through post-transcriptional regulation of RNA targets. Also, miRNAs were involved in the polarization of Th2 cells and pathogenesis of eosinophilic airway inflammation [36,37]. Similarly, recent studies also suggested that miRNAs, such as the miR-17–92 cluster of miRNAs [21], miR-146a-5p [22,33] and miR-155 [19,20], are important regulators of the homoeostasis and function of ILC2s in allergic airway inflammation. We then characterized the miRNA signatures of MSC-sEV in our study. We found that antagonization of miR-146a-5p in MSC-sEV abrogated their immunoregulatory effects in ILC2-dominant allergic airway inflammation, while the effects were observed in Fb-sEV from cells transfected with miR-146a-5p mimics. Our data, for the first time, demonstrated that transferring of miR-146a-5p in MSC-sEV to ILC2s was partially associated with the therapeutic effects of iPSC-MSC-sEV in ILC2-dominant allergic airway inflammation. However, MSC-sEV contain abundant of miRNAs, proteins and lipids, etc., in addition to miR-146a-5p. These components undoubtedly are bioactive and exhibiting overall therapeutic effects as an ensemble on allergic airway inflammation.
There are some limitations in our study. First, wild-type C57/BL6 mice were used to develop the ILC2-dominant allergic airway inflammation. In these mice, T and B cells are present and might have interacted with the function of ILC2s. As a future alternative, T/B cell-deficient Rag1−/- mice could be developed as an ILC2-dominant mouse model. Additionally, we only focused on the direct effects of MSC-sEV on immunoregulation of ILC2s. It cannot be excluded the possibility that MSC-sEV took effects through interacted indirectly with other immunocytes, such as myeloid cells like DCs. We acknowledged that the high expression of the liver-specific and foetal bovine serum-superabundant miR-122 maybe because of the possible leftover from the previous CCM media or because of some unknown operation in the experiments. It will be better to use serum-free CCM for culture of MSCs in the future.
Conclusions
In summary, we successfully optimized scalable anion-exchange chromatography for isolation of MSC-sEV. We found that the isolated MSC-sEV modulated human ILC2 function in vitro and ILC2-dominant allergic airway inflammation in an experimental mouse model, in part due to transfer of miR-146a-5p to ILC2s. Our study supports further development of MSC-sEV could as a novel cell-free strategy for the treatment of allergic airway inflammation.
Supplementary Material
Acknowledgments
We sincerely thank Guangzhou Blood Center for kindly providing us with buffy coats of human volunteers. We would like to give our sincere thanks to Dr. Kenneth W. Witwer from Johns Hopkins University School of Medicine for his useful discussion and constructive comments for our manuscript.
Author contributions
SBF and HYZ helped in collection and/or assembly of data and manuscript writing; CW, BXH, XQL, XCM, YQP, ZBX and XLF helped in the collection of data. DC helped in the collection of samples from the patients. ZJW prepared the sEV. LZ performed the experiments using Nanosight. SGZ helped in data analysis. QLF helped in concept and design, data analysis, manuscript writing and final approval of the manuscript.
Disclosure of interest
The authors report no conflict of interest.
Supplementary material
Supplemental data for this article can be accessed here.
References
- [1].Bousquet J, Khaltaev N, Cruz AA, et al. Allergic Rhinitis and its Impact on Asthma (ARIA) 2008 update (in collaboration with the World Health Organization, GA(2)LEN and AllerGen). Allergy. 2008;63(Suppl 86):8–19. [DOI] [PubMed] [Google Scholar]
- [2].Carr TF, Zeki AA, Kraft M.. Eosinophilic and Noneosinophilic Asthma. Am J Respir Crit Care Med. 2018;197(1):22–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Halim TY, Steer C, Mathä L, et al. Group 2 innate lymphoid cells are critical for the initiation of adaptive T helper 2 cell-mediated allergic lung inflammation. Immunity. 2014;40(3):425–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Drake LY, Iijima K, Kita H.. Group 2 innate lymphoid cells and CD4+ T cells cooperate to mediate type 2 immune response in mice. Allergy. 2014;69(10):1300–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Chang YJ, Kim HY, Albacker LA, et al. Innate lymphoid cells mediate influenza-induced airway hyper-reactivity independently of adaptive immunity. Nat Immunol. 2011;12(7):631–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Yu QN, Guo YB, Li X, et al. ILC2 frequency and activity are inhibited by glucocorticoid treatment via STAT pathway in patients with asthma. Allergy. 2018;73(9):1860–1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Zhong H, Fan XL, Yu QN, et al. Increased innate type 2 immune response in house dust mite-allergic patients with allergic rhinitis. Clin Immunol. 2017;183:293–299. [DOI] [PubMed] [Google Scholar]
- [8].Smith SG, Chen R, Kjarsgaard M, et al. Increased numbers of activated group 2 innate lymphoid cells in the airways of patients with severe asthma and persistent airway eosinophilia. J Allergy Clin Immunol. 2016;137(1):75–86. e8. [DOI] [PubMed] [Google Scholar]
- [9].Liu S, Verma M, Michalec L, et al. Steroid resistance of airway type 2 innate lymphoid cells from patients with severe asthma: the role of thymic stromal lymphopoietin. J Allergy Clin Immunol. 2018;141(1):257–268. e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Fu QL, Chow YY, Sun SJ, et al. Mesenchymal stem cells derived from human induced pluripotent stem cells modulate T-cell phenotypes in allergic rhinitis. Allergy. 2012;67(10):1215–1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Gao WX, Sun Y-Q, Shi J, et al. Effects of mesenchymal stem cells from human induced pluripotent stem cells on differentiation, maturation, and function of dendritic cells. Stem Cell Res Ther. 2017;8(1):48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Fang SB, Zhang HY, Jiang A-Y, et al. Human iPSC-MSCs prevent steroid-resistant neutrophilic airway inflammation via modulating Th17 phenotypes. Stem Cell Res Ther. 2018;9(1):147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Sun YQ, Deng MX, He J, et al. Human pluripotent stem cell-derived mesenchymal stem cells prevent allergic airway inflammation in mice. Stem Cells. 2012;30(12):2692–2699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Gao F, Chiu SM, Motan DAL, et al. Mesenchymal stem cells and immunomodulation: current status and future prospects. Cell Death Dis. 2016;7:e2062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Colombo M, Raposo G, Thery C. Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu Rev Cell Dev Biol. 2014;30:255–289. [DOI] [PubMed] [Google Scholar]
- [16].Lai RC, Yeo RW, Lim SK. Mesenchymal stem cell exosomes. Semin Cell Dev Biol. 2015;40:82–88. [DOI] [PubMed] [Google Scholar]
- [17].de Castro LL, Xisto DG, Kitoko JZ, et al. Human adipose tissue mesenchymal stromal cells and their extracellular vesicles act differentially on lung mechanics and inflammation in experimental allergic asthma. Stem Cell Res Ther. 2017;8(1):151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Cruz FF, Borg ZD, Goodwin M, et al. Systemic administration of human bone marrow-derived mesenchymal stromal cell extracellular vesicles ameliorates aspergillus hyphal extract-induced allergic airway inflammation in immunocompetent mice. Stem Cells Transl Med. 2015;4(11):1302–1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Knolle MD, Chin SB, Rana BMJ, et al. MicroRNA-155 protects group 2 innate lymphoid cells from apoptosis to promote type-2 immunity. Front Immunol. 2018;9:2232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Johansson K, Malmhäll C, Ramos-Ramírez P, et al. MicroRNA-155 is a critical regulator of type 2 innate lymphoid cells and IL-33 signaling in experimental models of allergic airway inflammation. J Allergy Clin Immunol. 2017;139(3):1007–1016. e9. [DOI] [PubMed] [Google Scholar]
- [21].Singh PB, Pua HH, Happ HC, et al. MicroRNA regulation of type 2 innate lymphoid cell homeostasis and function in allergic inflammation. J Exp Med. 2017;214(12):3627–3643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Lyu B, Wei Z, Jiang L, et al. MicroRNA-146a negatively regulates IL-33 in activated group 2 innate lymphoid cells by inhibiting IRAK1 and TRAF6. Genes Immun. 2019. [DOI] [PubMed] [Google Scholar]
- [23].Thery C, Amigorena S, Raposo G, et al. Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Curr Protoc Cell Biol. 2006;Unit 3 22 Chapter 3. [DOI] [PubMed] [Google Scholar]
- [24].Lian Q, Zhang Y, Zhang J, et al. Functional mesenchymal stem cells derived from human induced pluripotent stem cells attenuate limb ischemia in mice. Circulation. 2010;121(9):1113–1123. [DOI] [PubMed] [Google Scholar]
- [25].Sun YQ, Zhang Y, Li X, et al. Insensitivity of human iPS cells-derived mesenchymal stem cells to interferon-gamma-induced HLA expression potentiates repair efficiency of hind limb ischemia in immune humanized NOD scid gamma mice. Stem Cells. 2015;33(12):3452–3467. [DOI] [PubMed] [Google Scholar]
- [26].Thery C, Witwer KW, Aikawa E, et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): a position statement of the international society for extracellular vesicles and update of the MISEV2014 guidelines. J Extracell Vesicles. 2018;7(1):1535750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Kim DK, Nishida H, An SY, et al. Chromatographically isolated CD63+CD81+ extracellular vesicles from mesenchymal stromal cells rescue cognitive impairments after TBI. Proc Natl Acad Sci U S A. 2016;113(1):170–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Wallrapp A, Riesenfeld SJ, Burkett PR, et al. The neuropeptide NMU amplifies ILC2-driven allergic lung inflammation. Nature. 2017;549(7672):351–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Martinez-Gonzalez I, Mathä L, Steer C, et al. Allergen-experienced group 2 innate lymphoid cells acquire memory-like properties and enhance allergic lung inflammation. Immunity. 2016;45(1):198–208. [DOI] [PubMed] [Google Scholar]
- [30].Misharin AV, Morales-Nebreda L, Reyfman PA. Monocyte-derived alveolar macrophages drive lung fibrosis and persist in the lung over the life span. The Journal of Experimental Medicine. 2017;214(8):2387–2404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Witwer KW, EI Buzás, LT Bemis, et al. Standardization of sample collection, isolation and analysis methods in extracellular vesicle research. J Extracell Vesicles. 2013;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Zhang J, Li S, Li L, et al. Exosome and exosomal microRNA: trafficking, sorting, and function. Genomics Proteomics Bioinformatics. 2015;13(1):17–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Han S, Ma C, Bao L, et al. miR-146a mimics attenuate allergic airway inflammation by impacted group 2 innate lymphoid cells in an ovalbumin-induced asthma mouse model. Int Arch Allergy Immunol. 2018;177(4):302–310. [DOI] [PubMed] [Google Scholar]
- [34].Spees JL, Lee RH, Gregory CA. Mechanisms of mesenchymal stem/stromal cell function. Stem Cell Res Ther. 2016;7(1):125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Yao Y, Fan X-L, Jiang D, et al. Connexin 43-mediated mitochondrial transfer of iPSC-MSCs alleviates asthma inflammation. Stem Cell Reports. 2018;11(5):1120–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Simpson LJ, Patel S, Bhakta NR, et al. A microRNA upregulated in asthma airway T cells promotes TH2 cytokine production. Nat Immunol. 2014;15(12):1162–1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Pua HH, Ansel KM. MicroRNA regulation of allergic inflammation and asthma. Curr Opin Immunol. 2015;36:101–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.