

We present a modular approach to control the small molecule–mediated degradation of cellular proteins of interest using light.
Abstract
PROTACs (PROteolysis TArgeting Chimeras) are bifunctional molecules that target proteins for ubiquitylation by an E3 ligase complex and subsequent degradation by the proteasome. They have emerged as powerful tools to control the levels of specific cellular proteins. We now introduce photoswitchable PROTACs that can be activated with the spatiotemporal precision that light provides. These trifunctional molecules, which we named PHOTACs (PHOtochemically TArgeting Chimeras), consist of a ligand for an E3 ligase, a photoswitch, and a ligand for a protein of interest. We demonstrate this concept by using PHOTACs that target either BET family proteins (BRD2,3,4) or FKBP12. Our lead compounds display little or no activity in the dark but can be reversibly activated with different wavelengths of light. Our modular approach provides a method for the optical control of protein levels with photopharmacology and could lead to new types of precision therapeutics that avoid undesired systemic toxicity.
INTRODUCTION
Protein levels in cells result from a tightly controlled balance between synthesis and degradation. A wide range of small molecules have been identified that interfere with these processes. Most of them do not address specific proteins as they broadly inhibit the machinery necessary for transcription, translation, trafficking, or degradation. In recent years, PROteolysis TArgeting Chimeras (PROTACs) have emerged as a new principle of pharmacology (1–3). These bifunctional molecules combine a ligand for an E3 ubiquitin ligase with a second one that targets a protein of interest (POI), thereby promoting the physical interaction of the proteins, the polyubiquitylation of the POI, and its consequent proteasomal degradation. Both ends of the PROTAC are connected via a linker, the exact nature of which needs to be carefully chosen to ensure efficacy, cell permeability, and biodistribution.
First-generation PROTACs used peptides to recruit POIs to E3 ligases (4), but subsequent ones have relied on smaller and more cell-permeable synthetic ligands. These include hydroxyproline derivatives and molecules derived from thalidomide, which bind the von Hippel–Lindau (VHL) protein (5, 6) and cereblon (CRBN) (7, 8), respectively. VHL and CRBN are the substrate receptors of two cullin-RING ubiquitin ligase (CRL) complexes, namely, CRL2VHL and CRL4CRBN. Proteins that have been successfully targeted for degradation through these ubiquitin ligases include the androgen and estrogen receptors (9–12), the BET family epigenetic readers BRD2–4 (7, 8, 13), and FKBP12 and its fusion proteins (7, 14, 15). Soluble kinases, such as cyclin-dependent kinase 9 (CDK9) (16) and BCR-ABL (17), and receptor tyrosine kinases, such as epidermal growth factor receptor (EGFR) (18) and Bruton's tyrosine kinase (BTK) (19, 20), have also been amenable to this approach. Covering a broad spectrum from membrane proteins to nuclear hormone receptors, PROTACs have proven to be a highly versatile approach.
PROTACs do not merely inhibit the activity of their targets, like conventional drugs; rather, they decrease the levels of the targets by promoting their proteolysis. The transition from inhibition of proteins to their catalytic degradation enables the targeting of previously undruggable proteins. However, this mechanistic difference with conventional drugs also poses certain risks when applied systemically, since the POI is degraded and disappears with all of its functions in both cancer and normal cells. For instance, inhibition of BET bromodomains is tolerated, but a complete loss of BRD2 and BRD4 is lethal (21, 22). Therefore, it would be advantageous to locally activate PROTACs in tissues and cells where their effects (e.g., cytotoxicity) are desirable while avoiding deleterious effects elsewhere.
One method to localize the effect of drugs and achieve higher selectivity is to control their activity with light. In recent years, the usefulness of light to precisely regulate biological pathways has become increasingly apparent. Optical control can be achieved in a variety of ways: with caged compounds (23), with genetically engineered photoreceptors (optogenetics) (24), or with synthetic photoswitches whose activity can be changed through a combination of photochemical isomerization and thermal relaxation (photopharmacology) (25, 26).
Here, we report the application of photopharmacology to targeted protein degradation. By incorporating azobenzene photoswitches into PROTACs, we have designed photoswitchable versions that we named PHOTACs (PHOtochemically TArgeting Chimeras). These molecules show little or no proteolytic activity in the dark but can be activated with blue-violet light (380 to 440 nm). They can be used to degrade a variety of targets, including BRD2–4 and FKBP12, by binding to the CRL4CRBN complex and promoting proteolysis in a light-dependent fashion. This translates to the optical control of protein levels and, in the case of BRD2–4, of cell proliferation, survival, and viability.
RESULTS
Design, synthesis, and photophysical characterization
The design of our PHOTACs was guided by a desire to render our molecules as diversifiable and modular as possible while ensuring efficient synthetic access. To test the concept, we chose to target CRBN, which, together with VHL, accounts for most of the PROTAC platforms used to date. Accordingly, we focused on thalidomide derivatives, such as pomalidomide and lenalidomide, as CRBN ligands. As for the photoswitch, we decided to use azobenzenes, which are known for their fatigue resistance, large and predictable geometrical changes, and easily tunable photothermal properties. Azobenzenes are also among the smallest photoswitches and do not substantially increase the molecular weight of pharmaceuticals upon substitution. Ideal PHOTACs would be inactive in the dark and lead to efficient degradation of the POI upon irradiation. However, the impact of linker conformation on PROTAC activity is not fully understood. We therefore explored both regular azobenzenes (more stable in their trans form) and diazocines (more stable in cis) (27, 28).
Several approaches are conceivable for the incorporation of these photoswitches into PHOTACs: (i) They could be part of the ligand for the E3 ligase and change the affinity at this end of the chimera. (ii) The photoswitches could mostly reside in the tether, changing the length and orientation of this segment. (iii) The azobenzenes could be part of a POI ligand, controlling the affinity of the PHOTAC to the POI. It would be difficult, if not impossible, however, to define a strict boundary between ligands and linker, and combinations of all three modes are possible. As for our first POI ligand, we chose (+)-JQ1, a high-affinity inhibitor of BET proteins BRD2–4 and BRDT. PROTACs featuring this ligand, such as dBET1 (Fig. 1B), have proven to be particularly effective and have been developed for a variety of E3 ligases.
Fig. 1. PROTACs and PHOTACs.
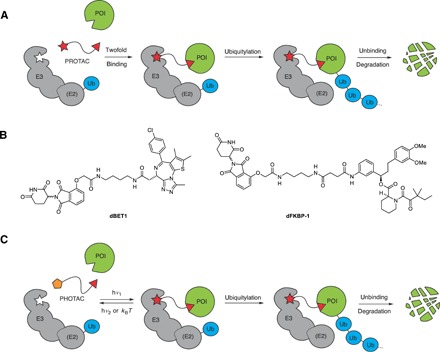
(A) Schematic depiction of a PROTAC. Formation of a ternary complex between an E3 ligase, a PROTAC, and a POI leads to degradation of the POI. (B) Chemical structures of PROTACs dBET1 and dFKBP-1. (C) Schematic depiction of a PHOTAC. The molecules toggle between an inactive form (yellow pentagon) and an active form (red star) upon irradiation.
The small library of PHOTACs that resulted from these considerations is depicted in Fig. 2A. Among these, PHOTAC-I-3 emerged as one of the most effective. Its synthesis started with the diazotization of lenalidomide and coupling of the resulting diazonium ion 1 to 2,6-dimethoxyphenol, which yielded azobenzene 2 (Fig. 2B). Alkylation with tert-butyl bromoacetate and subsequent deprotection then afforded the key intermediate 3. Amide coupling of this carboxylic acid with N-Boc-butane-1,4-diamine and deprotection then yielded 4, which underwent another deprotection, followed by peptide coupling with the free acid of (+)-JQ1 (5) to afford PHOTAC-I-3. HATU coupling of 3 to diaminoalkanes of different length provided easy access to the library of PHOTACs with varying linker length (i.e., PHOTAC-I-1,2,4,5). PHOTAC-I-6–8, which lack two methoxy groups on the azobenzene core, were synthesized analogously. PHOTAC-I-9 bears a different substitution pattern and was prepared via Baeyer-Mills coupling (see the Supplementary Materials). PHOTAC-I-10–13, which have the photoswitch more in the center of the molecule, were synthesized from 4-hydroxy thalidomide and azobenzene building blocks via alkylation and amide couplings (see the Supplementary Materials).
Fig. 2. Structure and synthesis of PHOTACs.
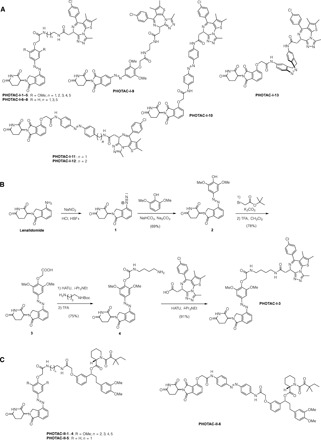
(A) Members of the PHOTAC-I series targeting BRDs. (B) Synthesis of PHOTAC-I-3 starting from lenalidomide. (C) Members of the PHOTAC-II series targeting FKBP12.
The photoswitching and thermal relaxation properties of one of our lead compounds, PHOTAC-I-3, are shown in Fig. 3 (A to E). The optimal wavelength to switch to the cis isomer is 390 nm, but similar photostationary states (PSSs) can be obtained between 380 and 400 nm (Fig. 3C). At 390 nm, a PSS of >90% cis could be obtained. Rapid cis to trans isomerization could be achieved by irradiation with wavelengths >450 nm, achieving PSS of ca. >70% trans (Fig. 3C). In the absence of light, cis–PHOTAC-I-3 slowly isomerized back to its trans form with a half-life of 8.8 hours at 37°C in dimethyl sulfoxide (DMSO) (Fig. 3D). Multiple cycles of photochemical isomerization are possible in keeping with the fatigue resistance of azobenzene photoswitches (Fig. 3E). Structurally related PHOTACS showed similar photophysical and thermal properties (see the Supplementary Materials).
Fig. 3. Photophysical properties, switching, and bistability of PHOTACs.
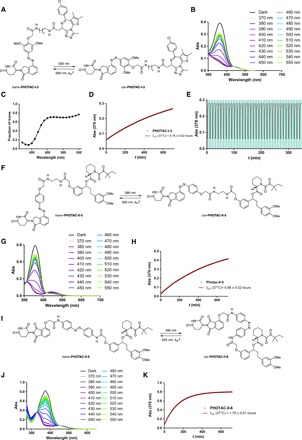
(A) Switching of PHOTAC-I-3 between the trans isomer (left) and the cis isomer (right). (B) Ultraviolet-visible (UV-vis) spectra PHOTAC-I-3 following irradiation with different wavelengths for 5 min. (C) Fraction of trans–PHOTAC-I-3 in the PSS. (D) Thermal relaxation of cis–PHOTAC-I-3 at 37°C in DMSO. (E) Reversible switching and photochemical stability of PHOTAC-I-3. (F) Switching of PHOTAC-II-5 between the trans isomer (left) and the cis isomer (right). (G) UV-vis spectra PHOTAC-II-5 following irradiation with different wavelengths for 5 min. (H) Thermal relaxation of cis–PHOTAC-II-5 at 37°C in DMSO. (I) Switching of PHOTAC-II-6 between the trans isomer (left) and the cis isomer (right). (J) UV-vis spectra PHOTAC-II-6 following irradiation with different wavelengths for 5 min. (K) Thermal relaxation of cis–PHOTAC-II-6 at 37°C in DMSO.
Optical control of BRD2–4 with PHOTACs
To assess the biological activity of our PHOTACs, we tested their effect on the viability of RS4;11 lymphoblast cells. Cells were treated in a 96-well plate with increasing concentrations of PHOTACs and were either irradiated with 390-nm light pulses (100 ms every 10 s) for 72 hours or incubated with the compound in the dark. Subsequently, we performed cell viability assays (Promega MTS), as previously described. PHOTAC-I-3 showed a promising activity difference upon irradiation (Fig. 4A). The median effective concentration (EC50) was determined to be 88.5 nM when irradiated with 390-nm light and 631 nM in the dark, resulting in a 7.1-fold EC50 difference. This indicates that cytotoxicity increases upon irradiation and that PHOTAC-I-3 is less toxic in the dark. Similar trends were observed for PHOTAC-I-1,2,4–8,10, all of which were more active in viability assays following pulse irradiation (fig. S3). By contrast, PHOTAC-I-9,11–13 showed no light-dependent differences in activity (fig. S3). In a control experiment, the BET inhibitor (+)-JQ1 alone showed no light-dependent toxicity either (Fig. 4B).
Fig. 4. Light-dependent viability of RS4;11 cells.
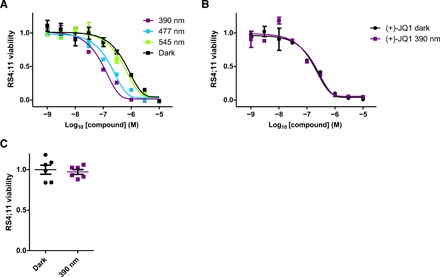
(A) Viability of RS4;11 acute lymphoblastic leukemia cells after treatment with PHOTAC-I-3 for 72 hours in the dark or under pulsed (100 ms every 10 s) 390-, 477-, or 545-nm irradiation. (B) Viability of RS4;11 after (+)-JQ1 treatment for 72 hours in the dark or under pulsed (100 ms every 10 s) 390-nm irradiation. (C) Viability of RS4;11 cells after 72 hours in the dark or under pulsed (100 ms every 10 s) 390-nm irradiation.
Next, we analyzed the light dependence of targeted protein degradation in RS4;11 cells by Western blot analysis of the BET proteins BRD2–4 (Fig. 5). To this end, we treated cells with increasing concentrations of our lead compound, PHOTAC-I-3, for 4 hours and pulse irradiated with 390-nm light (100 ms every 10 s). We observed a pronounced decrease in BRD4 levels in the presence of PHOTAC-I-3 (particularly between 100 nM and 3 μM) when irradiated with 390-nm light, but not in the dark (Fig. 5A). At 10 μM, we observed less degradation, which is consistent with the “hook effect” commonly observed with PROTACs (5, 19, 29). BRD3 levels were also greatly reduced upon exposure to concentrations in the range of 100 nM to 3 μM of PHOTAC-I-3 when irradiated with violet light, but not in the dark. In comparison, BRD2 was degraded to a lesser extent and within a narrower concentration range. Application of PHOTAC-I-3 (1 μM) together with the CRL inhibitor MLN4924 (2.5 μM), which inhibits neddylation and, consequently, the activity of all cellular CRLs (including CRL4CRBN), rescued BRD2–4 levels upon irradiation. The CRBN-dependent degradation was confirmed by a competition experiment (Fig. 5E) and further validated by small interfering RNA (siRNA) knockdown of CRBN (fig. S7). Methylation of the glutarimide prevented the degradation of BRD4 as previously demonstrated (fig. S8) (30).
Fig. 5. Optical control of BRD2–4 levels.

(A) Immunoblot analysis after treatment of RS4;11 cells with PHOTAC-I-3 for 4 hours at different concentrations. Cells were either irradiated with 100-ms pulses of 390-nm light every 10 s (left) or kept in the dark (right). MLN (MLN4924) was added as an additional control. (B) Time course of BRD2–4 degradation, c-MYC levels, and PARP1 cleavage assayed by immunoblotting. RS4;11 cells were treated with PHOTAC-I-3 (1 μM) and collected at the indicated time points. PHOTAC-I-3 has no effect on BRD2–4 levels in the dark over several hours. (C) Immunoblot of a rescue experiment demonstrating the reversibility of degradation promoted by PHOTAC-I-3 through thermal relaxation (left) or optical inactivation by 525-nm pulsed irradiation (right, 100 ms every 10 s). (D) Color dosing: Wavelength dependence of BRD2/4 degradation promoted by 300 nM PHOTAC-I-3. (E) Immunoblot analysis after treatment of RS4;11 cells with PHOTAC-I-3 and combinations with lenalidomide or (+)-JQ1 for 4 hours to confirm a CRBN-based mechanism. Cells were either irradiated with 100-ms pulses of 390-nm light every 10 s (left) or kept in the dark (right). (F) Optical degradation of BRD4 with the thalidomide derivative PHOTAC-I-10. Immunoblot analysis of RS4;11 cells after treatment with PHOTAC-I-10 for 4 hours at different concentrations, which were either irradiated with 100-ms pulses of 390-nm light every 10 s (left) or kept in the dark (right).
As expected from photoactivatable degraders and inhibitors of BRD4, c-MYC levels were also affected (13, 31, 32). Down-regulation of this transcription factor, which is a notoriously difficult target for pharmacological intervention (33), was more pronounced when cells were pulse irradiated in the presence of low concentrations of PHOTAC-I-3 than in the dark. The light-dependent degradation of BRD4 by PHOTAC-I-3 was also confirmed in two breast cancer cell lines (MB-MDA-231 and MB-MDA-468) (fig. S4, A and B). Immunoblot analyses of additional PHOTACs are presented in fig. S5.
The time dependence of BRD degradation is shown in Fig. 5B. BRD2 and BRD3 are largely absent after 1.5 hours of exposure and irradiation, whereas BRD4 is degraded more slowly. We also observed sustained degradation and c-MYC down-regulation over 24 hours. In the dark, PHOTAC-I-3 had no effect on BRD levels and relatively little effect on c-MYC levels. The slight effect on c-MYC can be explained by inhibition of BRD4 with the (+)-JQ1 derivative PHOTAC-I-3 in the absence of targeted degradation. Following sustained pulse irradiation, we also observed increasing cleavage of PARP-1 (Fig. 5B). This indicator of apoptosis (34) correlates to the cell viability assay shown in Fig. 4A. Persistent degradation of BRD4 in the dark could be achieved following a brief activation of PHOTAC-I-3 for 1 min (fig. S4C).
One of the principal advantages of photoswitches over caged compounds is their reversibility. Azobenzene photoswitches can thermally relax to an inactive form or be isomerized back photochemically. We demonstrated the photochemical reversibility with a rescue experiment wherein PHOTAC-I-3 was continuously irradiated for 1 min with the activating wavelength (390 nm) and then pulse irradiated with the deactivating wavelength (525 nm). Under these conditions, cellular BRD2 levels initially decreased but, subsequently, recovered faster than when left in the dark (Fig. 5C).
Another characteristic feature of photopharmacology is “color dosing” (i.e., the ability to control the concentration of the active species with the color of the incident light) (26, 35, 36). The PSS (i.e., the ratio between the two isomers) is a function of the wavelength. Figures 3A and 5D show that this principle can also be applied to PHOTACs. Cell viability assays gave left-shifted curves as the color gradually approached 390 nm (Fig. 4A). Western blots showed maximum degradation at 390 nm and gradually increasing BRD4 levels as the wavelength of the incident light increased (Fig. 5D). At 370 nm, we observed slightly increased protein levels, in agreement with the PSSs measured at different wavelengths, which are maximized toward the active cis isomer at the slightly longer wavelength of 390 nm (Fig. 3C).
The effect of PHOTAC-I-10, which is derived from thalidomide and has the photoswitch positioned deeper in the linker, on BRD4 levels, is shown in Fig. 5E. We found robust photodegradation even with 10 nM PHOTAC-I-10. A clear hook effect was observed. Once again, the thermally less stable cis azobenzene promoted ubiquitylation and degradation.
Optical control of FKBP12 with PHOTACs
To test whether the PHOTAC approach is generalizable, we turned to the prolyl cis-trans isomerase FKBP12. The structures of the corresponding PHOTACs are shown in Fig. 2C. PHOTACs of this series consist of a CRBN-targeting glutarimide, an azobenzene photoswitch in different positions, a linker, and SLF (synthetic ligand of FKBP) that binds to native FKBP12 (7, 14, 37, 38). The synthesis of PHOTAC-II-1–6 is detailed in the Supplementary Materials. The photophysical and thermal characterization of PHOTAC-II-5 and PHOTAC-II-6 are shown in Fig. 3 (F to H and I to K, respectively).
Among the molecules tested, PHOTAC-II-5 and PHOTAC-II-6 turned out to be the most useful, and our biological investigations have been focused on these molecules (for PHOTAC-II-1–4, see fig. S10). PHOTAC-II-5, which has the azobenzene switch in the same position as PHOTAC-I-1–8, had a pronounced effect on FKBP12 levels upon pulse irradiation (Fig. 6A). The degradation was slower than in the case of BET proteins, but between 6 and 12 hours, the protein was largely absent from our cell lysates (Fig. 6B). Again, no degradation could be observed in the dark. PHOTAC-II-6, wherein the photoswitch was moved further into the linker region, also elicited light-dependent degradation (Fig. 6C). The time course of FKBP12 degradation by PHOTAC-II-6 was similar to the one observed with PHOTAC-II-5 (Fig. 6D). In this case, however, we also observed slight dark activity at the 24-hour time point. Both PHOTACs showed a pronounced hook effect and were inactive in the presence of MLN4924.
Fig. 6. Optical control FKBP12 degradation.
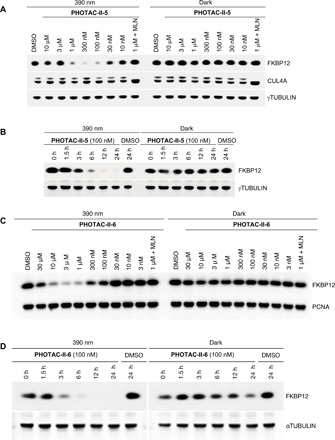
(A) Immunoblot analysis of FKBP12 after treatment of RS4;11 cells with PHOTAC-II-5 for 4 hours at different concentrations. Cells were either irradiated with pulses of 390-nm light (left, 100 ms every 10 s) or kept in the dark (right). (B) Time course of FKBP12 degradation visualized by immunoblotting. RS4;11 cells were treated with PHOTAC-II-5 (100 nM) and collected at the indicated time points. (C) Immunoblot analysis of FKBP12 after treatment of RS4;11 cells with PHOTAC-II-6 for 4 hours at different concentrations. Cells were either irradiated with pulses of 390-nm light (left, 100 ms every 10 s) or kept in the dark (right). (D) Time course of FKBP12 degradation visualized by immunoblotting. RS4;11 cells were treated with PHOTAC-II-6 (100 nM) and collected at the indicated time points.
DISCUSSION
By incorporating photoswitches into PROTACs, we have delineated a general strategy to control targeted protein degradation with the temporal and spatial precision that light affords. As such, we have applied the concept of photopharmacology to an important new target class, i.e., E3 ubiquitin ligases, and have added a highly useful functional feature to existing PROTACs.
As a proof of principle, we developed PHOTACs that combine CRBN ligands with azobenzene photoswitches and ligands for either BET proteins (BRD2,3,4) or FKBP12. The modularity of our approach should enable the straightforward development of PHOTACs that target many other classes of proteins. For instance, existing PROTACs that target CDK4/6 (30), CDK9 (16), BTK (19, 20), ABL (17, 39), ALK (anaplastic lymphoma kinase) (40), MET (18), MDM2 (murine double minute 2) (41), and Tau (42) could be adapted to become light activatable. Our PHOTACs for BRD2,3,4 may enable new insights into epigenetic pathways and potentially serve as precision tools in medicine. Color dosing and reversibility could be particularly useful in this regard.
Our PHOTACs for FKBP12 could enable the optical degradation of not only wild-type prolyl cis-trans isomerases but also of proteins that are tagged with this domain. This would substantially increase the utility of dTAGs (degradation tags), which have emerged as a broadly applicable technology to influence the homeostasis of fusion proteins (14, 15). In addition, PROTACs that link thalidomide derivatives to an alkyl halide could be modified to optically degrade proteins fused to a HaloTag domain (43). Note that some of our photoswitchable thalidomide derivatives, such as synthetic intermediates 2 to 4, and derivatives thereof, could function as dimerizers that recruit Ikaros (IKZF1) and Aiolos (IKZF3) to CRBN in a light-dependent fashion (44).
The PHOTACs introduced herein have several features that make them useful for biological studies: They are inactive as degraders in the dark and become active upon irradiation. Following activation, they gradually lose their activity through thermal relaxation. Alternatively, they can be quickly inactivated photochemically. In any scenario, their inactivation is much less dependent on dilution, clearance, or metabolism. In the case of compounds of the PHOTAC-I series, they still function as inhibitors of BET proteins, which explains the cytotoxicity observed in the dark. The concentrations needed for maximum photo effect are low (nanomolar range), substantially reducing possible off-target effects. The light needed for photoactivation is not cytotoxic, given the low intensities needed for photoisomerization and the pulse protocol used (26, 36, 45).
In principle, light activation of PROTACs could also be achieved with a caging strategy. Caged PROTACs have very recently emerged, complementing PHOTACs (46). The advantage of the latter lies in their reversibility, the low intensities of light needed to trigger the photochemistry, the ease with which the active concentration can be tuned, and the avoidance of potentially toxic by-products. Fast relaxing PHOTACs may also provide advantages in terms of localization and temporal control.
Genetically encoded degrons fused to a photosensitive LOV2 (light oxygen voltage-sensing) domain have been reported as an optogenetic approach to protein degradation (47, 48). Although this approach works well in vivo and provides a powerful method to study biological pathways, it requires transfection with a gene of interest, which limits its therapeutic applicability and can potentially create unphysiological protein levels and distributions within a cell. PHOTACs, by contrast, operate like drugs in native tissues.
An important question to address is how exactly the (E) and (Z) isomers of PHOTACs influence ternary complex formation and target degradation. Although an effect of photoswitching on pharmacokinetics cannot be ruled out entirely, the increased activity as a degrader seems to be primarily driven by pharmacodynamics. We consistently observed a hook effect and found no activity when we down-regulated CRBN or in the presence of the neddylation inhibitor MLN4924, indicating that PHOTACs function as true PROTACs. Whether the photoswitch primarily affects binding to CRBN or the relative positioning of the E3 ligase and the POI remains to be determined. Molecular modeling suggests that the (E) configuration of the photoswitch cannot be accommodated as well by CRBN as the (Z) configuration (see fig. S6). The importance of the exact nature, length, and orientation of the linker in PROTACs has been noted (“linkerology”) (19, 49, 50). In keeping with this, we also observed a pronounced effect of the diamine spacers on our systems. In the series targeting BET proteins, PHOTAC-I-3, which bears a 1,4-diamino butane spacer, showed the largest difference between light and dark activity in MTS assays. Related compounds that feature shorter or longer spacers showed smaller or no differences (see fig. S3). PHOTAC-I-10, wherein the photoswitch is positioned more centrally, also showed a light-dependent behavior, whereas analogs with different linker lengths did not. Among compounds targeting FKBP12, the lenalidomide derivative PHOTAC-II-5 and the thalidomide derivative PHOTAC-II-6, which feature different photoswitch incorporation modes, showed the largest light activation. In the case of derivative PHOTAC-II-5, longer linkers were detrimental to optical control (figs. S9 and S10). A satisfying explanation of these observations will require detailed biophysical and structural investigations.
The future development of PHOTACs can be taken into many different directions. The incorporation of red-shifted and faster-relaxing photoswitches could further improve the temporal and spatial precision of dark-inactive compounds (51). Our synthetic approaches are well suited to increase photoswitch diversity. Different ligands for E3 ligases should be explored, for instance, compounds that bind to VHL or the RING ligase MDM2. The optimal position of the photoswitches will depend on the exact nature of the system. Here, we have explored two different modes of photoswitch incorporation, but the third variety (photoswitch in the POI ligand), and combinations thereof, should also be considered. The POI ligand should ideally bind to its target without interfering with its function to cleanly distinguish between degradation and inhibition and to avoid unwanted toxicity.
We anticipate that PHOTACs will be useful tools in cell biology, but we believe that their clinical potential is also worthy of consideration. Since PROTACs operate in a catalytic fashion and enable systemic protein knockdown, their toxicity is a major concern (52, 53). PHOTACs of the type described herein could be activated with light within a tissue or before administration. They would then lose their activity with a given rate, which can be determined through engineering of the switch, or they could be actively turned off with a second wavelength. Moreover, PHOTACs locally activated by light would also lose efficacy by dilution, diffusing away from the point of irradiation. The usefulness of light in medicine is well established, and the combination of light and molecules has been studied for decades, e.g., in photodynamic therapy (PDT) (54). PDT has been applied with encouraging results to treat non–small cell lung cancer, dermatological cancers, and premalignant lesions of the upper digestive tract and is currently in clinical trials for the treatment of a large variety of other malignancies, including prostate, brain, and breast cancers (54). In the past, we have used PDT both in cultured cells and in mouse models to induce death of prostate cancer cells in a calcium-dependent manner (55). However, from a molecular point of view, conventional PDT, while effective, is unspecific. PHOTACs, by contrast, can be reversibly activated with the temporal and spatial precision that light affords and target specific proteins whose elimination would promote cell death. Therefore, we and others (56) believe that PHOTACs may provide a promising new direction in photomedicine.
MATERIALS AND METHODS
Synthesis
The reagents and solvents used in this study were bought from ABCR, Acros Organics, Alfa Aesar, Ark Pharm, Combi-Blocks, Oakwood, Oxchem, Sigma-Aldrich, Strem, and Toronto Research Chemicals and were used as purchased. Dry solvents used in reactions performed under inert atmosphere were obtained by passing the degassed solvents through activated alumina columns. Column chromatography was carried out on silica gel (pore size, 60 Å; 40 to 63 μm; Merck KGaA) using a Teledyne ISCO CombiFlash EZ Prep flash purification system.
Determination of photophysical properties
For ultraviolet-visible (UV-vis) spectroscopy on the Varian Cary 60 UV-Visible spectrophotometer, samples were stored and prepared under red light to avoid the formation of the (Z) isomers. Stock solutions (10 mM) were prepared in the dark and diluted to a final concentration of 25 μM for measurement. UV-vis spectra of PHOTACs were recorded following irradiation with different wavelengths for 5 min using a monochromator. Measurement was started from the dark adapted state followed by 370 nm irradiation and incrementally increased the wavelength. By increasing the wavelength from low to high, we could observe how much we could switch from (Z) to (E) isomers, whereas going from high to low wavelength, the PSS might not have been attained due to low absorptivity above 500 nm. PHOTAC-I-1–8 were measured in phosphate-buffered saline (PBS) with 10% DMSO, whereas PHOTAC-II-1–6 were measured in DMSO. Thermal relaxation was measured by preirradiating PHOTACs with 390-nm light and observing the absorption at 370 nm over 12 hours at 37°C in DMSO in tightly sealed cuvettes.
The reversible switching and photochemical stability of PHOTAC-I-3 were demonstrated in DMSO, cycling the irradiation of the monochromator between 390 nm (3 min) and 500 nm (7 min). The separated spectra of the (Z) and (E) isomers could be obtained from the internal UV-vis detector of the liquid chromatography–mass spectrometry (LC-MS) by irradiating the sample before injection and were normalized at the isosbestic point. In the case of nonsolvochromic photoswitches, PSSs were calculated in the region of the largest absorption difference between 330 and 390 nm from the separated spectra obtained by LC-MS and the spectra obtained following irradiation with different wavelengths for 5 min, all normalized to the isosbestic point.
LED illumination
For illumination of the cells, we used the cell disco system as previously described in the literature (36). Light-emitting diodes (LEDs) (5 mm) [370 nm (XSL-370-5E), 390 nm (VL390-5-15), 410 nm (VL410-5-15), 430 nm (VL430-5-15), 450 nm (ELD-450-525), 465 nm (RLS-B465), 477 nm (RLS-5B475-5), 490 nm (LED490-03), 505 nm (B5-433-B505), 525 nm (B5-433-B525), 545 nm (LED545-04), 572 nm (B5-433-20), and 590 nm (CY5111A-WY)] were purchased from Roithner Lasertechnik. For experiments using 390 nm, cells were preirradiated for 1 min at 390 nm to quickly switch the photoswitches in the active state. Pulsed irradiation was performed using 100-ms pulses every 10 s in 96- or 6-well plates, controlled by an Arduino system.
Cell culture
The human acute lymphoblastic leukemia RS4;11 (ATCC CRL1873) cell line was purchased from the American Type Culture Collection (ATCC) and cultured in RPMI 1640 medium (Gibco) with 10% fetal bovine serum and 1% penicillin/streptomycin in a humidified incubator at 37°C with 5% CO2 in air. For the experiments, compounds were serially diluted in RPMI 1640 without the dye phenol red (Gibco) to reduce the influence of the dye by absorption. Azobenzene stocks and dilutions were strictly kept in the dark and prepared under red light conditions.
For immunoblotting analysis, cells (2 × 106 for RS4:11) were incubated for the indicated times with PHOTACs, placed in a light-proof box, and preirradiated for 1 min at 390 nm followed by 100-ms pulses every 10 s or were kept in the dark for 4 hours. After incubation, cells were collected in the dark by centrifugation (150g, 5 min) at 4°C, and the pellets were washed twice with ice-cold PBS (1 ml).
Colorimetric MTS assays
The activity of dehydrogenase enzymes in metabolically active cells, as a quantitative measurement for cytotoxicity and proliferation, was determined by colorimetric measurement of the reduction of MTS to the formazan derivative. The absorbance of formazan at 490 nm was measured on a FLUOstar Omega microplate reader (BMG Labtech). Cells were treated with our compounds at different concentrations, ranging from 10 μM to 1 nM/1 pM, (from 10 μM to 1 nM/1 pM) in triplicates or sextuplicates, using 1% DMSO as cosolvent, and incubated on a 96-well plate for 72 hours. They were placed in light-proof boxes and exposed to the lighting conditions specified in the experiment for 72 hours. Next, 10 μl of Promega CellTiter 96 AQueous One Solution Reagent was added to each well and incubated for further 4 to 7 hours at 37°C. The absorbance at 490 nm was then recorded with a 96-well plate reader.
Data were analyzed using GraphPad Prism version 5.01 (GraphPad Software Inc.) and fitted using the sigmoidal dose-response (variable slope) fit. Results represent the mean viability ± SEM relative to the 1% DMSO–treated control.
Immunoblotting analysis
Cells were lysed in radioimmunoprecipitation assay buffer containing protease and phosphatase inhibitor, and protein concentration was determined using BCA (Thermo Fisher Scientific). Immunoblotting was performed as previously described (57). Briefly, samples were resolved under denaturing and reducing conditions using 4 to 12% bis-tris gels (NuPAGE) and transferred to a polyvinylidene fluoride membrane (Immobilon-P, Millipore). Membranes were blocked with 5% nonfat dried milk and incubated with primary antibodies overnight at 4°C. After washing the membranes, secondary antibodies coupled with horseradish peroxidase were applied (Amersham-GE). Immunoreactive bands were visualized by enhanced chemiluminescence reagent (Thermo Fisher Scientific), and signal was acquired using ImageQuant LAS 400 (GE) (Table 1).
Table 1. Antibodies used in this study.
PCNA, proliferating cell nuclear antigen; HRP, horseradish peroxidase; IgG, immunoglobulin G.
| Antibodies | Source | Identifier |
| β-Actin | Cell Signaling Technology |
#4970 |
| BRD2 | Bethyl Laboratories | A302-583A |
| BRD3 | Bethyl Laboratories | A302-368A-1 |
| BRD4 | Cell Signaling Technology |
#13440 |
| c-MYC | Cell Signaling Technology |
#5605 |
| PARP1 | Cell Signaling Technology |
Catalog no. 9542S |
| PCNA | Dako | Catalog no. M0879 |
| αTUBULIN | Sigma-Aldrich | T6557 |
| γTUBULIN | Sigma-Aldrich | T6074 |
| FKBP12 | Santa Cruz Biotechnology |
Catalog no. sc-133067 |
| CUL4A | Bethyl Laboratories | A300-739A |
| MCM2 | Santa Cruz Biotechnology |
Catalog no. sc-9839 |
| Anti-rabbit IgG, peroxidase-linked antibody |
Thermo Fisher Scientific |
NA934 |
| Anti-mouse IgG, peroxidase-linked antibody |
Thermo Fisher Scientific |
NA931 |
| Anti-goat IgG-HRP | Santa Cruz Biotechnology |
Catalog no. sc-2354 |
siRNA knockdown of CRBN
Real-time polymerase chain reaction (PCR) was performed as previously described (55). For knockdown of CRBN, MB-MDA-231 cells were seeded 18 hours before transfection. The following ON-TARGETplus siRNA oligos from Dharmacon were transfected with Lipofectamine for 8 hours according to the manufacturer’s instructions (RNAiMAX, Thermo Fisher Scientific): ON-TARGETplus human CRBN SMARTpool (L-021086-00-0005) and ON-TARGETplus nontargeting control siRNA (D-001810-01-05). The medium was replaced, and cells were incubated for 40 hours, upon which they were resubjected to another cycle of siRNA transfection. Medium was replaced, and after 1 hour, the cells were treated with PHOTAC-I-3 or vehicle for 18 hours under pulsed irradiation at 390 nm or in the dark, upon which the cells were collected and subjected to immunoblotting analysis as described above. Gene silencing was validated by reverse transcription PCR (RT-PCR), isolating total RNA using Qiagen’s RNeasy kit (catalog no. 74104). The RT of the mRNA was carried out using 3 μg of total RNA using random hexamers and oligo(dT)18 primers with SMART MMLV Reverse Transcriptase (RNA to cDNA EcoDry Premix, Takara, catalog no. 639549) according to the manufacturer’s instruction. The RT-PCR was carried out using PowerUp SYBR Green (Thermo Fisher, catalog no. A25742) with the Thermo Fisher QuantStudio 3 Real-Time PCR system in a 96-well format. Bar graphs represent the relative ratio of CRBN to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) values. The following primers were used for RT-PCR: human CRBN (primer set 1) (58) (forward: 5′-CCAGTCTGCCGACATCACAT-3′, reverse: 5′-GTCATCGTGCAAAGTCCTGC-3′), human CRBN (primer set 2) (59) (forward: 5′-CAGTCTGCCGACATCACATAC-3′, reverse: 5′-GCACCATACTGACTT CTTGAGGG-3′), and human GAPDH (forward: 5′-TGCACCACCAACTGCTTAGC-3′, reverse: 5′-GGCATGGACTGTGGTCATGAG-3′).
Supplementary Material
Acknowledgments
We are indebted to T. J. Wandless for the gift of SLF ligand. M.P. is grateful to T. M. Thor for continuous support. Funding: We thank New York University for financial support. Nuclear magnetic resonance spectra were acquired using the TCI cryoprobe supported by the NIH (OD016343). This work was partially funded by grant R01-CA76584 from the NIH to M.P. and a fellowship from the T32-CA009161 (Levy) grant to A.M. M.P. is an investigator with the Howard Hughes Medical Institute. Author contributions: D.T., M.R., and B.S.M. conceived the study. M.R., B.S.M., M.B., D.S, and A.M. designed the experiments and analyzed the data. D.T. and M.P. supervised the experiments. D.T., M.R., and B.S.M. wrote the paper with input from all authors. Competing interests: M.R., B.S.M., M.B., and D.T. are inventors on a patent application on PHOTACs. M.P. is a member of the scientific advisory boards of CullGen Inc. and Kymera Therapeutics and a consultant for BeyondSpring Pharmaceutical. All other authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. The compounds prepared in this study can be provided by the authors pending scientific review and a completed material transfer agreement. Requests for the PHOTACs should be submitted to D.T. (dirktrauner@nyu.edu) or M.P. (Michele.Pagano@nyulangone.org). Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/8/eaay5064/DC1
Fig. S1. UV-vis characterization.
Fig. S2. Thermal relaxation.
Fig. S3. Viability of RS4;11 acute lymphoblastic leukemia cells after treatment with PHOTAC-I for 72 hours in the dark or under pulsed (100 ms every 10 s) 390-nm irradiation.
Fig. S4. Immunoblot analysis of PHOTAC-I-3.
Fig. S5. Immunoblot analysis of BRD4 after treatment of RS4;11 cells with PHOTACs.
Fig. S6. Model of the photoswitch-cereblon interaction.
Fig. S7. CRBN knockdown control.
Fig. S8. Me-PHOTAC-I-3 control.
Fig. S9. FKBP12 Immunoblots in RS4;11 cells.
Fig. S10. Immunoblot analysis of FKBP12 after treatment of RS4;11 cells.
General information
Synthetic Procedures and Characterization
REFERENCES AND NOTES
- 1.Skaar J. R., Pagan J. K., Pagano M., SCF ubiquitin ligase-targeted therapies. Nat. Rev. Drug Discov. 13, 889–903 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lai A. C., Crews C. M., Induced protein degradation: An emerging drug discovery paradigm. Nat. Rev. Drug Discov. 16, 101–114 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Burslem G. M., Crews C. M., Small-Molecule Modulation of Protein Homeostasis. Chem. Rev. 117, 11269–11301 (2017). [DOI] [PubMed] [Google Scholar]
- 4.Sakamoto K. M., Kim K. B., Kumagai A., Mercurio F., Crews C. M., Deshaies R. J., Protacs: Chimeric molecules that target proteins to the Skp1–Cullin–F box complex for ubiquitination and degradation. Proc. Natl. Acad. Sci. U.S.A. 98, 8554–8559 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bondeson D. P., Mares A., Smith I. E. D., Ko E., Campos S., Miah A. H., Mulholland K. E., Routly N., Buckley D. L., Gustafson J. L., Zinn N., Grandi P., Shimamura S., Bergamini G., Faelth-Savitski M., Bantscheff M., Cox C., Gordon D. A., Willard R. R., Flanagan J. J., Casillas L. N., Votta B. J., den Besten W., Famm K., Kruidenier L., Carter P. S., Harling J. D., Churcher I., Crews C. M., Catalytic in vivo protein knockdown by small-molecule PROTACs. Nat. Chem. Biol. 11, 611–617 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zengerle M., Chan K.-H., Ciulli A., Selective small molecule induced degradation of the BET bromodomain protein BRD4. ACS Chem. Biol. 10, 1770–1777 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Winter G. E., Buckley D. L., Paulk J., Roberts J. M., Souza A., Dhe-Paganon S., Bradner J. E., Phthalimide conjugation as a strategy for in vivo target protein degradation. Science 348, 1376–1381 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lu J., Qian Y., Altieri M., Dong H., Wang J., Raina K., Hines J., Winkler J. D., Crew A. P., Coleman K., Crews C. M., Hijacking the E3 ubiquitin ligase cereblon to efficiently target BRD4. Chem. Biol. 22, 755–763 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sakamoto K. M., Kim K. B., Verma R., Ransick A., Stein B., Crews C. M., Deshaies R. J., Development of protacs to target cancer-promoting proteins for ubiquitination and degradation. Mol. Cell. Proteomics 2, 1350–1358 (2003). [DOI] [PubMed] [Google Scholar]
- 10.Schneekloth A. R., Pucheault M., Tae H. S., Crews C. M., Targeted intracellular protein degradation induced by a small molecule: En route to chemical proteomics. Bioorg. Med. Chem. Lett. 18, 5904–5908 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Han X., Wang C., Qin C., Xiang W., Fernandez-Salas E., Yang C.-Y., Wang M., Zhao L., Xu T., Chinnaswamy K., Delproposto J., Stuckey J., Wang S., Discovery of ARD-69 as a highly potent proteolysis targeting chimera (PROTAC) degrader of androgen receptor (AR) for the treatment of prostate cancer. J. Med. Chem. 62, 941–964 (2019). [DOI] [PubMed] [Google Scholar]
- 12.Hu J., Hu B., Wang M., Xu F., Miao B., Yang C.-Y., Wang M., Liu Z., Hayes D. F., Chinnaswamy K., Delproposto J., Stuckey J., Wang S., Discovery of ERD-308 as a highly potent proteolysis targeting chimera (PROTAC) degrader of estrogen receptor (ER). J. Med. Chem. 62, 1420–1442 (2019). [DOI] [PubMed] [Google Scholar]
- 13.Yang C.-Y., Qin C., Bai L., Wang S., Small-molecule PROTAC degraders of the Bromodomain and Extra Terminal (BET) proteins — A review. Drug Discov. Today Technol. 31, 43–51 (2019). [DOI] [PubMed] [Google Scholar]
- 14.Nabet B., Roberts J. M., Buckley D. L., Paulk J., Dastjerdi S., Yang A., Leggett A. L., Erb M. A., Lawlor M. A., Souza A., Scott T. G., Vittori S., Perry J. A., Qi J., Winter G. E., Wong K.-K., Gray N. S., Bradner J. E., The dTAG system for immediate and target-specific protein degradation. Nat. Chem. Biol. 14, 431– 441 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Boija A., Klein I. A., Sabari B. R., Dall’Agnese A., Coffey E. L., Zamudio A. V., Li C. H., Shrinivas K., Manteiga J. C., Hannett N. M., Abraham B. J., Afeyan L. K., Guo Y. E., Rimel J. K., Fant C. B., Schuijers J., Lee T. I., Taatjes D. J., Young R. A., Transcription factors activate genes through the phase-separation capacity of their activation domains. Cell 175, 1842–1855.e16 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Olson C. M., Jiang B., Erb M. A., Liang Y., Doctor Z. M., Zhang Z., Zhang T., Kwiatkowski N., Boukhali M., Green J. L., Haas W., Nomanbhoy T., Fischer E. S., Young R. A., Bradner J. E., Winter G. E., Gray N. S., Pharmacological perturbation of CDK9 using selective CDK9 inhibition or degradation. Nat. Chem. Biol. 14, 163–170 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lai A. C., Toure M., Hellerschmied D., Salami J., Jaime-Figueroa S., Ko E., Hines J., Crews C. M., Modular PROTAC design for the degradation of oncogenic BCR-ABL. Angew. Chem. Int. Ed. 55, 807–810 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Burslem G. M., Smith B. E., Lai A. C., Jaime-Figueroa S., McQuaid D. C., Bondeson D. P., Toure M., Dong H., Qian Y., Wang J., Crew A. P., Hines J., Crews C. M., The advantages of targeted protein degradation over inhibition: An RTK case study. Cell Chem. Biol. 25, 67–77.e3 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zorba A., Nguyen C., Xu Y., Starr J., Borzilleri K., Smith J., Zhu H., Farley K. A., Ding W., Schiemer J., Feng X., Chang J. S., Uccello D. P., Young J. A., Garcia-Irrizary C. N., Czabaniuk L., Schuff B., Oliver R., Montgomery J., Hayward M. M., Coe J., Chen J., Niosi M., Luthra S., Shah J. C., El-Kattan A., Qiu X., West G. M., Noe M. C., Shanmugasundaram V., Gilbert A. M., Brown M. F., Calabrese M. F., Delineating the role of cooperativity in the design of potent PROTACs for BTK. Proc. Natl. Acad. Sci. U.S.A. 115, E7285–E7292 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Buhimschi A. D., Armstrong H. A., Toure M., Jaime-Figueroa S., Chen T. L., Lehman A. M., Woyach J. A., Johnson A. J., Byrd J. C., Crews C. M., Targeting the C481S ibrutinib-resistance mutation in Bruton’s tyrosine kinase using PROTAC-mediated degradation. Biochemistry 57, 3564–3575 (2018). [DOI] [PubMed] [Google Scholar]
- 21.Houzelstein D., Bullock S. L., Lynch D. E., Grigorieva E. F., Wilson V. A., Beddington R. S. P., Growth and early postimplantation defects in mice deficient for the bromodomain-containing protein Brd4. Mol. Cell. Biol. 22, 3794–3802 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shang E., Wang X., Wen D., Greenberg D. A., Wolgemuth D. J., Double bromodomain-containing gene Brd2 is essential for embryonic development in mouse. Dev. Dyn. 238, 908–917 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Silva J. M., Silva E., Reis R. L., Light-triggered release of photocaged therapeutics - Where are we now? J. Control. Release 298, 154–176 (2019). [DOI] [PubMed] [Google Scholar]
- 24.Fenno L., Yizhar O., Deisseroth K., The development and application of optogenetics. Annu. Rev. Neurosci. 34, 389–412 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lerch M. M., Hansen M. J., van Dam G. M., Szymanski W., Feringa B. L., Emerging targets in photopharmacology. Angew. Chem. Int. Ed. 55, 10978–10999 (2016). [DOI] [PubMed] [Google Scholar]
- 26.Hüll K., Morstein J., Trauner D., In vivo photopharmacology. Chem. Rev. 118, 10710–10747 (2018). [DOI] [PubMed] [Google Scholar]
- 27.Siewertsen R., Neumann H., Buchheim-Stehn B., Herges R., Näther C., Renth F., Temps F., Highly efficient reversible Z−E photoisomerization of a bridged azobenzene with visible light through resolved S1(nπ*) absorption bands. J. Am. Chem. Soc. 131, 15594–15595 (2009). [DOI] [PubMed] [Google Scholar]
- 28.Trads J. B., Hüll K., Matsuura B. S., Laprell L., Fehrentz T., Görldt N., Kozek K. A., Weaver C. D., Klöcker N., Barber D. M., Trauner D., Sign inversion in photopharmacology: Incorporation of cyclic azobenzenes in photoswitchable potassium channel blockers and openers. Angew. Chem. Int. Ed. 131, 15567–11574 (2019). [DOI] [PubMed] [Google Scholar]
- 29.Douglass E. F. Jr., Miller C. J., Sparer G., Shapiro H., Spiegel D. A., A comprehensive mathematical model for three-body binding equilibria. J. Am. Chem. Soc. 135, 6092–6099 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brand M., Jiang B., Bauer S., Donovan K. A., Liang Y., Wang E. S., Nowak R. P., Yuan J. C., Zhang T., Kwiatkowski N., Müller A. C., Fischer E. S., Gray N. S., Winter G. E., Homolog-selective degradation as a strategy to probe the function of CDK6 in aml. Cell Chem. Biol. 26, 300–306.e9 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Delmore J. E., Issa G. C., Lemieux M. E., Rahl P. B., Shi J., Jacobs H. M., Kastritis E., Gilpatrick T., Paranal R. M., Qi J., Chesi M., Schinzel A. C., McKeown M. R., Heffernan T. P., Vakoc C. R., Bergsagel P. L., Ghobrial I. M., Richardson P. G., Young R. A., Hahn W. C., Anderson K. C., Kung A. L., Bradner J. E., Mitsiades C. S., BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell 146, 904–917 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stathis A., Bertoni F., BET proteins as targets for anticancer treatment. Cancer Discov. 8, 24–36 (2018). [DOI] [PubMed] [Google Scholar]
- 33.Chen H., Liu H., Qing G., Targeting oncogenic Myc as a strategy for cancer treatment. Signal Transduct. Target. Ther. 3, 5 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chaitanya G. V., Alexander J. S., Babu P. P., PARP-1 cleavage fragments: signatures of cell-death proteases in neurodegeneration. Cell Commun. Signal 8, 31 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rullo A., Reiner A., Reiter A., Trauner D., Isacoff E. Y., Woolley G. A., Long wavelength optical control of glutamate receptor ion channels using a tetra-ortho-substituted azobenzene derivative. Chem. Commun. 50, 14613–14615 (2014). [DOI] [PubMed] [Google Scholar]
- 36.Borowiak M., Nahaboo W., Reynders M., Nekolla K., Jalinot P., Hasserodt J., Rehberg M., Delattre M., Zahler S., Vollmar A., Trauner D., Thorn-Seshold O., Photoswitchable inhibitors of microtubule dynamics optically control mitosis and cell death. Cell 162, 403–411 (2015). [DOI] [PubMed] [Google Scholar]
- 37.Amara J. F., Clackson T., Rivera V. M., Guo T., Keenan T., Natesan S., Pollock R., Yang W., Courage N. L., Holt D. A., Gilman M., A versatile synthetic dimerizer for the regulation of protein–protein interactions. Proc. Natl. Acad. Sci. U.S.A. 94, 10618–10623 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Banaszynski L. A., Chen L. C., Maynard-Smith L. A., Lisa Ooi A. G., Wandless T. J., A rapid, reversible, and tunable method to regulate protein function in living cells using synthetic small molecules. Cell 126, 995–1004 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shibata N., Miyamoto N., Nagai K., Shimokawa K., Sameshima T., Ohoka N., Hattori T., Imaeda Y., Nara H., Cho N., Naito M., Development of protein degradation inducers of oncogenic BCR-ABL protein by conjugation of ABL kinase inhibitors and IAP ligands. Cancer Sci. 108, 1657–1666 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Powell C. E., Gao Y., Tan L., Donovan K. A., Nowak R. P., Loehr A., Bahcall M., Fischer E. S., Jänne P. A., George R. E., Gray N. S., Chemically induced degradation of anaplastic lymphoma kinase (ALK). J. Med. Chem. 61, 4249–4255 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li Y., Yang J., Aguilar A., McEachern D., Przybranowski S., Liu L., Yang C.-Y., Wang M., Han X., Wang S., Discovery of MD-224 as a first-in-class, highly potent, and efficacious proteolysis targeting chimera murine double minute 2 degrader capable of achieving complete and durable tumor regression. J. Med. Chem. 62, 448–466 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Silva M. C., Ferguson F. M., Cai Q., Donovan K. A., Nandi G., Patnaik D., Zhang T., Huang H.-T., Lucente D. E., Dickerson B. C., Mitchison T. J., Fischer E. S., Gray N. S., Haggarty S. J., Targeted degradation of aberrant tau in frontotemporal dementia patient-derived neuronal cell models. eLife 8, e45457 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Buckley D. L., Raina K., Darricarrere N., Hines J., Gustafson J. L., Smith I. E., Miah A. H., Harling J. D., Crews C. M., HaloPROTACS: Use of small molecule PROTACs to induce degradation of HaloTag fusion proteins. ACS Chem. Biol. 10, 1831–1837 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chamberlain P. P., Hamann L. G., Development of targeted protein degradation therapeutics. Nat. Chem. Biol. 15, 937–944 (2019). [DOI] [PubMed] [Google Scholar]
- 45.A. Griesbeck, M. Oelgemöller, F. Ghetti, CRC Handbook of Organic Photochemistry and Photobiology (CRC Press, ed. 3, 2012); www.crcpress.com/CRC-Handbook-of-Organic-Photochemistry-and-Photobiology-Third-Edition/Griesbeck-Oelgemoller-Ghetti/p/book/9781439899335.
- 46.Naro Y., Darrah K., Deiters A., Optical control of small molecule-induced protein degradation. J. Am. Chem. Soc. 142, 2193–2197 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Renicke C., Schuster D., Usherenko S., Essen L.-O., Taxis C., A LOV2 domain-based optogenetic tool to control protein degradation and cellular function. Chem. Biol. 20, 619–626 (2013). [DOI] [PubMed] [Google Scholar]
- 48.Bonger K. M., Rakhit R., Payumo A. Y., Chen J. K., Wandless T. J., General method for regulating protein stability with light. ACS Chem. Biol. 9, 111–115 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cyrus K., Wehenkel M., Choi E.-Y., Han H.-J., Lee H., Swanson H., Kim K.-B., Impact of linker length on the activity of PROTACs. Mol. BioSyst. 7, 359–364 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nowak R. P., DeAngelo S. L., Buckley D., He Z., Donovan K. A., An J., Safaee N., Jedrychowski M. P., Ponthier C. M., Ishoey M., Zhang T., Mancias J. D., Gray N. S., Bradner J. E., Fischer E. S., Plasticity in binding confers selectivity in ligand-induced protein degradation. Nat. Chem. Biol. 14, 706–714 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dong M., Babalhavaeji A., Samanta S., Beharry A. A., Woolley G. A., Red-shifting azobenzene photoswitches for in vivo use. Acc. Chem. Res. 48, 2662–2670 (2015). [DOI] [PubMed] [Google Scholar]
- 52.Raina K., Lu J., Qian Y., Altieri M., Gordon D., Rossi A. M. K., Wang J., Chen X., Dong H., Siu K., Winkler J. D., Crew A. P., Crews C. M., Coleman K. G., PROTAC-induced BET protein degradation as a therapy for castration-resistant prostate cancer. Proc. Natl. Acad. Sci. U.S.A. 113, 7124–7129 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sun X., Wang J., Yao X., Zheng W., Mao Y., Lan T., Wang L., Sun Y., Zhang X., Zhao Q., Zhao J., Xiao R.-P., Zhang X., Ji G., Rao Y., A chemical approach for global protein knockdown from mice to non-human primates. Cell Discov. 5, 10 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Van Straten D., Mashayekhi V., De Bruijn H. S., Oliveira S., Robinson D. J., Oncologic photodynamic therapy: Basic principles, current clinical status and future directions. Cancer 9, 19 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kuchay S., Giorgi C., Simoneschi D., Pagan J., Missiroli S., Saraf A., Florens L., Washburn M. P., Collazo-Lorduy A., Castillo-Martin M., Cordon-Cardo C., Sebti S. M., Pinton P., Pagano M., PTEN counteracts FBXL2 to promote IP3R3- and Ca2+-mediated apoptosis limiting tumour growth. Nature 546, 554–558 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pfaff P., Samarasinghe K. T. G., Crews C. M., Carreira E. M., Reversible spatiotemporal control of induced protein degradation by bistable PhotoPROTACs. ACS Cent. Sci. 5, 1682–1690 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Marzio A., Puccini J., Kwon Y., Maverakis N. K., Arbini A., Sung P., Bar-Sagi D., Pagano M., The F-Box domain-dependent activity of EMI1 regulates PARPi sensitivity in triple-negative breast cancers. Mol. Cell 73, 224–237.e6 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dimopoulos K., Helbo A. S., Munch-Petersen H. F., Sjö L., Christensen J., Kristensen L. S., Asmar F., Hermansen N. E. U., O’Connel C., Gimsing P., Liang G., Grønbæk K., Dual inhibition of DNMTs and EZH2 can overcome both intrinsic and acquired resistance of myeloma cells to IMiDs in a cereblon-independent manner. Mol. Oncol. 12, 180–195 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Xu Q., Hou Y. X., Langlais P., Erickson P., Zhu J., Shi C.-X., Luo M., Zhu Y., Xu Y., Mandarino L. J., Stewart K., Chang X. B., Expression of the cereblon binding protein argonaute 2 plays an important role for multiple myeloma cell growth and survival. BMC Cancer 16, 297 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Fischer E. S., Böhm K., Lydeard J. R., Yang H., Stadler M. B., Cavadini S., Nagel J., Serluca F., Acker V., Lingaraju G. M., Tichkule R. B., Schebesta M., Forrester W. C., Schirle M., Hassiepen U., Ottl J., Hild M., Beckwith R. E. J., Harper J. W., Jenkins J. L., Thomä N. H., Structure of the DDB1–CRBN E3 ubiquitin ligase in complex with thalidomide. Nature 512, 49–53 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.J. Bradner, D. Buckley, G. Winter, Methods to Induce Targeted Protein Degradation Through Bifunctional Molecules, U.S. Patent US2016176916 (2016); https://worldwide.espacenet.com/publicationDetails/biblio?FT=D&date=20160623&DB=EPODOC&locale=en_EP&CC=US&NR=2016176916A1&KC=A1&ND=4.
- 62.Jörg M., Glukhova A., Abdul-Ridha A., Vecchio E. A., Nguyen A. T. N., Sexton P. M., White P. J., May L. T., Christopoulos A., Scammells P. J., Novel irreversible agonists acting at the A1 adenosine receptor. J. Med. Chem. 59, 11182–11194 (2016). [DOI] [PubMed] [Google Scholar]
- 63.Stein M., Breit A., Fehrentz T., Gudermann T., Trauner D., Optical control of TRPV1 channels. Angew. Chem. Int. Ed. 52, 9845–9848 (2013). [DOI] [PubMed] [Google Scholar]
- 64.Samanta S., Qin C., Lough A. J., Woolley G. A., Bidirectional photocontrol of peptide conformation with a bridged azobenzene derivative. Angew. Chem. Int. Ed. 51, 6452–6455 (2012). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/8/eaay5064/DC1
Fig. S1. UV-vis characterization.
Fig. S2. Thermal relaxation.
Fig. S3. Viability of RS4;11 acute lymphoblastic leukemia cells after treatment with PHOTAC-I for 72 hours in the dark or under pulsed (100 ms every 10 s) 390-nm irradiation.
Fig. S4. Immunoblot analysis of PHOTAC-I-3.
Fig. S5. Immunoblot analysis of BRD4 after treatment of RS4;11 cells with PHOTACs.
Fig. S6. Model of the photoswitch-cereblon interaction.
Fig. S7. CRBN knockdown control.
Fig. S8. Me-PHOTAC-I-3 control.
Fig. S9. FKBP12 Immunoblots in RS4;11 cells.
Fig. S10. Immunoblot analysis of FKBP12 after treatment of RS4;11 cells.
General information
Synthetic Procedures and Characterization