

Abstract
Objective
Duchenne muscular dystrophy (DMD) is a progressive muscular disease characterized by chronic cycles of inflammatory and necrotic processes. Prostaglandin D2 (PGD2) is produced by hematopoietic PGD synthase (HPGDS), which is pathologically implicated in muscle necrosis. This randomized, double‐blind, placebo‐controlled early phase 2 study (NCT02752048) aimed to assess the efficacy and safety of the novel selective HPGDS inhibitor, TAS‐205, with exploratory measures in male DMD patients aged ≥5 years.
Methods
Patients were randomized 1:1:1 to receive low‐dose TAS‐205 (6.67–13.33 mg/kg/dose), high‐dose TAS‐205 (13.33–26.67 mg/kg/dose), or placebo. The primary endpoint was the change from baseline in a 6‐minute walk distance (6MWD) at Week 24.
Results
Thirty‐six patients were enrolled, of whom 35 patients were analysed for safety. The mean (standard error) changes from baseline to Week 24 in 6MWD were −17.0 (17.6) m in the placebo group (n = 10), −3.5 (20.3) m in the TAS‐205 low‐dose group (n = 11), and −7.5 (11.2) m in the TAS‐205 high‐dose group (n = 11). The mean (95% confidence interval) difference from the placebo group was 13.5 (−43.3 to 70.2) m in the TAS‐205 low‐dose group and 9.5 (−33.3 to 52.4) m in the TAS‐205 high‐dose group. No obvious differences were observed in the incidences of adverse events between treatment groups. No adverse drug reactions specific to TAS‐205 treatment were observed.
Interpretation
The HPGDS inhibitor TAS‐205 showed a favorable safety profile in DMD patients. Further research is required to examine the effectiveness of TAS‐205 in a larger trial.
Introduction
Duchenne muscular dystrophy (DMD) is a rare and severe X‐linked muscular disease characterized by progressive muscle weakening caused by a lack of dystrophin protein due to a mutation in the dystrophin gene.1 As the patient ages, the pathology of DMD is characterized by aggravated inflammatory and immune responses.2 However, the precise mechanism for DMD progression is not yet fully understood.
The DMD care consideration guidelines have been updated due to recent advances in the diagnosis of DMD and the emergence of novel treatments, including genetic and molecular therapies.3, 4, 5, 6, 7, 8, 9, 10 Currently, DMD is treated with orally administered steroids, which suppress the infiltration of inflammatory cells into the muscle.11, 12 However, there are several problems with steroidal treatments, particularly with their safety profile in paediatrics.
Hematopoietic prostaglandin D synthase (HPGDS) is the enzyme that catalyses the production of prostaglandin D2 (PGD2), an inflammatory mediator.13 The overproduction of PGD2 by HPGDS is implicated in muscle necrosis and has been shown to aggravate inflammation and exacerbate muscle tissue damage.14, 15, 16 Clinical research on PGD2 activity shows that HPGDS is expressed in myonecrotic areas in DMD patients and that PGD2‐mediated inflammation is associated with the development of muscle necrosis over time.17 In a DMD mouse model, a PGD2 inhibitor decreased the excretion of the PGD2 urinary metabolite, tetranor‐PGDM (tPGDM), and inhibited myonecrosis.16 Urinary tPGDM is the primary metabolite of PGD2 in both mice and humans, and therefore, can be used as a marker of PGD2 production in vivo.18 It has been reported that the urinary excretion of tPGDM increased in DMD patients compared with age‐matched healthy subjects or children with other diseases.17, 19 Therefore, PGD2‐mediated inflammation is suggested to be involved in the pathology of DMD, and inhibition of PGD2 production via HPGDS inhibition may be an effective therapeutic modality.
TAS‐205 was initially shown to be a highly selective HPGDS inhibitor, which improved locomotor activity and reduced the area of necrotic muscle fibres produced over time in dystrophin‐deficient mice.20 In a phase 1 study, we evaluated the safety, tolerability, and pharmacokinetic profile of TAS‐205 after administration of single and repeated doses for 7 days in DMD patients.21 In that study, there were dose‐dependent reductions in the urinary excretion of tPGDM after the administration of TAS‐205. Therefore, we planned this 24‐week early phase 2 study to examine the efficacy, safety, and pharmacodynamics of TAS‐205 with exploratory measures in patients with DMD.
Methods
Study design
This study was an exploratory, randomized, double‐blind, placebo‐controlled, early phase 2 trial conducted from February 2016 to August 2017 across 11 sites in Japan. The study protocol was reviewed and approved by the independent institutional review board for each center and conducted in accordance with Good Clinical Practice guidelines, as well as applicable regulatory requirements in Japan. This study also adhered to the ethical principles of the Declaration of Helsinki. This trial was registered with ClinicalTrials.gov, under the identification number NCT02752048.
Informed consent was obtained from the legal representative of the patient, and all attempts were made to obtain informed assent from the patient, within 14 days before enrolment. This was followed by eligibility confirmation. Patients were randomized 1:1:1 to the TAS‐205 low‐dose group (6·67–13·33 mg/kg/dose), the TAS‐205 high‐dose group (13·33 to 26·67 mg/kg/dose), or the placebo group via an Interactive Web Response System using the dynamic randomization method. Adjustment factors for stratification included age (5 to <7 years, or ≥7 years) and prior use of steroids (yes/no). The dosage of the study drug by body weight is described in Data S1. All study and sponsor personnel and caregivers were blinded during the course of the study.
Patients
Male patients were enrolled according to the following major inclusion criteria: age ≥5 years at the time of consent; a bodyweight of ≥7.5 kg and <60 kg within 14 days before enrollment; diagnosis of dystrophinopathy as determined by a dystrophin genetic test or muscle pathology findings; symptoms or signs characteristic of DMD (proximal muscular weakness, waddling gait, and/or Gowers’ sign), and progressive walking difficulty; ability to rise from the floor, or a chair; ability to walk by themselves; and a 6‐minute walk distance (6MWD) of ≥75 m. The major exclusion criteria are provided in Data S1.
Outcomes
The primary endpoint was the change in motor function as measured by the 6MWD test from baseline to Week 24. The 6MWD assessment was measured at baseline, Week 12, and Week 24.
Secondary efficacy endpoints included the change from baseline in time in the rise from the floor test, timed up and go test (described in Data S1), and 10‐m walk/run test. The evaluation of motor function was performed in a single day in the following order: 6MWD test, rise from the floor test, timed up and go test, and 10‐m walk/run test. For these procedures, the order of execution and intervals between evaluations were defined in the procedure manual. These procedures were performed at each center by a certified evaluator.
We also assessed muscle volume in the middle part of the thigh (right and left) and the lower leg (right and left) using the muscle volume index (MVI),22 and the change from baseline in the percentage of MVI (%MVI) was included in the secondary efficacy endpoints. MVI is the estimated muscle volume evaluated using skeletal muscle computed tomography (CT).22, 23 The assessment of skeletal muscle volume using CT are provided in Data S1.
We also evaluated the effect of TAS‐205 on the urinary excretion of tPGDM and tetranor‐prostaglandin E2 metabolite (tPGEM) in pooled urine samples at baseline, Day 4, Week 12, and Week 24 using a previously reported method.21 Pharmacodynamic endpoints were percent changes from baseline in urinary tPGDM/creatinine (Cre) concentration ratio, total urinary tPGDM excretion, urinary tPGEM/Cre concentration ratio, and total urinary tPGEM excretion.
The safety endpoints included analyses of cardiac ultrasonography, 12‐lead electrocardiography, vital signs (blood pressure, pulse rate, and body temperature), adverse events (AEs), and adverse drug reactions (ADRs).
Statistical analysis
Sample size was based on patient recruitment feasibility as DMD is a rare disease. This study was an early phase 2 trial that examined efficacy with exploratory measures without defining significance level and statistical power for comparison. The target sample size was 33 patients (11 patients per group). The safety analysis was performed on all patients who received the study drug at least once in each treatment group (safety population). The efficacy analyses were performed on all treated patients who were assessed for the primary endpoint according to the protocol (efficacy population).
The summary statistics of the change in 6MWD from baseline to Week 24 were calculated and assessed as the primary analysis. To estimate the effect of TAS‐205 treatment in an exploratory manner, a two‐sample t‐test was performed between each treatment group and the placebo group. Moreover, a mixed‐effect model for repeated measures (MMRM) that included treatment, visit, baseline, and the interaction of treatment and visit as explanatory variables was performed. For the secondary efficacy endpoints and pharmacodynamics endpoints, summary statistics of the change from baseline to Week 24 were calculated and a two‐sample t‐test was performed between each treatment group and the placebo group.
Statistical software (SAS version 9.4) was used to conduct these analyses.
Results
Thirty‐six patients were enrolled, of whom 35 patients received the study drug at least once and were analysed for safety (safety population) (Fig. 1). One patient in the placebo group was untreated because the patient or guardian withdrew consent. Thirty‐two patients were assessed for the primary endpoint (efficacy population). Characteristics of patients included in the efficacy population (Table 1) and randomized to each of the three groups were comparable at baseline except for a slight imbalance in motor function measurements.
Figure 1.
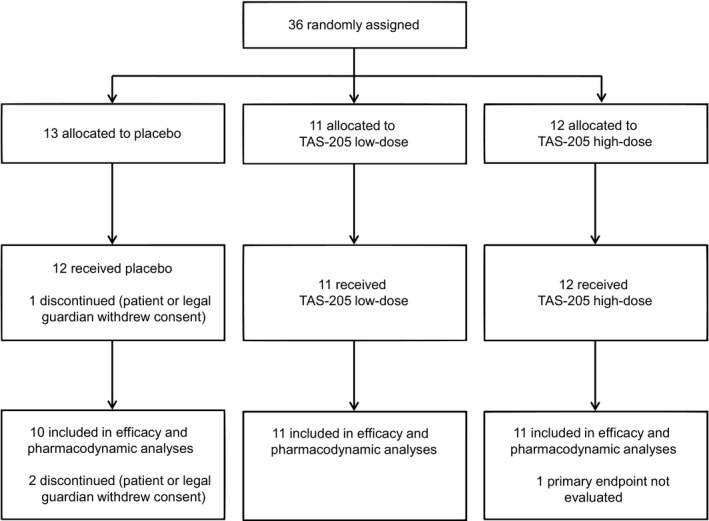
Patient disposition.
Table 1.
Patient characteristics (efficacy population).
| Placebo | TAS‐205 low‐dose | TAS‐205 high‐dose | |
|---|---|---|---|
| (n = 10) | (n = 11) | (n = 11) | |
| Age (years) | 8.4 (1.6) | 7.9 (2.0) | 8.3 (2.9) |
| Age category (years) | |||
| ≥5 to < 7 | 1 (10%) | 2 (18%) | 2 (18%) |
| ≥7 | 9 (90%) | 9 (82%) | 9 (82%) |
| Height (cm) | 122.0 (8.3) | 117.6 (8.1) | 119.5 (9.5) |
| Weight (kg) | 24.9 (6.4) | 25.0 (5.8) | 25.1 (8.6) |
| Prior steroid therapy | |||
| No | 1 (10%) | 2 (18%) | 3 (27%) |
| Yes | 9 (90%) | 9 (82%) | 8 (73%) |
| Baseline 6MWD | |||
| ≥350 | 7 (70%) | 4 (36%) | 8 (73%) |
| <350 | 3 (30%) | 7 (64%) | 3 (27%) |
| Motor functional characteristics | |||
| 6MWD (m) | 375.9 (72.8) | 327.6 (54.4) | 377.8 (93.2) |
| Rise from the floor test (s) | 7.07 (4.28) | 8.07 (3.52) | 5.56 (3.75) |
| Timed up and go test (s) | 9.97 (5.05) | 9.65 (1.78) | 8.69 (2.36) |
| 10‐m walk/run test (s) | 6.39 (2.15) | 6.55 (1.56) | 5.71 (1.95) |
Data are mean (SD) or number (%).
6MWD, 6‐minute walk distance; SD, standard deviation.
The mean (standard deviation [SD]) study drug administration rate was 93.1 (18.4) % in the placebo group, 96.8 (5.1) % in the TAS‐205 low‐dose group, and 95.5 (8.1) % in the TAS‐205 high‐dose group. The incidence of concomitant steroid use for the treatment of DMD during this study was 83.3% (10/12 patients) in the placebo group, 81.8% (9/11 patients) in the TAS‐205 low‐dose group, and 75.0% (9/12 patients) in the TAS‐205 high‐dose group. Therefore, there were no observable imbalances in the study drug administration rate or concomitant steroid use between groups during this study.
By analysing the change in 6MWD from baseline to Week 24 for each treatment group, we observed that the mean (standard error [SE]) changes at Week 24 were −17.0 (17.6) m in the placebo group, −3.5 (20.3) m in the TAS‐205 low‐dose group, and −7.5 (11.2) m in the TAS‐205 high‐dose group (Fig. 2). The mean difference from the placebo group (95% confidence interval [CI], P value by 2‐sample t‐test) was 13.5 m (−43.3 to 70.2 m, P = 0.625) in the TAS‐205 low‐dose group and 9.5 m (−33.3 to 52.4 m, P = 0.646) in the TAS‐205 high‐dose group. The estimated changes in 6MWD from baseline at Week 24 using MMRM are described in Data S2. Overall, the decline in 6MWD tended to be suppressed in both TAS‐205 treatment groups compared with the placebo group.
Figure 2.
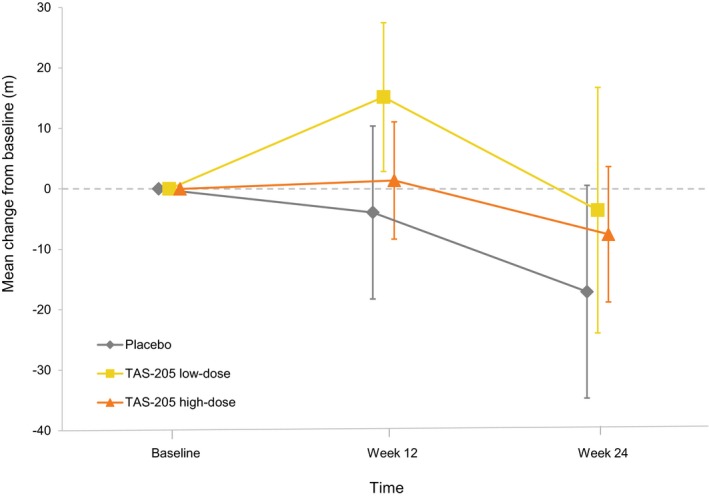
Change in 6‐minute walk distance from baseline to Week 24. Bars indicate standard error.
We analysed the changes in the timed motor function endpoints from baseline at Week 24, including the times measured in the rise from the floor test, the timed up and go test, and the 10‐m walk/run test (Table 2). The time required in all three motor function tests was increased in both TAS‐205 treatment groups in comparison with the placebo group, except for the rise from the floor test in the TAS‐205 high‐dose group.
Table 2.
Change from baseline in timed motor function endpoints at Week 24.
| Placebo | TAS‐205 low‐dose | TAS‐205 high‐dose | |
|---|---|---|---|
| Rise from floor (s) | |||
| Number of patients | 8 | 11 | 9 |
| Mean change (SD) | 2.63 (4.25) | 4.01 (6.21) | 1.28 (2.55) |
| Difference with placebo | |||
| Mean (95% CI) | − | 1.38 (−4.00 to 6.76) | −1.35 (−4.92 to 2.22) |
| P value* | − | 0.596 | 0.433 |
| Timed up and go (s) | |||
| Number of patients | 9 | 11 | 10 |
| Mean change (SD) | 0.06 (1.63) | 0.59 (2.16) | 0.29 (1.63) |
| Difference with placebo | |||
| Mean (95% CI) | − | 0.53 (−1.30 to 2.36) | 0.23 (−1.35 to 1.80) |
| P value* | − | 0.549 | 0.767 |
| 10‐m walk/run (s) | |||
| Number of patients | 9 | 11 | 11 |
| Mean change (SD) | 0.63 (1.27) | 1.11 (1.14) | 1.03 (1.30) |
| Difference with placebo | |||
| Mean (95% CI) | − | 0.48 (−0.65 to 1.62) | 0.40 (−0.82 to 1.61) |
| P value* | − | 0.382 | 0.501 |
SD, standard deviation; CI, confidence interval.
2‐sample t‐test.
The changes in %MVI from baseline at Week 24, as measured by skeletal muscle CT, in the right thigh, left thigh, right lower leg, and left lower leg were also analyzed (Table 3). The %MVI in the right thigh and left thigh was reduced in the TAS‐205 low‐dose group in comparison with the placebo group, while a reduction in %MVI was suppressed in the TAS‐205 high‐dose group. However, a reduction in %MVI in the right lower leg and left lower leg was suppressed in both TAS‐205 treatment groups in comparison with the placebo group.
Table 3.
Change from baseline in %MVI by skeletal muscle CT at Week 24.
| Placebo | TAS‐205 low‐dose | TAS‐205 high‐dose | |
|---|---|---|---|
| %MVI (%) in the right thigh | |||
| Number of patients | 9 | 10 | 11 |
| Mean change (SD) | −4.12 (2.43) | −4.23 (3.02) | −3.17 (2.93) |
| Difference with placebo | |||
| Mean (95% CI) | − | −0.11 (−2.78 to 2.56) | 0.95 (−1.62 to 3.52) |
| P value* | − | 0.933 | 0.447 |
| %MVI (%) in the left thigh | |||
| Number of patients | 9 | 10 | 11 |
| Mean change (SD) | −3.89 (2.55) | −4.28 (2.97) | −2.96 (3.01) |
| Difference with placebo | |||
| Mean (95% CI) | − | −0.39 (−3.09 to 2.31) | 0.93 (−1.74 to 3.59) |
| P value* | − | 0.763 | 0.474 |
| %MVI (%) in the right lower leg | |||
| Number of patients | 9 | 10 | 11 |
| Mean change (SD) | −3.69 (2.96) | −2.25 (3.10) | −1.21 (2.16) |
| Difference with placebo | |||
| Mean (95% CI) | − | 1.44 (−1.51 to 4.38) | 2.48 (0.07 to 4.89) |
| P value* | − | 0.317 | 0.044 |
| %MVI (%) in the left lower leg | |||
| Number of patients | 9 | 10 | 11 |
| Mean change (SD) | −3.30 (2.22) | −1.01 (3.13) | −2.65 (2.62) |
| Difference with placebo | |||
| Mean (95% CI) | − | 2.29 (−0.37 to 4.95) | 0.65 (−1.66 to 2.97) |
| P value* | − | 0.087 | 0.559 |
%MVI, percentage of muscle volume index; CT, computed tomography; SD, standard deviation; CI, confidence interval.
2‐sample t‐test.
Regarding the percent changes in the pharmacodynamic endpoints from baseline to Week 24 (Fig. 3A–D), the difference in the mean urinary tPGDM/Cre concentration ratio (95% CI, p value by 2‐sample t‐test) compared with the placebo group at Week 24 was −16.5% (−41.4 to 8.4%, P = 0.182) in the TAS‐205 low‐dose group and −25.5% (−50.6 to 0.4%, P = 0.047) in the TAS‐205 high‐dose group (Fig. 3A). Additionally, the difference in the mean total urinary excretion of tPGDM in the TAS‐205 treatment groups compared with placebo at Week 24 was −31.2% (−67.3 to 4.8%, P = 0.086) in the TAS‐205 low‐dose group and −25.7% (−72.8 to 21.3%, P = 0.266) in the TAS‐205 high‐dose group (Fig. 3B). For the urinary tPGEM/Cre concentration ratio, the difference in the mean value compared with the placebo group at Week 24 was 27.7% (−47.5 to 103.0%, P = 0.450) in the TAS‐205 low‐dose group and − 17.9% (−64.6 to 28.9%, P = 0.434) in the TAS‐205 high‐dose group (Fig. 3C). For the total urinary excretion of tPGEM, the difference in the mean value compared with the placebo group at Week 24 was −17.3% (−62.9 to 28.3%, P = 0.437) in the TAS‐205 low‐dose group and −29.6% (−80.1 to 21.0%, P = 0.236) in the TAS‐205 high‐dose group (Fig. 3D). The urinary tPGDM/Cre concentration ratio and the total urinary excretion of tPGDM tended to be reduced in both TAS‐205 treatment groups as compared with the placebo group. The total urinary excretion of tPGEM also tended to be reduced in both TAS‐205 treatment groups when compared with the placebo group, however there was no consistency observed in the urinary tPGEM/Cre concentration ratios.
Figure 3.
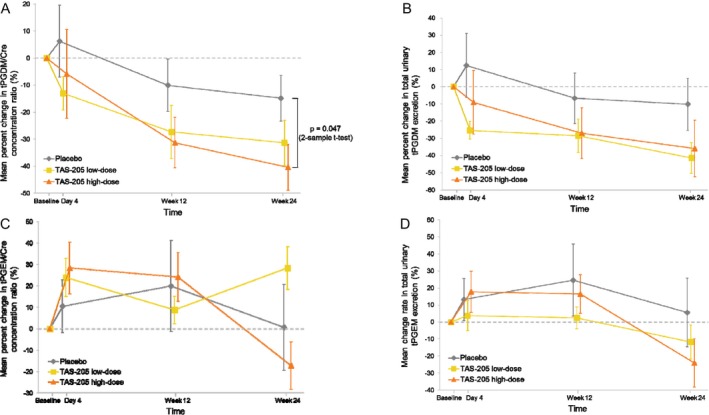
Percent change in pharmacodynamic endpoints from baseline to Week 24; (A) urinary tPGDM/Cre concentration ratio, (B) total urinary tPGDM excretion, (C) urinary tPGEM/Cre concentration ratio and (D) total urinary tPGEM excretion. Bars indicate standard error. Abbreviations: tPGDM, tetranor prostaglandin D2 metabolite; Cre, creatinine; tPGEM, tetranor prostaglandin E2 metabolite.
The incidences of AEs in both TAS‐205 treatment groups were similar to the incidence of AEs in the placebo group (Table 4). All AEs were either mild or moderate except for one serious AE (asthma) reported in the TAS‐205 high‐dose group, which was judged to be unrelated to TAS‐205 treatment. No ADR occurred in the placebo group and no differences were observed in the incidence of ADRs between both TAS‐205 treatment groups (Data S2). All observed ADRs were either mild or moderate in severity. There were no clinically significant changes in vital signs, cardiac ultrasonography, or 12‐lead electrocardiogram in both TAS‐205 groups (data not shown).
Table 4.
Summary of adverse events (safety population).
| Placebo | TAS‐205 low‐dose | TAS‐205 high‐dose | |
|---|---|---|---|
| (n = 12) | (n = 11) | (n = 12) | |
| Any adverse event | 10 (83.3) | 9 (81.8) | 9 (75.0) |
| Infections and infestations | |||
| Bronchitis | 2 (16.7) | 1 (9.1) | 1 (8.3) |
| Gastroenteritis | 0 (0.0) | 2 (18.2) | 3 (25.0) |
| Influenza | 2 (16.7) | 0 (0.0) | 0 (0.0) |
| Upper respiratory tract infection | 1 (8.3) | 1 (9.1) | 2 (16.7) |
| Viral upper respiratory tract infection | 2 (16.7) | 0 (0.0) | 1 (8.3) |
| Injury, poisoning, and procedural complications | |||
| Chilblains | 2 (16.7) | 0 (0.0) | 0 (0.0) |
| Investigations | |||
| Urobilinogen urine increased | 0 (0.0) | 0 (0.0) | 2 (16.7) |
| Musculoskeletal and connective tissue disorders | |||
| Arthralgia | 2 (16.7) | 1 (9.1) | 1 (8.3) |
| Pain in extremity | 2 (16.7) | 2 (18.2) | 1 (8.3) |
| Respiratory, thoracic, and mediastinal disorders | |||
| Upper respiratory tract inflammation | 0 (0.0) | 2 (18.2) | 3 (25.0) |
| Skin and subcutaneous tissue disorders | |||
| Urticaria | 0 (0.0) | 2 (18.2) | 1 (8.3) |
Data are number (%).
Discussion
This is the first study to assess the clinical efficacy of the novel selective HPGDS inhibitor, TAS‐205, in patients with DMD. In this study, the primary efficacy endpoint was the change in 6MWD from baseline to Week 24. Previous studies have concluded that the 6MWD test is safe and effective and can detect clinically meaningful changes in ambulatory function.24, 25 In a phase 3 study of ataluren, which is the dystrophin gene‐targeted drug and has been approved in the EU26 as a treatment option for DMD, the least‐squares mean (95% CI) change for ataluren versus placebo in 6MWD from baseline to Week 48 was reported 13·0 (−7·4 to 33·4) m in the intention‐to‐treat population and 42·9 (11·8–74·0) m in the group with a baseline 6MWD of 300 m or more to less than 400 m.6 In a small phase 2 study of eteplirsen, which is another dystrophin gene‐targeted drug and has been approved in the United States,27 the adjusted mean (SE) change using MMRM in 6MWD from baseline to Week 24 was reported −25.8 (30.6) m for the placebo cohort and −0.3 (31.2) m for the high‐dose eteplirsen cohort.10 In our study, the difference from placebo for TAS‐205 in 6MWD was approximately 10 m at Week 24. Although TAS‐205 does not target dystrophin directly, it is possible that by inhibiting inflammation, it achieved the same anti‐inflammatory level as drugs that restore dystrophin expression. To assess the benefits of long‐term dosing of TAS‐205 on disease progression, additional analyses in larger studies are needed.
With regard to the timed motor function tests for each patient’s functional mobility, there was no notably favorable tendency across the three tests. In this study, the timed motor function tests were performed after the 6MWD test and because these tests were effort‐dependent, the order of tests may have contributed to the observed increased interpatient variability. Additionally, evaluation of the efficacy of TAS‐205 was not achieved for some efficacy endpoints because the changes from baseline at Week 24 were too small in the placebo group.
Evaluating muscle volume as a marker of muscle dystrophy is clinically important as it has previously been shown that reductions in muscle volume in the thigh and lower leg correlate with age in patients with DMD.28 In our study, the MVI in the thigh and the lower leg was measured using CT images.22, 23 Magnetic resonance imaging (MRI) is generally preferable to CT imaging for the evaluation of inflammation. However, young DMD patients, especially patients younger than 6 years of age, often have difficulty staying still inside the MRI scanner to undergo the test owing to young age claustrophobia or other causes. Therefore, skeletal muscle CT was used, as it can obtain muscle images in a short time. Furthermore, CT imaging is a reliable method for muscle volumetry. Additionally, using this method made it possible to perform the evaluation in a standard manner across multiple sites. Godi et al showed that in older DMD patients, MVI in the lower leg was greater than in the thigh because as the patient ages, the progression of muscle necrosis is faster in the thigh than in the lower leg.28 Although the treatment period of this study was relatively short, the reduction in %MVI from baseline in the placebo group was greater in the thigh than in the lower leg. The effect of TAS‐205 in maintaining thigh muscle may be limited because the reduction in thigh muscle volume is faster than in muscles in the lower leg.
Nakagawa et al.17 and Takeshita et al.19 previously reported that urinary Cre excretion could be a reliable index of muscle mass and that urinary Cre‐normalized tPGDM levels were considered to reflect PGD2 production in DMD‐associated muscle atrophy. Therefore, we evaluated total urinary excretion of tPGDM (and tPGEM) and urinary tPGDM/Cre (and tPGEM/Cre) concentration ratio in this study. The results of this pharmacodynamic analysis suggest that TAS‐205 selectively inhibits PGD2 production throughout the treatment period.
When considering the results of these analyses, there were potential indications that as a result of TAS‐205 administration, PGD2 production was inhibited, which subsequently reduced muscle deterioration in the lower legs thus resulting in the suppression of further declines in 6MWD in DMD patients. However, further research is needed to clarify this association.
TAS‐205 showed a favorable safety profile in DMD patients. Although 80% of patients concomitantly received steroid treatment during this study, there were no observable issues associated with the concomitant use of TAS‐205 and steroids. Additionally, there were no differences in the safety analyses observed between low‐dose or high‐dose TAS‐205 treatments, and there were no tolerability issues in any of the dose groups.
This study is limited by the small number of East Asian patients enrolled and the short observation period. The study was exploratory and was not powered for efficacy. We are therefore planning a next study to investigate the long‐term dosing benefits of TAS‐205 in a greater DMD patient population. Furthermore, it has been previously reported that steroid therapy does not alter levels of tPGDM/Cre in DMD patients even though steroid treatment has been shown to inhibit prostaglandin biosynthesis.17, 19 Therefore, the interaction between steroid treatment and the selective HPGDS inhibitor, TAS‐205, requires more consideration.
It is expected that TAS‐205 could potentially be used by all DMD patients, including those who are candidates for dystrophin gene‐targeted therapies, as its mechanism of action does not depend on specific gene mutations.
Conflicts of Interest
HK reports grants from Taiho Pharmaceutical Co., Ltd. during the conduct of the study. TN reports grants from National Centre of Neurology and Psychiatry (NCNP) and Japan Agency for Medical Research and Development (AMED); and personal fees from Taiho Pharmaceutical Co., Ltd. during the conduct of the study. YM, HA, YI, HY, ST, TM, KS, AN, SK, KO, KI, SS, MF, and SK have nothing to disclose in relation to this manuscript.
Author Contributions
HK contributed to the conception and design of the study, and the acquisition, analysis, and interpretation of data for the study; YM, TM, KS, HA, AN, SK, KO, KI, SS, and MF contributed to the acquisition of data for the study; SK, TN, YI, and HY contributed to the conception and design of the study; and ST contributed to the conception and design of the study, and the analysis and interpretation of the data. All authors contributed to the drafting and reviewing of the manuscript and gave final approval for submission. All authors agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Supporting information
Data S1. Supplementary Methods.
Data S2. Supplementary Results.
Acknowledgments
This study was funded by Taiho Pharmaceutical Co., Ltd. We thank the participating patients and their families; and all the investigators, site staff, and operation staff who participated in the study. We also thank Professor Emeritus J. Patrick Barron, Tokyo Medical University, and James Graham, PhD, of Edanz Medical Writing for providing medical writing support.
Trial registration: ClinicalTrials.gov: NCT02752048
Funding Information
This study was funded by Taiho Pharmaceutical Co., Ltd.
Funding Statement
This work was funded by Taiho Pharmaceutical grant .
References
- 1. Hoffman E, Brown R, Kinkel L. Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell 1987;51:919–928. [DOI] [PubMed] [Google Scholar]
- 2. Evans N, Misyak S, Robertson J, et al. Dysregulated intracellular signaling and inflammatory gene expression during initial disease onset in Duchenne muscular dystrophy. Am J Phys Med Rehabil 2009;88:502–522. [DOI] [PubMed] [Google Scholar]
- 3. Birnkrant D, Bushby K, Bann C, et al. Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and neuromuscular, rehabilitation, endocrine, and gastrointestinal and nutritional management. Lancet Neurol 2018;17:251–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Birnkrant D, Bushby K, Bann C, et al. Diagnosis and management of Duchenne muscular dystrophy, part 2: respiratory, cardiac, bone health, and orthopaedic management. Lancet Neurol 2018;17:347–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Birnkrant D, Bushby K, Bann C, et al. Diagnosis and management of Duchenne muscular dystrophy, part 3: primary care, emergency management, psychosocial care, and transitions of care across the lifespan. Lancet Neurol 2018;17:445–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. McDonald C, Campbell C, Torricelli R, et al. Ataluren in patients with nonsense mutation Duchenne muscular dystrophy (ACT DMD): a multicentre, randomised, double‐blind, placebo‐controlled, phase 3 trial. Lancet 2017;390:1489–1498. [DOI] [PubMed] [Google Scholar]
- 7. Bushby K, Finkel R, Wong B, et al. Ataluren treatment of patients with nonsense mutation dystrophinopathy. Muscle Nerve 2014;50:477–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Goemans N, Mercuri E, Belousova E, et al. A randomized placebo‐controlled phase 3 trial of an antisense oligonucleotide, drisapersen, in Duchenne muscular dystrophy. Neuromuscul Disord 2018;28:4–15. [DOI] [PubMed] [Google Scholar]
- 9. Voit T, Topaloglu H, Straub V, et al. Safety and efficacy of drisapersen for the treatment of Duchenne muscular dystrophy (DEMAND II): an exploratory, randomised, placebo‐controlled phase 2 study. Lancet Neurol 2014;13:987–996. [DOI] [PubMed] [Google Scholar]
- 10. Mendell J, Rodino‐Klapac L, Sahenk Z, et al. Eteplirsen for the treatment of Duchenne muscular dystrophy. Ann Neurol 2013;74:637–647. [DOI] [PubMed] [Google Scholar]
- 11. Falzarano M, Scotton C, Passarelli C, Ferlini A. Duchenne muscular dystrophy: from diagnosis to therapy. Molecules 2015;20:18168–18184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Verhaart IEC, Aartsma‐Rus A. Therapeutic developments for Duchenne muscular dystrophy. Nat Rev Neurol 2019;15:373–386. [DOI] [PubMed] [Google Scholar]
- 13. Ricciotti E, FitzGerald G. Prostaglandins and inflammation. Arterioscler Thromb Vasc Biol 2012;31:986–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Urade Y, Hayaishi O. Prostaglandin D synthase: structure and function. Vitam Horm 2000;58:89–120. [DOI] [PubMed] [Google Scholar]
- 15. Matsuoka T, Hirata M, Tanaka H, et al. Prostaglandin D2 as a mediator of allergic asthma. Science 2000;287:2013–2017. [DOI] [PubMed] [Google Scholar]
- 16. Mohri I, Aritake K, Taniguchi H, et al. Inhibition of prostaglandin D synthase suppresses muscular necrosis. Am J Pathol 2009;174:1735–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Nakagawa T, Takeuchi A, Kakiuchi R, et al. A prostaglandin D2 metabolite is elevated in the urine of Duchenne muscular dystrophy patients and increases further from 8 years old. Clinica Chemica Acta 2013;423:10–14. [DOI] [PubMed] [Google Scholar]
- 18. Song W, Wang M, Ricciotti E, et al. Tetranor PGDM, an abundant urinary metabolite reflects biosynthesis of prostaglandin d2 in mice and humans. J Biol Chem 2008;283:1179–1188. [DOI] [PubMed] [Google Scholar]
- 19. Takeshita E, Komaki H, Tachimori H, et al. Urinary prostaglandin metabolites as Duchenne muscular dystrophy progression markers. Brain Dev 2018;40:918–925. [DOI] [PubMed] [Google Scholar]
- 20. Tanaka K, Aritake K, Tayama M, et al. Novel inhibitor of hematopoietic prostaglandin D synthase improves the muscle disorder in an experimental model of Duchenne muscular dystrophy. Neuromuscul Disord 2014;24:821 (Abstract). [Google Scholar]
- 21. Takeshita E, Komaki H, Shimizu‐Motohashi Y, et al. A phase I study of TAS‐205 in patients with Duchenne muscular dystrophy. Ann Clin Transl Neurol 2018;5:1338–1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kuru S, Sakai M, Tanaka N, et al. Natural course of muscular involvement assessed by a new computed tomography method in Duchenne muscular dystrophy. Neurol Clin Neurosci 2013;1:63–68. [Google Scholar]
- 23. Nakayama T, Kuru S, Okura M, et al. Estimation of net muscle volume in patients with muscular dystrophy using muscle CT for prospective muscle volume analysis: an observational study. BMJ Open 2013;3:e003603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. McDonald C, Henricson E, Abresch R, et al. The 6‐minute walk test and other endpoints in Duchenne muscular dystrophy: longitudinal natural history observations over 48 weeks from a multicenter study. Muscle Nerve 2013;48:343–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. McDonald C, Henricson E, Abresch R, et al. The 6‐minute walk test and other clinical endpoints in Duchenne muscular dystrophy: reliability, concurrent validity, and minimal clinically important differences from a multicentre study. Muscle Nerve 2013;48:357–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Haas M, Vlcek V, Balabanov P, et al. European medicines agency review of ataluren for the treatment of ambulant patients aged 5 years and older with Duchenne muscular dystrophy resulting from a nonsense mutation in the dystrophin gene. Neuromuscul Disord 2015;25:5–13. [DOI] [PubMed] [Google Scholar]
- 27. Drugs FDA. Drug approval package: Exondys 51 injection (eteplirsen). https://www.accessdata.fda.gov/drugsatfda_docs/nda/2016/206488_TOC.cfm. (accessed Feb 12, 2017).
- 28. Godi C, Ambrosi A, Nicastro F, et al. Longitudinal MRI quantification of muscle degeneration in Duchenne muscular dystrophy. Ann Clin Transl Neurol 2016;16:607–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Supplementary Methods.
Data S2. Supplementary Results.