

Abstract
Pseudomonas aeruginosa is one of the leading causes of nosocomial pneumonia and its associated mortality. Moreover, extensively drug-resistant high-risk clones are globally widespread, presenting a major challenge to the healthcare systems. Despite this, no vaccine is available against this high-concerning pathogen. Here we tested immunogenicity and protective efficacy of an experimental live vaccine against P. aeruginosa pneumonia, consisting of an auxotrophic strain which lacks the key enzyme involved in D-glutamate biosynthesis, a structural component of the bacterial cell wall. As the amounts of free D-glutamate in vivo are trace substances in most cases, blockage of the cell wall synthesis occurs, compromising the growth of this strain, but not its immunogenic properties. Indeed, when delivered intranasally, this vaccine stimulated production of systemic and mucosal antibodies, induced effector memory, central memory and IL-17A-producing CD4+ T cells, and recruited neutrophils and mononuclear phagocytes into the airway mucosa. A significant improvement in mice survival after lung infection caused by ExoU-producing PAO1 and PA14 strains was observed. Nearly one third of the mice infected with the XDR high-risk clone ST235 were also protected. These findings highlight the potential of this vaccine for the control of acute pneumonia caused by this bacterial pathogen.
Author summary
Pseudomonas aeruginosa is an opportunistic bacterium and one of the most common causes of healthcare-associated diseases, including acute pneumonia, causing high mortality within immunocompromised hosts. Most of these infections are strikingly difficult to treat using conventional antibiotic therapies, since this microorganism displays high intrinsic resistance to a wide range of antibiotics. Moreover, to date, no vaccine is available for prevention. Here we used a mutated bacterial strain, which is unable to replicate in vivo and to cause disease, as a live vaccine against acute pneumonia caused by this pathogen. When applied intranasally, this vaccine induced immunity both at local and distant body sites, activating immune cells which were recruited into the airway mucosa. This evoked immune response reduced the number of non-surviving mice after infection with two cytotoxic P. aeruginosa strains causing acute lung infection. Some protection was also observed against an internationally disseminated cytotoxic strain. These data indicate that this is a promising vaccine candidate against P. aeruginosa-pneumonia.
Introduction
Pseudomonas aeruginosa (PA) is one of the most common gram-negative pathogens causing pneumonia in immunocompromised patients[1]. Moreover, the mortality rate of ventilator-associated pneumonia (VAP) due to PA is higher than that due to other pathogens[2]. Acute nosocomial pneumonias due to PA are typically the result of direct trauma, such as damage to the epithelium due to mechanical ventilation in VAP patients[3].
During acute infection, PA secretes exotoxins into the environment that damage host tissue. Namely, type III secretion system (TTSS) works as a molecular syringe, delivering toxins into the cytosol of target eukaryotic cells. ExoU, one of these toxins, disrupts the integrity of the lipid membrane causing epithelial cell damage and lung injury. It also contributes to the release of inflammatory mediators that give rise to inflammation and septic shock. Indeed, high cytotoxicity, severity of lung epithelial injury, and bacterial dissemination into the circulation correlated with the exoU genotype in different PA strains[4].
The resistance rates of PA are increasing in many parts of the world: recent studies reported the widespread presence of extreme drug-resistant (XDR) high-risk clones in healthcare settings[5]. Therefore, several evolving translational strategies are being explored for the control and therapy of PA, including immunotherapy[6], phage therapy[7] and vaccination. Several types of vaccines have been developed and tested in Phases I–III clinical trials[8]; however, currently there is no approved option for use in humans.
Respiratory tract infections usually occur or initiate at a mucosal surface like the one lining the lumen of the lung[9]. At the airway surface, inhaled PA become trapped in the viscous mucous layer. Then, flagella, lipopolysaccharide, and type 4 pili are recognized by host pattern recognition receptors such as TLRs thus initiating an inflammatory response[9]. After recognition, activated alveolar macrophages as well as neutrophils recruited by IL-8 phagocytose and kill PA. Dendritic cells (DCs) sample the lung lumen and trigger the adaptive response by presenting antigens to T cells. Therefore, vaccine-based immunity against PA should ideally induce local immunity at the lung mucosa.
Mucosal vaccination has superior ability to induce local mucosal immune responses over systemic vaccination[10]. In addition, immunization at one mucosal site can result in antibody secretion systemically, as well as at other selected mucosal sites[10]. These features can prevent the establishment and dissemination of an infection, particularly if it initiates at a mucosal surface.
We previously characterized an experimental live vaccine against PA (α-PA vaccine), composed of a D-glutamate (D-Glu) auxotrophic strain with inactivated Glutamate racemase (MurI), a key enzyme that provides the D-Glu required for peptidoglycan biosynthesis[11]. This live vaccine has a self-limited growth in vivo, elicits functional and cross-reactive antibodies, triggers cellular immunity and confers protection against disease caused by different PA strains in a murine sepsis model. The robust protection observed in immunized mice–~100% vaccine efficacy–, and the so far scarce description of effective prototype vaccines prompted us to evaluate whether this D-Glu auxotroph holds the potential to be immunogenic and effective against lethal pneumonia caused by PA.
In the present study, we explored a mucosal vaccination route to deliver our live α-PA vaccine, having found that it elicits a multifactorial immunity, involving T cells and systemically and mucosally-produced antibodies. This multifactorial immunity was correlated with partial, but considerable protection against acute fatal lung infection caused by two highly virulent ExoU-positive cytotoxic PA strains, and mild protection against a XDR disseminated clone, also ExoU-positive. Some constrains posed by the mild dose-dependent toxic effect observed, prompted us to test parenteral routes for immunization also, and their effect on mucosal immunity against acute pneumonia. Intramuscular, intradermal and subcutaneous immunization conferred significant improvement in survival of PA14-infected mice, however, transient adverse reactions were observed with the last two. Intramuscular immunization conferred only moderate protection, however, when applied in combination with intranasal route, it allowed using lower and safer doses of vaccine intranasally resulting in considerable efficacy.
We conclude that our D-Glu auxotrophic strain enables local, as well as systemic immunity providing heterogeneous but considerable protection against lethal pneumonia caused by cytotoxin-producing strains, thus it should be considered as a promising prototype vaccine against PA in the future.
Results
Characterization of α-PA vaccine-induced antibody responses after intranasal immunization
PA PAO1 was previously genetically manipulated to obtain a D-Glu auxotrophic strain, PAO1 ΔmurI, by eliminating the MurI coding gene, murI[11]. Mice were inoculated intranasally (IN) with PAO1 ΔmurI (2×108 CFU)–live α-PA vaccine–on days 0 and 14. An extra immunization was performed on day 8, when using a three-dose schedule. Control mice were inoculated saline. PAO1-specific antibody levels were determined in sera obtained at different time points. When using the two-dose schedule (Fig 1A), significantly elevated levels of IgM were present on day 7, while increased IgG1, IgG2a, IgG2b and IgG3 titers were detected on days 20 and 42. IgM levels were significantly reduced on day 42, indicating isotype switching.
Fig 1. α-PA vaccine generates cross-reactive antibodies and heterogeneous protection against PA-pneumonia.

(A) Immunization schedule and antibody titers against PAO1 in vaccinated (n = 7) and control (n = 7) mice after one or two IN immunizations with α-PA vaccine (2×108 CFU), or saline administration, respectively. (B) IgG titers against different live and Hk PA strains in vaccinated (n = 6–7) and control (n = 5–7) mice after two IN immunizations with α-PA vaccine (2×108 CFU), or saline administration, respectively. (C) Immunization schedule and antibody titers against PAO1 in vaccinated (n = 5) and control (n = 6–7) mice after three IN immunizations with α-PA vaccine (2×108 CFU), or saline administration, respectively. (A-C) S, saline; D, day. (A, B) *P<0.05 (Kruskal-Wallis test), compared with saline and between the indicated groups. (B) #P<0.05 (Mann-Whitney U test), compared with saline group. (C) *P<0.05 (Mann-Whitney U test), compared with saline group. (D) Immunization schedule, mice survival and assessment of murine disease severity score (MDSS) after IN administration of α-PA vaccine (2×108 CFU), or saline and IN challenge with PAO1 ExoU+ (7×105 CFU, n = 7), PA14 (1×106 CFU, n = 8), ST235 (3×107 CFU, n = 7) and PAO1 (2×106 CFU, n = 7–8). (E) Mice survival after IN challenge with different doses of PAO1 (n = 3–5 mice/group). (F) Immunization schedule and mice survival after IN administration of α-PA vaccine (2×108 CFU, n = 6), or saline (n = 7) and IN challenge with PA14 (1×106 CFU). (G) Mice survival after IN immunization with α-PA vaccine (2×108 CFU, n = 8), inactivated PAO1 (2×108 CFU, n = 8) or saline administration (n = 15) and IN challenge with PA14 (1×106 CFU), according to schedule (D). (D, F and G) *P<0.05 (log-rank test), compared with saline group. (D) #P<0.05 (Mann-Whitney U test). (G) #P<0.05 (log-rank test), compared with mice administered inactivated PAO1.
Cross-reactivity of the total IgG raised by using the two-dose schedule was determined against 11 heterologous PA strains (S1 Table and Fig 1B). The levels of anti- PA14 (ExoU-producing strain), -ST235 (XDR high-risk clone), -ST175 (XDR high-risk clone), -LES400 and -12142 (epidemic strains from CF patients), -51442390 (MemR mucoid isolate from a CF patient), -29606 and -28757 (mucoid isolates from CF patients), -LES431 (epidemic strain from a non-CF patient), -51441321 and -28562 (MDR/mucoid isolates from bronchiectasis patients) IgG’s were significantly elevated on day 42, compared with the saline group. The mucoid phenotype is associated with chronic infections from cystic fibrosis patients[12]. When using a three-dose schedule, IN vaccination also resulted in significantly elevated PAO1-specific antibody levels of all assessed isotypes (Fig 1C). IgG titers against boiled PAO1 and PA14 (which should destroy heat-labile protein epitopes) (Fig 1B) were similar to those recognizing whole bacteria, indicating that IgG antibodies raised by immunization mostly recognize heat-stable antigens, which might include O antigen.
Assessing protective efficacy against acute lung infection using α-PA IN vaccination
We assessed the survival of mice IN-challenged with three cytotoxin-producing PA strains causative of acute lung infection for testing the cross-protection of the α-PA vaccine and the robustness of the pneumonia model (Fig 1D).
PAO1 ExoU+ is a derivative from PAO1 carrying exoU gene. Introduction of this gene in PAO1 confers a cytotoxic phenotype and increased virulence in a murine model of acute pneumonia and systemic spread[13]. PA14 is a highly cytotoxic exoU+ reference strain[14,15] representing the most common clonal group worldwide[16]. PA ST235 is an international XDR high-risk clone, which is among the most disseminated XDR clones in hospitals worldwide[17]. ST111 and ST175 are likely the other more widespread ones[5,18,19]. Here we used the previously described ST235 strain 8[15], since it is able to cause mortality in a peritonitis/sepsis model in mice, in contrast to ST111 and ST175. Moreover, ST235 contains the exoU-encoded exotoxin[17].
Survival rates of vaccinated mice using a two-dose vaccination schedule were 86, 88 and 29% after challenge with PAO1 ExoU+, PA14 and ST235, respectively; whereas control mice presented 0, 13 and 0% survival (Fig 1D). Despite this heterogeneous protection, the differences in survival were significant for the three assays, indicating a general reduction in the severity of disease in vaccinated animals.
We also tested vaccine efficacy against the wild-type PAO1, as ExoU-negative strains typically require higher challenge doses to cause 100% lethality, thus giving a better assessment of the potency of the vaccine. Indeed, whereas inoculation of 7×105 CFU of PAO1 ExoU+ caused 100% mortality (Fig 1D), challenge doses as high as 4×107 CFU of PAO1 were required to observe 100% death (Fig 1E). But because infected mice showed indicators of pain, suffering and illness, a murine disease severity score (MDSS) was used for monitoring surrogate endpoints and determining disease severity after challenge with a lower dose. Five criteria were assessed to provide an overall score between 1 and 10, which subsequently dictated the clinical condition and/or euthanasia (HEP, human endpoint) (S2 Table). Succumbed mice and HEP were given a score of 11. Compared to vaccinated mice, sham-immunized mice had a significantly higher mean score (0.71 vs 10, respectively) (Fig 1D). Moreover, 2 out of 8 control mice died, whereas 3 presented scores compatible for HEP, thus indicating a reduction in the severity of infection. Using a three-dose immunization schedule, survival rates of vaccinated mice were 83%, after challenge with PA14, whereas all control mice died (Fig 1F). Considering that using an extra immunization dose did not boost the protective effect of the vaccine substantially, we kept the two-dose schedule for the posterior IN immunizations. To further investigate whether our auxotrophic strain has any benefit as a vaccine over inactivated bacteria, we compared its protective efficacy with formalin-inactivated PAO1. As shown in Fig 1G, only one out of 8 mice administered α-PA vaccine and infected with PA14 died, whereas 93% of control mice succumbed to acute pneumonia within 84 h. In contrast with live vaccine, 75% of mice administered formalin-inactivated PAO1 succumbed to infection. The protection conferred by α-PA vaccine was significantly higher when compared with formalin-inactivated PAO1. These results show that IN immunization with α-PA vaccine offers an advantage over wild-type inactivated bacteria in the improvement of survival.
Cellular immune response induced by α-PA IN vaccination before and after infection
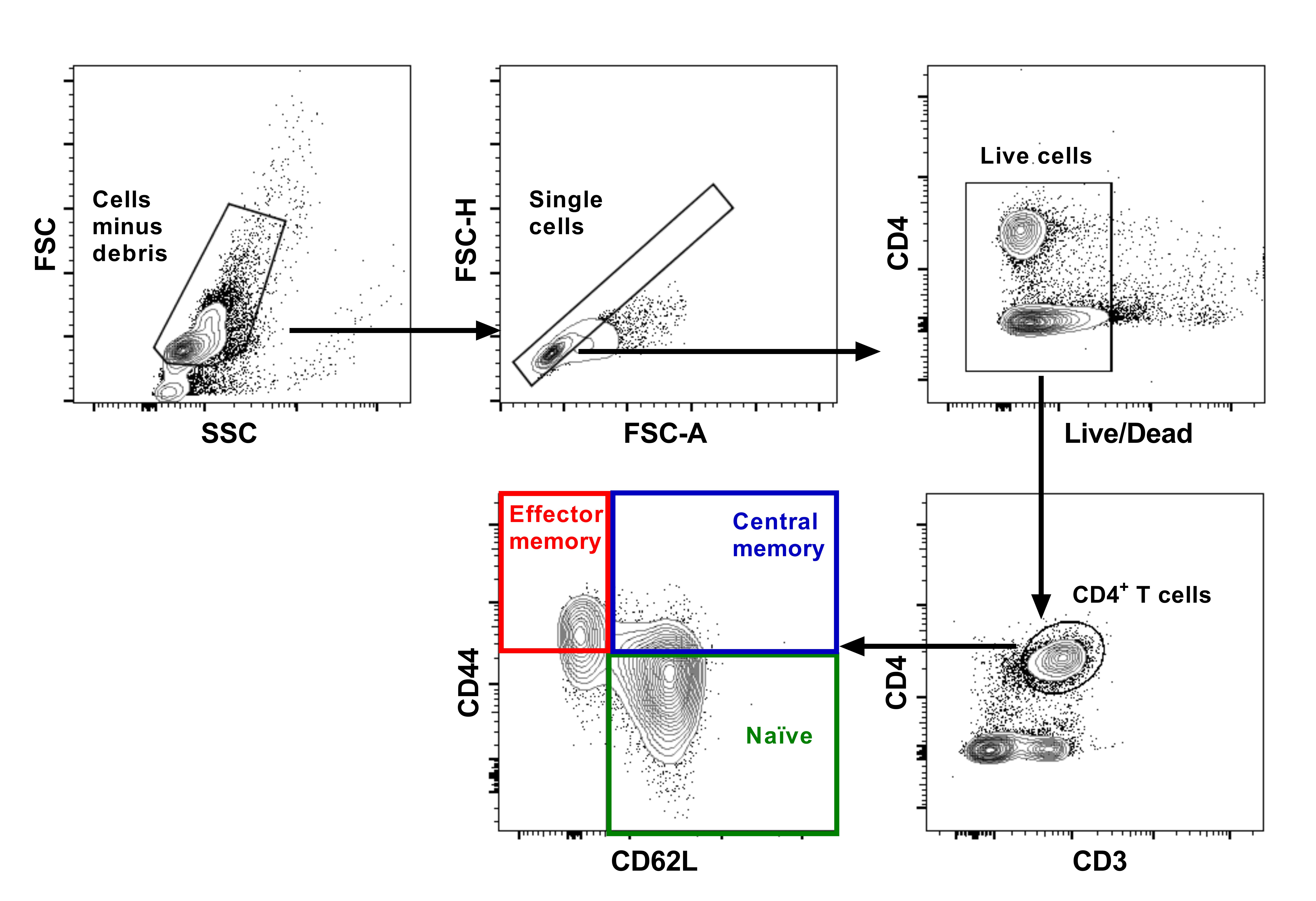
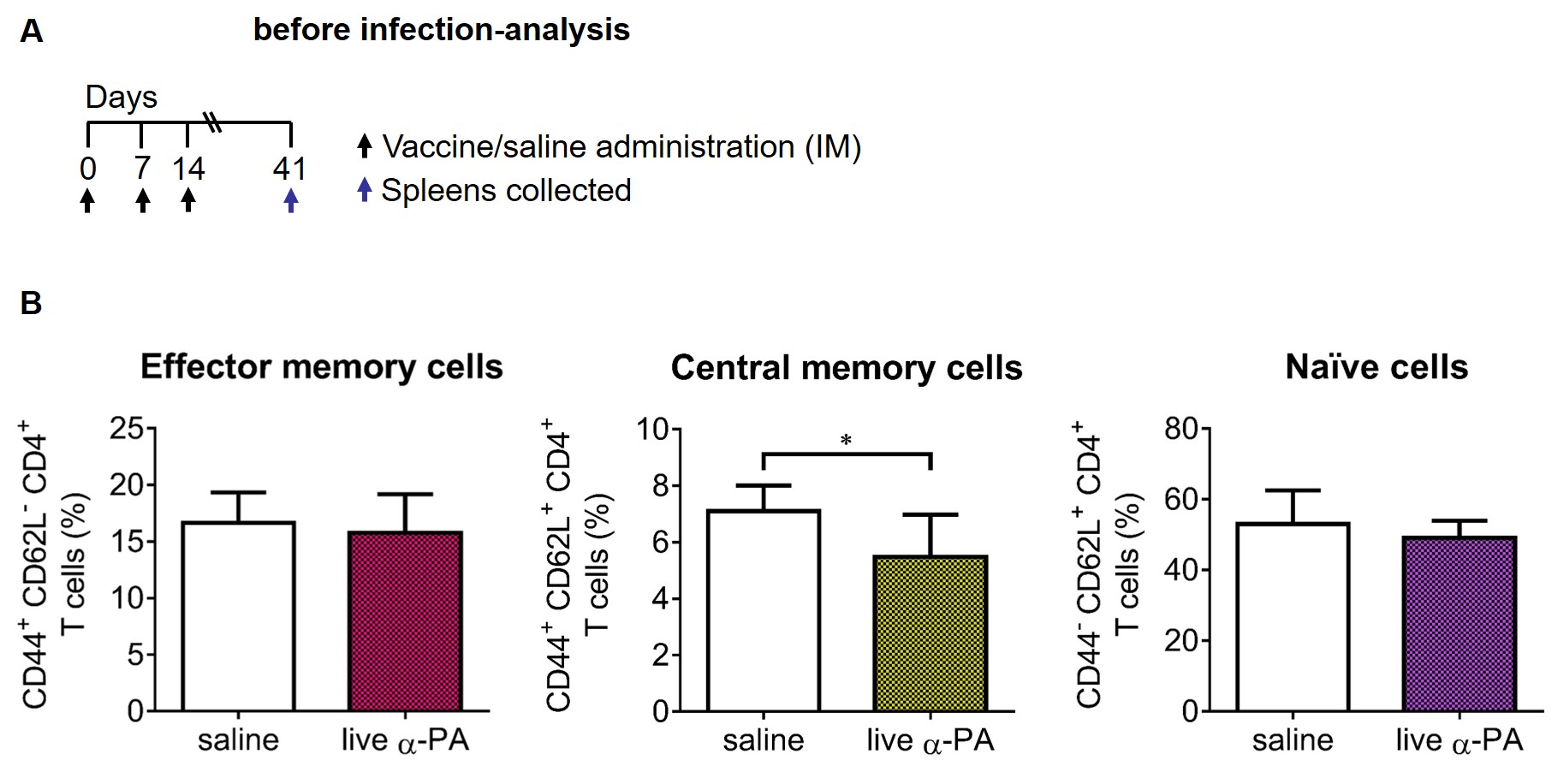
As shown in Fig 2, the proportion of CD4+ T cells displaying the CD44+CD62L- and CD44+CD62L+ phenotypes were slightly but significantly increased in the spleens of α-PA vaccinated mice as compared to controls, which respectively indicates the stimulation of effector and central memory CD4+ T cells. The gating strategy used to discriminate memory cells by flow cytometry is presented in S1 Fig.
Fig 2. Production of memory CD4+ T cells in vaccinated mice.

(A) Mice (n = 6/group) were immunized with α-PA vaccine (2×108 CFU) or administered saline, according to the schedule; then spleens were collected. (B) Percentage of spleen effector memory (CD44+ CD62L-), central memory (CD44+ CD62L+) and naïve (CD44- CD62L+) CD4+ T cells. Contour plots correspond to a representative example of CD4-gated T cells of the analyzed samples. Analysis regions were set according to FMO and isotype control-stained samples. Numbers inside plot regions represent means ± SD of the frequency of cells due to respective cell staining. Bars represent mean ± SD of data. *P<0.05 (t-test).
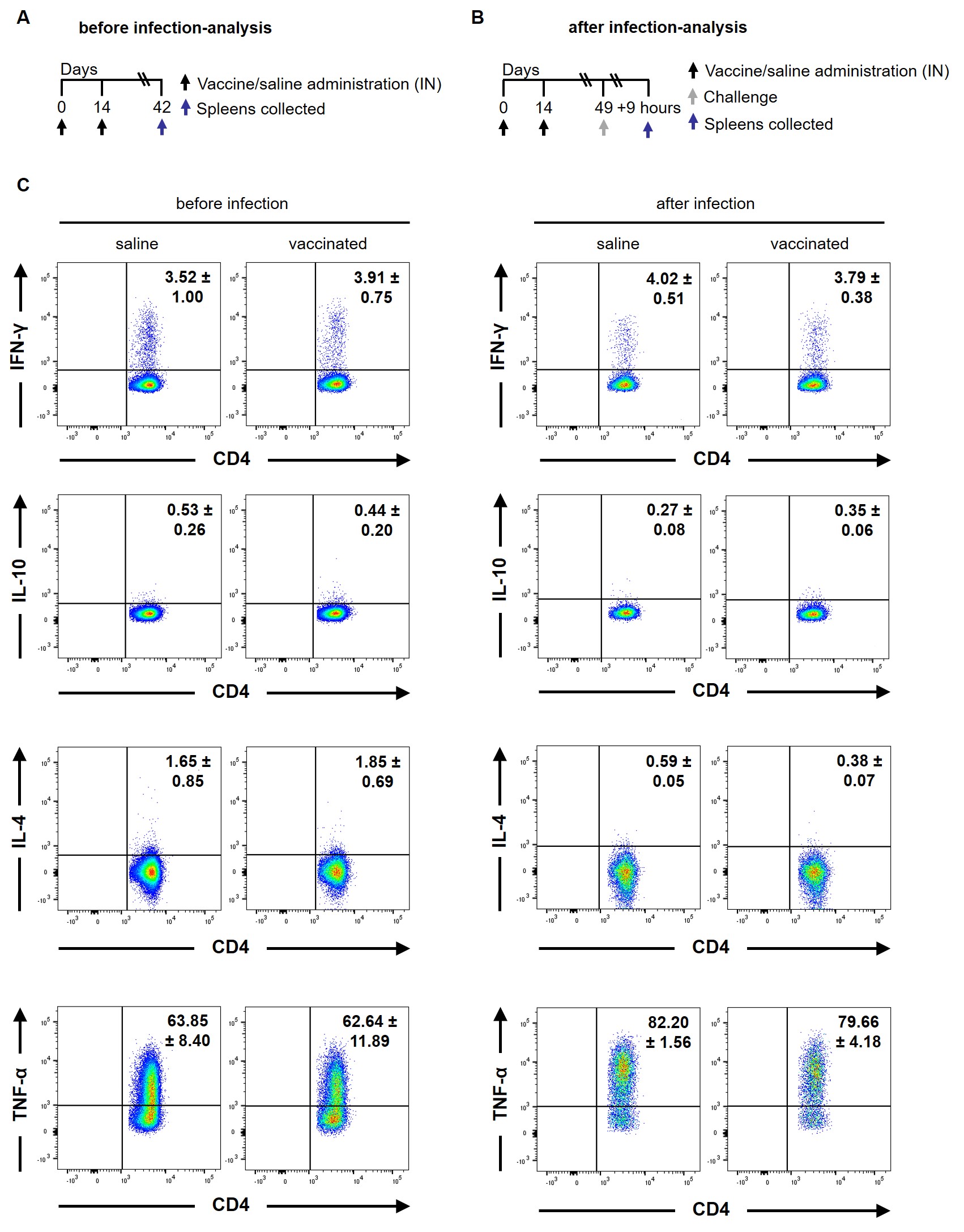
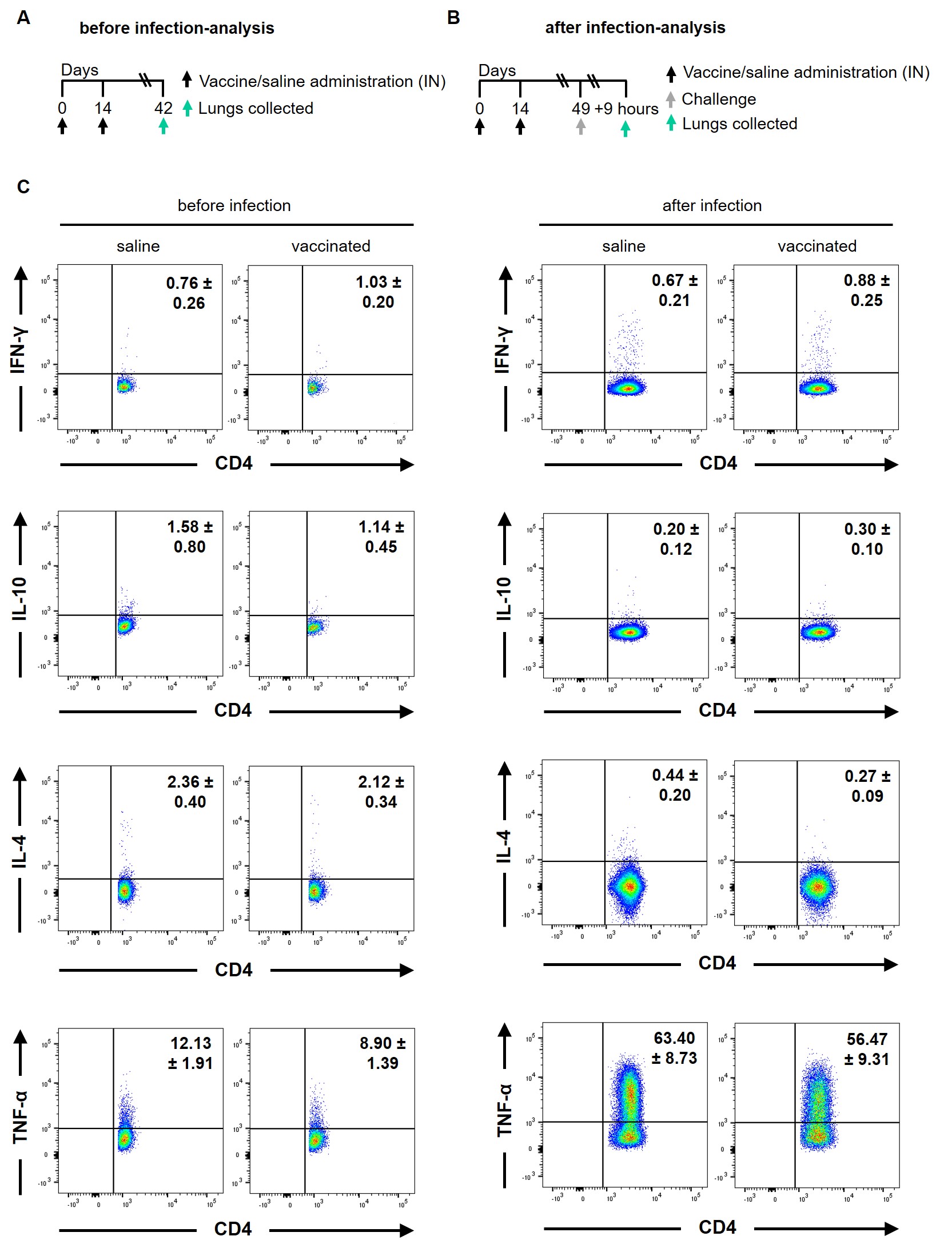
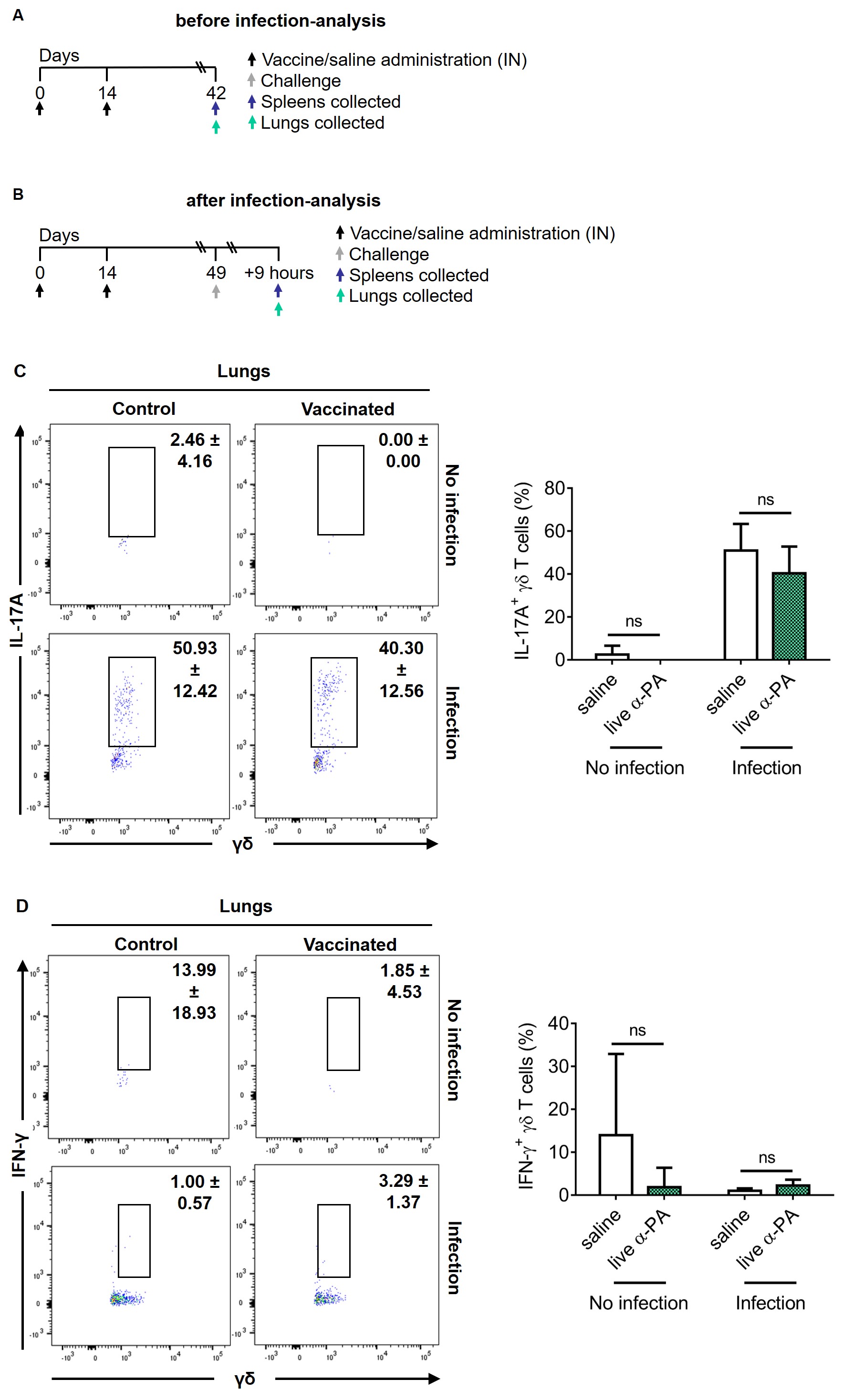
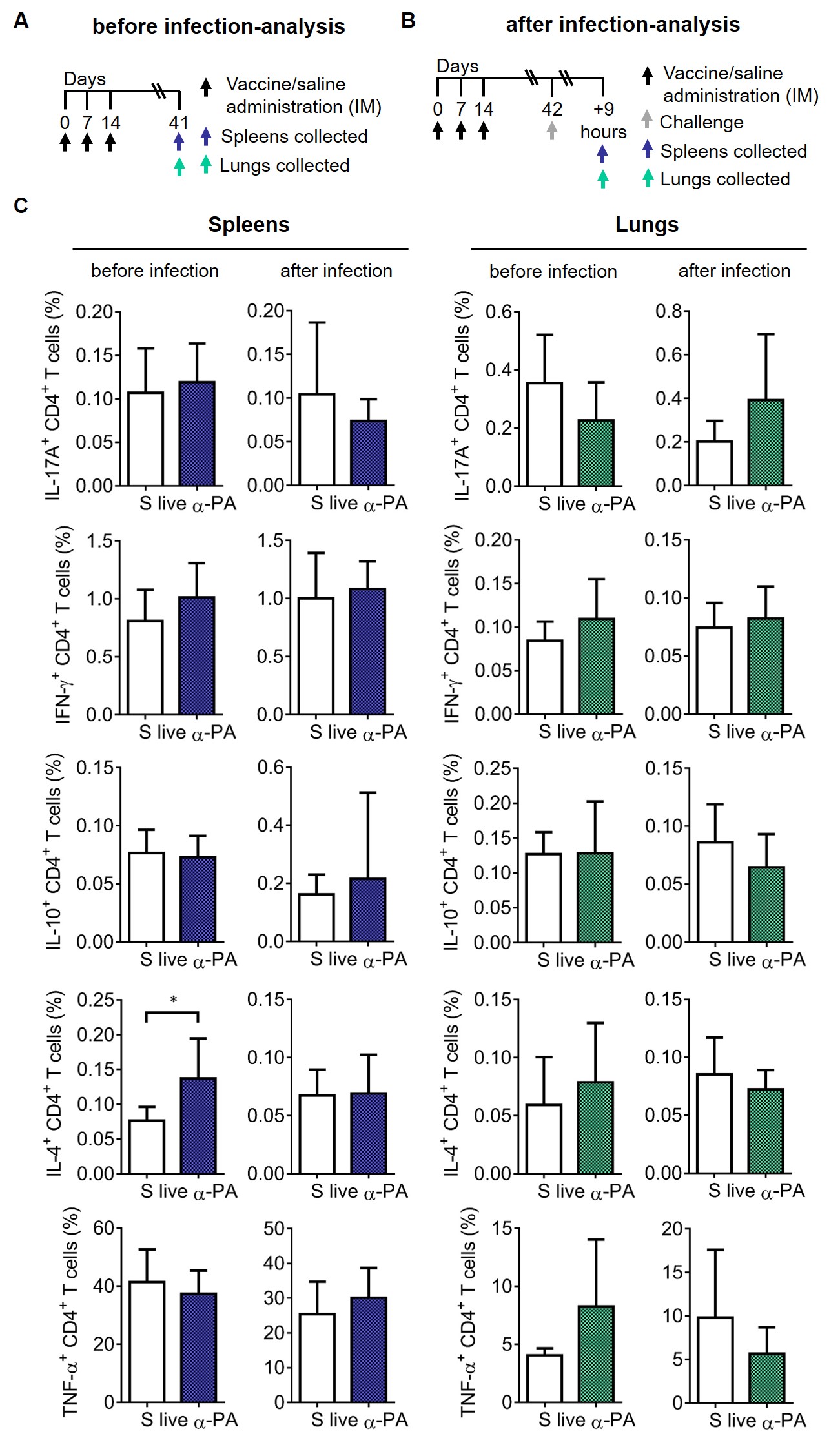
To better elucidate the effector function of CD4+ T cells induced by vaccination, the proportions of splenic and lung CD4+ T cells producing IL-17A (Fig 3), IFN-γ, IL-10, IL-4 and TNF-α (S2 Fig) was determined by flow cytometry analysis in both infected and non-infected mice. Representative dot plots showing different cellular populations are presented in Fig 3 and S3 and S4 Figs. The corresponding gating strategy is presented in S5 Fig. An increased frequency of splenic CD4+ T cells expressing IL-17A was observed for vaccinated mice compared to controls, both in infected and non-infected mice (Fig 3C). The same was observed for CD4+ T cells expressing IL-17A from lungs (Fig 3D). The frequencies of splenic CD4+ T cells expressing IFN-γ, IL-10 and TNF-α were found similar in vaccinated and control mice, whereas a reduction of CD4+IL-4+ cells frequency was observed upon infection in vaccinated mice (S2 Fig). In the lungs, no different proportions of cells producing any of these cytokines were found between vaccinated mice and controls, except for a decreased frequency of CD4+ T cells expressing TNF-α in non-infected vaccinated mice (S2 Fig). γδ T cells have also been shown to be a potent source of IL-17 and, in some cases, to produce more IL-17 than αβ T cells[20]. As shown in S6 Fig, a considerable increase of IL-17A+ γδ T cells was detected in the lungs upon infection, that was however not different between vaccinated and control mouse groups. Also, no significant differences in IFN-γ+ γδ T cells were observed between vaccinated and control mice (the gating strategy is shown in S7 Fig). This suggests that protection conferred by our vaccine is not dependent on these T cell populations.
Fig 3. Increased production of IL-17A by CD4+ T cells of vaccinated mice.

(A, B) Mice (n = 6/group) were immunized with α-PA vaccine (2×108 CFU) or administered saline, according to the schedule; then spleens and lungs were collected. In (B), mice (n = 6/group) were infected with PA14 (1×106 CFU), 9 h before the spleens and lungs were obtained. (C, D) Representative examples and percentage of CD4-gated T cells expressing IL-17A obtained from spleens (C) and lungs (D) of infected and non-infected mice, detected by intracellular staining after stimulation with PMA/ionomycin. Analysis regions were set according to FMO and isotype control-stained samples. Numbers inside dot plot regions represent means ± SD of the frequency of cells due to respective cytokine staining. Bars represent mean ± SD of data. *P<0.05, **P<0.01 (t-test).
To confirm the type of T cell response generated by the α-PA vaccine, IFN-γ, IL-17A, IL-10 and IL-4 levels were evaluated in culture supernatants of antigen-stimulated splenocytes obtained from vaccinated and non-vaccinated mice prior to and after being infected with PA14. As shown in S8 Fig, splenocytes recovered from vaccinated-infected mice responded to heat-killed (Hk) PA14 by producing significantly higher amounts of pro-inflammatory cytokines IL-17A and IFN-γ than sham-immunized controls. Nevertheless, the results from splenocyte cultures of non-infected mice indicate that immunization preferentially induces commitment of responding cells for IL-17A production, as the PA14 antigen induced IFN-γ production also in cells from non-immunized animals. Higher production of counterinflammatory cytokine IL-10 was also observed for cells obtained from vaccinated mice, however, at lower amounts than IL-17A and IFN-γ. IL-4 was not detected in any assessed condition.
These results indicate that α-PA vaccine triggers a cellular immune response polarized to the production of IL-17A in spleens and lungs, suggesting that this cytokine mediates the protective effect of the vaccine against the acute lung infectious process.
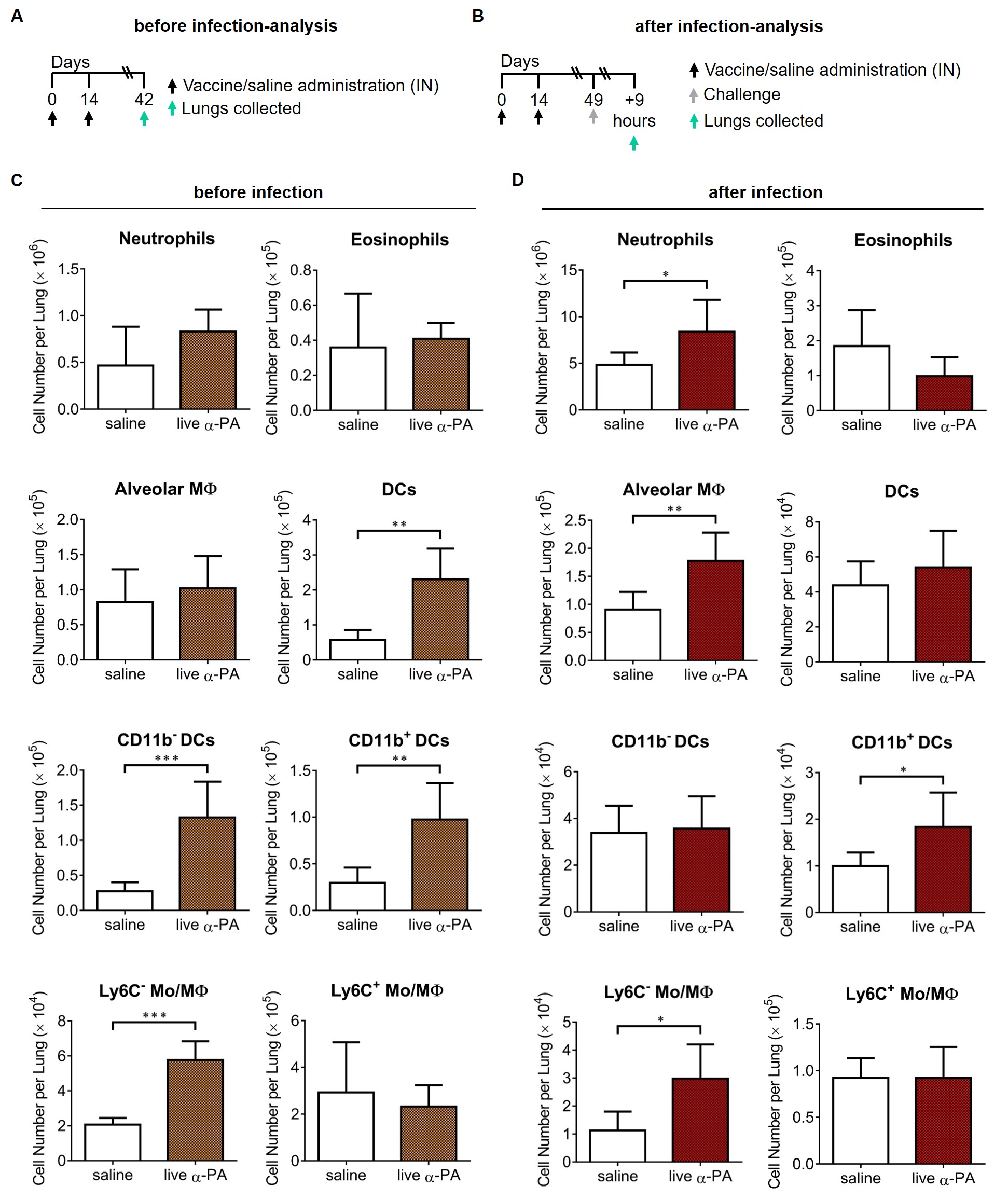
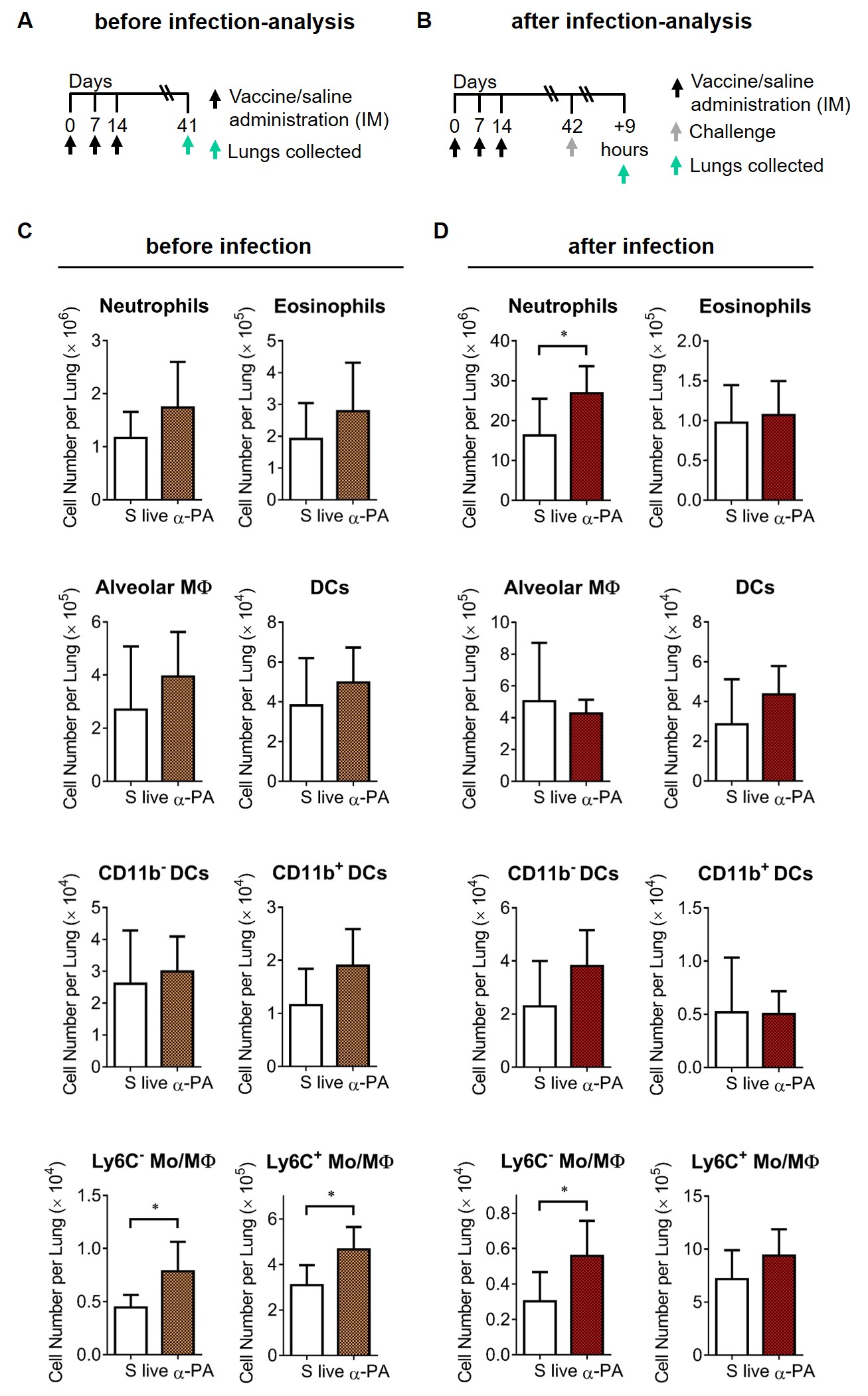
Bacteria clearance requires phagocyte activity and neutrophils and other phagocytes are essential innate cellular effectors for immunity against PA-pneumonia[9,21–23]. Thus, the numbers of myeloid cells of different populations were determined in the lungs of α-PA vaccinated and control mice, before and after PA infection, using flow cytometry (S9 Fig). Neutrophils, eosinophils, alveolar macrophages (AM), recruited monocytes and dendritic cell (DC) populations were defined as shown in the gating strategy presented in S10 Fig. IN vaccination with α-PA led to a significant increase in the number of lung DCs of both CD11b- and CD11b+ subsets and of Ly6C- Monocytes/Macrophages (Mo/Mφ). CD11b+ increase is especially relevant as they include professional antigen presenting cells (APCs). Upon infection, the numbers of CD11b+ DC in vaccinated mice remained above controls. Ly6C- Monocytes/Macrophages (Mo/Mφ), often recognised as resident monocytes that are the precursors of tissue macrophages[24], were still elevated upon infection. Alveolar macrophages and neutrophils were also found at increased numbers in infected-vaccinated mice when compared to controls. These results suggest that vaccination increased the recruitment/proliferation of mononuclear and polymorphonuclear cells into the lungs, which might contribute to more bacterial clearance.
Intramuscular (IM) immunization with α-PA vaccine stimulates humoral immunity but offers reduced protection against infection
Because different vaccine candidates against PA infections have been tested in clinical trials using IM administration[25,26] we tested the immune response generated with α-PA vaccine using this route. Mice were IM-inoculated with α-PA vaccine (3×107 CFU) on days 0, 7 and 14. Control mice were sham-immunized with saline. To assess the humoral response elicited by the immunization, the levels of PAO1-specific IgM and IgG isotypes were determined in sera (Fig 4A). The cross-reactivity of the IgG obtained after finalizing the vaccination schedule was also determined as before. Serum antibody levels were comparable to those obtained when using the IN route on day 42 (Figs 4B and 1A), with the exception of IgM, which remained elevated after IM immunization. IM immunization also resulted in increased anti-PA IgG1, IgG2a, IgG2b and IgG3 levels. A similar trend was seen for the cross-reactivity of serum IgG obtained and for antibodies recognizing heat-stable antigens (Fig 4C). Thus, it is unlikely that different immunization routes may induce IgG antibodies with clearly different specificities, such as protein vs carbohydrate antigens.
Fig 4. IM immunization with α-PA vaccine generates systemic and cross-reactive antibodies but reduced protection against pneumonia.

(A) Immunization schedule for (B) and (C). (B) Antibody titers against PAO1 in vaccinated (n = 6) and control (n = 6) mice after IM immunization with α-PA vaccine (3×107 CFU), or saline administration, respectively. (C) IgG titers against different live and Hk PA strains in vaccinated (n = 6) and control (n = 6) mice after IM immunization with α-PA vaccine (3×107 CFU), or saline administration, respectively. (B, C) S, saline; D, day. *P<0.05 (Mann-Whitney U test), compared with saline group. (D) Immunization schedule and mice survival after IM immunization with α-PA vaccine (3×107 CFU, n = 8), or saline administration (n = 9) and challenge with PAO1 ExoU+ (1×105 CFU). (E) Immunization schedule and mice survival after IM immunization with α-PA vaccine (3×107 CFU, n = 8), or saline administration (n = 8) and challenge with PA14 (9×105 CFU). (F) Immunization schedule and mice survival after IM immunization with α-PA vaccine (3×107 CFU, n = 9), or saline administration (n = 9) and challenge with ST235 (5×106 CFU). (D-F) *P<0.05 (log-rank test).
We next evaluated whether the immune response induced by IM immunization reduces the severity of disease at the same level as IN immunization. Although different vaccination schedules were used for IN and IM delivery, we established this comparison because antibody levels and cross-reactivity obtained was similar between them. After challenging mice with PAO1 ExoU+, IM vaccination resulted in 100% survival (Fig 4D). This result was superior to that obtained with IN immunization (86% survival, Fig 1D). However, when the challenge was performed with PA14, survival of IM-vaccinated mice was 38% (Fig 4E), contrasting with the 88% survival observed for IN-vaccinated mice (Fig 1D). Finally, when using the ST235 strain, all mice receiving the IM-α-PA vaccine did not survive lethal challenge (Fig 4F), in contrast with the 29% survival presented by IN-vaccinated mice (Fig 1D). This suggests that α-PA vaccine is less effective against pneumonia caused by cytotoxin-producing strains when applied through IM route, offering reduced protection.
To elucidate whether the lower protection against the LPS-heterologous strain PA14 after IM immunization could be related with the lack of cellular immune responses, we performed flow cytometry analysis and cytokine quantification in the culture supernatants as previously made for IN-vaccinated mice. In contrast to IN-immunized mice, non-infected IM-vaccinated mice presented no increased numbers of effector and central memory CD4+ T cells, thus indicating that this route of immunization does not stimulate an adaptive response at the same level as the IN-route (S11 Fig). In the spleens of IM-immunized mice, the frequencies of CD4+ T cells expressing IL-17A, IFN-γ, IL-10 and TNF-α were found similar when compared to controls, both before and after infection. The only exception was a significant increased frequency of CD4+ IL-4+ T cells in non-infected mice. In the lungs, no differences in CD4+ T cells populations were found (S12 Fig). On this occasion, none of the assessed cytokines were significantly produced by IM-vaccinated mice, except for IL-17A (S13 Fig). These IL-17A levels, however, were much lower when compared to IN-vaccination (mean 127 vs 2489 pg/mL, before infection; mean 422 vs 4782 pg/mL, after infection; 1:1 stimuli). In contrast to IN vaccination, no significant increase in the number of lung DCs (neither CD11b- nor CD11b+ subsets) was observed after IM immunization (S14 Fig). This might support the observation that T cell differentiation was essentially not different between IM-vaccinated and control mice. Moreover, the increase in Ly6C- Monocytes/Macrophages (Mo/Mφ) was modest when compared to IN vaccination (S14 Fig, panels A and C). Somehow surprisingly, Ly6C+ Monocytes/Macrophages (Mo/Mφ) recruitment was stimulated after IM immunization, but not after IN immunization. Although Ly6C+ monocytes might differentiate into Ly6C- monocytes or moDCs, no increased numbers of cells of such populations were detected upon infection. Upon infection, a significantly higher number of Ly6C- Mo/Mφ and neutrophils were found in IM-vaccinated mice when compared to controls, as previously observed in IN-vaccinated mice, but not of alveolar macrophages (S14 Fig, panels B and D). In short, these results might indicate that IM immunization with live α-PA vaccine does not promote DCs accumulation in the lungs at the same level as IN vaccination, and suggest a minor effect of this immunization route in the numbers of lung myeloid populations.
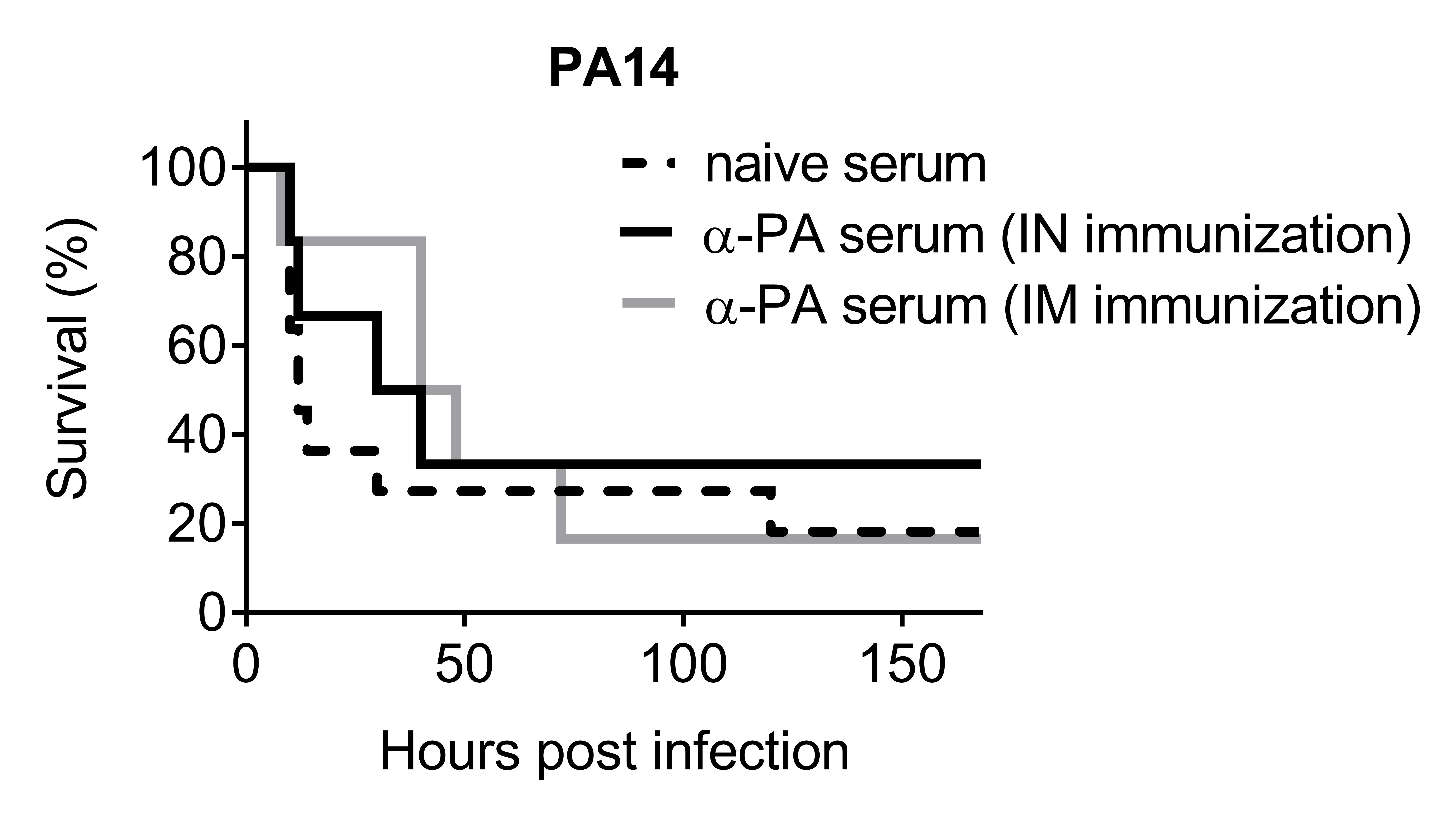
We also compared the different specificities of the antisera from IN- and IM-immunized mice against PA14, using passive immunization experiments. However, mice given IgG-enriched anti-PA serum from immunized donors presented no significant differences in survival when compared with mice given naïve serum (0.3% vs 0.2% vs 0.2% survival, respectively) (S15 Fig). These results suggest that the improvement in survival conferred by IN vaccination is not dependent on serum IgG antibodies and thus the lower protection observed with the IM route cannot be associated with different specificities of the systemic response.
Safety assessment of α-PA vaccine
α-PA vaccine or vehicle (saline) were administered IN and IM to mice, approximately according to the previous regimens and schedules. As clinical toxicological signs can be difficult to discern by visual examination, we recorded mice body weight prior to and during the immunization regimens (S16 Fig). When using the IN route (2×108 CFU), a mean 13% weight loss was observed after the first administration of α-PA vaccine, on day 2. After the second administration (day 16), mice presented 15% weight loss. These differences were significant when using mixed ANOVA with “study day” as within-subjects factor. Althought significant weight differences with respect to controls were observed until completion of the experiment, these animals recovered their body weight at pre-treatment on days 8 and 22 (S16A Fig). In contrast to IN immunization, no body weight loss was observed for IM-vaccinated mice (S16B Fig).
Considering that IM immunization was performed using a lower vaccine dose (3×107 CFU), we tested the same dosing regimen, but using the IN route (S16C Fig). On this occasion, significant weight loss was only seen after the first immunization (13%, day 2), moreover, differences in body weight when compared to controls were transient. This might indicate that administration of α-PA vaccine through IN route induces a mild to moderate dose-dependent toxic effect. When using IN doses of the live α-PA vaccine lower than 3×107 CFU (8×105 CFU and 7×106 CFU, n = 8 mice/group), vaccinated mice presented similar outcomes as controls after challenge with PA14, thus suggesting no protective effect (S17 Fig).
We also tested a combination of IN plus IM immunization with 3×107 CFU of α-PA vaccine (S16D Fig): one IN plus IM administration on days 0 and 14, and extra IM immunization on day 7. Here, we observed some weight loss (not significant) after the first and second vaccine administrations, concurring with IN immunizations, but not after IM administration, on day 7. This might support that combining these two immunization routes does not deepen vaccine toxicity. Again, differences in body weight in vaccinated mice were transient when compared to controls.
Further, we evaluated if the α-PA vaccine could colonize and multiply in the host, after IN or IM administration. Thus, we measured α-PA vaccine loads in the blood, lungs and livers of IN- (n = 4) and IM- (n = 4) vaccinated mice (3×107 CFU). No CFUs were detected in the blood of mice as soon as 2 h after vaccination. Also, lungs and livers obtained on day 8 were sterile. Thus, it seems unlikely that α-PA vaccine strain can colonize the host.
Combination of IN plus IM immunization with α-PA vaccine confers long-term protection against pneumonia
Body weight monitoring showed that using a two-dose regimen with 3×107 CFU of α-PA vaccine administered IN can be less toxic than using 2×108 CFU. To test the efficacy of this alternative dosing regimen, we challenged mice with PAO1 ExoU+ using the previous model of lethal lung infection. We simultaneously tested protection in the long term, challenging mice 6 months after the last immunization (Fig 5A). As shown, survival of vaccinated mice (IN route) was 100%, whereas 86% of control mice died. Thus, IN immunization using 3×107 CFU of α-PA vaccine offered considerable protection in the long-term. Thus, we used this dose thereafter. When the vaccine was administered IM (using 3×107 CFU) or combining IN plus IM routes, 88% and 100% mice survival was observed, respectively, being in line with the previous findings suggesting that IM route is not as protective as the IN one. In this experiment, we also performed a macroscopic observation of lungs and livers collected from mice at two different time points. The first analyzed group were those mice which succumbed to infection between 16 and 24 h after challenge. On this occasion, all mouse lungs showed a visible and disseminated bloody appearance, indicating severe congestion and intra-alveolar hemorrhage. In turn, livers were paler than normal, suggesting inflammation. The second group were surviving mice, which were sacrificed 7 days after challenge. Lungs from these animals had no noticeable signs of haemorrhages and livers had no visible signs of inflammation.
Fig 5. Combination of IN plus IM immunization with α-PA vaccine and protection against pneumonia.

(A) Immunization schedule and mice survival after IN plus IM immunization with α-PA vaccine (3×107 CFU) (IN+IM, n = 7), IM immunization with α-PA vaccine (3×107 CFU, n = 8), IN immunization with α-PA vaccine (3×107 CFU) (IN, n = 5), or saline administration (IM, n = 8; IN, n = 8) and challenge with PAO1 ExoU+ (4×105 CFU). *P<0.05 (log-rank test). Images show lungs and livers collected from saline mice which succumbed to infection 16 h after challenge and from random vaccinated mice, sacrificed one week after challenge. Dashed squares indicate pulmonary hemorrhages and pale zones in livers. (B) Immunization schedule and mice survival after IN plus IM immunization with α-PA vaccine (3×107 CFU) (IN+IM, n = 9), IN immunization with α-PA vaccine (3×107 CFU) (IN, n = 9), or saline administration (IN+IM, n = 9) and challenge with PA14 (1×106 CFU). *P<0.05 (log-rank test). (C) Immunization schedule and mice survival after IN plus IM immunization with α-PA vaccine (3×107 CFU) (IN+IM, n = 9), IM immunization with α-PA vaccine (3×107 CFU, n = 9), IN immunization with α-PA vaccine (3×107 CFU) (IN, n = 9), or saline administration (IN+IM, n = 8; IM, n = 9; IN, n = 9) and challenge with PA14 (1×106 CFU). *P<0.05 (log-rank test). Images show lungs and livers collected 12 days after challenge from random vaccinated mice. (D) Immunization schedule and bacterial loads in lungs and livers of mice after IN plus IM immunization with α-PA vaccine (3×107 CFU) (IN+IM, n = 8), IM immunization with α-PA vaccine (3×107 CFU, n = 10), IN immunization with α-PA vaccine (3×107 CFU) (IN, n = 8), or saline administration (IN+IM, n = 8; IN, n = 8) and challenge with PA14 (1×106 CFU). *P<0.05 (Kruskal-Wallis and Mann-Whitney U test were used for 3- and 2-group comparison, respectively).
Then, we compared the protective effect of α-PA vaccine using the IN, and the combination of IN plus IM routes against PA14 lethal pneumonia. As shown in Fig 5B, IN-vaccinated mice showed only 67% survival, whereas 100% of mice administered the second regimen survived. This suggests that IN vaccination alone is not as effective against PA14 as the IN plus IM combination. Next, we performed the same experiment, but using also the IM route and challenging mice on day 113 (~3 months after the last immunization) (Fig 5C). A substantial decrease on survival was observed for IN and IM routes of administration; 33 and 44%, respectively. Still, IN plus IM administration of α-PA vaccine resulted in 78% survival. Lungs and livers randomly collected from vaccinated mice which survived to challenge neither presented PA14 bacterial counts nor visible pathology (Fig 5C, right panel).
Further, we measured PA14 bacterial loads in the lungs and livers of vaccinated and control mice challenged ~3 months after the last immunization, in order to find a correlation between bacterial burden and the survival observed in the previous experiment (Fig 5C). As shown in Fig 5D, high CFU counts were obtained in the lungs of all mice, but with slight reduction in vaccinated mice (IM and IN+IM). Vaccination resulted in all cases in a more pronounced reduction in liver bacterial loads, with undetected levels in some vaccinees. No clear correlation could be established between CFU and protection observed in the long-term with the different immunization routes, however, it seems likely that vaccination reduces the local and systemic bacterial burden.
Histological analysis of lungs after α-PA vaccination and PA14 infection
We assessed the differential outcomes of non-infected and infected mice after immunization (IN and IN plus IM administration) and control mice, by histological analysis of lungs (Fig 6). As shown in Fig 6C, the lungs of mice given α-PA vaccine showed some perivascular and periarteriolar foci of inflammatory infiltrate of the mononuclear type (arrows), evidencing mild to moderate inflammation, but with overall preservation of alveolar and airway architecture. Non-infected control mice showed no signals of inflammation. Lung histopathology of vaccinated and non-vaccinated mice subsequently infected with PA14 (Fig 6D) showed extensive polymorphonuclear (PMN) infiltration dispersed throughout the pulmonary parenchyma (arrows). This is consistent with recruitment of inflammatory cells and the role of neutrophils as essential mediators of host defense in the lungs against PA infection[27]. However, whereas sham-immunized mice presented PMN infiltrates filling most of the alveoli (arrowheads), vaccinated mice showed scarce alveolar infiltration, which might indicate less severe disease due to the reduced presence of tissue-injuring neutrophils[28] in these areas (alveoli preservation).
Fig 6. Lung histopathology caused by PA-pneumonia in vaccinated and naive mice.

(A) Immunization and lung collection schedule for (C). (B) Immunization, challenge and lung collection schedule for (D). (C) Representative photomicrographs of histological sections of lungs obtained from mice after IN (IN) immunization with α-PA vaccine (3×107 CFU) (n = 3), IN plus IM (IN+IM) immunization with α-PA vaccine (3×107 CFU) (n = 3) and saline administration (IN+IM) (n = 3). (C) Arrows show perivascular and periarteriolar foci of inflammatory infiltrate of the mononuclear type in the parenchyma. (D) Representative photomicrographs of histological sections of lungs obtained from mice after IN (IN) immunization with α-PA vaccine (3×107 CFU) (n = 6), IN plus IM (IN+IM) immunization with α-PA vaccine (3×107 CFU) (n = 5), saline administration (IN+IM) (n = 5) and challenge with PA14 (1×106 CFU). (D) Arrows show a severe inflammatory infiltrate of the polymorphonuclear type dispersed throughout the pulmonary parenchyma. Arrowheads show polymorphonuclear infiltrate filling most of the alveoli (saline), areas of scarce inflammatory mononuclear infiltrate (IN-immunized mice) and areas of inflammatory infiltrate of the polymorphonuclear type leukocyte with intrabronchial location (IN+IM-immunized mice), respectively. (C, D) The columns show images with ×25 (left), ×100 (center left), ×200 (center right) and ×400 (right) magnifications.
Overall, these results suggest that nasal vaccination with α-PA vaccine protects animals from the extensive bacterial dissemination in the lungs triggered by infection, thus it might reduce the damaging effects caused by neutrophil invasion of alveoli.
Mucosal antibody responses after immunization with α-PA vaccine
We investigated the local and distal mucosal responses to α-PA vaccine after immunization of mice via IN, IN plus IM and IM routes. IgA antibodies were quantified in bronchoalveolar lavages (BAL) and vaginal lavage fluids (VLF) (Fig 7A and 7B). Significantly increased IgA levels were detected in the BAL of IN- and IN plus IM-vaccinated mice, with the higher amounts being produced after IN immunization. IN immunization stimulated IgA production also in VLF. IM immunization elicited a slight increase in VLF IgA antibodies. Therefore, we hypothesize that intranasal vaccination might influence mucosal immunity via IgA secretion.
Fig 7. Live α-PA vaccine generates mucosal antibody responses and functional IgG in blood.

(A) Immunization schedule and IgA titers in BAL after IN plus IM immunization with α-PA vaccine (3×107 CFU; IN+IM, n = 9), IM immunization with α-PA vaccine (3×107 CFU; IM, n = 7), IN immunization with α-PA vaccine (3×107 CFU; IN, n = 8), or saline administration (IN+IM, n = 9; IM, n = 9; IN, n = 9). Titers were normalized respective to 50 μL. (B) Immunization schedule and IgA titers in VLF after IN plus IM immunization with α-PA vaccine (3×107 CFU; IN+IM, n = 8), IM immunization with α-PA vaccine (3×107 CFU; IM, n = 7), IN immunization with α-PA vaccine (3×107 CFU; IN, n = 8), or saline administration (IN+IM, n = 8; IM, n = 8; IN, n = 8). (A, B) S, saline. *P<0.05 (Kruskal-Wallis test), between the indicated groups. (C) Immunization schedule and in vitro OPKA assay, representing percent killing of PAO1 (2×104 CFU) and PA14 (2×104 CFU) by α-PA serum (or naïve serum) in the presence of human PMNs. α-PA and naïve sera were obtained from the blood of mice after IN immunization with α-PA vaccine (3×107−2×108 CFU, n = 6) or saline administration (n = 6), respectively. *P<0.05 (Mann-Whitney U test).
IN immunization with α-PA vaccine induces functional antibodies against LPS-homologous serogroup
We previously observed that α-PA-IN-vaccinated mice challenged with a PAO1 sepsis presented 88.9% survival, whereas all control mice died[11]. Based on this precedent, we hypothesized that antimicrobial activities of serum antibodies generated by IN immunization might also present complement-mediated killing and opsonophagocytosis against PA. Thus, we measured PAO1 and PA14-specific opsonophagocytic killing antibody (OPKA) activities, using α-PA serum obtained from the blood of mice after IN immunization and naïve serum from control mice. As illustrated in Fig 7C, there was significant killing of PAO1 in the presence of α-PA serum and human PMNs, but not of PA14. For PAO1, no significant killing was observed without PMNs (0 vs 19% killing; naïve vs α-PA serum, respectively). These data are consistent with the previous observed lack of passively-transferred protection (S15 Fig), and suggests that an antibody-independent mechanism might be responsible for the α-PA vaccine-mediated protection against the LPS-heterologous strain PA14.
Exploring alternative routes of immunization for mucosal immunity
We also tested the protective mucosal immunity generated by intradermal (ID) and subcutaneous (SC) delivery, considering that these immunization routes evoke both systemic and mucosal immunity. No body weight loss was observed for mice vaccinated using ID and SC routes (S18A and S18C Fig), resembling the previous results obtained with IM immunization. However, 6 days after the first ID injection of α-PA vaccine (1.5×108 CFU), all mice presented skin lesions at the inoculation site (S18B Fig). These lesions were transient, not being appreciable beyond day 9. Likewise, 2 out of 8 mice developed lesions in the neck area 6 days after SC immunization (1.5×108 CFU) (S18D Fig). Again, these lesions remitted 3 days after. These results suggest that α-PA vaccine might elicit transient local adverse reactions when injected ID and SC at high doses as 1.5×108 CFU.
Then, mice were challenged IN with PA14 (S19 Fig). As shown in S19B Fig, all mice ID-administered 1.5×108 CFU of α-PA vaccine survived infection, whereas all control mice succumbed to acute pneumonia. Mice administered 3×107 CFU of α-PA vaccine presented 67% survival. Thus, it seems likely that α-PA vaccine administered ID confers a significant protection against acute lung infection caused by PA14, being apparently more effective when using higher doses as 1.5×108 CFU. Increased IgG levels were observed in all immunized mice (S19C Fig). IgG titers of 3×107 CFU-vaccinated mice were not lower than when 1.5×108 CFU were used.
Then, we tested vaccine efficacy using SC route. As illustrated in S7E Fig, mice administered 1.5×108 CFU and 3×107 CFU of α-PA vaccine presented 88 and 25% survival, respectively, suggesting that SC route might not elicit the same level of protection as the ID route. Again, increased IgG levels were present in all immunized mice (S19F Fig), but no differences were observed between 3×107 CFU and 1.5×108 CFU-vaccinated mice. This observation is consistent with the results obtained when using ID immunization, supporting that mucosal protection observed may not depend on systemic antibodies alone.
Discussion
PA is among the several highly resistant gram-negative pathogens identified as emerging pathogens in both the US and other parts of the world, a group named ESKAPE[29]. It is also a leading pathogen causative of nosocomial infections, frequently originating acute pneumonia associated with mechanical ventilation (VAP)[30]. PA-derived VAP has a high attributable mortality, ranging from 13.5%[31] to 38%[32]. The worldwide increasing prevalence of MDR and XDR-resistant PA is of grave concern, making it imperative to develop a vaccine to protect against pneumonia. In our previous study, a D-Glu auxotrophic strain was selected and tested as a vaccine prototype against PA-derived sepsis using intraperitoneal immunization[11]. Nevertheless, injection of substances into the peritoneal cavity is rarely used in larger mammals and humans. Now, this work aims at identifying whether this D-Glu auxotroph holds the potential to be immunogenic and effective in a model of lethal pneumonia. Although other routes were tested (IM, SC and ID), we focused on IN immunization, as nasal mucosa provides a universal entry portal for PA, inducing local mucosal immune responses[33]. When administered by this route, our live vaccine elicited both humoral and cellular immunity and reduced the number of non-surviving mice considerably after infection with two ExoU-positive PA strains. Some protection against the ST235 XDR disseminated clone was also observed. Although ExoU-positive strains are not responsible for the majority of hospital-acquired PA infections, we selected these for challenge as mice are more likely to die than those inoculated with ExoU-negative strains. Moreover, ExoU is the most potent cytotoxin, present in approximately 30% of strains associated with the most severe infections, including septicemia [34,35]. Thus, the relative virulence associated with this type III effector protein may have more important prognostic implications for patients infected with PA.
Despite protection levels for PAO1 ExoU+ and PA14 were below 100% in some occasions, a trend toward reducing disease severity was observed when using IN immunization. However, none of the immunological determinants investigated could explain the unequal protection against ST235. According to our observation that IN immunization using a lower vaccine dose (3×107 CFU) resulted in 67% survival of mice infected with PA14 in contrast to the 88% observed when using 2×108 CFU, it might be hypothesized that using higher vaccine doses can compensate for the low vaccine efficacy observed against ST235 strain; however, we could not test this hypothesis due to the safety margin for IN administration.
Numerous studies have shown a host protective role of IL-17A in PA pneumonia[21,36,37]. IL-17-producing γδ T cells have been shown to promote neutrophil chemotaxis, eliminating PA bacteria during acute pulmonary infection[38] and to be involved in B cell activation[39]. However, our results appear to indicate that CD4+ T helper cells, rather than γδ T cells are stimulated by the α-PA vaccine to produce IL-17A as the IN-immunized mice presented increased frequencies of IL-17A+CD4+ cells in lungs than controls, while IL-17-producing γδ T cells were found at similar frequencies in both groups. IL-17A produced by Th17 cells is therefore likely implicated in the protective effect of the α-PA vaccine against acute lung infection. Indeed, this T cell population has been previously implicated in vaccination-induced protection against PA[21,36] and other bacteria causative of pulmonary infections[40].
The protection conferred by IL-17A has been associated with a rapid recruitment of neutrophils to the airways and the subsequent reduction of bacterial load[41]. Accordingly, vaccinated mice presented higher numbers of neutrophils in the lungs than controls. Despite having a marked neutrophil infiltration in the pulmonary parenchyma, vaccinated mice nevertheless presented markedly lower transmigration of neutrophils into the alveolar airspace, which is associated with damage to alveolar epithelium and impaired alveolar function[28]. Mononuclear CD11b+ cells, present at elevated numbers in the lungs of vaccinated mice might contribute to the lower inflammatory pathology in these mice as it has been previously shown in a condition of excessive lung inflammation[42]. Macrophages, which were found more abundant in the lungs of vaccinated mice might also contribute for protection against excessive inflammation. Indeed, host tissue-damaging inflammation in mice infected with PA was associated with lower macrophage bactericidal activity[43]. The mononuclear cell response that was observed in vaccinated mice, but not in controls, might thus explain the higher host protection at lower inflammatory cost.
Serum obtained from IN-vaccinated mice showed opsonic killing activity against PAO1. Thus, serum antibodies raised by immunization might thus mediate antimicrobial functions through opsonization, either by promoting complement deposition and cell lysis, and/or by promoting phagocytosis and killing by innate immune cells via Fc receptor recognition. This could help reduce or prevent bacterial dissemination into the circulation, acting synergistically with IgA in the lungs to prevent disease exacerbation. However, the absence of opsonic killing activity against PA14 suggests that an antibody-independent mechanism as T-cell help could be necessary to promote effective killing of the PA14 strain by phagocytic cells.
IM immunization with α-PA vaccine evoked titers of serum antibody comparable to the ones detected upon IN immunization. However, reduced protection against PA14 strain was observed. Taken together the results from passive immunization experiments, OPKA, flow cytometry and cytokine quantification, it is likely that the lower protection observed after IM immunization is associated with lower activation of cellular immunity (including lower Th17 activation). This hypothesis is further supported by recent findings describing an association between local IgA responses and Th17 establishment in the lungs after airway immunization [44]. No IgA was detected in BAL of IM-immunized mice, accordingly. Indeed, increased susceptibility to PA-pneumonia was observed in patients with secondary IgA deficiency[45].
Despite IN administration of 2×108 CFU of α-PA vaccine offered better improvement in mice survival than IM route, it nevertheless induced significant body weight loss and mild inflammation in the lungs. This may be due to intrinsic pathogenicity of the PA strain PAO1[46], emphasizing the need for using low doses. Indeed, when using a lower dose of α-PA vaccine (3×107 CFU), body weight losses were transient and no more present one week after each immunization. This effect was considered of mild to moderate toxicological relevance, and appeared to be dose-dependent. Thus, the IN immunization route using 3×107 CFU was combined with IM delivery, the last one conferring reduced protection but no significant body weight loss. The combined IN plus IM administrations with α-PA vaccine was apparently more effective in reducing disease severity that IN vaccination alone, as survival of mice was superior. Therefore, combining the two immunization routes could be an option to overcome the toxic effect of IN administration.
We also tested the protective effect generated by ID and SC delivery as these vaccination routes were reported to induce both systemic and mucosal immune responses[47,48]. ID and SC administration of α-PA vaccine offered significant protection against PA14 when using high vaccine doses, however, transient local adverse reactions were observed and this side-effect should be considered in the future.
The results obtained in this study are still limited due to the novelty of this D-Glu auxotrophic vaccine, impeding an accurate comparison with other reported α-PA vaccines. Indeed, the most similar vaccine candidates described are live-attenuated aroA deletants of PAO1[49] and PA14[21], but none of these strategies rely on using bacterial auxotrophy for D-Glu. Perhaps, some similarities can be established with BPZE1, a genetically-modified Bordetella pertussis strain which was developed as a live attenuated nasal pertussis vaccine by genetically eliminating or detoxifying 3 toxins[50]. BPZE was shown to be safe in humans[51], able to transiently colonize the human nasopharynx and to induce antibody responses, when using 103, 105 and 107 CFU through IN delivery. However, despite dose optimization regarding α-PA vaccine should be considered in future studies, it may be reasonable to hypothesize that this vaccine may similarly provide an acceptable level of efficacy without compromising safety. Moreover, refinement of this prototype vaccine through deletion of virulence genes could be explored to abrogate the toxic effect observed. An additional limitation is that in vivo studies were performed in BALB/c mice only, and differences in immunity may exist if other mouse strains are used.
While several candidates to receive α-PA vaccination have been discussed[8], those patients scheduled for cardiac surgery[52] and those undergoing mechanical ventilation[53] appear to be the most obvious target individuals. Another important infection group are those undergoing chemotherapy[54] or AIDS patients[55]. But because live vaccines are generally contraindicated for patients with severe immunocompromised status, some debate exists about targeting these individuals. However, as live attenuated Salmonella expressing PA-O antigen was immunogenic and effective in immunocompromised mice [56], it seems reasonable to consider the use of auxotrophic live vaccines in the future, after further virulence attenuation.
In summary, these findings suggest that the live α-PA vaccine described herein could be a promising approach to achieve mucosal immunity against pneumonia caused by highly pathogenic PA strains and possibly those that are refractory to broad-spectrum antibiotics. Immunization using the IN route seems favorable in comparison to conventional parenteral delivery routes, enabling local as well as systemic immunity and conferring an improvement in survival in a model of lethal pneumonia. However, further studies on the effects of eliminating toxicity genes on the PAO1 vaccine-strain background are needed to consider nasal administration.
Materials and methods
Ethics Statement
All animal experiments were done with the approval of Comité Ético de Experimentación Animal of Complejo Hospitalario Universitario A Coruña, Spain, under project license number 15002/2016/08 and in compliance with the Real Decreto 53/2013, of 1st february, which establishes the basic rules applicable for the protection of animals used in experimentation and other scientific purposes, including teaching.
Bacterial strains and culture conditions
All bacterial strains and plasmids used in this study are listed in S1 Table. All PA strains were grown in Luria-Bertani broth (LB) or on LB agar at 37°C. When indicated, D-Glu was used at 10 mM.
Animals
All mice were maintained in the specific pathogen-free facility at the Centro Tecnológico de Formación de la Xerencia de Xestión Integrada A Coruña. Male and female BALB/c mice were bred in our colony and used for first time procedures between the ages of 6 and 8 weeks. Studies were not blinded.
Immunization and lethal pneumonia challenge
To prepare inocula for active immunizations and infections, bacteria were cultured at 37°C under agitation (180–210 rpm) until reaching OD600nm = 0.7. Cultures were harvested by centrifugation (4,000 g, 20 min, 4°C), pellets washed twice, suspended and adjusted in sterile NaCl 0.9% to different doses (vaccine vehicle was always NaCl 0.9%—saline). Prior to mice inoculation, bacterial inocula were quantified by CFU enumeration in agar plates. In detail, PA and PA ΔmurI (α-PA vaccine) strains were cultured in LB and LB with D-Glu, respectively, at 37°C and 180 rpm, harvested by centrifugation, washed twice with LB and finally adjusted with saline as above. To prepare formalin-inactivated PAO1, cultures were harvested by centrifugation, washed with saline and inactivated in 1% (w/v) paraformaldehyde in PBS by incubation at 37°C under agitation (180 rpm) for 2 h. The bacteria were then washed twice with NaCl 0.9% and adjusted as before. Killing of bacteria was checked by plating on LB agar to determine the absence of colonies after overnight incubation. For active immunization assays, mice were inoculated IN with α-PA vaccine (or formalin-inactivated PAO1) with a total volume of 20 μL. Control mice were administrated saline. For IM, SC and ID injections of α-PA vaccine, we used a total volume of 100 (half volume per leg), 200 and 40 μL, respectively. Four weeks after the last immunization (at least), mice were anesthetized and IN-challenged (20 μL) with virulent PA strains. To facilitate migration of the inoculum to the alveoli, mice were held in a head-up vertical position for 2 min. Then, mice were monitored for mortality.
Sampling of mice
Blood samples were collected from the submandibular vein of anesthetized mice, and sera were separated from the blood cells by centrifugation (1,500 g, 15 min). To assess bacterial burden in lungs and livers, mice were euthanized at indicated time points, tissues were extracted aseptically, homogenized in sterile NaCl 0.9% and CFU enumerated by plating 10−2-fold serial dilutions in agar plates. VLF was obtained by washing the vagina of female mice twice with 50 μL sterile NaCl 0.9%, then VLF was centrifuged and the clear supernatant was used for the assays. BAL was performed by insertion of a catheter in the trachea of terminally anesthetized mice, through which 5–8 mL saline sterile solution was instilled into the bronchioles and gently retracted to maximize fluid retrieval.
ELISA
Quantification of IgM, IgG, IgG1, IgG2a, IgG2b and IgG3 in mouse sera was performed using an indirect ELISA as described in[11]. Hk PAO1 and PA14 were obtained by growing cells in LB at 37°C under agitation (180 rpm) until reaching OD600nm = 1.0. Then, bacteria were harvested by centrifugation (3,900 g, 20 min, 4°C), washed, suspended in sterile NaCl 0.9% and heat-inactivated by incubation at 100°C for 2 h. Killing of bacteria was checked by plating on LB agar and overnight incubation at 37°C. Quantification of IgA antibodies in VLF and BAL was performed by indirect ELISA according to[57], using secondary antibody alkaline phosphatase-conjugated anti-mouse IgA diluted 1:500. Plates were “coated” with formalin-inactivated PAO1, which was fixed to the bottom of the wells after overnight incubation at 4°C in 100 mM carbonate-bicarbonate buffer, pH 9.6. IgA titers in BAL were normalized respective to 50 μL, the same volume as those tested for VLF samples.
Cytokine detection in splenocyte cultures
Spleen single cell suspensions suspended in RPMI-1640 complete medium were plated in round-bottom 96-well plates (5×105/well) and stimulated with Hk PA14 (5×105 CFU and 5×104 CFU) at splenocytes:bacteria ratios of 1:1 and 1:0.1 for 3 days at 37°C and 5% CO2. Hk PA14 was obtained as described above, but growing cells in LB until reaching OD600nm = 0.7. Then, bacteria were heat-inactivated as before. Control splenocytes were added RPMI-1640 only (no stimuli). Samples from all animals were used in duplicate wells. The concentrations of IFN-γ, IL-17A, and IL-4 in cell culture supernatants were quantified using Ready-SET-Go! Mouse IFN-γ, IL-17A and IL-4 ELISA kits (eBioscience), respectively. IL-10 concentration was determined using the Mouse IL-10 DuoSet ELISA development system (R&D Systems).
Flow cytometry analysis
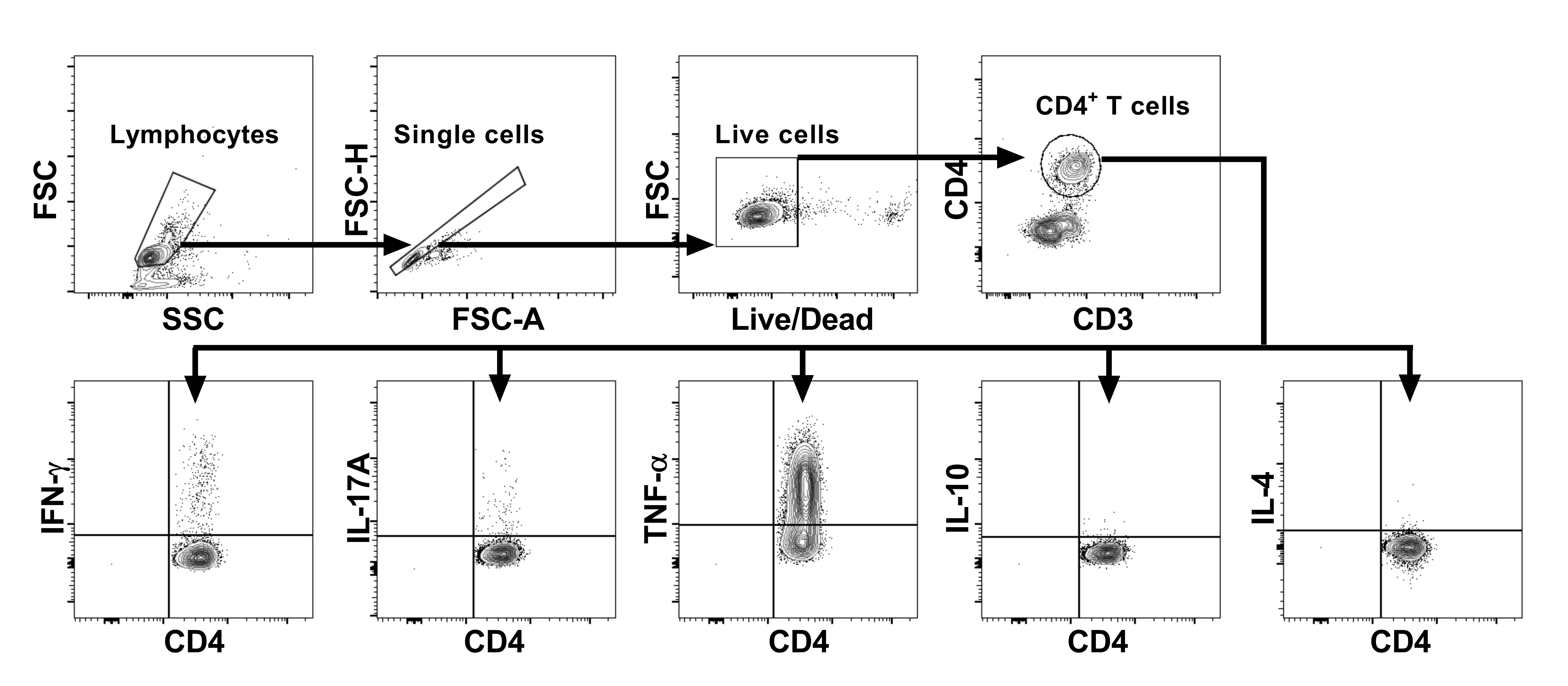
The assessment of cell surface lineage or activation markers on different splenic and lung leukocyte populations was performed by flow cytometry analysis (fluorescence-activated cell sorter [FACS] analysis), as described in Supporting Information, S1 Material and methods.
Passive immunization
Mice were given IgG-enriched antiserum (100 μL IP, 400 μg IgG/mouse), or naïve serum, 4 hours prior to the challenge with PA14. IgG-enriched antiserum was prepared using purified antibodies obtained from IN- and IM-immunized mice using the previous vaccination regimens (Figs 1A and 4A), which were then diluted in naïve serum. Antibody purification was carried out using the Protein G GraviTrap gravity-flow columns (GE Healthcare Life Science, Inc., USA). Amicon Ultra-15 filter devices (Millipore, Inc., USA) were used thereafter for desalting and concentration of eluted antibodies.
Histopathology
Lungs were harvested and fixed in 4% paraformaldehyde pH 7.0 for a minimum of 24h. Fixed tissues were then routinely processed and embedded in paraffin, cut in 3 μm sections and stained with hematoxylin and eosin (HE) after dewaxing in xylene and rehydration in decreasing ethanol concentrations, for histologic evaluation by a pathologist. The lung sections throughout the entire organ were microscopically evaluated to assess the distribution and character of pathologic alterations, as well as the lung inflammation pattern. All examinations were performed by a trained veterinary experimental pathologist blinded to the experimental conditions.
Opsonophagocytic killing assay (OPKA)
OPKA was assessed against PAO1 as previously[11], using α-PA serum obtained from the blood of mice after IN immunization and naïve serum from control mice. The target strains, 5×105 polymorphonuclear leukocytes (PMNs) from human volunteers, 2.5% complement serum from rabbit and test sera (1:8 final dilution) were all prepared in RPMI 1640 medium containing 10% FBS. PMNs were obtained from whole blood using a single Percoll density gradient[58].
Statistics
Data presented in box plots were analyzed using Mann Whitney test for 2 experimental groups-comparison and Kruskal-Wallis for 3 or more groups-comparison (with Dunn’s multiple comparison test). Otherwise, means were compared by using Student’s t test. Survival data were compared using the log-rank test. Statistical significance for mice weight loss was assessed by mixed ANOVA with “study day” as within-subjects factor and “treatment group” as between-subjects factor, followed by Bonferroni’s Multiple Comparison Test. Sphericity was checked via Mauchly’s test. Differences were considered significant when P<0.05. Significant changes within groups due to mice body weight gain along the time are not shown for clarity.
Supporting information
(DOCX)
Debris were excluded (Cell minus debris) based on FSC and SSC parameters and doublets were excluded (Single cells) in FSC-H versus FSC-A plots. Viable cells (Live cells) were gated as fixable viability dye negative cells. CD4 T cells were defined as CD3+CD4+ and quadrants were set, based on FMO stainings on contour plots, to define naïve (CD62L+CD44-), central memory (CD62L+CD44+) and effector memory (CD62L-CD44+) cells.
(JPG)
{kind=link}
(A) BALB/c mice (n = 6/group) were immunized with live α-PA vaccine (2×108 CFU) or administered saline, according to the schedule; then spleens and lungs were collected on the day indicated. (B) BALB/c mice (n = 6/group) were immunized with live α-PA vaccine (2×108 CFU) or administered saline, according to the schedule; then mice were infected with PA14 (1×106 CFU) on the day indicated. Spleens and lungs were collected 9 hours after. (C) Frequency (percentage) of splenic and lung CD4-gated T cells expressing IFN-γ, IL-10, IL-4 and TNF-α of infected and non-infected mice, detected by intracellular staining after stimulation with PMA/ionomycin. Bars represent mean ± SD of data. *P<0.05, **P<0.01, ***P<0.001 (t-test), compared with saline group. ns, not significant.
(JPG)
{kind=link}
(A) BALB/c mice (n = 6/group) were immunized with live α-PA vaccine (2×108 CFU) or administered saline, according to the schedule; then spleens were collected on the day indicated. (B) BALB/c mice (n = 6/group) were immunized with live α-PA vaccine (2×108 CFU) or administered saline, according to the schedule; then mice were infected with PA14 (1×106 CFU) on the day indicated. Spleens were collected 9 hours after. (C) Representative examples of CD4-gated T cells of spleens obtained from infected and non-infected mice, detected by intracellular staining, after stimulation with PMA/ionomycin. Analysis regions were set according to FMO and isotype control-stained samples. Numbers inside dot plot regions represent means ± SD of the frequency of cells due to respective cytokine staining.
(JPG)
{kind=link}
(A) BALB/c mice (n = 6/group) were immunized with live α-PA vaccine (2×108 CFU) or administered saline, according to the schedule; then lungs were collected on the day indicated. (B) BALB/c mice (n = 6/group) were immunized with live α-PA vaccine (2×108 CFU) or administered saline, according to the schedule; then mice were infected with PA14 (1×106 CFU) on the day indicated. Lungs were collected 9 hours after. (C) Representative examples of CD4-gated T cells of lungs obtained from infected and non-infected mice, detected by intracellular staining, after stimulation with PMA/ionomycin. Analysis regions were set according to FMO and isotype control-stained samples. Numbers inside dot plot regions represent means ± SD of the frequency of cells due to respective cytokine staining.
(JPG)
{kind=link}
Cells were stimulated with PMA/ionomycin in the presence of Brefeldin A for 4 h, followed by lineage marker surface staining. Produced cytokines were labelled by intracellular staining. Lymphocytes were selected based on FSC and SSC parameters, doublets were then excluded (Single cells) and viable cells (Live cells) were gated as fixable viability dye negative cells. CD4 T cells were defined as CD3+CD4+ and quadrants were set, based on FMO staining on contour plots, to define cytokine-producing cells. Representative contour plots of splenic CD4 T cells producing IFN-γ, IL-17A, IL-10, and IL-4, are shown.
(JPG)
{kind=link}
(A) BALB/c mice (n = 6/group) were immunized with live α-PA vaccine (2×108 CFU) or administered saline, according to the schedule; then spleens and lungs were collected on the day indicated. (B) BALB/c mice (n = 6/group) were immunized with live α-PA vaccine (2×108 CFU) or administered saline, according to the schedule; then mice were infected with PA14 (1×106 CFU) on the day indicated. Spleens and lungs were collected 9 hours after. (C, D) Representative examples and frequency (percentage) of lung γδ-gated T cells expressing IL-17A (C) and IFN-γ (D) of infected and non-infected mice, detected by intracellular staining after stimulation for 4 h with PMA/ionomycin in the presence of Brefeldin A. Analysis regions were set according to FMO and isotype control-stained samples. Numbers inside dot plot regions represent means ± SD of the frequency of cells due to respective cytokine staining. Bars represent mean ± SD of data. *P<0.05, **P<0.01 (t-test), compared with saline group.
(JPG)
{kind=link}
Cells were stimulated with PMA/ionomycin in the presence of Brefeldin A for 4 h, followed by lineage marker surface staining. Produced cytokines were labelled by intracellular staining. Debris were excluded (Cell minus debris), and lymphocytes were selected based on FSC and SSC parameters. Doublets were then excluded (Single cells), followed by dead cell exclusion (Live cells). γδ T cells were defined as γδ TCR+ and quadrants were set, based on FMO stainings to define cytokine-producing cells. Representative contour plots of lung γδ T cells producing IFN-γ and IL-17A are shown.
(JPG)
{kind=link}
(A, B) Mice (n = 6-7/group) were immunized with α-PA vaccine (2×108 CFU) or administered saline, according to the schedules; then spleens were collected. In (B), mice were challenged with PA14 (1×106 CFU), 9 h before the spleens were obtained. (A, B) Graphics show IFN-γ, IL-17A and IL-10 concentration in the supernatants of splenocyte cultures of vaccinated and non-vaccinated mice after being stimulated ex vivo with Hk PA14 (5×105 CFU and 5×104 CFU for 1:1 and 1:0.1 ratio of splenocytes:bacteria, respectively) for 3 days. No stimuli were added to control splenocytes. Mean ± SD. *P<0.05, (Mann-Whitney U test).
(JPG)
{kind=link}
(A, B) Mice (n = 6/group) were immunized with α-PA vaccine (2×108 CFU) or administered saline, according to the schedule; then lungs were collected. In (B), mice (n = 6/group) were infected with PA14 (1×106 CFU), 9 h before the lungs were obtained. (C, D) Total number of cells in the lungs of non-infected (C) and infected (D) mice, detected by flow cytometry upon surface staining, according to the schedules indicated in (A) and (B), respectively. Analysis regions were set according to FMO control-stained samples. Bars represent mean ± SD of data. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001 (t-test).
(JPG)
{kind=link}
Cells isolated from collagenase-digested mouse lungs were stained with a mix of fluorophore conjugated antibodies after a pre-incubation step with anti-mouse CD16/CD32 (FcBlock). A sequential gating strategy was used to identify cell populations based on their surface expression of specific markers. After exclusion of debris (Cell minus debris) and doublets (Single cells), immune cells were gated based on CD45 expression (CD45+ cells), followed by dead cell exclusion (Live cells). Neutrophils were defined as CD11b+ Ly6G+ cells; dendritic cells (DC) were identified as Ly6G- CD11c+ F4/80- MHC class II+, that could be further divided into CD11b+ and CD11b- DC subsets; alveolar macrophages (AM) were defined as Ly6G- CD11c+ F4/80+ MHC class II- and interstitial macrophages (IM) were set as Ly6G- CD11c+ F4/80+ MHC class II+; within the Ly6G- CD11c- CD11b+ F4/80+ population, cells were divided based on their Ly6C expression and side-scatter characteristics: Ly6C+ monocytes/macrophages (Ly6C+ Mo/MΦ) were defined as Ly6G- CD11c- CD11b+ F4/80+ Ly6C+, Ly6C- Mo/MΦ as Ly6G- CD11c- CD11b+ F4/80+ and SSClo and eosinophils as Ly6G- CD11c- CD11b+ F4/80+ and SSChi.
(JPG)
{kind=link}
(A) Mice (n = 6/group) were immunized with α-PA vaccine (3×107 CFU) or administered saline, according to the schedule; then spleens were collected. (B) Percentage of spleen effector memory (CD44+ CD62L-), central memory (CD44+ CD62L+) and naïve (CD44- CD62L+) CD4+ T cells. Mean ± SD of data. *P<0.05 (t-test).
(JPG)
{kind=link}
(A) BALB/c mice (n = 6/group) were immunized with live α-PA vaccine (3×107 CFU) or administered saline, according to the schedule; then spleens and lungs were collected on the day indicated. (B) BALB/c mice (n = 6/group) were immunized with live α-PA vaccine (3×107 CFU) or administered saline, according to the schedule; then mice were infected with PA14 (1×106 CFU) on the day indicated. Spleens and lungs were collected 9 hours after. (C) Frequency (percentage) of splenic and lung CD4-gated T cells expressing IL-17A, IFN-γ, IL-10, IL-4 and TNF-α of infected and non-infected mice, detected by intracellular staining after stimulation with PMA/ionomycin. Bars represent mean ± SD of data. *P<0.05 (t-test).
(JPG)
{kind=link}
(A, B) Mice (n = 6/group) were immunized with α-PA vaccine (3×107 CFU) or administered saline, according to the schedules; then spleens were collected. In (B), mice were challenged with PA14 (1×106 CFU), 9 h before the spleens were obtained. (A, B) Graphics show IFN-γ, IL-17A, IL-10 and IL-4 concentrations in the supernatants of splenocyte cultures of vaccinated and non-vaccinated mice after being stimulated ex vivo with Hk PA14 (5×105 CFU and 5×104 CFU for 1:1 and 1:0.1 ratio of splenocytes:bacteria, respectively) for 3 days. No stimuli were added to control splenocytes. Mean ± SD. *P<0.05, (Mann-Whitney U test).
(JPG)
{kind=link}
(A, B) Mice (n = 6/group) were immunized with α-PA vaccine (3×107 CFU) or administered saline, according to the schedule; then lungs were collected. In (B), mice (n = 6/group) were infected with PA14 (1×106 CFU), 9 h before the lungs were obtained. (C, D) Total number of cells in the lungs of non-infected (C) and infected (D) mice, detected by flow cytometry upon surface staining, according to the schedules indicated in (A) and (B), respectively. Analysis regions were set according to FMO control-stained samples. Bars represent mean ± SD of data. *P<0.05 (t-test).
(JPG)
{kind=link}
Mice survival after passive immunization with IgG-enriched mouse antiserum obtained from IN- and IM-immunized mice (live α-PA vaccine), or naïve serum and challenge with PA14 (3×106 CFU) (n = 6–11 mice/group).
(JPG)
{kind=link}
(A) Percentage of mice weight change after IN immunization with α-PA vaccine (2×108 CFU, n = 8) and saline administration (n = 8). (B) Percentage of mice weight change after IM immunization with α-PA vaccine (3×107 CFU, n = 7) and saline administration (n = 7). (C) Percentage of mice weight change after IN immunization with α-PA vaccine (3×107 CFU, n = 9) and saline administration (n = 8). (D) Percentage of mice weight change after IN plus IM immunization with α-PA vaccine (3×107 CFU, n = 9) and saline administration (n = 8). (C, D) The same control group–mice administered saline through IN plus IM route–was used for comparison. (A-D) Mean ± SD. *P<0.05 (pairwise comparison of weights within vaccinated mice, mixed ANOVA). #P<0.05, (pairwise comparison of weights between groups, mixed ANOVA).
(JPG)
{kind=link}
(A) Mice were IN-immunized with α-PA vaccine (8×105 CFU, n = 8; 7×106 CFU, n = 8) or administered saline (n = 8), according to the schedule; then mice were challenged with PA14 (1×106 CFU) on the day indicated. The MDSS system was applied for determining disease severity and surrogate endpoints, 16 hours after infection. Bacterial loads in the lungs were determined immediately after. (B) Percentage of mice weight change after IN immunization with α-PA vaccine or saline administration. Mean ± SD. *P<0.05, 7×106 CFU-vaccinated group (pairwise comparison of weights within groups, mixed ANOVA). #1P<0.05, saline vs 8×105 CFU, 8×105 vs 7×106 CFU; #2P<0.05, saline vs 7×106 CFU, 8×105 vs 7×106 CFU (pairwise comparison of weights between groups, mixed ANOVA). (C) MDSS after immunization with the different doses of α-PA vaccine and challenge with PA14. (D) Bacterial loads in the lungs of mice after immunization with the different doses of α-PA vaccine and challenge with PA14.
(JPG)
{kind=link}
(A) Percent of mice weight change after ID immunization with live α-PA vaccine (1.5×108 CFU, n = 8; 3×107 CFU, n = 9) or saline administration (n = 8). (B) Skin lesion produced by ID injection of live α-PA vaccine (1.5×108 CFU). (C) Percent of mice weight change after SC immunization with live α-PA vaccine (1.5×108 CFU, n = 8; 3×107 CFU, n = 8) or saline administration (n = 8). (D) Skin lesion produced by SC injection of live α-PA vaccine (1.5×108 CFU). (A, C) Mean ± SD.
(JPG)
{kind=link}
(A) Immunization schedule using ID route, sampling of mice and lung infection challenge. (B) Mice survival after ID immunization with live α-PA vaccine (1.5×108 CFU, n = 7; 3×107 CFU, n = 9) or saline administration (n = 7) and challenge with PA14 (1×106 CFU), causing acute pneumonia. (C) Titers of PAO1-specific antibodies after ID immunization with live α-PA vaccine (3×107 CFU, n = 6; 1.5×108 CFU, n = 7) or saline administration (n = 5). (D) Immunization schedule using SC route, sampling of mice and lung infection challenge. (E) Mice survival after SC immunization with live α-PA vaccine (1.5×108 CFU, n = 8; 3×107 CFU, n = 8) or saline administration (n = 8) and challenge with PA14 (1×106 CFU), causing acute pneumonia. (F) Titers of PAO1-specific antibodies after SC immunization with live α-PA vaccine (3×107 CFU, n = 7; 1.5×108 CFU, n = 8) or saline administration (n = 8). (B, E) *P<0.05 (log-rank test), compared with saline group. (C, F) S, saline. *P<0.05 (Kruskal-Wallis test), compared with saline group.
(JPG)
{kind=link}
(DOCX)
(DOCX)
Acknowledgments
We thank M. Fernández for technical assistance, B. Fernández for the provision of clinical P. aeruginosa strains, C. Winstanley (University of Liverpool) for LES400 and LES431, J. Blázquez (IBIS) for PA14, F. Chaves (Hospital 12 de Octubre) for PA ST175 and C. Peña (Hospital Universitari de Bellvitge) for PA ST235.
Data Availability
All relevant data are within the manuscript and its Supporting Information files.
Funding Statement
This work was supported by the Project PI15/00860 and PI18/00501 – ISCIII, REIPI RD16/0016/0006 and “Innova Saúde” – SERGAS – Galician Healthcare Service. MPC was partially supported by SEIMC. AC was supported by an FCT grant (SFRH/BPD/91623/2012). AP was supported by the Juan de la Cierva Program (MINECO, IJCI-2016-29524) and SEIMC. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Quartin AA, Scerpella EG, Puttagunta S, Kett DH. A comparison of microbiology and demographics among patients with healthcare-associated, hospital-acquired, and ventilator-associated pneumonia: a retrospective analysis of 1184 patients from a large, international study. BMC Infect Dis. 2013. December 27;13(1):561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fagon JY, Chastre J, Hance AJ, Montravers P, Novara A, Gibert C. Nosocomial pneumonia in ventilated patients: a cohort study evaluating attributable mortality and hospital stay. Am J Med. 1993. March;94(3):281–8. 10.1016/0002-9343(93)90060-3 [DOI] [PubMed] [Google Scholar]
- 3.Williams BJ, Dehnbostel J, Blackwell TS. Pseudomonas aeruginosa: Host defence in lung diseases. Respirology. 2010. August 16;15(7):1037–56. 10.1111/j.1440-1843.2010.01819.x [DOI] [PubMed] [Google Scholar]
- 4.Sawa T. The molecular mechanism of acute lung injury caused by Pseudomonas aeruginosa: from bacterial pathogenesis to host response. J intensive care. 2014;2(1):10 10.1186/2052-0492-2-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cabot G, Ocampo-Sosa AA, Domínguez MA, Gago JF, Juan C, Tubau F, et al. Genetic markers of widespread extensively drug-resistant Pseudomonas aeruginosa high-risk clones. Antimicrob Agents Chemother. 2012. December;56(12):6349–57. 10.1128/AAC.01388-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Le HN, Tran VG, Vu TTT, Gras E, Le VTM, Pinheiro MG, et al. Treatment Efficacy of MEDI3902 in Pseudomonas aeruginosa Bloodstream Infection and Acute Pneumonia Rabbit Models. Antimicrob Agents Chemother. 2019. August;63(8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pires DP, Vilas Boas D, Sillankorva S, Azeredo J. Phage Therapy: a Step Forward in the Treatment of Pseudomonas aeruginosa Infections. J Virol. 2015. August 1;89(15):7449–56. 10.1128/JVI.00385-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Priebe GP, Goldberg JB. Vaccines for Pseudomonas aeruginosa: a long and winding road. Expert Rev Vaccines. 2014. April;13(4):507–19. 10.1586/14760584.2014.890053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gellatly SL, Hancock REW. Pseudomonas aeruginosa: new insights into pathogenesis and host defenses. Pathog Dis. 2013. April;67(3):159–73. 10.1111/2049-632X.12033 [DOI] [PubMed] [Google Scholar]
- 10.Holmgren J, Czerkinsky C. Mucosal immunity and vaccines. Nat Med. 2005. April;11(4s):S45–53. [DOI] [PubMed] [Google Scholar]
- 11.Cabral MP, García P, Beceiro A, Rumbo C, Pérez A, Moscoso M, et al. Design of live attenuated bacterial vaccines based on D-glutamate auxotrophy. Nat Commun. 2017. May 26;8:15480 10.1038/ncomms15480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pritt B, O’Brien L, Winn W. Mucoid Pseudomonas in cystic fibrosis. Am J Clin Pathol. 2007;128(1):32–4. 10.1309/KJRPC7DD5TR9NTDM [DOI] [PubMed] [Google Scholar]
- 13.Allewelt M, Coleman FT, Grout M, Priebe GP, Pier GB. Acquisition of expression of the Pseudomonas aeruginosa ExoU cytotoxin leads to increased bacterial virulence in a murine model of acute pneumonia and systemic spread. Infect Immun. 2000. July;68(7):3998–4004. 10.1128/iai.68.7.3998-4004.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mikkelsen H, McMullan R, Filloux A. The Pseudomonas aeruginosa Reference Strain PA14 Displays Increased Virulence Due to a Mutation in ladS. Cornelis P, editor. One PLoS. 2011. December 22;6(12):e29113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gómez-Zorrilla S, Juan C, Cabot G, Camoez M, Tubau F, Oliver A, et al. Impact of multidrug resistance on the pathogenicity of Pseudomonas aeruginosa: in vitro and in vivo studies. Int J Antimicrob Agents. 2016;47(5):368–74. 10.1016/j.ijantimicag.2016.02.010 [DOI] [PubMed] [Google Scholar]
- 16.Wiehlmann L, Wagner G, Cramer N, Siebert B, Gudowius P, Morales G, et al. Population structure of Pseudomonas aeruginosa. Proc Natl Acad Sci. 2007. May 8;104(19):8101–6. 10.1073/pnas.0609213104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Treepong P, Kos VN, Guyeux C, Blanc DS, Bertrand X, Valot B, et al. Global emergence of the widespread Pseudomonas aeruginosa ST235 clone. Clin Microbiol Infect. 2018. March 1;24(3):258–66. 10.1016/j.cmi.2017.06.018 [DOI] [PubMed] [Google Scholar]
- 18.Mulet X, Cabot G, Ocampo-Sosa AA, Domínguez MA, Zamorano L, Juan C, et al. Biological markers of Pseudomonas aeruginosa epidemic high-risk clones. Antimicrob Agents Chemother. 2013. November 1;57(11):5527–35. 10.1128/AAC.01481-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.García-Castillo M, Del Campo R, Morosini MI, Riera E, Cabot G, Willems R, et al. Wide dispersion of ST175 clone despite high genetic diversity of carbapenem-nonsusceptible Pseudomonas aeruginosa clinical strains in 16 Spanish hospitals. J Clin Microbiol. 2011. August;49(8):2905–10. 10.1128/JCM.00753-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lockhart E, Green AM, Flynn JL. IL-17 production is dominated by gammadelta T cells rather than CD4 T cells during Mycobacterium tuberculosis infection. J Immunol. 2006. October 1;177(7):4662–9. 10.4049/jimmunol.177.7.4662 [DOI] [PubMed] [Google Scholar]
- 21.Priebe GP, Walsh RL, Cederroth TA, Kamei A, Coutinho-Sledge YS, Goldberg JB, et al. IL-17 is a critical component of vaccine-induced protection against lung infection by lipopolysaccharide-heterologous strains of Pseudomonas aeruginosa. J Immunol. 2008. October 1;181(7):4965–75. 10.4049/jimmunol.181.7.4965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lavoie EG, Wangdi T, Kazmierczak BI. Innate immune responses to Pseudomonas aeruginosa infection. Microbes Infect. 2011. December;13(14–15):1133–45. 10.1016/j.micinf.2011.07.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kamei A, Wu W, Traficante DC, Koh AY, Van Rooijen N, Pier GB, et al. Collaboration between macrophages and vaccine-induced CD4+ T cells confers protection against lethal Pseudomonas aeruginosa pneumonia during neutropenia. J Infect Dis. 2013. January 1;207(1):39–49. 10.1093/infdis/jis657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Geissmann F, Jung S, Littman DR. Blood monocytes consist of two principal subsets with distinct migratory properties. Immunity. 2003. July;19(1):71–82. 10.1016/s1074-7613(03)00174-2 [DOI] [PubMed] [Google Scholar]
- 25.Rello J, Krenn C-G, Locker G, Pilger E, Madl C, Balica L, et al. A randomized placebo-controlled phase II study of a Pseudomonas vaccine in ventilated ICU patients. Crit Care. 2017. December 4;21(1):22 10.1186/s13054-017-1601-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Döring G, Meisner C, Stern M. A double-blind randomized placebo-controlled phase III study of a Pseudomonas aeruginosa flagella vaccine in cystic fibrosis patients. Proc Natl Acad Sci U S A. 2007. June 26;104(26):11020–5. 10.1073/pnas.0702403104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Koh AY, Priebe GP, Ray C, Van Rooijen N, Pier GB. Inescapable need for neutrophils as mediators of cellular innate immunity to acute Pseudomonas aeruginosa pneumonia. Infect Immun. 2009. December 1;77(12):5300–10. 10.1128/IAI.00501-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang J. Neutrophils in tissue injury and repair. Cell Tissue Res. 2018. March;371(3):531–9. 10.1007/s00441-017-2785-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Boucher HW, Talbot GH, Bradley JS, Edwards JE, Gilbert D, Rice LB, et al. Bad bugs, no drugs: no ESKAPE! An update from the Infectious Diseases Society of America. Clin Infect Dis. 2009;48(1):1–12. 10.1086/595011 [DOI] [PubMed] [Google Scholar]
- 30.Lambert M-L, Suetens C, Savey A, Palomar M, Hiesmayr M, Morales I, et al. Clinical outcomes of health-care-associated infections and antimicrobial resistance in patients admitted to European intensive-care units: a cohort study. Lancet Infect Dis. 2011. January;11(1):30–8. 10.1016/S1473-3099(10)70258-9 [DOI] [PubMed] [Google Scholar]
- 31.Rello J, Jubert P, Vallés J, Artigas A, Rué M, Niederman MS. Evaluation of outcome for intubated patients with pneumonia due to Pseudomonas aeruginosa. Clin Infect Dis. 1996. November;23(5):973–8. 10.1093/clinids/23.5.973 [DOI] [PubMed] [Google Scholar]
- 32.Crouch Brewer S, Wunderink RG, Jones CB, Leeper K V. Ventilator-associated pneumonia due to Pseudomonas aeruginosa. Chest. 1996. Apr;109(4):1019–29. 10.1378/chest.109.4.1019 [DOI] [PubMed] [Google Scholar]
- 33.Neutra MR, Kozlowski PA. Mucosal vaccines: the promise and the challenge. Nat Rev Immunol. 2006. February 1;6(2):148–58. 10.1038/nri1777 [DOI] [PubMed] [Google Scholar]
- 34.Berthelot P, Attree I, Plésiat P, Chabert J, de Bentzmann S, Pozzetto B, et al. Genotypic and phenotypic analysis of type III secretion system in a cohort of Pseudomonas aeruginosa bacteremia isolates: evidence for a possible association between O serotypes and exo genes. J Infect Dis. 2003. August 15;188(4):512–8. 10.1086/377000 [DOI] [PubMed] [Google Scholar]
- 35.Finck-Barbançon V, Goranson J, Zhu L, Sawa T, Wiener-Kronish JP, Fleiszig SM, et al. ExoU expression by Pseudomonas aeruginosa correlates with acute cytotoxicity and epithelial injury. Mol Microbiol. 1997. August;25(3):547–57. 10.1046/j.1365-2958.1997.4891851.x [DOI] [PubMed] [Google Scholar]
- 36.Wu W, Huang J, Duan B, Traficante DC, Hong H, Risech M, et al. Th17-stimulating protein vaccines confer protection against Pseudomonas aeruginosa pneumonia. Am J Respir Crit Care Med. 2012. September 1;186(5):420–7. 10.1164/rccm.201202-0182OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li Y, Wang Z, Liu X, Tang J, Peng B, Wei Y. X-ray Irradiated Vaccine Confers protection against Pneumonia caused by Pseudomonas Aeruginosa. Sci Rep. 2016;6(April 2015):18823 10.1038/srep18823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu J, Qu H, Li Q, Ye L, Ma G, Wan H. The responses of γδ T-cells against acute Pseudomonas aeruginosa pulmonary infection in mice via interleukin-17. Pathog Dis. 2013. July;68(2):44–51. 10.1111/2049-632X.12043 [DOI] [PubMed] [Google Scholar]
- 39.Pan T, Tan R, Li M, Liu Z, Wang X, Tian L, et al. IL17-Producing γδ T Cells May Enhance Humoral Immunity during Pulmonary Pseudomonas aeruginosa Infection in Mice. Front Cell Infect Microbiol. 2016. December 6;6:170 10.3389/fcimb.2016.00170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Khader SA, Bell GK, Pearl JE, Fountain JJ, Rangel-Moreno J, Cilley GE, et al. IL-23 and IL-17 in the establishment of protective pulmonary CD4+ T cell responses after vaccination and during Mycobacterium tuberculosis challenge. Nat Immunol. 2007. April;8(4):369–77. 10.1038/ni1449 [DOI] [PubMed] [Google Scholar]
- 41.Liu J, Feng Y, Yang K, Li Q, Ye L, Han L, et al. Early production of IL-17 protects against acute pulmonary Pseudomonas aeruginosa infection in mice. FEMS Immunol Med Microbiol. 2011;61(2):179–88. 10.1111/j.1574-695X.2010.00764.x [DOI] [PubMed] [Google Scholar]
- 42.Eldredge LC, Treuting PM, Manicone AM, Ziegler SF, Parks WC, McGuire JK. CD11b(+) Mononuclear Cells Mitigate Hyperoxia-Induced Lung Injury in Neonatal Mice. Am J Respir Cell Mol Biol. 2016. February;54(2):273–83. 10.1165/rcmb.2014-0395OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wilson KR, Napper JM, Denvir J, Sollars VE, Yu HD. Defect in early lung defence against Pseudomonas aeruginosa in DBA/2 mice is associated with acute inflammatory lung injury and reduced bactericidal activity in naive macrophages. Microbiology. 2007. April 1;153(Pt 4):968–79. 10.1099/mic.0.2006/002261-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Christensen D, Mortensen R, Rosenkrands I, Dietrich J, Andersen P. Vaccine-induced Th17 cells are established as resident memory cells in the lung and promote local IgA responses. Mucosal Immunol. 2017. January 6;10(1):260–70. 10.1038/mi.2016.28 [DOI] [PubMed] [Google Scholar]
- 45.Robak OH, Heimesaat MM, Kruglov AA, Prepens S, Ninnemann J, Gutbier B, et al. Antibiotic treatment-induced secondary IgA deficiency enhances susceptibility to Pseudomonas aeruginosa pneumonia. J Clin Invest. 2018. August 1;128(8):3535–45. 10.1172/JCI97065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bartell JA, Blazier AS, Yen P, Thøgersen JC, Jelsbak L, Goldberg JB, et al. Reconstruction of the metabolic network of Pseudomonas aeruginosa to interrogate virulence factor synthesis. Nat Commun. 2017. March 7;8:14631 10.1038/ncomms14631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Locher CP, Witt SA, Ashlock BM, Levy JA. Evaluation of Genetic Immunization Adjuvants to Improve the Effectiveness of a Human Immunodeficiency Virus Type 2 (HIV-2) Envelope DNA Vaccine. DNA Cell Biol. 2004. February;23(2):107–10. 10.1089/104454904322759911 [DOI] [PubMed] [Google Scholar]
- 48.Lima FA, Miyaji EN, Quintilio W, Raw I, Ho PL, Oliveira MLS. Pneumococcal Surface Protein A does not affect the immune responses to a combined diphtheria tetanus and pertussis vaccine in mice. Vaccine. 2013. May 7;31(20):2465–70. 10.1016/j.vaccine.2013.03.026 [DOI] [PubMed] [Google Scholar]
- 49.Priebe GP, Meluleni GJ, Coleman FT, Goldberg JB, Pier GB. Protection against fatal Pseudomonas aeruginosa pneumonia in mice after nasal immunization with a live, attenuated aroA deletion mutant. Infect Immun. 2003. March;71(3):1453–61. 10.1128/IAI.71.3.1453-1461.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mielcarek N, Debrie AS, Raze D, Bertout J, Rouanet C, Younes A Ben, et al. Live attenuated B. pertussis as a single-dose nasal vaccine against whooping cough. PLoS Pathog. 2006;2(7):0662–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Thorstensson R, Trollfors B, Al-Tawil N, Jahnmatz M, Bergström J, Ljungman M, et al. A phase I clinical study of a live attenuated Bordetella pertussis vaccine—BPZE1; a single centre, double-blind, placebo-controlled, dose-escalating study of BPZE1 given intranasally to healthy adult male volunteers. PLoS One. 2014. Jan;9(1):e83449 10.1371/journal.pone.0083449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Allou N, Kermarrec N, Muller C, Thabut G, Philip I, Lucet J-C, et al. Risk factors and prognosis of post-operative pneumonia due to Pseudomonas aeruginosa following cardiac surgery. J Antimicrob Chemother. 2010. April 1;65(4):806–7. 10.1093/jac/dkq030 [DOI] [PubMed] [Google Scholar]
- 53.Hoang S, Georget A, Asselineau J, Venier A-G, Leroyer C, Rogues AM, et al. Risk factors for colonization and infection by Pseudomonas aeruginosa in patients hospitalized in intensive care units in France. PLoS One. 2018;13(3):e0193300 10.1371/journal.pone.0193300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ohmagari N, Hanna H, Graviss L, Hackett B, Perego C, Gonzalez V, et al. Risk factors for infections with multidrug-resistant Pseudomonas aeruginosa in patients with cancer. Cancer. 2005. July 1;104(1):205–12. 10.1002/cncr.21115 [DOI] [PubMed] [Google Scholar]
- 55.Mendelson MH, Gurtman A, Szabo S, Neibart E, Meyers BR, Policar M, et al. Pseudomonas aeruginosa Bacteremia in Patients with AIDS. Clin Infect Dis. 1994. June 1;18(6):886–95. 10.1093/clinids/18.6.886 [DOI] [PubMed] [Google Scholar]
- 56.Scarff JM, Goldberg JB. Vaccination against Pseudomonas aeruginosa pneumonia in immunocompromised mice. Clin Vaccine Immunol. 2008. February;15(2):367–75. 10.1128/CVI.00419-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ferreirinha P, Dias J, Correia A, Pérez-Cabezas B, Santos C, Teixeira L, et al. Protective effect of intranasal immunization with Neospora caninum membrane antigens against murine neosporosis established through the gastrointestinal tract. Immunology. 2014;141(2):256–67. 10.1111/imm.12191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Harbeck RJ, Hoffman AA, Redecker S, Biundo T, Kurnick J. The isolation and functional activity of polymorphonuclear leukocytes and lymphocytes separated from whole blood on a single Percoll density gradient. Clin Immunol Immunopathol. 1982;23(3):682–90. 10.1016/0090-1229(82)90331-2 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(DOCX)
Debris were excluded (Cell minus debris) based on FSC and SSC parameters and doublets were excluded (Single cells) in FSC-H versus FSC-A plots. Viable cells (Live cells) were gated as fixable viability dye negative cells. CD4 T cells were defined as CD3+CD4+ and quadrants were set, based on FMO stainings on contour plots, to define naïve (CD62L+CD44-), central memory (CD62L+CD44+) and effector memory (CD62L-CD44+) cells.
(JPG)
(A) BALB/c mice (n = 6/group) were immunized with live α-PA vaccine (2×108 CFU) or administered saline, according to the schedule; then spleens and lungs were collected on the day indicated. (B) BALB/c mice (n = 6/group) were immunized with live α-PA vaccine (2×108 CFU) or administered saline, according to the schedule; then mice were infected with PA14 (1×106 CFU) on the day indicated. Spleens and lungs were collected 9 hours after. (C) Frequency (percentage) of splenic and lung CD4-gated T cells expressing IFN-γ, IL-10, IL-4 and TNF-α of infected and non-infected mice, detected by intracellular staining after stimulation with PMA/ionomycin. Bars represent mean ± SD of data. *P<0.05, **P<0.01, ***P<0.001 (t-test), compared with saline group. ns, not significant.
(JPG)
(A) BALB/c mice (n = 6/group) were immunized with live α-PA vaccine (2×108 CFU) or administered saline, according to the schedule; then spleens were collected on the day indicated. (B) BALB/c mice (n = 6/group) were immunized with live α-PA vaccine (2×108 CFU) or administered saline, according to the schedule; then mice were infected with PA14 (1×106 CFU) on the day indicated. Spleens were collected 9 hours after. (C) Representative examples of CD4-gated T cells of spleens obtained from infected and non-infected mice, detected by intracellular staining, after stimulation with PMA/ionomycin. Analysis regions were set according to FMO and isotype control-stained samples. Numbers inside dot plot regions represent means ± SD of the frequency of cells due to respective cytokine staining.
(JPG)
(A) BALB/c mice (n = 6/group) were immunized with live α-PA vaccine (2×108 CFU) or administered saline, according to the schedule; then lungs were collected on the day indicated. (B) BALB/c mice (n = 6/group) were immunized with live α-PA vaccine (2×108 CFU) or administered saline, according to the schedule; then mice were infected with PA14 (1×106 CFU) on the day indicated. Lungs were collected 9 hours after. (C) Representative examples of CD4-gated T cells of lungs obtained from infected and non-infected mice, detected by intracellular staining, after stimulation with PMA/ionomycin. Analysis regions were set according to FMO and isotype control-stained samples. Numbers inside dot plot regions represent means ± SD of the frequency of cells due to respective cytokine staining.
(JPG)
Cells were stimulated with PMA/ionomycin in the presence of Brefeldin A for 4 h, followed by lineage marker surface staining. Produced cytokines were labelled by intracellular staining. Lymphocytes were selected based on FSC and SSC parameters, doublets were then excluded (Single cells) and viable cells (Live cells) were gated as fixable viability dye negative cells. CD4 T cells were defined as CD3+CD4+ and quadrants were set, based on FMO staining on contour plots, to define cytokine-producing cells. Representative contour plots of splenic CD4 T cells producing IFN-γ, IL-17A, IL-10, and IL-4, are shown.
(JPG)
(A) BALB/c mice (n = 6/group) were immunized with live α-PA vaccine (2×108 CFU) or administered saline, according to the schedule; then spleens and lungs were collected on the day indicated. (B) BALB/c mice (n = 6/group) were immunized with live α-PA vaccine (2×108 CFU) or administered saline, according to the schedule; then mice were infected with PA14 (1×106 CFU) on the day indicated. Spleens and lungs were collected 9 hours after. (C, D) Representative examples and frequency (percentage) of lung γδ-gated T cells expressing IL-17A (C) and IFN-γ (D) of infected and non-infected mice, detected by intracellular staining after stimulation for 4 h with PMA/ionomycin in the presence of Brefeldin A. Analysis regions were set according to FMO and isotype control-stained samples. Numbers inside dot plot regions represent means ± SD of the frequency of cells due to respective cytokine staining. Bars represent mean ± SD of data. *P<0.05, **P<0.01 (t-test), compared with saline group.
(JPG)
Cells were stimulated with PMA/ionomycin in the presence of Brefeldin A for 4 h, followed by lineage marker surface staining. Produced cytokines were labelled by intracellular staining. Debris were excluded (Cell minus debris), and lymphocytes were selected based on FSC and SSC parameters. Doublets were then excluded (Single cells), followed by dead cell exclusion (Live cells). γδ T cells were defined as γδ TCR+ and quadrants were set, based on FMO stainings to define cytokine-producing cells. Representative contour plots of lung γδ T cells producing IFN-γ and IL-17A are shown.
(JPG)
(A, B) Mice (n = 6-7/group) were immunized with α-PA vaccine (2×108 CFU) or administered saline, according to the schedules; then spleens were collected. In (B), mice were challenged with PA14 (1×106 CFU), 9 h before the spleens were obtained. (A, B) Graphics show IFN-γ, IL-17A and IL-10 concentration in the supernatants of splenocyte cultures of vaccinated and non-vaccinated mice after being stimulated ex vivo with Hk PA14 (5×105 CFU and 5×104 CFU for 1:1 and 1:0.1 ratio of splenocytes:bacteria, respectively) for 3 days. No stimuli were added to control splenocytes. Mean ± SD. *P<0.05, (Mann-Whitney U test).
(JPG)
(A, B) Mice (n = 6/group) were immunized with α-PA vaccine (2×108 CFU) or administered saline, according to the schedule; then lungs were collected. In (B), mice (n = 6/group) were infected with PA14 (1×106 CFU), 9 h before the lungs were obtained. (C, D) Total number of cells in the lungs of non-infected (C) and infected (D) mice, detected by flow cytometry upon surface staining, according to the schedules indicated in (A) and (B), respectively. Analysis regions were set according to FMO control-stained samples. Bars represent mean ± SD of data. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001 (t-test).
(JPG)
Cells isolated from collagenase-digested mouse lungs were stained with a mix of fluorophore conjugated antibodies after a pre-incubation step with anti-mouse CD16/CD32 (FcBlock). A sequential gating strategy was used to identify cell populations based on their surface expression of specific markers. After exclusion of debris (Cell minus debris) and doublets (Single cells), immune cells were gated based on CD45 expression (CD45+ cells), followed by dead cell exclusion (Live cells). Neutrophils were defined as CD11b+ Ly6G+ cells; dendritic cells (DC) were identified as Ly6G- CD11c+ F4/80- MHC class II+, that could be further divided into CD11b+ and CD11b- DC subsets; alveolar macrophages (AM) were defined as Ly6G- CD11c+ F4/80+ MHC class II- and interstitial macrophages (IM) were set as Ly6G- CD11c+ F4/80+ MHC class II+; within the Ly6G- CD11c- CD11b+ F4/80+ population, cells were divided based on their Ly6C expression and side-scatter characteristics: Ly6C+ monocytes/macrophages (Ly6C+ Mo/MΦ) were defined as Ly6G- CD11c- CD11b+ F4/80+ Ly6C+, Ly6C- Mo/MΦ as Ly6G- CD11c- CD11b+ F4/80+ and SSClo and eosinophils as Ly6G- CD11c- CD11b+ F4/80+ and SSChi.
(JPG)
(A) Mice (n = 6/group) were immunized with α-PA vaccine (3×107 CFU) or administered saline, according to the schedule; then spleens were collected. (B) Percentage of spleen effector memory (CD44+ CD62L-), central memory (CD44+ CD62L+) and naïve (CD44- CD62L+) CD4+ T cells. Mean ± SD of data. *P<0.05 (t-test).
(JPG)
(A) BALB/c mice (n = 6/group) were immunized with live α-PA vaccine (3×107 CFU) or administered saline, according to the schedule; then spleens and lungs were collected on the day indicated. (B) BALB/c mice (n = 6/group) were immunized with live α-PA vaccine (3×107 CFU) or administered saline, according to the schedule; then mice were infected with PA14 (1×106 CFU) on the day indicated. Spleens and lungs were collected 9 hours after. (C) Frequency (percentage) of splenic and lung CD4-gated T cells expressing IL-17A, IFN-γ, IL-10, IL-4 and TNF-α of infected and non-infected mice, detected by intracellular staining after stimulation with PMA/ionomycin. Bars represent mean ± SD of data. *P<0.05 (t-test).
(JPG)
(A, B) Mice (n = 6/group) were immunized with α-PA vaccine (3×107 CFU) or administered saline, according to the schedules; then spleens were collected. In (B), mice were challenged with PA14 (1×106 CFU), 9 h before the spleens were obtained. (A, B) Graphics show IFN-γ, IL-17A, IL-10 and IL-4 concentrations in the supernatants of splenocyte cultures of vaccinated and non-vaccinated mice after being stimulated ex vivo with Hk PA14 (5×105 CFU and 5×104 CFU for 1:1 and 1:0.1 ratio of splenocytes:bacteria, respectively) for 3 days. No stimuli were added to control splenocytes. Mean ± SD. *P<0.05, (Mann-Whitney U test).
(JPG)
(A, B) Mice (n = 6/group) were immunized with α-PA vaccine (3×107 CFU) or administered saline, according to the schedule; then lungs were collected. In (B), mice (n = 6/group) were infected with PA14 (1×106 CFU), 9 h before the lungs were obtained. (C, D) Total number of cells in the lungs of non-infected (C) and infected (D) mice, detected by flow cytometry upon surface staining, according to the schedules indicated in (A) and (B), respectively. Analysis regions were set according to FMO control-stained samples. Bars represent mean ± SD of data. *P<0.05 (t-test).
(JPG)
Mice survival after passive immunization with IgG-enriched mouse antiserum obtained from IN- and IM-immunized mice (live α-PA vaccine), or naïve serum and challenge with PA14 (3×106 CFU) (n = 6–11 mice/group).
(JPG)
(A) Percentage of mice weight change after IN immunization with α-PA vaccine (2×108 CFU, n = 8) and saline administration (n = 8). (B) Percentage of mice weight change after IM immunization with α-PA vaccine (3×107 CFU, n = 7) and saline administration (n = 7). (C) Percentage of mice weight change after IN immunization with α-PA vaccine (3×107 CFU, n = 9) and saline administration (n = 8). (D) Percentage of mice weight change after IN plus IM immunization with α-PA vaccine (3×107 CFU, n = 9) and saline administration (n = 8). (C, D) The same control group–mice administered saline through IN plus IM route–was used for comparison. (A-D) Mean ± SD. *P<0.05 (pairwise comparison of weights within vaccinated mice, mixed ANOVA). #P<0.05, (pairwise comparison of weights between groups, mixed ANOVA).
(JPG)
(A) Mice were IN-immunized with α-PA vaccine (8×105 CFU, n = 8; 7×106 CFU, n = 8) or administered saline (n = 8), according to the schedule; then mice were challenged with PA14 (1×106 CFU) on the day indicated. The MDSS system was applied for determining disease severity and surrogate endpoints, 16 hours after infection. Bacterial loads in the lungs were determined immediately after. (B) Percentage of mice weight change after IN immunization with α-PA vaccine or saline administration. Mean ± SD. *P<0.05, 7×106 CFU-vaccinated group (pairwise comparison of weights within groups, mixed ANOVA). #1P<0.05, saline vs 8×105 CFU, 8×105 vs 7×106 CFU; #2P<0.05, saline vs 7×106 CFU, 8×105 vs 7×106 CFU (pairwise comparison of weights between groups, mixed ANOVA). (C) MDSS after immunization with the different doses of α-PA vaccine and challenge with PA14. (D) Bacterial loads in the lungs of mice after immunization with the different doses of α-PA vaccine and challenge with PA14.
(JPG)
(A) Percent of mice weight change after ID immunization with live α-PA vaccine (1.5×108 CFU, n = 8; 3×107 CFU, n = 9) or saline administration (n = 8). (B) Skin lesion produced by ID injection of live α-PA vaccine (1.5×108 CFU). (C) Percent of mice weight change after SC immunization with live α-PA vaccine (1.5×108 CFU, n = 8; 3×107 CFU, n = 8) or saline administration (n = 8). (D) Skin lesion produced by SC injection of live α-PA vaccine (1.5×108 CFU). (A, C) Mean ± SD.
(JPG)
(A) Immunization schedule using ID route, sampling of mice and lung infection challenge. (B) Mice survival after ID immunization with live α-PA vaccine (1.5×108 CFU, n = 7; 3×107 CFU, n = 9) or saline administration (n = 7) and challenge with PA14 (1×106 CFU), causing acute pneumonia. (C) Titers of PAO1-specific antibodies after ID immunization with live α-PA vaccine (3×107 CFU, n = 6; 1.5×108 CFU, n = 7) or saline administration (n = 5). (D) Immunization schedule using SC route, sampling of mice and lung infection challenge. (E) Mice survival after SC immunization with live α-PA vaccine (1.5×108 CFU, n = 8; 3×107 CFU, n = 8) or saline administration (n = 8) and challenge with PA14 (1×106 CFU), causing acute pneumonia. (F) Titers of PAO1-specific antibodies after SC immunization with live α-PA vaccine (3×107 CFU, n = 7; 1.5×108 CFU, n = 8) or saline administration (n = 8). (B, E) *P<0.05 (log-rank test), compared with saline group. (C, F) S, saline. *P<0.05 (Kruskal-Wallis test), compared with saline group.
(JPG)
(DOCX)
(DOCX)
Data Availability Statement
All relevant data are within the manuscript and its Supporting Information files.