

Abstract
Neuromyelitis optica spectrum disorders (NMOSD) is a heterogeneous group of neuroinflammatory conditions associated with demyelination primarily in spinal cord and optic nerve, and to a lesser extent in brain. Most NMOSD patients are seropositive for IgG autoantibodies against aquaporin‐4 (AQP4‐IgG), the principal water channel in astrocytes. There has been interest in establishing experimental animal models of seropositive NMOSD (herein referred to as NMO) in order to elucidate NMO pathogenesis mechanisms and to evaluate drug candidates. An important outcome of early NMO animal models was evidence for a pathogenic role of AQP4‐IgG. However, available animal models of NMO, based largely on passive transfer to rodents of AQP4‐IgG or transfer of AQP4‐sensitized T cells, often together with pro‐inflammatory maneuvers, only partially recapitulate the clinical and pathological features of human NMO, and are inherently biased toward humoral or cellular immune mechanisms. This review summarizes current progress and shortcomings in experimental animal models of seropositive NMOSD, and opines on the import of advancing animal models.
Keywords: animal models, autoimmunity, neuroinflammation, NMO
Introduction
Neuromyelitis optica spectrum disorders (NMOSD), as currently defined 62, consist of a heterogeneous group of diseases characterized by neuroinflammation and demyelination that can produce motor, visual and other neurological deficits. Most NMOSD patients are seropositive for IgG autoantibodies against astrocyte water channel aquaporin‐4 (AQP4), called AQP4‐IgG (or NMO‐IgG). This review is focused on experimental animal models of AQP4‐IgG‐seropositive NMOSD (herein called NMO). Having AQP4 as a well‐defined immune target in NMO affords a rational basis to establish animal models. The development of animal models of NMO is motivated by their potential utility in studying NMO disease pathogenesis mechanisms and testing therapeutic candidates.
An ideal animal model of NMO would closely recapitulate the major features of NMO disease in humans. NMO in humans is a spontaneously occurring autoimmune condition, with relapses and disease‐inactive periods, and clinical findings consequent to inflammatory demyelinating lesions in spinal cord and optic nerve, and to a lesser extent in brain. There are often characteristic clinical features of optic neuritis and transverse myelitis, such as vision loss in one or both eyes, weakness or paralysis in the legs or arms, painful spasms, sensory loss, vomiting and bladder dysfunction, with a typical relapsing course. A characteristic radiographic feature of NMO is longitudinally extensive spinal cord lesions on T2‐weighted MRI extending over three or more vertebral segments 16, 61. Pathological features of active NMO include astrocytopathy with loss of AQP4 and glia fibrillary acidic protein (GFAP), inflammation with granulocyte and macrophage infiltration and microglial activation, complement activation, blood–brain barrier disruption and demyelination 12, 31, 41. These processes can lead to neuronal loss and tissue scarring. Clinical and pathological studies in human NMO have suggested a central role for complement activation in NMO pathogenesis, though various other potential primary mechanisms have been proposed such as antibody‐dependent cellular cytotoxicity, glutamate excitotoxicity, AQP4 cellular internalization, AQP4 water transport inhibition and blood–brain barrier disruption 41.
We review here reported experimental animal models of NMO, many of which involve passive transfer of AQP4‐IgG to rodents using a variety of methods and conditions, with some models involving T cell administration. Table 1 summaries the various models that are discussed below. We discuss the scientific advances that have emerged from animal models, and comment on the debatable need for their continued advancement.
Table 1.
Summary of papers reporting experimental animal models of NMO. Abbreviation: ND = not reported.
| Reference | Animal | Model | Sites of pathology | AQP4 loss | GFAP loss | Infiltration cell type(s) | Complement deposition | Myelin loss | Time point (s) examined | Behavioral phenotype |
|---|---|---|---|---|---|---|---|---|---|---|
| Bradl et al 5 | Lewis rats | EAE (+AQP4‐IgG) | Spinal cord | Yes | Yes | T cells | Yes | No | 24 hours | ND |
| Macrophages | ||||||||||
| Bennett et al 4 | Lewis rats | EAE (+AQP4‐IgG) | Spinal cord | ND | Yes | No | Yes | No | 30 hours | ND |
| Kinoshita et al 20 | Lewis rats | EAE (+AQP4‐IgG) | Spinal cord | Yes | Yes | Macrophages | Yes | No | 4 days | Clinical score increase |
| Neutrophils | ||||||||||
| Saadoun et al 45 | CD1 mice | Intracerebral injection | Brain | Yes | Yes | CD45+ cells | Yes | Yes | 12 hours‐7 days | Right‐turning at 7 days |
| Pohl et al 37 | Lewis rats | T cell delivery | Brain | Yes | ND | T cells | Yes | No | 24 hours | ND |
| Spinal cord | Macrophages | |||||||||
| Ratelade et al 39 | CD1 mice | Intravenous injection | Area postrema | Yes | Yes | ND | ND | Yes | 5 days | ND |
| Zhang et al 71 | CD1 mice | Ex vivo spinal cord culture | Spinal cord slice | Yes | Yes | Neutrophils | Yes | Yes | 7 days | ND |
| Macrophages | ||||||||||
| Saini et al 48 | C57/BL6 mice | EAE (+AQP4‐IgG) | Spinal cord | Yes | ND | Neutrophils | ND | Yes | 60 days | Clinical worsening |
| Optic nerve | Eosinophils | |||||||||
| Zhang et al 72 | CD1 mice | Intracerebral infusion | Brain | Yes | Yes | Eosinophils | Yes | Yes | 3 days | ND |
| Spinal cord | Neutrophils | |||||||||
| Asavapanumas et al 2 | Lewis rats | Intracerebral injection | Brain | Yes | Yes | Neutrophils | Yes | Yes | 5 days | ND |
| Macrophages | ||||||||||
| Asavapanumas et al 1 | CD1 mice | Intrcranial infusion | Optic nerve | Yes | Yes | Neutrophils | Yes | Yes | 3 days | ND |
| Macrophages | ||||||||||
| Asavapanumas et al 3 | Lewis rats | Intraperitoneal injection | Brain | Yes | Yes | Neutrophils | Yes | Yes | 5 days | ND |
| Macrophages | ||||||||||
| Matsumoto Y et al 28 | SD rats | Injection under optic nerve sheath | Optic nerve | Yes | Yes | CD11 + cells | ND | Yes | 7‐14 days | ND |
| Retina | ||||||||||
| Zhang et al 73 | CD59−/− mice | Intrathecal injection | Spinal cord | Yes | Yes | CD45+ cells | Yes | Yes | 2 days | Hind limb weakness |
| Geis et al 11 | Lewis rats | Intrathecal injection | Spinal cord | Yes | Yes | Macrophages | No | No | 19 days | Progressive motor deficit |
| Jones et al 17 | C57/BL6 mice | T cell delivery | Brain | No | ND | T cells | ND | Yes | 21 days | Weight loss |
| Spinal cord | Hind limb weakness | |||||||||
| Optic nerve | ||||||||||
| Kurosawa 21 | Lewis rats | EAE | Spinal cord | Yes | Yes | Neutrophils | Yes | No | 2 days | Weakness |
| Brainstem | ||||||||||
| Optic chiasm | ||||||||||
| Zeka et al 68 | Lewis rats | T cell delivery | Brain | Yes | Yes | T cells | Yes | ND | 7 days | Tail paralysis |
| Spinal cord | macrophages | |||||||||
| Felix et al 10 | rats | Intravitreal injection | Retina | Yes | No | CD45+ cells | Yes | Yes | 6 hours–30 days | ND |
| Marignier et al 27 | OFA rats | Intraventricular infusion | Brain | Yes | Yes | No | No | Yes | 7 days | Impaired motor behavior |
| Spinal cord | ||||||||||
| Optic nerve | ||||||||||
| Sagan et al 47 | C57/BL6 mice | T cell delivery | Spinal Cord | ND | ND | T cells | ND | No | 2 days | Paraplegia |
| Optic nerve | ||||||||||
| Retina | ||||||||||
| Zeka et al 69 | Lewis rats | T cell delivery | Optic nerve | Yes | Yes | T cells | Yes | Yes | 5 days | ND |
| Retina | macrophages | |||||||||
| Zhang et al 70 | SD rats | Injection under optic nerve sheath | Optic nerve | Yes | Yes | CD68+ cells | ND | Yes | 7 days | Reduced visual evoked |
| potentials & pupillary light reflex | ||||||||||
| Hillebrand et al 14 | Lewis rats RNU rats | Intraperitoneal injection | Brain | Yes | Yes | Neutrophils | Yes | ND | 1‐5 days | Impaired balance |
| Spinal cord | macrophages | Hind limb weakness | ||||||||
| Lee et al 22 | C57/BL6 mice | EAE (+AQP4‐IgG) | Brain | Yes | Yes | ND | Yes | ND | 21 days | Clinical score increasing |
| Spinal cord | ||||||||||
| Yao et al 64 | SD rats | Focused ultrasound | Brain | Yes | Yes | CD45+ cells | Yes | Yes | 5 days | ND |
| Spinal cord |
Aquaporin‐4, the Target of IgG Autoantibodies in NMO
AQP4 is a water‐selective transport protein expressed at the plasma membrane of astrocytes throughout the CNS, particularly in astrocyte foot processes 31. AQP4 is expressed as well outside of the CNS in skeletal muscle and in a subset of epithelial cells in the kidney, airways, stomach and exocrine glands. Studies in AQP4 knockout mice have elucidated its function in water movement (brain edema), neuroexcitation, astrocyte migration and glial scarring, and neuroinflammation [reviewed in Ref. 32]. AQP4 is considered to be an attractive drug target for various neurological conditions such as ischemic stroke, though at present there are no validated AQP4 inhibitors 59.
AQP4 is the target of AQP4‐IgG autoantibodies in NMO 23. Several studies have addressed the nature of the extracellular epitopes in AQP4 that bind AQP4‐IgGs 29, 58. Though aquaporins in general assemble in membrane in tetramers, AQP4 is unique as its tetramers further aggregate at the cell plasma membrane as orthogonal arrays of particles (OAPs), which are square supramolecular clusters that have been visualized by freeze‐fracture electron microscopy, and more recently by super‐resolution microscopy 44, 49, 63. Of relevance to NMO pathogenesis and animal models, AQP4 assembly in OAPs is necessary for AQP4‐IgG pathogenicity, as most AQP4‐IgGs bind to OAPs much better than to separated AQP4 tetramers 7, and a multivalent interaction between membrane‐bound AQP4‐IgG with C1q, the initial component of complement, is essential for complement activation 34 (Figure 1). As in humans, AQP4 is assembled into OAPs in rodents, which is a pre‐requisite for models of NMO based on passive transfer of AQP4‐IgG.
Figure 1.
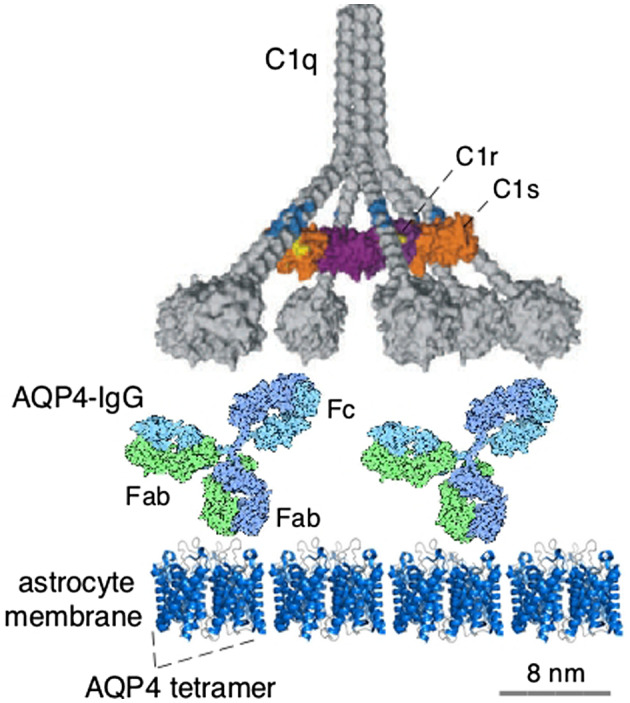
Complement activation following AQP4‐IgG binding to AQP4 aggregates on astrocyte membrane, showing multivalent interaction of AQP4‐IgG with C1q, the complement protein involved in activation of the classical complement pathway. Relative sizes of the proteins are shown to scale. Adapted from Ref. 34.
Early Models Involving Experimental Autoimmune Encephalomyelitis (EAE)
EAE is an animal model of CNS inflammation and demyelination that is produced by immunization with various myelin‐related peptides or other materials, resulting in a T cell‐mediated autoimmune response. Early NMO models involved administration of AQP4‐IgG to rodents with preexisting EAE, in which there is underlying CNS inflammation and injury to the blood–CNS barrier. One study involved intraperitoneal injections of AQP4‐IgG (purified from NMO patient sera) for 4 days in rats with EAE induced by active immunization with myelin basic protein (MBP) 20. The spinal cord showed infiltration of macrophages, neutrophils and eosinophils around blood vessels in gray matter, with loss of GFAP and AQP4, and perivascular deposition of human IgG and C5b‐9. Similar spinal cord pathology, as well as lesions in the fourth ventricle and optic chiasm, was found when AQP4‐IgG was administered to rats with EAE produced by passive transfer of MBP‐activated T cells 5. In a third related study, retrobulbar administration of a recombinant AQP4‐IgG in rats with EAE produced by active immunization with MBP showed astrocyte injury with complement deposition in spinal cord 4. However, each of the studies showed little demyelination and axonal injury, important pathological feature in NMO. The acknowledged weakness of these studies is that preexisting EAE confounds interpretation of the effects of AQP4‐IgG, as NMO is not a disease of myelin‐targeted T cells. More recently, a longer term EAE model of NMO in mice was created by active immunization with myelin oligodendrocyte glycoprotein peptide (MOG35‐55) for 60 days followed by AQP4‐IgG administration 48. AQP4‐IgG penetration into spinal cord was seen in this model, with clinical disability over 2 months, and unlike the early models there was significant demyelination and axonal injury.
Another more recent EAE‐based NMO model involved MBP‐immunized Lewis rats with intraperitoneal injection of purified human IgG from a seropositive NMO patient or a high‐affinity anti‐AQP4 monoclonal antibody 21. Pathological features of NMO were found, including large lesions extending longitudinally from medulla oblongata to spinal cord. Most recently, a model of MOG‐induced EAE with repetitive intrathecal injection of AQP4‐IgG and human complement showed astrocyte injury in brain and spinal cord, with complement deposition 22.
Passive Transfer Models of NMO
Passive transfer models of NMO involve administration of AQP4‐IgG, from NMO patient sera or as recombinant monoclonal antibody, into animals, which at present are limited to mice and rats. As summarized in Figure 2, the routes of administration have included direct injection or infusion of AQP4‐IgG into various CNS tissues, sometimes together with human complement, or injection of AQP4‐IgG peripherally (intraperitoneal or intravenous) to mimic seropositivity in humans. The goal has been to reproduce the major pathological features of human NMO, and in doing so afford the opportunity to study disease pathogenesis mechanisms and test drug candidates. The various models as described below have recapitulated many of the major features of NMO, albeit with differing degrees of fidelity and robustness.
Figure 2.
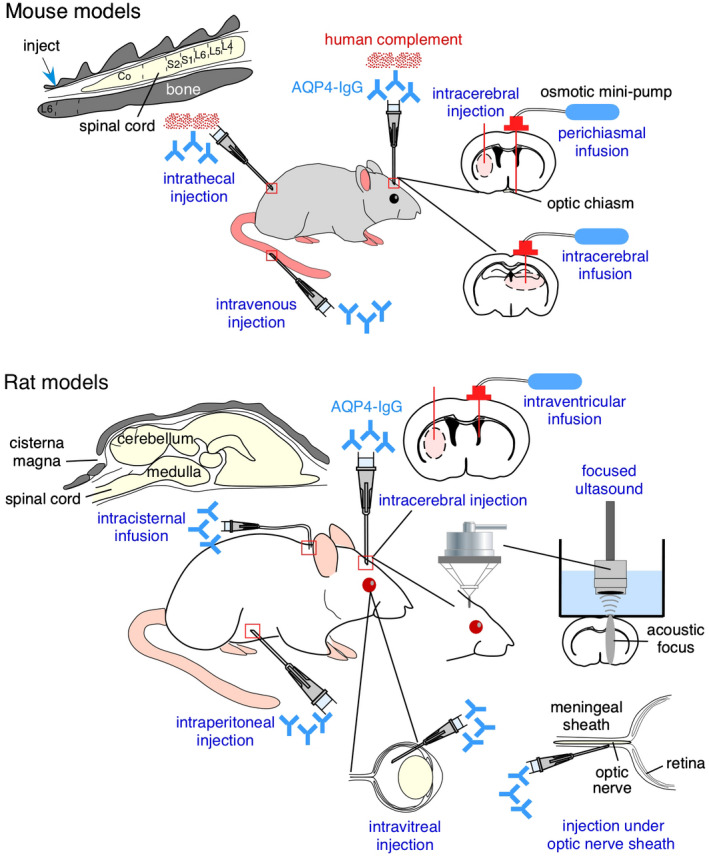
Passive transfer models of NMO in mice (top) and rats (bottom) involving direct administration of AQP4‐IgG, sometimes together with human complement. As diagrammed, routes of administration have included direct injection into CNS tissues or peripherally, sometimes together with maneuvers to permeabilize the blood‐brain barrier. See text for explanations.
Models involving AQP4‐IgG delivery to brain
The first passive transfer model of NMO, without preexisting EAE, involved direct intracerebral injection of AQP4‐IgG (IgG purified from NMO patient sera) and human complement (non‐heat‐inactivated human serum) into mouse brain 45. Within 12 h after injection marked loss of AQP4 and GFAP immunoreactivity was seen together with astrocyte swelling, myelin breakdown and axonal injury, albeit with minimal inflammation. By 7 days extensive inflammatory cell infiltration and perivascular deposition of activated complement was seen, as well as marked demyelination and neuronal cell death. Behavioral studies showed a propensity for right turning in mice injected with AQP4‐IgG and complement in their right hemisphere. In control studies, pathological or behavioral changes were not seen in AQP4 knockout mice injected with AQP4‐IgG and complement, or in wild‐type mice injected with AQP4‐IgG alone or control (non‐NMO) human IgG together with complement. These results offered the first compelling evidence for a pathogenic role of AQP4‐IgG in seropositive NMO. This intracerebral injection model has been used widely as discussed further below.
In a variant of the original model, in a project designed to investigate the role of eosinophils in NMO and the potential utility of eosinophil‐targeted therapeutics 72, NMO pathology was produced in mice by continuous intracerebral infusion of AQP4‐IgG and human complement for 3 days. In contrast to single injection models, brain pathology in the continuous infusion model was associated with marked eosinophil infiltration.
Motivated by the need to inject human complement into mouse brain to produce NMO pathology, a study was done involving intracerebral injection of AQP4‐IgG into brain of naïve rats 2, as rats have human‐like complement activity and human IgG effectively activates the classical complement pathway in rats. A single intracerebral injection of AQP4‐IgG into brains of adult Lewis rats produced robust NMO pathology around the needle track in all rats, with marked loss of AQP4, GFAP and myelin at day 5, along with granulocyte and macrophage infiltration, microglial activation, complement deposition, blood–brain barrier disruption and neuron death (Figure 3). Interestingly, a distinct “penumbra” was seen around lesions characterized by loss of AQP4 but not of GFAP or myelin. No lesion or penumbra was seen in rats receiving control IgG. The penumbra, but not the central lesion, was seen in AQP4‐IgG‐injected rats made complement deficient by cobra venom factor or when rats were administered AQP4‐IgG lacking complement effector function, suggesting a complement‐independent pathogenic mechanism. Mechanistic studies implicated antibody‐dependent cellular cytotoxicity (ADCC) as responsible for the penumbra, as the penumbra was eliminated by injection of AQP4‐IgG lacking ADCC effector function. This study established a robust, passive transfer model of NMO that does not require preexisting neuroinflammation or complement administration. This model supported a central role of complement in NMO pathogenesis, though also suggested the involvement of complement‐independent pathogenic mechanisms.
Figure 3.
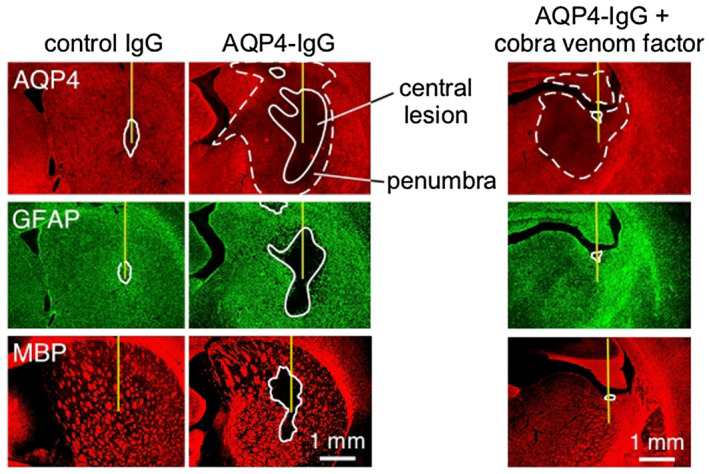
NMO pathology produced in rat brain following intracerebral injection of AQP4‐IgG. AQP4, GFAP and MPB immunofluorescence at 5 days after injection of control (non‐NMO) IgG or AQP4‐IgG in rats. Solid line shows central area of fluorescence loss and dashed line denotes penumbra region seen for AQP4. Where indicated (right), rats were pretreated with cobra venom factor to inactive complement. Adapted from Ref. 2.
Models involving AQP4‐IgG delivery to spinal cord
Following the success of passive transfer models based on intracerebral injection of AQP4‐IgG, various attempts were made to produce NMO pathology in spinal cord, a more relevant tissue than brain in human NMO. These studies generally involved injection of patient‐derived or recombinant AQP4‐IgG directly into the cerebrospinal space. In a study designed to investigate the role of complement regulator protein CD59 in NMO, AQP4‐IgG and human complement were delivered to mice by intrathecal injection at L5–L6 73. Though relatively minor pathology was found in wild‐type mice, robust, longitudinally extensive white matter lesions were seen in CD59‐deficient mice by day 2, with loss of AQP4 and GFAP, inflammation, deposition of activated complement deposition and demyelination. Hindlimb motor function was remarkably impaired as well. Interestingly, partial remyelination and recovery of motor function was seen at day 5. The results implicated CD59 as an important modulator of the immune response in NMO, and provided an animal model of NMO that recapitulated the longitudinally extensive spinal cord pathology seen in human NMO.
In another intrathecal injection model, AQP4‐IgG was repetitively injected into rats over 19 days using an indwelling catheter positioned at the atlanto‐occipital joint 11. The rats developed progressive motor deficit with impaired ambulation, with recovery in a few weeks after discontinuing injections. MRI of spinal cord showed longitudinal lesions and histology showed accumulation of human IgG with reduced AQP4 and GFAP, but without demyelination or axonal injury. Also, “penumbra‐like” lesions were seen with loss of AQP4 but preserved GFAP and lack of complement activation. Curiously, similar pathology was seen in rats pre‐treated with cobra venom factor, suggesting a complement‐independent mechanism. A related model involved continuous intraventricular infusion of patient‐derived AQP4‐IgG for 7 days by osmotic mini‐pump 27. Human IgG was seen in brain, spinal cord and optic nerve, with the loss of AQP4 and reactive astrocytes in spinal cord and optic nerve, and demyelination and axonal injury in spinal cord. In a study using two‐photon imaging of spinal cord to study early pathological events in NMO, astrocyte and axon injury were seen within 2 h after local application of AQP4‐IgG and complement, though the mechanism of the early axon injury was not established 13. Together, these various models support the conclusion that passive transfer of AQP4‐IgG can produce human‐like NMO pathology in spinal cord.
Models of NMO optic neuritis and retinitis
Various passive transfer models have been attempted to create NMO pathology in optic nerve, the other major site of pathology in NMO, as well as in retina. In an initial study 1, neither retrobulbar infusion nor intravitreal injection of AQP4‐IgG and human complement produced pathological changes in optic nerve in mice. However, a 3‐day continuous intracranial infusion of AQP4‐IgG, with needle tip inserted near the optic chiasm, produced characteristic NMO pathology in optic nerves, including loss of AQP4 and GFAP, granulocyte and macrophage infiltration, deposition of activated complement, demyelination and axonal injury. The injury was more pronounced, with retinal ganglion cell loss, in CD59 knockout mice or in wild‐type mice infused with a genetically modified AQP4‐IgG having enhanced complement effector function. Optic nerve pathology was not seen in AQP4 knockout mice receiving AQP4‐IgG and complement, or in wild‐type mice receiving control (non‐NMO) IgG and complement. The results supported the conclusion that similar NMO pathogenic mechanisms are involved in optic nerve as in brain and spinal cord, though the model is somewhat technically challenging and variable, as exposure of the optic nerves to AQP4‐IgG depends on precise needle positioning.
In another study, NMO patient serum was injected beneath the optic nerve sheath of Sprague Dawley rats after exposing the optic nerve by blunt dissection via a superior conjunctival approach 28. AQP4 and GFAP were reduced in optic nerves at day 7, with a marked inflammatory response and evidence of axonal injury at day 14, with loss of retinal ganglion cells. A recent study using same technique 70 demonstrated reduced amplitudes in visual evoked potentials at day 7, with reduced pupillary light reflex and retinal nerve fiber layer thickness. Optic nerve histology showed reduced AQP4, GFAP, MBP and NF in optic nerve at day 21, with an inflammatory response. The correlation between visual functional changes and optic nerve pathology supported a pathogenic role of AQP4‐IgG in NMO optic neuritis.
In a study done using rats that attempted to produce NMO optic neuritis or retinitis, AQP4‐IgG (without complement) was delivered to the eye by a single intravitreal injection 10. AQP4‐IgG deposition was seen on retinal Müller cells. At day 5, AQP4 expression was reduced and GFAP expression was increased, with mild retinal inflammation seen; at day 30 loss of retinal ganglion cells was seen with thinning of the ganglion cell complex. Interestingly, the loss of AQP4 was complement independent as evidenced by lack of complement deposition and by the similar retinal pathology in cobra venom factor‐treated rats receiving intravitreal AQP4‐IgG, and in normal rats receiving a mutated AQP4‐IgG lacking complement effector function. Intravitreal transfer of AQP4‐IgG thus produces primary, complement‐independent retinal pathology, which might contribute to retinal abnormalities seen in human NMO. Mechanistic studies in ex vivo retinal cultures showed reduced AQP4 expression after 24‐h exposure to AQP4‐IgG alone, which was largely prevented by inhibitors of endocytosis or lysosomal acidification. NMO pathogenesis mechanisms in retina may thus differ from those in optic nerve, spinal cord and brain.
Models involving systemic AQP4‐IgG delivery to mimic seropositivity
In our initial attempt to produce NMO pathology by intravenous administration of AQP4‐IgG to mice, AQP4‐IgG was strongly deposited on AQP4‐expressing cell membranes in kidney (collecting duct), skeletal muscle, trachea (epithelial cells) and stomach (parietal cells), but not in brain, spinal cord, optic nerve or retina, except for the area postrema in brain 39. Pharmacokinetic analysis showed that serum AQP4‐IgG decreased with t1/2 ∼18 h in wild‐type mice and ∼41 h in AQP4 knockout mice. Pathology was not seen in the CNS or peripheral organs, which in retrospect is not surprising, as mice have a very weak complement activation pathway 42. In a separate study in rats made seropositive by intraperitoneal injection of AQP4‐IgG, as in mice, AQP4‐IgG deposition was seen in peripheral AQP4‐expressing cells and area postrema in brain, but without significant pathological changes 3. However, one‐time puncture of brain parenchyma with a 28‐gauge needle in AQP4‐IgG‐seropositive rats produced robust NMO pathology around the needle track, with loss of AQP4 and GFAP, granulocyte and macrophage infiltration, and deposition of activated complement, with demyelination by 5 days. Pathology was not seen in rats receiving control human IgG or in AQP4‐IgG‐seropositive rats treated with cobra venom factor. AQP4‐IgG seropositivity alone is thus not sufficient to cause NMO pathology in rats, though a single intracerebral needle insertion, which disrupts the blood–brain barrier locally and may initiate an inflammatory response, was able to produce local NMO pathology.
Motivated by the observation that NMO pathology could be created in AQP4‐IgG‐seropositive rats by intracerebral needle injury, we tested whether focused ultrasound, which can transiently permeabilize the blood–CNS barrier, could produce NMO pathology in AQP4‐IgG‐seropositive rats 6. Prior literature indicated that focused pulsed ultrasound directed non‐invasively through intact skin, following intravenous delivery of microbubbles, could transiently permeabilize the blood–brain barrier with minimal tissue injury or inflammation 64. Application of pulsed ultrasound to rat brain using a 1 MHz transducer of 6 cm focal length allowed passage of IgG for 3–6 h in an approximately prolate ellipsoidal volume of diameter ∼3.5 mm and length ∼44 mm. Ultrasound treatment in brain in AQP4‐IgG‐seropositive rats produced localized NMO pathology, with characteristic astrocyte injury, inflammation and demyelination at day 5. NMO pathology was similarly created in cervical spinal cord in seropositive rats with ultrasound focused on spinal cord. Focused ultrasound offers a non‐invasive, targeted method to create NMO pathology in AQP4‐IgG‐seropositive rats. These findings suggest that blood–brain barrier permeabilization in AQP4‐IgG‐seropositive rats, without underlying injury or inflammation, is sufficient to create NMO pathology.
In a recent study, rats were injected multiple times with a highly pathogenic, monoclonal AQP4‐IgG via an intraperitoneal route 14. Various types of pathology were seen in brain and spinal cord as a presumed consequence of AQP4‐IgG entry into the CNS through circumventricular organs and meningeal vessels, which was exacerbated by administration of encephalitogenic T cells. However, the relevance of the findings to human NMO is not clear because of the large amounts of highly pathogenic AQP4‐IgG administered and the fact that humans can be seropositive without clinical disease prior to and in between disease exacerbations.
Ex vivo CNS organ culture models
Organ culture models of CNS tissues were developed to study NMO disease pathogenesis mechanisms, motivated by the limitations of cell culture models and the potential advantages of precise manipulation of putative effectors in disease‐relevant tissues 71. To create an ex vivo model of NMO in spinal cord, vibratome‐cut transverse spinal cord slices from adult mice of thickness ~300 µm were cultured for 7 days on transwell porous supports, after which they were exposed for 1–3 days to AQP4‐IgG and various effectors. The 7‐day slice cultures showed preserved cellular structures, including astrocytes, microglia, neurons and myelin (though axons are transected in transverse spinal cord slices). Slices exposed to AQP4‐IgG and complement showed marked loss of GFAP, AQP4 and myelin, with increased NMO pathology seen when slice cultures were incubated neutrophils or macrophages, or various soluble inflammatory factors including TNF‐α, IL‐6, IL‐1β and interferon‐γ. NMO pathology was similarly created in ex vivo optic nerve and hippocampal slice cultures incubated with AQP4‐IgG and complement.
In subsequent studies, we used spinal cord cultures to test NMO therapeutics 34, 57 and cerebellar slice cultures, in which axons remain largely intact, to study remyelination in NMO 65. Other labs have adapted mouse cerebellar slice cultures to study the sensitivity of neurons and oligodendrocytes to astrocyte injury by recombinant AQP4‐IgG and human complement 25 and to compare immunoreactivity of recombinant monoclonal antibodies cloned from cerebrospinal fluid plasmablasts of multiple sclerosis vs. NMO patients 24. It was reported that a multiple sclerosis antibody that bound to oligodendrocyte processes and myelinated axons caused complement injury to oligodendrocytes and demyelination, whereas AQP4‐IgG injured astrocytes with microglial activation, demyelination and neuronal death. It was concluded that myelin‐specific multiple sclerosis antibodies cause oligodendrocyte loss and demyelination, quite distinct from the AQP4‐targeted pathology in NMO.
T Cell Delivery Models of NMO
Motivated by the early work using EAE‐based models, several groups studied the potential role of AQP4‐specific T cells in NMO pathogenesis. A study involving immunization of C57BL/6 mice with AQP422‐36 or AQP4289‐303 peptide, without AQP4‐IgG administration, showed no pathology 18. In another study, AQP4‐specific T cells were generated by immunization of Lewis rats with AQP4207‐232 peptide from which peptide‐specific T cells lines were isolated from lymph nodes 37. Rats that received AQP4‐specific T cells showed inflammation in brain and spinal cord, but without astrocyte or myelin injury. However, transfer of AQP4207‐232‐specific T cells together with AQP4‐IgG derived from NMO patient sera produced greater pathology with parenchymal infiltration of T cells and macrophages, and complement deposition, though no demyelination. It was concluded that AQP4‐specific T cells together with AQP4‐IgG can produce NMO‐like pathology in the CNS, which supported a role for encephalitogenic AQP4‐specific T cells in NMO. In follow‐on work, highly encephalitogenic, AQP4268‐285‐specific T cells were generated in Lewis rats 68, which deeply infiltrated into brain parenchyma when transferred intraperitoneally to rats. Administration of AQP4 following the T cells produced loss of AQP4 and GFAP in spinal cord and brain, while optic nerve showed inflammation without loss of AQP4. In another study from the same group, pathogenic T cell lines combined with AQP4‐IgG injection produced damage in optic nerve and retina 69.
In mice, AQP4‐reactive T cells were generated in AQP4 knockout C57BL/6 mice immunized with AQP4 peptide 17. This approach obviated the potential immunological tolerance of T cells generated using wild‐type mice. After inducing Th17 polarization in vitro, intravenous transfer of the T cells in wild‐type mice produced weight loss and tail and hind limb weakness, with demyelination and perivascular T cell infiltration in spinal cord, optic nerve and brain. However, loss of AQP4, a hallmark of NMO, was not seen despite widespread inflammation and demyelination. A similar study compared the pathogenic capability of Th1‐ and TH17‐polarized AQP4‐specific T cells from AQP4 null mice 47. Th17‐polarized T cells induced more severe clinical disease in recipient wild‐type mice, which was associated with T cell infiltration in spinal cord and optic nerve. Retinal swelling and increased thickness of inner retinal layers was seen by optical coherence tomography. Together, these models support an encephalitogenic role of AQP4‐specific T cells, as some clinical features of myelitis and optic neuritis are reproduced. However, the relevance of these findings to human NMO is unclear, as mice lack an active complement system and AQP4‐IgG was not administered in the mouse models.
Scientific Advances Utilizing NMO Animal Models
Important metrics of success of NMO animal models in disease pathogenesis mechanisms include whether major questions in NMO pathogenesis have been answered and new mechanisms identified. Major questions include: Is AQP4‐IgG pathogenic? Why is NMO pathology restricted to spinal cord and optic nerve more than brain, with little pathology in peripheral tissues where AQP4 is also expressed? What is the relative role of humoral vs. cellular immune mechanisms in NMO? What are the triggers of AQP4‐IgG autoimmunity and clinical disease? Why are some NMO patients seropositive for many years prior to their first clinical attack? Important metrics of success of NMO models in therapeutics include discovery and validation of new therapeutic strategies and their translation to human clinical trials.
Pathogenicity of AQP4‐IgG
The pathogenicity of AQP4‐IgG had been hypothesized prior to animal data, based on the prediction that an IgG1 antibody, when bound to a target, could activate complement and cellular cytotoxicity mechanisms though its Fc receptor effector functions. Cytotoxicity by CDC and ADCC mechanisms had already been demonstrated in various AQP4‐expressing cell cultures 15, 60. Passive transfer of AQP4‐IgG in rodents confirmed early astrocyte cytotoxicity, and demonstrated that the various major pathological feature of human NMO could be recapitulated, including inflammation with granulocyte and macrophage predominance, centrovascular deposition of activated complement, blood–brain barrier disruption and demyelination 2, 45. While such information cannot quantify the relative role of complement vs. non‐complement mechanisms in human NMO, the finding that AQP4‐IgG can produce NMO‐like pathology in animals has been taken as compelling evidence for its pathogenicity.
Complement bystander mechanism
Human NMO pathology, data from passive transfer animal models of NMO, and the demonstrated efficacy of complement inhibition in human clinical trials implicate a central role for complement in NMO disease pathogenesis 26, 36. Animal models have supported a novel complement bystander mechanism, which was hypothesized based on the early and marked demyelination and neuronal injury seen in NMO, features that are hard to explain by secondary injury to oligodendrocytes, neurons and other cell types in the CNS. The mechanism of complement bystander killing involves local diffusion of activated, soluble complement proteins following AQP4‐IgG binding to astrocyte AQP4, which leads to deposition of the complement membrane attack complex (MAC) on nearby non‐AQP4‐expressing cells (Figure 4A). In an initial study, primary cocultures of rat astrocytes and mature oligodendrocytes exposed to AQP4‐IgG and complement showed early death of oligodendrocytes in close contact with astrocytes, which was not seen in pure oligodendrocyte cultures or when astrocytes were damaged by a complement‐independent mechanism 55. Remarkably, astrocyte–oligodendrocyte cocultures exposed to AQP4‐IgG and complement showed prominent MAC deposition on oligodendrocytes in contact with astrocytes, whereas C1q, the initiating protein in the classical complement pathway, was deposited only on astrocytes. In rats receiving an intracerebral injection of AQP4‐IgG, complement and a fixable dead cell stain, oligodendrocyte injury with MAC deposition was seen by 90 min, supporting a complement bystander mechanism for early oligodendrocyte injury and demyelination in NMO. In a more recent study done on astrocyte–neuron cocultures and rat models, complement bystander killing was demonstrated in neurons, which, like oligodendrocytes, do not express AQP4 or the complement regulator protein CD59 8.
Figure 4.
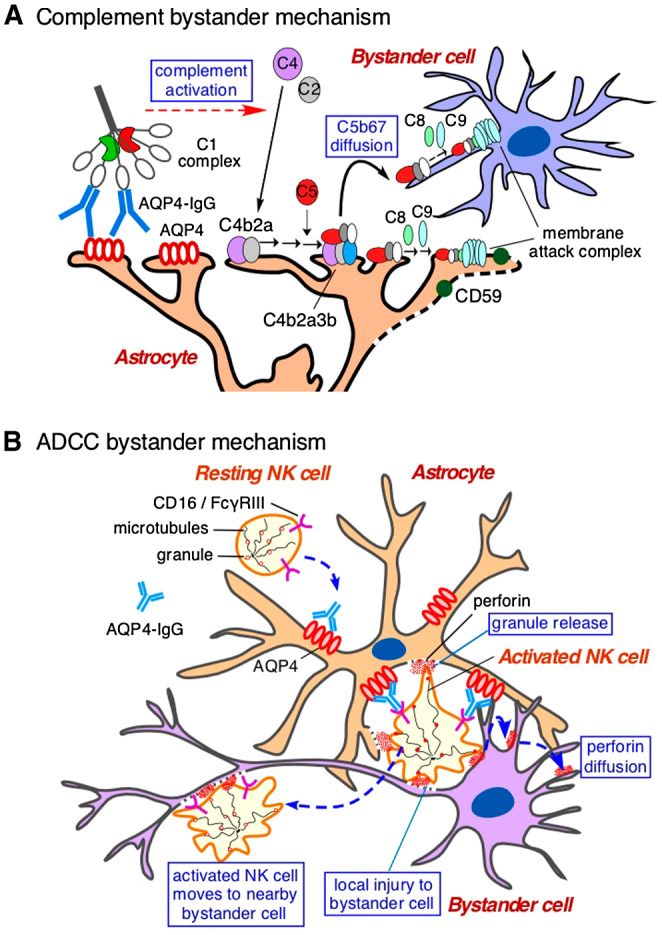
Bystander cell killing in NMO caused by complement and ADCC mechanisms. A. Complement bystander killing produced by AQP4‐IgG binding to AQP4 on astrocytes, involving activation of the classical complement pathway through C1q interaction, and diffusion of activated complement complex to nearby bystander cell. Adapted from Ref. 55. B. ADCC bystander killing produced by AQP4‐IgG binding to AQP4 on astrocytes, involving activation of leukocytes (NK cell shown) through Fcγ receptor interaction, and killing of bystander cells by release of toxic granules. Adapted from Ref. 9.
Antibody‐dependent cellular cytotoxicity (ADCC) in NMO
While complement activation clearly plans a major role in NMO disease pathogenesis, the involvement of ADCC in NMO pathogenesis has been less clear. ADCC had been demonstrated in cell culture models and is expected from interaction of the Fc region of an IgG1 antibody with Fc gamma receptors on leukocytes, resulting in their activation, degranulation and killing of target cells. In an animal model study of ADCC in NMO it was found that ADCC in the absence of complement can produce NMO‐like pathology 43. Injection of AQP4‐IgG and natural killer (NK) cells in mouse brain caused loss of AQP4 and GFAP, though minimal loss of myelin. Pathology was not seen with injection of control IgG and NK cells in wild‐type mice, or injection of AQP4‐IgG and NK cells in AQP4 knockout mice. In another study, mice were administered, by intracerebral injection, human complement plus AQP4‐IgGs containing Fc mutations to enhance or abolish complement and/or ADCC effector functions 38. As anticipated, the mutated AQP4‐IgG lacking complement effector function (but with 10‐fold enhanced ADCC effector function) produced little pathology. However, unexpectedly, a mutated antibody lacking ADCC effector function (but with ninefold enhanced complement effector function), produced much less pathology than the original AQP4‐IgG. These findings, taken together with the observation of a penumbra lesion in rats as mentioned above 2, support a role for ADCC in NMO pathogenesis and suggested ADCC as a potential therapeutic target. In a follow‐on study, in vivo evidence was reported for an “ADCC bystander” mechanism of killing involving leukocyte activation by astrocyte‐bound AQP4‐IgG and killing of nearby cells (Figure 4B) 9.
Role of complement regulator proteins
Complement regulator proteins in astrocytes, such as CD55 and CD59, can in principle modulate the action of complement by preventing MAC formation; CD55 can in addition inhibit anaphylatoxin generation. Animal models have supported a role of complement regulators in NMO. Using a mouse model of NMO produced by intracerebral or intrathecal injection of AQP4‐IgG and human complement, NMO pathology was much greater in CD59 knockout mice than in wild‐type mice 73. Motivated by the finding that rats, unlike mice, have human‐like complement activity 42, CD59 knockout rats were generated. Intracerebral injection of AQP4‐IgG in CD59 knockout rats produced much greater NMO pathology than in wild‐type rats (Figure 5A); a single, intracisternal injection of AQP4‐IgG in CD59 knockout rats produced hindlimb paralysis by 3 days, with inflammation and deposition of activated complement in spinal cord, optic nerves and brain periventricular and surface matter 67. Remarkably, CD59 rats made seropositive by intraperitoneal injection of AQP4‐IgG developed marked weakness by 24 h with creatine phosphokinase > 900‐fold greater than in seropositive wild‐type rats 66. Pathology in the CD59 knockout rats showed marked injury to organs where CD59 is normally expressed at high levels, including skeletal muscle, with inflammation and deposition of activated complement (Figure 5B). The results suggested a major protective role of CD59 outside of the central nervous system in seropositive NMO, and hence offer an explanation as to why peripheral, AQP4‐expressing cells are largely unaffected in NMO. The results also implicate complement regulator proteins as new targets in NMO in which their upregulation is predicted to protect against complement‐induced injury. Motivated by these observations, drug screening identified statins as transcriptional enhancers of CD55 expression in astrocyte cell culture models and in mice, and reported that oral atorvastatin significantly reduced NMO pathology in a passive transfer mouse model of NMO 52.
Figure 5.
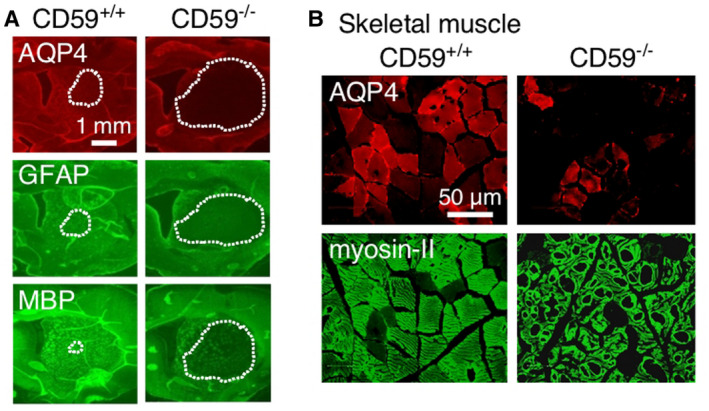
CD59 knockout in rats greatly amplifies NMO pathology following passive transfer of AQP4‐IgG. A. AQP4, GFAP and MBP immunofluorescence of rat brain at 7 days after intracerebral injection of AQP4‐IgG in wild‐type (CD59+/+) and knockout (CD59−/−) rats. Dotted line denotes areas of loss of fluorescence. Adapted from Ref. 67. B. AQP4 and myosin‐II immunofluorescence at 24 h after intraperitoneal injection of AQP4‐IgG in CD59+/+ and CD59−/− rats. Adapted from Ref. 66.
Proof‐of‐concept testing of novel therapeutics
The AQP4‐IgG‐passive transfer models of NMO in rodents have been applied to test various therapeutic strategies [reviewed in Ref. 30]. Motivated by the presence of granulocytes in human NMO lesions, studies were done to investigate the role of neutrophils and eosinophils in NMO pathogenesis. Using the mouse model involving intracerebral injection of AQP4‐IgG and human complement, NMO pathology was significantly reduced in neutropenic mice, and increased in mice made neutrophilic by granulocyte colony stimulating factor 46. Remarkably, NMO pathology was much reduced by the neutrophil elastase inhibitor Sivelestat, which motivated a small clinical trial. Studies were also done on eosinophils, which are abundant in inflammatory demyelinating lesions in NMO 34. In vitro studies showed eosinophil‐induced cytotoxicity to AQP4‐expressing cells and spinal cord slice cultures when added with AQP4‐IgG, without or with complement. In mice, demyelinating NMO lesions with marked eosinophil infiltration were produced by continuous intracerebral infusion of AQP4‐IgG and complement. NMO pathology was increased in transgenic hypereosinophilic mice, and reduced in mice made hypoeosinophilic by anti‐IL‐5 antibody or gene deletion. The second‐generation antihistamine cetirizine, which has eosinophil‐stabilizing actions, greatly reduced NMO‐IgG/eosinophil‐dependent cytotoxicity in vitro and NMO pathology in mice, which motivated a small human clinical trial 19.
Animal models of NMO were also used to investigate the possibility of remyelinating therapy in NMO 65. The FDA‐approved drug clobetasol, which was found to promote differentiation in oligodendrocyte precursor cell (OPC) and cerebellar slice cultures, was tested in mice using the intracerebral injection model. Intraperitoneal administration of 2 mg/kg/day clobetasol reduced myelin loss by ~60%, with increased numbers of mature oligodendrocytes within lesions. These data offered proof‐of‐concept for the potential utility of a remyelinating approach in NMO. Animal models of NMO have also been used to demonstrate efficacy of C1q‐targeted 35 and Fc hexamer 53 complement therapeutics, aquaporumab antibody 57 and small molecule 56 blockers of AQP4‐IgG/AQP4 binding, IgG‐inactivating enzymes 51, 54, IVIG 40 and a statin upregulator of CD55 52, as well as the lack of efficacy of various proposed therapeutics including C1‐esterase inhibitor 50.
Perspective
Though experimental animal models of NMO have advanced in the past decade and, as reviewed herein, been applied to study specific disease pathogenesis mechanisms and therapeutic approaches, there remain major shortcomings. No model has achieved spontaneous AQP4 autoimmunity with pathology in optic nerve and spinal cord. Models involving passive transfer of AQP4‐IgG, often with human complement, conclude, not surprisingly, that humoral immune mechanisms can explain fully, or at least in large part, NMO disease pathogenesis, and so are arguably intrinsically biased. Passive transfer models involving direct injection of AQP4‐IgG into CNS tissues, which generally create astrocyte injury, inflammation and demyelination around the injection site, can be confounded by an inflammatory response and injury to the blood‐CNS barrier caused by needle insertion. Passive transfer models involving systemic AQP4‐IgG administration generally require concomitant CNS inflammation or injury. Models involving transfer of sensitized T cells have generally poorly recapitulated human NMO pathology and largely ignore the central role of humoral immune mechanisms and complement, as T cell models often use mice, a species lacking a functional classical complement activation pathway. T cell models have not been useful in testing of specific hypotheses on NMO disease pathogenesis or therapeutics.
An important consideration in advancing NMO models is tailoring the model to the contemplated application, such as testing a proposed disease pathogenesis mechanism or evaluating a new therapy. For example, whereas existing passive transfer models may be informative in evaluating a complement‐targeted drug therapy, they are not appropriate to study B cell‐targeted or tolerizing therapeutics. Underlying limitations in rodent models of NMO include the very different astrocyte‐to‐neuron ratio in rodents compared to humans, and differences in AQP4 expression and distribution. Perhaps alternative animal models may better recapitulate the human NMO disease, as might creative adaptation of advances in immune cell molecular genetics involving reconstitution of key components of human AQP4 autoimmunity.
Given the shortcomings of available animal models of NMO and the absence of a clear path forward to a model of spontaneous AQP4 autoimmunity with human‐like pathology in optic nerve and spinal cord, the question arises whether further research is warranted to advance experimental animal models of NMO. It can be argued that recent advances in NMO animal models are largely incremental and have not clarified the roles of humoral vs. cellular immune mechanisms or of complement vs. cellular cytotoxicity to astrocytes, nor have they clarified the sites of pathology or the clinical course of human NMO disease. Animal models did not played a role in advancement of three drugs, eculizumab, satralizumab and inebelizumab, that have recently shown benefit in NMO clinical trials and may be approved for the NMO indication 33. Nevertheless, notwithstanding these caveats, breakthrough animal models of NMO would be most welcomed that substantially clarify human disease mechanisms and accelerate therapeutics developments.
Conflicts of Interest
The authors declare no conflicts of interest.
Acknowledgments
Supported by the National Institutes of Health (EY13574), the Guthy‐Jackson Charitable Foundation, and the Focused Ultrasound Foundation.
References
- 1. Asavapanumas N, Ratelade J, Papadopoulos MC, Bennett JL, Levin MH, Verkman AS (2014) Experimental mouse model of optic neuritis with inflammatory demyelination produced by passive transfer of neuromyelitis optica‐immunoglobulin G. J Neuroinflammation 11:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Asavapanumas N, Ratelade J, Verkman AS (2014) Unique neuromyelitis optica pathology produced in naive rats by intracerebral administration of NMO‐IgG. Acta Neuropathol 127:539–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Asavapanumas N, Verkman AS (2014) Neuromyelitis optica pathology in rats following intraperitoneal injection of NMO‐IgG and intracerebral needle injury. Acta Neuropathol Commun 2:48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bennett JL, Lam C, Kalluri SR, Saikali P, Bautista K, Dupree C et al (2009) Intrathecal pathogenic anti‐aquaporin‐4 antibodies in early neuromyelitis optica. Ann Neurol 66:617–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bradl M, Misu T, Takahashi T, Watanabe M, Mader S, Reindl M et al (2009) Neuromyelitis optica: pathogenicity of patient immunoglobulin in vivo. Ann Neurol 66:630–643. [DOI] [PubMed] [Google Scholar]
- 6. Chen KT, Wei KC, Liu HL (2019) Theranostic strategy of focused ultrasound induced blood‐brain barrier opening for CNS disease treatment. Front Pharmacol 10:86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Crane JM, Lam C, Rossi A, Gupta T, Bennett JL, Verkman AS (2011) Binding affinity and specificity of neuromyelitis optica autoantibodies to aquaporin‐4 M1/M23 isoforms and orthogonal arrays. J Biol Chem 286:16516–16524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Duan T, Smith AJ, Verkman AS (2018) Complement‐dependent bystander injury to neurons in AQP4‐IgG seropositive neuromyelitis optica. J Neuroinflammation 15:294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Duan T, Smith AJ, Verkman AS (2019) Complement‐independent bystander injury in AQP4‐IgG seropositive neuromyelitis optica produced by antibody‐dependent cellular cytotoxicity. Acta Neuropathol Commun 7:112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Felix CM, Levin MH, Verkman AS (2016) Complement‐independent retinal pathology produced by intravitreal injection of neuromyelitis optica immunoglobulin G. J Neuroinflammation 13:275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Geis C, Ritter C, Ruschil C, Weishaupt A, Grunewald B, Stoll G et al (2015) The intrinsic pathogenic role of autoantibodies to aquaporin 4 mediating spinal cord disease in a rat passive‐transfer model. Exp Neurol 265:8–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Graber DJ, Levy M, Kerr D, Wade WF (2008) Neuromyelitis optica pathogenesis and aquaporin 4. J Neuroinflammation 5:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Herwerth M, Kalluri SR, Srivastava R, Kleele T, Kenet S, Illes Z et al (2016) In vivo imaging reveals rapid astrocyte depletion and axon damage in a model of neuromylitis optica‐related pathology. Ann Neurol 79:794–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hillebrand S, Schanda K, Nigritinou M, Tsymala I, Böhm D, Peschl P et al (2019) Circulated AQP4‐specific auto‐antibodies alone can induce neuromyelitis optica spectrum disorder in the rat. Acta Neuropathol 137:467–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hinson SR, McKeon A, Fryer JP, Apiwattanakul M, Lennon VA, Pittock SJ (2009) Prediction of neuromyelitis optica attack severity by quantitation of complement‐mediated injury to aquaporin‐4‐expressing cells. Arch Neurol 66:1164–1167. [DOI] [PubMed] [Google Scholar]
- 16. Huda S, Whittam D, Bhojak M, Chamberlain J, Noonan C, Jacob A (2019) Neuromyelitis optica spectrum disorders. Clin Med (Lond) 19:169–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Jones MV, Huang H, Calabresi PA, Levy M (2015) Pathogenic aquaporin‐4 reactive T cells are sufficient to induce mouse model of neuromyelitis optica. Acta Neuropathol Commun 3:28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kalluri SR, Rothhammer V, Staszewski O, Srivastava R, Petermann F, Prinz M et al (2011) Functional characterization of aquaporin‐4 specific T cells: towards a model for neuromyelitis optica. PLoS ONE 6:e16083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Katz Sand I, Fabian MT, Telford R, Kraus TA, Chehade M, Masilamani M et al (2018) Open‐label, add‐on trial of cetirizine for neuromyelitis optica. Neurol Neuroimmunol Neuroinflamm 5:e441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kinoshita M, Nakatsuji Y, Kimura T, Moriya M, Takata K, Okuno T et al (2009) Neuromyelitis optica: Passive transfer to rats by human immunoglobulin. Biochem Biophys Res Commun 386:623–627. [DOI] [PubMed] [Google Scholar]
- 21. Kurosawa K, Misu T, Takai Y, Sato DK, Takahashi T, Abe Y et al (2015) Severely exacerbated neuromyelitis optica rat model with extensive astrocytopathy by high affinity anti‐aquaporin‐4 monoclonal antibody. Acta Neuropathol Commun 3:82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lee CL, Wang KC, Chen SJ, Chen CM, Tsai CP, Chen SY (2019) Repetitive intrathecal injection of human NMO‐IgG with complement exacerbates disease severity with NMO pathology in experimental allergic encephalomyelitis mice. Mult Scler Relat Disord 30:225–230. [DOI] [PubMed] [Google Scholar]
- 23. Lennon VA, Kryzer TJ, Pittock SJ, Verkman AS, Hinson SR (2005) IgG marker of optic‐spinal multiple sclerosis binds to the aquaporin‐4 water channel. J Exp Med 202:473–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Liu Y, Given KS, Owens GP, Macklin WB, Bennett JL (2018) Distinct patterns of glia repair and remyelination in antibody‐mediated demyelination models of multiple sclerosis and neuromyelitis optica. Glia 66:2575–2588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Liu Y, Harlow DE, Given KS, Owens GP, Macklin WB, Bennett JL (2016) Variable sensitivity to complement‐dependent cytotoxicity in murine models of neuromyelitis optica. J Neuroinflammation 13:301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lucchinetti CF, Mandler RN, McGavern D, Bruck W, Gleich G, Ransohoff RM et al (2002) A role for humoral mechanisms in the pathogenesis of Devic's neuromyelitis optica. Brain 125:1450–1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Marignier R, Ruiz A, Cavagna S, Nicole A, Watrin C, Touret M et al (2016) Neuromyelitis optica study model based on chronic infusion of autoantibodies in rat cerebrospinal fluid. J Neuroinflammation 13:111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Matsumoto Y, Kanamori A, Nakamura M, Takahashi T, Nakashima I, Negi A (2014) Sera from patients with seropositive neuromyelitis optica spectral disorders caused the degeneration of rodent optic nerve. Exp Eye Res 119:61–69. [DOI] [PubMed] [Google Scholar]
- 29. Owens GP, Ritchie A, Rossi A, Schaller K, Wemlinger S, Schumann H et al (2015) Mutagenesis of the aquaporin 4 extracellular domains defines restricted binding patterns of pathogenic neuromyelitis optica IgG. J Biol Chem 290:12123–12134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Papadopoulos MC, Bennett JL, Verkman AS (2014) Treatment of neuromyelitis optica: state‐of‐the‐art and emerging therapies. Nat Rev Neurol 10:493–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Papadopoulos MC, Verkman AS (2012) Aquaporin 4 and neuromyelitis optica. Lancet Neurol 11:535–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Papadopoulos MC, Verkman AS (2013) Aquaporin water channels in the nervous system. Nat Rev Neurosci 14:265–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Paul F, Murphy O, Pardo S, Levy M (2018) Investigational drugs in development to prevent neuromyelitis optica relapses. Expert Opin Investig Drugs 27:265–271. [DOI] [PubMed] [Google Scholar]
- 34. Phuan PW, Ratelade J, Rossi A, Tradtrantip L, Verkman AS (2012) Complement‐dependent cytotoxicity in neuromyelitis optica requires aquaporin‐4 protein assembly in orthogonal arrays. J Biol Chem 287:13829–13839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Phuan PW, Zhang H, Asavapanumas N, Leviten M, Rosenthal A, Tradtrantip L, Verkman AS (2013) C1q‐targeted monoclonal antibody prevents complement‐dependent cytotoxicity and neuropathology in in vitro and mouse models of neuromyelitis optica. Acta Neuropathol 125:829–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Pittock SJ, Berthele A, Fujihara K, Kim HJ, Levy M, Palace J et al (2019) Eculizumab in Aquaporin‐4‐positive neuromyelitis optica spectrum disorder. N Engl J Med 381:614–625. [DOI] [PubMed] [Google Scholar]
- 37. Pohl M, Fischer MT, Mader S, Schanda K, Kitic M, Sharma R et al (2011) Pathogenic T cell responses against aquaporin 4. Acta Neuropathol 122:21–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ratelade J, Asavapanumas N, Ritchie AM, Wemlinger S, Bennett JL, Verkman AS (2013) Involvement of antibody‐dependent cell‐mediated cytotoxicity in inflammatory demyelination in a mouse model of neuromyelitis optica. Acta Neuropathol 126:699–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ratelade J, Bennett JL, Verkman AS (2011) Intravenous neuromyelitis optica autoantibody in mice targets aquaporin‐4 in peripheral organs and area postrema. PLoS ONE 6:e27412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ratelade J, Smith AJ, Verkman AS (2014) Human immunoglobulin G reduces the pathogenicity of aquaporin‐4 autoantibodies in neuromyelitis optica. Exp Neurol 255:145–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ratelade J, Verkman AS (2012) Neuromyelitis optica: aquaporin‐4 based pathogenesis mechanisms and new therapies. Int J Biochem Cell Biol 44:1519–1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ratelade J, Verkman AS (2014) Inhibitor(s) of the classical complement pathway in mouse serum limit the utility of mice as experimental models of neuromyelitis optica. Mol Immunol 62:104–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ratelade J, Zhang H, Saadoun S, Bennett JL, Papadopoulos MC, Verkman AS (2012) Neuromyelitis optica IgG and natural killer cells produce NMO lesions in mice without myelin loss. Acta Neuropathol 123:861–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Rossi A, Moritz TJ, Ratelade J, Verkman AS (2012) Super‐resolution imaging of aquaporin‐4 orthogonal arrays of particles in cell membranes. J Cell Sci 125:4405–4412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Saadoun S, Waters P, Bell BA, Vincent A, Verkman AS, Papadopoulos MC (2010) Intra‐cerebral injection of neuromyelitis optica immunoglobulin G and human complement produces neuromyelitis optica lesions in mice. Brain 133:349–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Saadoun S, Waters P, MacDonald C, Bell BA, Vincent A, Verkman AS, Papadopoulos MC (2012) Neutrophil protease inhibition reduces neuromyelitis optica‐immunoglobulin G‐induced damage in mouse brain. Ann Neurol 71:323–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Sagan SA, Winger RC, Cruz‐Herranz A, Nelson PA, Hagberg S, Miller CN et al (2016) Tolerance checkpoint bypass permits emergence of pathogenic T cells to neuromyelitis optica autoantigen aquaporin‐4. Proc Natl Acad Sci U S A 113:14781–14786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Saini H, Rifkin R, Gorelik M, Huang H, Ferguson Z, Jones MV, Levy M (2013) Passively transferred human NMO‐IgG exacerbates demyelination in mouse experimental autoimmune encephalomyelitis. BMC Neurol 13:104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Smith AJ, Jin BJ, Ratelade J, Verkman AS (2014) Aggregation state determines the localization and function of M1‐ and M23‐aquaporin‐4 in astrocytes. J Cell Biol 204:559–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Tradtrantip L, Asavapanumas N, Phuan PW, Verkman AS (2014) Potential therapeutic benefit of C1‐esterase inhibitor in neuromyelitis optica evaluated in vitro and in an experimental rat model. PLoS ONE 9:e106824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Tradtrantip L, Asavapanumas N, Verkman AS (2013) Therapeutic cleavage of anti‐aquaporin‐4 autoantibody in neuromyelitis optica by an IgG‐selective proteinase. Mol Pharmacol 83:1268–1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Tradtrantip L, Duan T, Yeaman MR, Verkman AS (2019) CD55 upregulation in astrocytes by statins as potential therapy for AQP4‐IgG seropositive neuromyelitis optica. J Neuroinflammation 16:57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Tradtrantip L, Felix CM, Spirig R, Morelli AB, Verkman AS (2018) Recombinant IgG1 Fc hexamers block cytotoxicity and pathological changes in experimental in vitro and rat models of neuromyelitis optica. Neuropharmacology 133:345–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Tradtrantip L, Ratelade J, Zhang H, Verkman AS (2013) Enzymatic deglycosylation converts pathogenic neuromyelitis optica anti‐aquaporin‐4 immunoglobulin G into therapeutic antibody. Ann Neurol 73:77–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Tradtrantip L, Yao X, Su T, Smith AJ, Verkman AS (2017) Bystander mechanism for complement‐initiated early oligodendrocyte injury in neuromyelitis optica. Acta Neuropathol 134:35–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Tradtrantip L, Zhang H, Anderson MO, Saadoun S, Phuan PW, Papadopoulos MC et al (2012) Small‐molecule inhibitors of NMO‐IgG binding to aquaporin‐4 reduce astrocyte cytotoxicity in neuromyelitis optica. FASEB J 26:2197–2208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Tradtrantip L, Zhang H, Saadoun S, Phuan PW, Lam C, Papadopoulos MC et al (2012) Anti‐aquaporin‐4 monoclonal antibody blocker therapy for neuromyelitis optica. Ann Neurol 71:314–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Tuller F, Holzer H, Schanda K, Aboulenein‐Djamshidian F, Hoftberger R, Khalil M et al (2016) Characterization of the binding pattern of human aquaporin‐4 autoantibodies in patients with neuromyelitis optica spectrum disorders. J Neuroinflammation 13:176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Verkman AS, Smith AJ, Phuan PW, Tradtrantip L, Anderson MO (2017) The aquaporin‐4 water channel as a potential drug target in neurological disorders. Expert Opin Ther Targets 21:1161–1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Vincent T, Saikali P, Cayrol R, Roth AD, Bar‐Or A, Prat A, Antel JP (2008) Functional consequences of neuromyelitis optica‐IgG astrocyte interactions on blood‐brain barrier permeability and granulocyte recruitment. J Immunol 181:5730–5737. [DOI] [PubMed] [Google Scholar]
- 61. Weinshenker BG, Wingerchuk DM (2017) Neuromyelitis spectrum disorders. Mayo Clin Proc 92:663–679. [DOI] [PubMed] [Google Scholar]
- 62. Wingerchuk DM, Banwell B, Bennett JL, Cabre P, Carroll W, Chitnis T et al (2015) International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology 85:177–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Wolburg H, Wolburg‐Buchholz K, Fallier‐Becker P, Noell S, Mack AF (2011) Structure and functions of aquaporin‐4‐based orthogonal arrays of particles. Int Rev Cell Mol Biol 287:1–41. [DOI] [PubMed] [Google Scholar]
- 64. Yao X, Adams MS, Jones PD, Diederich CJ, Verkman AS (2019) Noninvasive, targeted creation of neuromyelitis optica pathology in AQP4‐IgG seropositive rats by pulsed focused ultrasound. J Neuropathol Exp Neurol 78:47–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Yao X, Su T, Verkman AS (2016) Clobetasol promotes remyelination in a mouse model of neuromyelitis optica. Acta Neuropathol Commun 4:42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Yao X, Verkman AS (2017) Complement regulator CD59 prevents peripheral organ injury in rats made seropositive for neuromyelitis optica immunoglobulin G. Acta Neuropathol Commun 5:57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Yao X, Verkman AS (2017) Marked central nervous system pathology in CD59 knockout rats following passive transfer of neuromyelitis optica immunoglobulin G. Acta Neuropathol Commun 5:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Zeka B, Hastermann M, Hochmeister S, Kogl N, Kaufmann N, Schanda K et al (2015) Highly encephalitogenic aquaporin 4‐specific T cells and NMO‐IgG jointly orchestrate lesion location and tissue damage in the CNS. Acta Neuropathol 130:783–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Zeka B, Hastermann M, Kaufmann N, Schanda K, Pende M, Misu T et al (2016) Aquaporin 4‐specific T cells and NMO‐IgG cause primary retinal damage in experimental NMO/SD. Acta Neuropathol Commun 4:82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Zhang Y, Bao Y, Qiu W, Peng L, Fang L, Xu Y, Yang H (2018) Structural and visual functional deficits in a rat model of neuromyelitis optica spectrum disorders related optic neuritis. Exp Eye Res 175:124–132. [DOI] [PubMed] [Google Scholar]
- 71. Zhang H, Bennett JL, Verkman AS (2011) Ex vivo spinal cord slice model of neuromyelitis optica reveals novel immunopathogenic mechanisms. Ann Neurol 70:943–954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Zhang H, Verkman AS (2013) Eosinophil pathogenicity mechanisms and therapeutics in neuromyelitis optica. J Clin Invest 123:2306–2316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Zhang H, Verkman AS (2014) Longitudinally extensive NMO spinal cord pathology produced by passive transfer of NMO‐IgG in mice lacking complement inhibitor CD59. J Autoimmun 53:67–77. [DOI] [PMC free article] [PubMed] [Google Scholar]