

Abstract
Objective
SerpinA1 (alpha‐1 antitrypsin) is an acute inflammatory protein, which seems to play a role in neurodegeneration and neuroinflammation. In Alzheimer’s disease and synucleinopathies, SerpinA1 is overexpressed in the brain and the cerebrospinal fluid (CSF) showing abnormal patterns of its charge isoforms. To date, no comprehensive studies explored SerpinA1 CSF isoforms in Creutzfeldt–Jakob disease (CJD) and frontotemporal lobar degeneration (FTLD).
Methods
Using a capillary isoelectric focusing immunoassay, we analyzed CSF SerpinA1 isoforms in control cases (n = 31) and patients with a definite or probable diagnosis of CJD (n=77) or FTLD (n = 30), belonging to several disease subtypes.
Results
The overall SerpinA1 signal was significantly higher than in controls in CJD subtypes linked to abnormal prion protein (PrPSc) type 1, such as sporadic CJD (sCJD) MM(V)1, and in FTLD‐TDP. Moreover, CJD linked to PrPSc type 1 and FTLD‐TAU groups showed a significant relative increase of acidic and basic isoforms in comparison with controls, thereby forming two distinct SerpinA1 isoform profiles.
Interpretation
CJD linked to PrPSc type 1 and FTLD show a differential upregulation and post‐translational modifications of CSF SerpinA1. Further studies are needed to clarify whether these findings may reflect a common, albeit disease‐specific, pathogenetic mechanism related to neurodegeneration.
Introduction
SerpinA1, also known as alpha‐1 antitrypsin, is an acute inflammatory protein belonging to the superfamily of serpins, which acts as a serine protease inhibitor. With the exception of liver and blood cells, its expression is strictly downregulated throughout the body, including the brain.1 Nonetheless, several pieces of evidence suggest that SerpinA1 may play a role in neurodegeneration. In this regard, overexpression of the protein or its RNA has been described in the brains of patients with Alzheimer’s disease (AD)2 and frontotemporal lobar degeneration (FTLD),1 suggesting that SerpinA1 overexpression may be detrimental to neuronal function.1 Moreover, the protein may play an anti‐inflammatory role by attenuating microglial activation, causing a reduction of neuroinflammation.3, 4, 5 It is also known that the protein might diffuse out of venous blood and be released from the brain tissue into the cerebrospinal fluid (CSF).6 Previous studies documented a higher concentration of CSF SerpinA1 and/or different levels of its charge isoforms in AD, Parkinson’s disease (PD), Parkinson’s disease dementia (PDD), and dementia with Lewy bodies in comparison with controls.6, 7, 8, 9 However, to date, no studies specifically investigated CSF SerpinA1 isoforms in Creutzfeldt–Jakob disease (CJD) and FTLD, with the exception of a preliminary study in the latter.9
CJD and FTLD are highly heterogeneous neurodegenerative diseases from the clinical, genetic, and neuropathological points of view. CJD includes six major clinicopathological subtypes that are largely determined by the genotype at the methionine (M)/valine (V) polymorphic codon 129 of the PRNP gene and the type (1 or 2) of disease‐associated prion protein (PrPSc) accumulating in the brain.10 At variance, phenotypic heterogeneity in FTLD spectrum is mainly related to two major proteinopathies, namely FTLD with TDP43 (FTLD‐TDP) and tau pathology (FTLD‐TAU).11
To further study the role of SerpinA1 in neurodegenerative disorders, we tested CSF SerpinA1 in controls and patients with a definite or probable diagnosis of CJD and FTLD subtypes. To this aim, we took advantage of our previously developed capillary isoelectric focusing (CIEF) immunoassay for the analysis of SerpinA1.6 Moreover, we evaluated the possible correlations between SerpinA1, clinical variables, and the levels of currently established biomarkers of neurodegeneration.
Methods
Inclusion criteria and case classification
CSF samples were submitted for analysis to the Neuropathology Laboratory at the Institute of Neurological Sciences of Bologna (Italy) or to the Neurology Department of Ulm University Hospital (Germany) between 2010 and 2018. The cohort comprised 31 healthy controls, 77 patients with CJD, and 30 with FTLD. The study was conducted according to the revised Declaration of Helsinki and Good Clinical Practice guidelines. Informed consent was given by study participants or by their next of kin. The present study was approved by the ethics committees of “Area Vasta Emilia Centro” and Ulm University.
Classification of sporadic CJD (sCJD) was made according to the newly proposed criteria for CJD and related disorders (http://www.cjd.ed.ac.uk/sites/default/files/criteria_0.pdf). Specifically, the group of “definite” prion disease consisted of 52 prion‐positive cases at postmortem examination (27 sCJD MM(V)1, 13 VV2, 9 MV2K, 2 MM2C, and 1 VV1) and 5 cases carrying a pathogenic PRNP mutation [5 genetic CJD with E200K‐129M haplotype (gCJD E200K‐129M)] whereas the group of “probable” sCJD included 18 patients fulfilling the clinical criteria for possible sCJD and showing a positive prion real‐time quaking‐induced conversion (RT‐QuIC) assay. Molecular analysis of the PRNP gene, PrPSc typing, and CJD histotype classification were performed according to established methodologies and consensus criteria.12, 13, 14 In autopsied cases of CJD (n = 52), which were selected from a previous study,15 the assessment of AD copathology, performed according to the ABC score,16 showed either the absence or low level or AD‐related pathology in all but two cases demonstrating an intermediate level. Furthermore, there was no difference in the distribution of the severity of AD copathology among sCJD subtypes.
For the analysis according to the sCJD molecular subtypes, we merged the subjects with definite sCJD MM(V)1, VV2, MV2K, MM2C, and VV112 with those with a probable sCJD diagnosis and a high level of certainty for a given subtype (7 probable VV2, 11 probable MV2K, and 2 probable MM2C). In detail, the classification was determined by the consensus of two consultant neurologists (SAR and PP) after reviewing typical clinical features, disease duration at death or at last at follow‐up, the results of codon 129 genotype (MM, MV, and VV), CSF biomarkers, and brain magnetic resonance imaging (supplementary methods of Appendix S1). However, to exclude a bias related to possible misdiagnosis, we also analyzed the data after the exclusion of probable cases.
The FTLD group comprised cases with a pathological and/or genetic diagnosis of FTLD‐TDP (n = 24) or FTLD‐TAU (n = 6) as previously described.17 Specifically, the FTLD‐TDP group included patients with (1) a pathological diagnosis of TDP43 subtype B pathology (n = 1); (2) a pathogenic mutation in chromosome 9 open reading frame 72 gene (C9orf72) (n = 16, including the one with pathological diagnosis), progranulin (GRN) (n = 7), and TAR DNA‐binding protein 43 gene (TARDBP) (n = 1). Otherwise, the FTLD‐TAU group included patients with (1) a pathological diagnosis of primary tauopathy (1 progressive supranuclear palsy, 1 corticobasal degeneration) and (2) a pathogenic mutation in microtubule‐associated protein tau gene (MAPT) (n = 4). In all FTLD cases, the in vivo evidence of AD pathology was gathered using the AD core CSF biomarkers and in‐house calculated cutoff ratios.18 Specifically, a phosphorylated (p)‐tau/amyloid‐β (Aβ)42 ratio >0.108 (Bologna)17 or >0.08 (Ulm) was considered supportive for AD.
The control group included 31 subjects lacking any clinical or neuroradiologic evidence of central nervous system disease and having all three biomarkers total (t)‐tau, p‐tau and Aβ42 in the normal range.19
CSF biochemical analysis
In both centers, CSF samples were obtained by lumbar puncture (LP) at the L3/L4 or L4/L5 level following a standard procedure. All samples were centrifuged (multiple times in case of blood contamination); the supernatant was divided into aliquots and stored in polypropylene tubes at −80°C until analysis. Given the use of different tubes for storage in the two centers, we performed a SerpinA1 absorption property test. We could not detect significant differences in SerpinA1 signals for the same three samples aliquoted in two distinct storage tubes (data not shown). Furthermore, all samples were at maximum 3 times frozen and thawed. A previous stability analysis with samples four times frozen and thawed found no loss in signal intensity.6 For SerpinA1, each CSF sample was measured in duplicate. The mean intra‐assay coefficient of variation (CV) was 3.45% and the mean interassay CV 7.11% (calculated by repeating the analysis of two samples in four different runs). The CV of repeatability (two samples analyzed in one run in three different cycles) was 9.02%. The CSF SerpinA1 analysis was performed using a CIEF immunoassay platform (NanoPro 1000, ProteinSimple, Santa Clara, USA) as previously described.6 The reagents for the assay including pH gradient, fluorescent standards, capillaries were directly obtained from ProteinSimple. As primary antibody, a monoclonal mouse anti‐human SerpinA1 antibody was bought from R&D Systems (MAB1268, Minneapolis, USA).
CSF neurofilament light‐chain protein (NfL), t‐tau, p‐tau and Aβ42 were analyzed using commercially available enzyme‐linked immunosorbent assay kits (NF‐light, IBL, Hamburg, Germany; INNOTEST htau‐Ag, INNOTEST phosphorylated‐Tau181, INNOTEST Aβ1–42, Innogenetics/Fujirebio Europe, Ghent, Belgium) as previously described.20 PrPSc seeding activity was detected by RT‐QuIC as previously described.13 The mean intra‐ and interassays coefficients of variation (CVs) were ≤5% and ≤11%, respectively, for t‐tau and NfL as previously reported.20
Statistical analysis
Statistical analysis was performed using IBM SPSS Statistics version 21 (IBM, Armonk, NY) and GraphPad Prism 7 (GraphPad Software, La Jolla, CA) software. Based on the presence or not of a normal distribution of the values, data were expressed as mean ± standard deviation (SD) or median and interquartile range (IQR). For continuous variables, depending on the data distribution, the Mann–Whitney U test or the t test was used to test differences between two groups, while the Kruskal–Wallis test (followed by Dunn–Bonferroni’s post hoc test) or the one‐way analysis of variance (followed by Tukey’s post hoc test) was applied for multiple group comparisons. Chi‐square test was adopted for categorical variables. Spearman’s correlations were used to test the possible associations between analyzed variables. All reported P‐values have been adjusted for multiple comparison analyses. Differences were considered statistically significant at P < 0.05.
Results
Demographics and CSF biomarker results in the diagnostic groups
Demographic data concerning the diagnostic groups are shown in Table 1. There were no significant differences regarding age and sex distribution among patient groups. As expected, the time interval between onset and LP was significantly shorter in subjects with CJD than in those with FTLD (P < 0.001). Indeed, due to the frequent subacute onset and rapid progression, the former are usually evaluated early after the onset of symptoms. Times from onset to LP were 1.99 ± 1.82, 3.38 ± 1.16,7.66 ± 6.40, 13.25 ± 8.5 and 10 months for MM(V)1, VV2, MV2K, MM2C, and VV1 groups, respectively. Statistically significant differences were found between MM(V)1 and MV2K (P < 0.001) or VV2 (P < 0.001) groups and between MV2K and VV2 groups (P = 0.042). CSF t‐tau and NfL values for each group are shown in Table 1.
Table 1.
Demographic and biochemical data in the diagnostic groups.
| Diagnosis | CJD | FTLD | Controls |
|---|---|---|---|
| N | 77 | 30 | 31 |
| Age at LP (years ±SD) | 66.14 ± 7.78 | 63.19 ± 7.66 | 64.87 ± 9.85 |
| Female (%) | 48.1% | 53.3% | 47.2% |
| Time from onset to LP (months ± SD)1 | 4.50 ± 3.01 | 38.68 ± 35.00 | – |
| t‐tau (pg/mL) Median (IQR)2 | 4262 (2037–9173) | 307 (221–430) | 167 (136–227) |
| NfL (pg/mL) Median (IQR)3 | 8785 (4242–12225) | 4644 (3000–7075) | 601 (421–807) |
CJD versus FTLD P < 0.001.
CJD versus controls P < 0.001; CJD versus FTLD P < 0.001.
CJD versus controls P < 0.001; CJD versus FTLD P = 0.029; FTLD versus controls P < 0.001.
Distribution of CSF SerpinA1 isoforms across diagnostic groups
In all cases and independently from the disease group or subtype, we found at least six distinct SerpinA1 peaks with isoelectric points (pI) ranging from 4.3 to 4.7 (peaks 1–6). Moreover, we observed a seventh peak on the acidic side (peak 0) in several samples with CJD or FTLD pathology, as previously described for PDD6 (Fig. 1).
Figure 1.
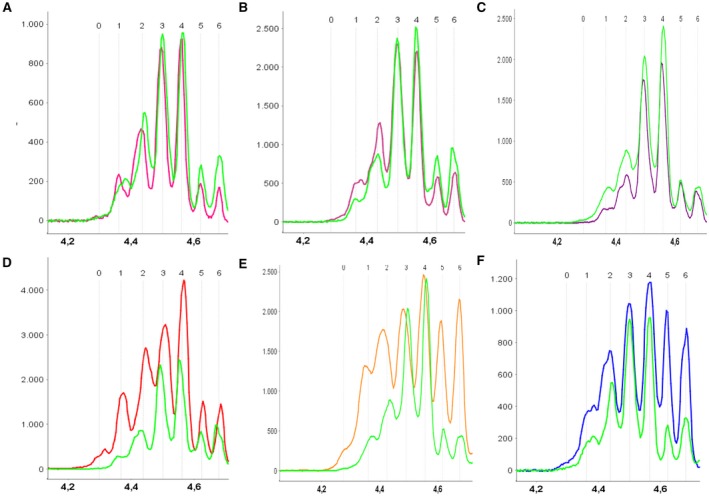
Typical CSF electropherograms of SerpinA1 isoforms. The y‐axis depicts the signal in arbitrary units; the x‐axis the isoelectric point (pI). (A) Overlay of a sCJD VV2 (pink) and a control (green) electropherogram. (B) Overlay of a sCJD MV2K (rose) and a control (green) electropherogram. (C) Overlay of a sCJD MM2C (purple) and a control (green) electropherogram. (D) Overlay of a sCJD MM(V)1 (red) and a control electropherogram (green). At variance with the VV2, MV2K, and MM2C profiles, the MM(V)1 electropherogram differs from the control one. Seven [MM(V)1] or six (control) distinct peaks around the pI of 4.5 were detected. Most notably is the difference in the abundance of the acidic isoforms. (E) Overlay of a sCJD VV1 (orange) and a control (green) electropherogram, showing, in the former, the same increase of the acidic isoforms detected in the MM(V)1 cases. (F) Overlay of a FTLD (blue) and a control (green) electropherogram. In the case of FTLD, the most significant difference is in the abundance of the two most basic isoforms. CJD, Creutzfeldt–Jakob disease; FTLD, frontotemporal lobar degeneration; MM(V)1, methionine homozygosity (valine) and disease‐associated prion protein type 1; MM2C, methionine homozygosity and disease‐associated prion protein type 2; MV2K, methionine–valine heterozygosity and disease‐associated prion protein type 2; sCJD, sporadic Creutzfeldt–Jakob disease; VV1, valine homozygosity and disease‐associated prion protein type 1; VV2, valine homozygosity and disease‐associated prion protein type 2.
For the evaluation of CIEF electropherograms, we calculated semiquantitatively the peak areas by analyzing the signal intensities for each SerpinA1 isoform individually and for total SerpinA1 as the sum of single peaks. We analyzed the following variables: (1) absolute area of total SerpinA1, calculated as the summed area of all peaks and (2) normalized area values, consisting of the ratio between the single peak area and the summed‐up area of all peaks as previously described.6
The overall SerpinA1 signal was significantly higher in FTLD cases compared to controls (P = 0.002), although no differences were detected between CJD and controls or FTLD (Fig. 2A). After stratification according to the three most representative sCJD molecular subtypes [MM(V)1, VV2, and MV2K], we detected a significant difference between controls and sCJD MM(V)1 (P = 0.026) but not between controls and the VV2 or MV2K groups (Fig. 2B). Similarly, CJD types linked to PrPSc type 1 [sCJD MM(V)1, VV1, and gCJD E200K‐129M] showed higher SerpinA1 total area compared to controls (P = 0.007) and cases associated with PrPSc type 2 (VV2, MV2K, and MM2C) (P = 0.014), whereas the values in the latter group were similar to those of the controls. Moreover, while the total area was significantly higher in the FTLD‐TDP group compared to controls (P = 0.005), the FTLD‐TAU group showed only a tendency toward an increased value (P = 0.02; adjusted post hoc P = 0.06), which is possibly explained by the small sample size (Fig. 2C).
Figure 2.

Results of absolute peak area analysis. (A) Comparison of the total absolute area of the three main cohorts (controls, CJD, FTLD). (B) Comparison between controls and sCJD patients after stratification into sCJD subtypes. (C) Comparison between controls and FTLD patients after stratification into the two FTLD subtypes. Box plots show median concentrations with 25% and 75% percentiles and Tukey whiskers. *P < 0.05, **P < 0.01. CJD, Creutzfeldt–Jakob disease; Con, controls; CU, chemiluminescence unit; FTLD, frontotemporal lobar degeneration; FTLD‐TAU, frontotemporal lobar degeneration with tau pathology; FTLD‐TDP, frontotemporal lobar degeneration with TDP43 pathology; MM(V)1, methionine homozygosity (valine) and disease‐associated prion protein type 1; MV2K, methionine/valine heterozygosity and disease‐associated prion protein type 2; sCJD, sporadic Creutzfeldt–Jakob disease; VV2, valine homozygosity and disease‐associated prion protein type 2.
In addition, the three diagnostic groups differed in the relative abundance of several charge isoforms (Fig. 3A). Specifically, in controls the most intense peak (peak 3) had a pI between 4.5 and 4.6 (controls vs. FTLD P = 0.010; controls vs. CJD P = 0.031), while the isoforms on its acidic and basic sides were less abundant. At variance, the CJD and FTLD groups were characterized by a relative increase of acidic (peak 0: P = 0.001; peak 1: P = 0.024) and basic isoforms (peak 6: P = 0.028) respectively in comparison with the controls (Fig. 3B–D). While the normalized areas of peak 4 and 5 were similar in the three diagnostic groups, the signals of peaks 2 (P = 0.029) and 6 (P = 0.007) differed between CJD and FTLD (Fig. 3D).
Figure 3.
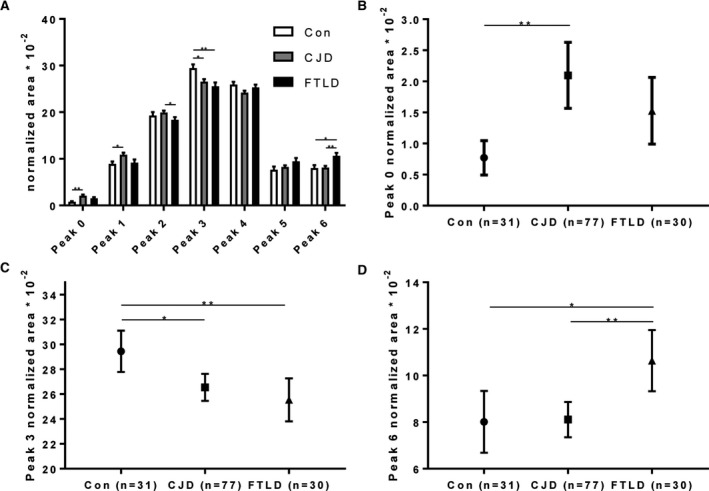
Results of relative peak area analysis. (A) Comparison of mean relative peak areas of all seven peaks of the three main cohorts (controls, CJD, FTLD). Error bars indicate SEM. (B) Normalized area results for peak 0. (C) Normalized area results for peak 3. (D) Normalized area results for peak 6. Symbols in B, C, and D indicate mean normalized area of peak 0, 3, and 6, respectively. Error bars show the 95% confidence interval. *P < 0.05, **P < 0.01. CJD, Creutzfeldt–Jakob disease; Cons, control; FTLD, frontotemporal lobar degeneration.
After stratification according to the CJD molecular subtype, we found that the differences between CJD and controls in single peak signals on the acidic side were driven by the MM(V)1 group (peak 0: P < 0.0001; peak 1: P = 0.006). Indeed, no differences in normalized areas were detected between VV2 and MV2K groups and controls (Fig. 4A). Similarly, electropherogram morphology in the few sCJD MM2C cases (Fig. 1C) resembled that of controls and VV2/MV2K cases (Fig. 1A and B), whereas in the single VV1 patient (Fig. 1E) looked similar to that of the MM(V)1 group (Fig. 1D). Indeed, the subanalysis based on PrPSc types confirmed that CJD cases linked to PrPSc type 1 showed different normalized areas of individual peaks on the acidic side compared to controls (peak 0: P < 0.001; peak 1: P = 0.002) and CJD linked to PrPSc type 2 (peak 0: P = 0.002; peak 1: P = 0.049). In addition, gCJD E200K‐129M and VV2 patients showed values of both total and normalized peak areas comparable, respectively, to those of the sCJD MM(V)1 and the MV2K groups. All the analyses after exclusion of probable sCJD cases confirmed the same findings (supplementary results of Appendix S1). In contrast, the relative increase of peak 6 in the FTLD group compared to controls was associated with the FTLD‐TAU group (P = 0.028) but not with FTLD‐TDP (Fig. 4B). Distinct FTLD genetic subgroups did not differ in both total area and single peak areas.
Figure 4.
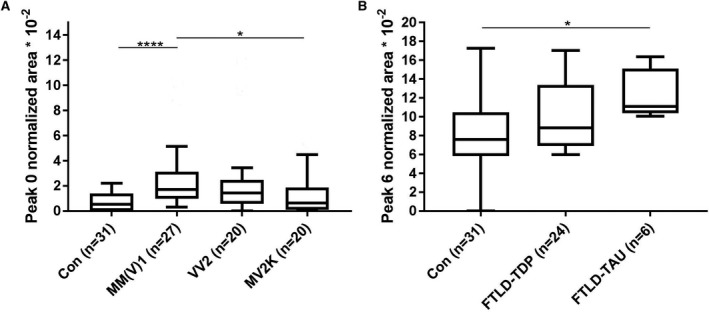
Relative peak area analysis for sCJD and FTLD subtypes. (A) Normalized peak area analysis of peak 0 for the sCJD subgroups. (B) Normalized peak area analysis of peak 6 for the FTLD subgroups. Box plots show median concentrations with 25% and 75% percentiles and Tukey whiskers. *P < 0.05, ****P < 0.0001. Con, controls; FTLD, frontotemporal lobar degeneration; FTLD‐TAU, frontotemporal lobar degeneration with tau pathology; FTLD‐TDP, frontotemporal lobar degeneration with TDP43 pathology; MM(V)1, methionine homozygosity (valine) and disease‐associated prion protein type 1; MV2K, methionine/valine heterozygosity and disease‐associated prion protein type 2; sCJD, sporadic Creutzfeldt–Jakob disease; VV2, valine homozygosity and disease‐associated prion protein type 2.
In both the CJD (even when limited to the single molecular subtypes) and the FTLD groups, there were no associations between age, time from onset to LP, t‐tau, NfL, and SerpinA1 total area. Further details regarding correlations between time from onset to LP and SerpinA1 variables in the CJD group are illustrated in supplementary results of Appendix S1.
Discussion
The results of the present study demonstrated that SerpinA1 is highly expressed in the CSF of patients with CJD linked to PrPSc type 1 and in a subgroup of those of the FTLD spectrum. Moreover, different isoform patterns of SerpinA1 characterize specific CJD and FTLD subtypes, as previously shown for AD, PDD, and PD.6, 7, 8, 9
The role of upregulated and post‐translationally modified SerpinA1 in neurodegeneration remains speculative. While protein abnormal post‐translational phosphorylation and glycosylation are recognized as pathogenetic mechanisms in neurodegenerative disorders,6 sialylation modifications have been less frequently reported, with SerpinA1 being one of the exceptions.9 On the one hand, SerpinA1 may play a protective role as a molecular chaperone that prevents amyloid fibril formation21 and its O‐linked sialylation could act as a suppressor of pathogenetic self‐hyperphosphorylation as sialylation and phosphorylation share similar motifs.6, 22 Alternatively, SerpinA1 post‐translational modifications might be detrimental by (1) blocking specific serine proteases and lead to impaired clearance of prion‐like proteins23 or (2) promoting the formation of a misfolded and self‐aggregate‐prone structure of SerpinA1.9, 21 On another issue, SerpinA1 is emerging as an important modulator of neuroinflammation.3, 4, 5
Independently from the hypothetical roles of SerpinA1, it seems that a different isoform‐overexpression characterizes some of the CJD and FTLD subtypes. On the other hand, the preservation of the most abundant peak (number 3) in controls could suggest a possible critical physiological role of this isoform.
Most interestingly, CJD cases linked to PrPSc type 1, such as sCJD MM(V)1, VV1, and gCJD E200K‐129M types, differed in total and single peak area from both controls and cases linked to PrPSc type 2 (VV2, MV2K, and MM2C groups). It is well established that the type of PrPSc largely determines the distinctive histopathological and molecular features of sCJD.24 In this respect, the different post‐translational modifications of SerpinA1 between sCJD subtypes may represent a PrPSc‐specific or a strain pathogenetic event. Accordingly, the lack of difference in SerpinA1 profile between sCJD MM(V)1 and gCJD E200K‐129M or between sCJD VV2 and sCJD MV2K are consistent with the notion that these couples of prion variants are, respectively, linked to the same PrPSc type and strain.25 Another possible hypothesis could relate the different SerpinA1 patterns to the kinetics of the disease even if we did not find significant associations between the time from onset to LP and SerpinA1 signal in each CJD molecular subgroup. Moreover, given that SerpinA1 might play an anti‐inflammatory role by attenuating microglial activity,3, 4, 5 the increase in total SerpinA1 and in some of its isoforms in sCJD MM(V)1 compared to sCJD linked to PrPSc type 2 could be considered a stronger early attempt to balance the developing neuroinflammation/neurodegeneration that progresses faster in the former group than in the latter. Possibly, SerpinA1 immunomodulation may attenuate during the slower disease course in VV2, MV2K, and MM2C cases. In this regard, a higher severity/amount of activated microglia has been described in the sCJD VV2 compared to the MM(V)1 subtype.26 Further studies which correlate SerpinA1 levels with the microglial load in CJD brain tissues and the kinetics of the pathology could clarify the role of SerpinA1 in the pathogenesis of prion disease.
On another issue, the difference in SerpinA1 between PrPSc type 1 and 2 CJD cases could be helpful to distinguish in vivo PrPSc types. However, the partial overlap in SerpinA1 levels and electropherogram profiles and the limited diagnostic accuracy (data not shown) represent a limit to its application at the individual level.
Similarly, in FTLD the highly expressed isoform corresponding to peak 6 seems to be a characteristic of the TAU group, even if this finding needs to be validated in a larger cohort. Thus, we speculate that the wide pathological heterogeneity in both CJD and FTLD could reflect a different expression of SerpinA1 isoforms. Accordingly, all the mentioned isoform patterns were different from those previously described by us in PD and PDD.6
On another issue, recent studies showed that SerpinA3, another member of the serpin family, is significantly upregulated in brains and CSF of patients with prion disease, both at the mRNA and at the protein level, although to a different extent for each pathological subtype.23, 27, 28 However, all these studies focused on the levels of total protein but did not evaluate isoforms with a very sensitive assay such as CIEF.
The major strength of our study is the inclusion of disease cases with certainty or high level of probability in predicting the underlying neuropathology (e.g., autopsy‐proven, genetic, and RT‐QuIC‐positive cases). In the present study, we did not investigate SerpinA1 isoforms with CIEF in brain tissue of our autopsy‐verified cases, due to the potential infective load of sCJD samples. As another limitation, we included only a few sporadic cases of FTLD, and so our findings might need confirmation in other cohorts of pathologically proven sporadic FTLD. Finally, the effect of preanalytical variables (e.g., tube material) deserves further investigations, if quantitative assays for the measurement of SerpinA1 should be developed in the future.
Taking our findings and previous data together, we speculate that upregulation and post‐translational modifications of SerpinA1, and to a large extent of all serpin proteins, could be common features of most neurodegenerative disorders. Nevertheless, further studies on experimental models and in other neurodegenerative disorders (e.g., amyotrophic lateral sclerosis, Huntington’s disease) are needed to clarify the specific functions of SerpinA1 and other members of the serpin family in neurodegeneration. In detail, studies in mouse and cell models in which SerpinA1 expression might be up‐ or downregulated will be helpful to investigate whether SerpinA1 could drive or mitigate the pathological hallmarks through the modulation of neuroinflammation in distinct CJD and FTLD subtypes, or whether its dysregulation is only one of several downstream effects of the underlying cause.
Author Contributions
Conception and design of the study (SAR, SH, PP, and MO), acquisition and analysis of the data (SAR, SH, PS, SAS, BP, ACL, SC, PP, and MO), and drafting the manuscript and figures (SAR, SH, PP, and MO). Study supervision (MO and PP).
Conflict of Interest
Nothing to report.
Supporting information
Appendix S1. Supplementary methods: Inclusion criteria and case classification for probable sporadic CJD cases. Supplementary results: Distribution of CSF SerpinA1 isoforms across diagnostic groups. Supplementary references.
Acknowledgments
The authors wish to thank Silvia Piras, B.Sc., and Benedetta Carlà, M.Sc., for their valuable technical assistance. This work was supported by grants from the German Federal Ministry of Education and Research (project: FTLDc 01GI1007A), the EU Joint Programme‐Neurodegenerative Disease Research (JPND) network, PreFrontAls (01ED1512), the foundation of the state of Baden‐Württemberg (D.3830), the Thierry Latran foundation, BIU (D.5009), and the Italian Ministry of Health (“Ricerca corrente”).
Funding Information
This work was supported by grants from the German Federal Ministry of Education and Research (project: FTLDc 01GI1007A), the EU Joint Programme‐Neurodegenerative Disease Research (JPND) network, PreFrontAls (01ED1512), the foundation of the state of Baden‐Württemberg (D.3830), the Thierry Latran foundation, BIU (D.5009), and the Italian Ministry of Health (“Ricerca corrente”).
Funding Statement
This work was funded by EU Joint Programme ‐ Neurodegenerative Disease Research (JPND) network grant ; PreFrontAls grant 01ED1512; the foundation of the state of Baden‐Württemberg grant D.3830; the Thierry Latran foundation grant ; German Federal Ministry of Education and Research grant FTLDc 01GI1007A; BIU grant D.5009; Italian Ministry of Health grant Ricerca corrente.
Contributor Information
Piero Parchi, Email: piero.parchi@unibo.it.
Markus Otto, Email: markus.otto@uni-ulm.de.
References
- 1. Ebbert MTW, Ross CA, Pregent LJ, et al. Conserved DNA methylation combined with differential frontal cortex and cerebellar expression distinguishes C9orf72‐associated and sporadic ALS, and implicates SERPINA1 in disease. Acta Neuropathol 2017;134:715–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Gollin PA, Kalaria RN, Eikelenboom P, et al. Alpha 1‐antitrypsin and alpha 1‐antichymotrypsin are in the lesions of Alzheimer's disease. Neuroreport 1992;3:201–203. [DOI] [PubMed] [Google Scholar]
- 3. Gold M, Dolga AM, Koepke J, et al. α1‐antitrypsin modulates microglial‐mediated neuroinflammation and protects microglial cells from amyloid‐β‐induced toxicity. J Neuroinflammation 2014;11:165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Yang S, Xian B, Li K, et al. Alpha 1‐antitrypsin inhibits microglia activation and facilitates the survival of iPSC grafts in hypertension mouse model. Cell Immunol 2018;328:49–57. [DOI] [PubMed] [Google Scholar]
- 5. Zhou T, Huang Z, Zhu X, et al. Alpha‐1 antitrypsin attenuates M1 microglia‐mediated neuroinflammation in retinal degeneration. Front Immunol 2018;9:1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Halbgebauer S, Nagl M, Klafki H, et al. Modified serpinA1 as risk marker for Parkinson's disease dementia: analysis of baseline data. Sci Rep 2016;6:26145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Puchades M, Hansson SF, Nilsson CL, et al. Proteomic studies of potential cerebrospinal fluid protein markers for Alzheimer's disease. Mol Brain Res 2003;118:140–146. [DOI] [PubMed] [Google Scholar]
- 8. Nielsen HM, Minthon L, Londos E, et al. Plasma and CSF serpins in Alzheimer disease and dementia with Lewy bodies. Neurology. 2007;69(16):1569-1579. [DOI] [PubMed] [Google Scholar]
- 9. Jesse S, Lehnert S, Jahn O, et al. Differential sialylation of serpin A1 in the early diagnosis of Parkinson's disease dementia. PLoS One 2012;7:e48783 10.1371/journal.pone.0048783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Parchi P, Giese A, Capellari S, et al. Classification of sporadic Creutzfeldt‐Jakob disease based on molecular and phenotypic analysis of 300 subjects. Ann Neurol 1999;46:224–233. [PubMed] [Google Scholar]
- 11. Mann DMA, Snowden JS. Frontotemporal lobar degeneration: pathogenesis, pathology and pathways to phenotype. Brain Pathol 2017;27:723–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Parchi P, de Boni L, Saverioni D, et al. Consensus classification of human prion disease histotypes allows reliable identification of molecular subtypes: an inter‐rater study among surveillance centres in Europe and USA. Acta Neuropathol 2012;124:517–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lattanzio F, Abu‐Rumeileh S, Franceschini A, et al. Prion‐specific and surrogate CSF biomarkers in Creutzfeldt‐Jakob disease: diagnostic accuracy in relation to molecular subtypes and analysis of neuropathological correlates of p‐tau and Aβ42 levels. Acta Neuropathol 2017;133:559–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Parchi P, Saverioni D. Molecular pathology, classification, and diagnosis of sporadic human prion disease variants. Folia Neuropathol 2012;50:20–45. [PubMed] [Google Scholar]
- 15. Rossi M, Kai H, Baiardi S, et al. The characterization of AD/PART co‐pathology in CJD suggests independent pathogenic mechanisms and no cross‐seeding between misfolded Aβ and prion proteins. Acta Neuropathol Commun 2019;7:53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Montine TJ, Phelps CH, Beach TG, et al. National Institute on Aging‐Alzheimer's association guidelines for the neuropathologic assessment of Alzheimer's disease: a practical approach. Acta Neuropathol 2012;123:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Abu‐Rumeileh S, Mometto N, Bartoletti‐Stella A, et al. Cerebrospinal fluid biomarkers in patients with frontotemporal dementia spectrum: a single‐center study. J Alzheimers Dis 2018;66:551–563. [DOI] [PubMed] [Google Scholar]
- 18. Lleó A, Irwin DJ, Illán‐Gala I, et al. A 2‐step cerebrospinal algorithm for the selection of frontotemporal lobar degeneration subtypes. JAMA Neurol 2018;75:738–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Jack CR Jr, Bennett DA, Blennow K, et al. NIA‐AA Research Framework: toward a biological definition of Alzheimer's disease. Alzheimers Dement 2018;14:535–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Abu‐Rumeileh S, Giannini G, Polischi B, et al. Revisiting the cerebrospinal fluid biomarker profile in idiopathic normal pressure hydrocephalus: the Bologna Pro‐Hydro Study. J Alzheimers Dis 2019;68:723–733. [DOI] [PubMed] [Google Scholar]
- 21. Zsila F. Inhibition of heat‐ and chemical‐induced aggregation of various proteins reveals chaperone‐like activity of the acute‐phase component and serine protease inhibitor human alpha(1)‐antitrypsin. Biochem Biophys Res Commun 2010;393:242–247. [DOI] [PubMed] [Google Scholar]
- 22. Yen HY, Liu YC, Chen NY, et al. Effect of sialylation on EGFR phosphorylation and resistance to tyrosine kinase inhibition. Proc Natl Acad Sci USA 2015;112:6955–6960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Vanni S, Moda F, Zattoni M, et al. Differential overexpression of SERPINA3 in human prion diseases. Sci Rep 2017;7:15637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Baiardi S, Rossi M, Capellari S, Parchi P. Recent advances in the histo‐molecular pathology of human prion disease. Brain Pathol 2019;29:278–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Rossi M, Baiardi S, Parchi P. Understanding prion strains: evidence from studies of the disease forms affecting humans. Viruses 2019;11:309 10.3390/v11040309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Franceschini A, Strammiello R, Capellari S, et al. Regional pattern of microgliosis in sporadic Creutzfeldt‐Jakob disease in relation to phenotypic variants and disease progression. Neuropathol Appl Neurobiol 2018;44:574–589. [DOI] [PubMed] [Google Scholar]
- 27. Miele G, Seeger H, Marino D, et al. Urinary alpha1‐antichymotrypsin: a biomarker of prion infection. PLoS One 2008;3(12):e3870 10.1371/journal.pone.0003870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bartoletti‐Stella A, Corrado P, et al. Analysis of RNA expression profiles identifies dysregulated vesicle trafficking pathways in Creutzfeldt‐Jakob disease. Mol Neurobiol 2019;56(7):5009–5024. 10.1007/s12035-018-1421-1 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1. Supplementary methods: Inclusion criteria and case classification for probable sporadic CJD cases. Supplementary results: Distribution of CSF SerpinA1 isoforms across diagnostic groups. Supplementary references.