

Abstract
Current electrophysiological or optical techniques cannot reliably perform simultaneous intracellular recordings from more than a few tens of neurons. Here, we report a nanoelectrode array that can simultaneously obtain intracellular recordings from thousands of connected mammalian neurons in vitro. The array consists of 4,096 platinum-black electrodes with nanoscale roughness fabricated on top of a silicon chip that monolithically integrates 4,096 microscale amplifiers, configurable into pseudo-current-clamp mode (for concurrent current injection and voltage recording) or into pseudo-voltage-clamp mode (for concurrent voltage application and current recording). We used the array in pseudo-voltage-clamp mode to measure the effects of drugs on ion-channel currents. In pseudo-current-clamp mode, the array recorded intracellular action potentials and post-synaptic potentials from over thousands of neurons. In addition, we mapped over 300 excitatory and inhibitory synaptic connections from over 1,700 neurons that were recorded for 19 mins. This high-throughput intracellular-recording technology could benefit functional connectome mapping, electrophysiological screening, and other functional interrogations of neuronal networks.
The patch clamp electrode is celebrated for its high-sensitivity intracellular recording that can measure not only action potential (AP) propagation in neurons but also subthreshold events such as postsynaptic potentials (PSPs). Dense, parallel execution of such high-sensitivity intracellular recording across a neuronal network is a significant technological pursuit in neurobiology1–4, but has never been achieved5. The patch clamp electrode itself is not well suited for scaling into a dense array, and only ~10 parallel patch recordings of neurons have been made to date6. Top-down fabricated nanoelectrode arrays have been developed in hopes of parallelizing intracellular recording7,8, but these intracellular interfaces exhibited fidelities much inferior to the patch clamp and could not be applied to neuronal networks.
Here we report a breakthrough neuroelectronic interface that is capable of massively parallel intracellular neuronal recording. It consists of 4,096 recording/stimulation sites, or “pixels,” of platinum black (PtB) electrodes, defined on top of a complementary metal-oxide-semiconductor (CMOS) chip—i.e., a silicon integrated circuit (IC)—that monolithically integrates 4,096 microscale amplifiers. The combination of the nanoscale-rough surface morphology of the PtB electrode and the on-site amplification, configurable between a pseudo current-clamp (pCC) and a pseudo voltage-clamp (pVC) mode with a continued electrode current injection, allows for stable, sensitive intracellular recording of subthreshold membrane potentials (pCC mode) and ion-channel currents (pVC mode) and for concurrent stimulation of neurons at each pixel. Its arrayed operation in the pCC mode then routinely records not only APs but also PSPs from thousands of connected mammalian neurons cultured in vitro. This unprecedented merger of parallelism for network coverage and intracellular recording for subthreshold sensitivity enables an explicit network-wide mapping of over 300 excitatory and inhibitory synaptic connections from over 1,700 neurons intracellularly recorded for only 19 minutes. This level of experimental throughput can transform a broad range of functional interrogations of neuronal networks, such as functional connectome mapping and high-throughput electrophysiological screening.
The capabilities brought by this CMOS-neuroelectronic interface (CNEI) can be highlighted in contrast to three other mainstream electrophysiology techniques. First, CMOS microelectrode arrays (MEAS)9–13, featuring as many as ~65,000 electrodes/circuit channels12, can monitor a large number of neurons, but they are an extracellular technique and so cannot measure subthreshold events that are critical for examining the synaptic connectivity of neurons5. In contrast, our CNEI subsumes the capability of the CMOS MEA (extracellular measurements of a large number of cells) and expands that parallelism to the intracellular measurements. Second, the microfluidic planar patch-clamp array14–16 is an electrophysiological drug screening tool that can perform parallel intracellular recording, but only from disconnected, non-neuronal cells due to its construct. In contrast, our CNEI can measure connected neurons (including induced pluripotent stem cell (iPSC) derived neurons) forming a network and can provide both an ion-channel assay (in the pVC mode) and a synaptic connection assay (in the pCC mode), directly measuring the effects of drugs on ion channel currents and network connectivity/dynamics. Third, optical electrophysiology, based on voltage sensitive dyes/proteins and Ca2+ indicators, features high signal-to-noise ratios, and when coupled with optical stimulation techniques, provides a promising path towards network-wide recording and stimulation17,18. To date, however, network-scale monitoring has been performed mostly with indirect Ca2+ imaging19–22, with membrane potential recordings being limited to tens of neurons17,18,23–25.
In the remainder of this Article we will give a detailed account of our development of the CNEI and the key experiments that highlight its performance.
The CMOS Neuroelectronic interface
CMOS electronics—that is, silicon ICs that form the heart of today’s computers—are known for the scalability that can integrate a massive number of transistors into a chip-scale footprint. The core of our neuroelectronic interface is one such CMOS IC, which we design (Fig. 1a, right). At the center of the IC is an array of 64 × 64 = 4,096 surface aluminum (Al) pads, each with the size of 10.5 μm × 10.5 μm and the pitch of 20 μm (Fig. 1b). On each pad, PtB-coated platinum (Pt) electrodes—another critical component of our neuroelectronic interface—are to be built. Each pad, which defines a unit recording/stimulation site, or a pixel, is wired to its own active circuit located outside the center pad array area (Supplementary Fig. 3a). We chose the pixel pad pitch of 20 μm so that each pixel can couple to one in vitro rat cortical neuron (soma diameter ~20 μm). The heart of each pixel’s active circuit is a low-noise voltage amplifier and a current injector: this active circuit block can be arranged into either the pCC or the pVC configuration (Supplementary Fig. 3b; see also Fig. 2b–c). We use the word ‘pseudo’ because our current and voltage clamp configurations end up fixing the current and the voltage of the electrode rather than those of the neuron due to the particular nature of our electrode-neuron interface, as discussed later. On each pad, we post-fabricate Pt electrodes of varying geometries such as vertical nanoneedles, vertical nanoneedles with pad edge lines, and hole electrodes using standard top-down technology (Supplementary Fig. 1a–d, Methods). We then package the CMOS IC with a microfluidic well into a chip carrier (Fig. 1a, left and Supplementary Fig. 1e) and place it on a custom-designed printed circuit board (Supplementary Fig. 1f). The final step of the CNEI making is the deposition of PtB on the Pt electrodes via electrodeposition26 (Fig. 1c–e, Supplementary Fig. 2a).
Fig. 1 |. The CMOS neuroelectronic interface (CNEI).

a, A CMOS-activated Pt black (PtB) electrode array packaged with a microfluidic well (left) in which neurons are cultured. Its CMOS IC (right) contains an array of 64×64 pixel pads (20 μm pitch) at its center. On each pad PtB-coated Pt electrodes are built: see (c-e). b, False colored scanning electron microscope (SEM) image of neurons cultured on top of the electrode array of an example CNEI device. Actual recording experiments are performed with much higher neuron densities containing 3-6 cell layers covering the entire electrode array (Supplementary Fig. 4). c-e, SEM images (top view) of the three different structures of Pt electrodes after PtB deposition, all of which have been demonstrated for intracellular access:(c) PtB-coated vertical Pt nanoneedles (dimensions before PtB coating: 9 Pt vertical nanoneedles per pad at a 3 μm pitch, ~100 nm diameter, 1 μm height);(d) PtB-coated vertical Pt nanoneedles with PtB-coated Pt pad edge electrodes (dimensions of the Pt pad edge electrode before PtB coating: ~10 μm long ridge along each pad edge, up to 1 μm height); and(e) PtB-coated Pt planar hole electrodes (each Pt hole has a 2-μm diameter opening and is 1.2-μm deep, as PtB is electrodeposited into the hole, a micrometer-scale mound of PtB builds). Since the electrode structure of (d) proved to be the most effective for highly parallel intracellular recording, the majority of the data presented in the manuscript and Supplementary Figures are with this electrode structure. The hole structure of (e), on the other hand, is fabricated with a much simplified process in comparison to the vertical nanoneedles and thus could be an attractive alternative in the future, once further optimized.
Fig. 2 |. Intracellular recording and stimulation of disassociated rat neurons using the pseudo current-clamp (pCC) and pseudo voltage-clamp (pVC) configurations.

a, A simplified small-signal model of the electrode-neuron interface is shown, including the membrane potential, Vm, membrane current, Im, junctional membrane resistance Rjm, seal resistance, Rs, and membrane resistance, Rm. b-c, Each pixel circuit can be arranged into either the pCC or pVC configuration. The pCC configuration consists of a current injector with output current Ie, and a high-input impedance (Zeq) voltage amplifier (Av ~ 30 V/V) that operate in parallel; the pVC configuration is a low-input impedance transimpedance amplifier (Rf ~ 750 MΩ). d, Extracellular measurement of a neuron transitions to intracellular measurement with Ie = −1 nA applied through the pixel in the pCC configuration, resulting in a ~twenty-fold improvement in AP signal amplitude. EPSPs and their triggering of an AP are also clearly visible. e, After attaining intracellular access in the pCC configuration, an effective positive stimulation current can be applied by adjusting Ie to a less negative value: +550 pA injections of 10 s are applied every 60 s (top), which excites the neuron to fire APs during the stimulation windows (bottom, S►APs). f, Intracellular access is gained in the pVC configuration by setting Ve to −0.65 V to pass Ie ~ -900 pA. No spontaneous activity is observed during this process due to the low input impedance of the pVC circuit. Voltage stimulations are used to activate the neuron’s ion-channels with characteristic negative Na+ spikes and positive K+ repolarization currents observed. g, Application of the ion-channel drugs tetrodotoxin (TTX, Na+ channel blocker, left) and tetraethylammonium (TEA, K+ channel blocker, right) confirm the origin of the measured Na+ and K+ currents and highlight the pVC’s capability for ion-channel drug screening applications.
The CNEI differs from our earlier CMOS-nanoelectrode array8, which could intracellularly record hundreds of cardiomyocytes but was not applicable to even a small number of neurons. We engineered the pixel’s passive electrodes and active circuit to enable stable and controlled intracellular recordings with the sensitivity to measure subthreshold PSPs (pCC mode) and ion-channel currents (pVC mode) of mammalian neurons. This had only been possible with patch-clamp electrodes.
One advantage is that the PtB coating that adds nanometer-scale roughness to the Pt electrode (Fig. 1c–e) increases its surface area and lowers its impedance (Ze of Fig. 2a) by more than ten times (the measured electrode impedance per pixel is ~300 kΩ at 5 kHz (~95 pF): Supplementary Fig. 2b, Methods). The reduced electrode impedance enhances the cell-to-amplifier signal transfer, and thus the recording sensitivity. This reduced electrode impedance also lowers the likelihood of the large (micrometer-scale) gas bubble generation during electrode current injection that is used to permeabilize the neuron’s cell membrane for intracellular access in both pCC and pVC configurations. This prevention of large bubble formation is critical because those bubbles could disrupt the membrane-electrode interface and possibly damage the neuron. For example, Pt vertical nanoneedles were able to gain intracellular access to neurons with current injection only when coated with PtB (Fig. 1c, Supplementary Fig. 5). Another advantage of the PtB nanoscale roughness, beside the reduced impedance, is its physical role in helping to form a tight/stable seal with the cell membrane27–30. For example, when the planar Pt hole is electrodeposited with PtB, a micrometer-scale mound of PtB with nanometer-scale surface roughness forms (Fig. 1e). While the overall structure is not as sharply protruding as the PtB-coated vertical nanoneedle (Fig. 1c–d), it still capable of intracellular access (Supplementary Fig. 5). This suggests that the nanoscale surface roughness of PtB itself may help establish a stable seal. Supplementary Table 1 and Supplementary Discussion 1 present in more details our investigation of the intracellular access using the various PtB-coated electrode structures of Fig. 1c–e. The majority of the data presented in the remainder of this Article and the Supplementary Information (all data except Fig. 2f–g, Supplementary Fig. 5, and Supplementary Fig. 13) are from the PtB-coated vertical nanoneedles with PtB-coated pad edge electrodes (Fig. 1d); the edge electrodes provide an extra current flow path, further reducing the chance of large gas bubbling, thus leading to intracellular coupling with the highest yield.
A second advantage is the configurability of the active pixel circuit between the pCC and pVC modes. For membrane potential measurement, the pixel circuit is arranged into the pCC configuration, consisting of a current injector that runs in parallel with a high-impedance voltage amplifier (Fig. 2b; see also Supplementary Fig. 3b, g–h). This configuration permits concurrent current (Ie) injection and voltage (Ve) amplification through the same electrode. Here the voltage amplifier is designed in a bandpass filter topology with a bandwidth from ~0.5 Hz to ~9.4 kHz (Supplementary Fig. 3c–d) to cover the spectral ranges of APs, PSPs, and membrane potential oscillations of neurons5.
For ion channel current measurement, the pixel circuit is arranged into the pVC configuration where the current injector is turned off and the high-impedance voltage amplifier mentioned above is turned into a low-impedance transimpedance amplifier with the feedback impedance Rf (~750 MΩ) switched on (Fig. 2c). This pVC configuration permits concurrent voltage (Ve) application and current (Ie) recording through the same electrode.
Another advantage is the ten-fold enhancement in the sensitivity of the active pixel circuits. The voltage amplifier in the pCC configuration achieves a ~20 μVrms measured input referred voltage noise at the electrode voltage (Ve) node (which is the amplifier input node) when integrated across 1 Hz to 4.7 kHz (Supplementary Fig. 3e–f, Methods). This noise is ten times lower than our previous design8 and is achieved by the aforementioned separation of the passive pixel pad array region and the active pixel circuit array region (Supplementary Fig. 3a). With such separation, despite the dense pixel pad pitch (20 μm), each pixel circuit can occupy a larger area (100 μm × 250 μm) so that larger transistors with lower noise can be accommodated. The transimpedance amplifier in the pVC configuration benefits from the same lower transistor noise: it achieves a ~1 pArms measured input referred current noise in the electrode current (Ie) path, when integrated across 1 Hz to 4.7 kHz (Supplementary Fig. 3n).
Fourth, both of the pCC and pVC configurations enable the injection of Faradaic current (Ie) through the electrode (Fig. 2b–c), which plays a critical role for intracellular access in our work. The pCC configuration (Fig. 2b) has an explicit current injector that directly controls the electrode current Ie. The pVC configuration (Fig. 2c) indirectly sets the electrode current Ie by applying the electrode voltage Ve. The pVC configuration records this injected current Ie and any modulation of it due to ion channel currents. This electrode current injection in both the pCC and pVC configurations is used to permeabilize the cell membrane and thus to create an intracellular access to neurons, either by electroporation, or through nanoscale bubbles generated from hydrolysis (not to be confused with the detrimental microscale bubbling), or by gating of the ion-channels in the membrane31 (Supplementary Discussion 2). We continue the Faradaic current injection even after the attainment of the intracellular access to maintain the membrane permeabilization, and also critically, to compensate for leakage current from within the neuron (and further, to manipulate the membrane potential for neuronal stimulation). Throughout this current injection, the pCC mode records the membrane potential (smaller than the real value due to signal attenuation: see below) and the pVC records the current injection itself and any modulation of it due to membrane currents (also smaller than the real value). In this way, the pixel can perform stable, sensitive, and controlled intracellular probing of subthreshold membrane potentials as well as ion channel currents. These pCC and pVC configurations with the electrode current injection differentiate the device from our previous CMOS-nanoelectrode array8, whose pixel combined a voltage stimulator for membrane permeabilization and a voltage amplifier for recording: those two building blocks could not run simultaneously as both deal with voltage, and the pixel was therefore unsuccessful in the intracellular recording of neurons.
Intracellular recording and stimulation of neurons
Figure 2 presents single-pixel recording and stimulation experiments with dissociated primary rat cortical neurons (cultured in vitro at high densities to form multiple cell layers, Supplementary Fig. 4). Every recorded voltage and current shown here and throughout this Article are the electrode voltage (amplifier input voltage) Ve and the electrode current Ie (Fig. 2b–c), which we directly measure.
At Ie = 0 A in the pCC configuration and by setting Ve at a proper DC value to cause no bias current into the electrode in the pVC configuration (Supplementary Fig. 6a), no membrane permeabilization occurs. In this non-intracellular mode, we measure extracellular signals with AP voltage amplitudes measured as ΔVe = 50 ~ 300 μV similar to traditional MEAs5 and current variations measured as ΔIe = 50 ~ 100 pA (Supplementary Fig. 6a). Our propensity to measure negative polarity extracellular voltage spikes 98% of the time (Supplementary Fig. 7a) indicates a tendency of the PtB electrodes to interface strongly with the neuron’s soma or axon (negative spikes) over the neuron’s dendrites (positive spikes)32.
For intracellular modes, we pass a Faradaic current (Ie ≠ 0 A) to induce membrane permeabilization. In the pCC mode that records Ve (Fig. 2a, b), the membrane potential Vm causes a change in Ve from its bias value (this voltage bias is set by the injected Ie), where the change in Ve is an attenuated version of Vm. Concretely, ΔVe = α·ΔVm where α = Rs/(Rjm+Rs) is the pCC attenuation factor with Rs and Rjm being the seal and junctional membrane resistance (Fig. 2a). The electrode impedance Ze does not appear here because the PtB coating has made Ze small enough to render the extra attenuation due to Ze negligible. In the pVC mode that records Ie (Fig. 2a, c), the membrane current (e.g., ion channel currents) Im causes a change in Ie from its bias value (this current bias is set by the applied voltage Ve and is what is injected into the electrode), where the change in Ie is an attenuated version of Im. Concretely, ΔIe = γ·ΔIm where γ = Rm/(Rjm+Rm) is the pVC attenuation factor with Rm being the membrane resistance (see Supplementary Discussion 2 for detailed discussions of these coupling parameters).
We first look at the Ve recording in the pCC configuration (Fig. 2d). At Ie = 0 A (no electrode current injection), no membrane permeabilization is caused and Rjm is much larger than Rs to put the attenuation factor α on the order of 10−4 ~ 10−3: this is the extracellular recording discussed earlier. To transition to intracellular access, we change Ie to a negative value in the range of -0.5 nA to -3.0 nA (for the determination of these current values see Supplementary Fig. 5). This current induces a gradual permeabilization of the cellular membrane and causes the cell-electrode coupling to change from extracellular to intracellular (over the time course of ~10 s to a few minutes). During this transition to intracellular coupling, Rjm is drastically reduced, greatly increasing α and thus the amplitude of recorded APs (Fig. 2d). Concretely, α is increased to the order of 10−2 ~ 10−1 from its extracellular value of 10−4 ~ 10-3. While this α is still smaller than the patch clamp’s α that is close to 1 (i.e., Rjm is still substantially larger than Rs in our case), it is large enough to enable clear measurements of subthreshold signals such as excitatory PSPs (EPSPs) (Fig. 2d) and inhibitory PSPs (IPSPs) (Supplementary Fig. 7b) well above the noise level. Similar intracellular access with subthreshold PSP sensitivity was also achieved using iPSC-derived glutamatergic neurons (Supplementary Fig. 7c), which may provide a better model system for drug screening for human neurological diseases and/or personalized medicine.
Importantly, the injection of the negative Ie is sustained throughout the pCC intracellular recording (Fig. 2d) and contrasts other substrate-based electrode work that use electroporation7,8,13,27,28,35,36: in previous substrate-based work, electroporation was performed using a voltage application and their signal was recorded using a voltage amplifier, and thus electroporation and recording couldn’t be concurrent. The purpose of our continued current injection is twofold. Firstly, it serves to maintain the membrane permeabilization it initiated. Secondly, a portion (1 ~ 10 %) of the Ie enters the neuron through Rjm to compensate the leakage from within the cell, where such leakage is an important side effect of membrane permeabilization required for intracellular access (the remainder of Ie that does not enter the neuron through Rjm flows through Rs). In essence, after the membrane has become permeabilized, the current injection plays the role similar to the holding current during the patch-clamp measurements, keeping the neuron’s membrane potential at a value near the normal resting potential. Without this compensation, the neuronal membrane potential can be depolarized to an unhealthy value (as in the case of previous intracellular nanoelectrode works7,8,39–41,27–30,35–38), affecting or even preventing normal electrophysiological function. In Fig. 2d, we observe that the neuron firing pattern does not change by intracellular access (Supplementary Fig. 8, bottom), indicating successful leakage compensation via current injection.
This intracellular recording with the sustained current injection eventually stops on its own (Fig. 2d, Supplementary Fig. 10). This is either due to the gradually decreasing Rjm with the Ie injection (Supplementary Fig. 11), which makes the leakage compensation imperfect to eventually hyperpolarize the neuron and stop its activity, or due possibly to the loss of the seal that eventually decreases the recorded amplitude (Supplementary Discussion 2). While the intracellular recording of Fig. 2d lasts for ca. 1 min, this is an example on the shorter end: in the array-wide measurements discussed shortly, the intracellular coupling overall lasts for a median duration of 8 min (Supplementary Fig. 10 is an example of longer intracellular recording).
The ability to compensate the current leakage can be further exploited to enable direct modulation of Vm for neuronal stimulation in the pCC configuration (Fig. 2e): Vm is related to Ie by Vm ∝ β·Ie where β = Rs·Rm/(Rjm+Rm) (see Supplementary Discussion 2). By changing Ie to a less negative value, from −1.1 nA to −550 pA, we can inject an effective positive current into the neuron, thus causing depolarization and the firing of APs. Returning Ie to −1.1 nA brings the neuron’s Vm below threshold, inhibiting APs. We can modulate the neuron’s firing rate as well by changing the Ie with various stimulation patterns (Supplementary Fig. 9).
The pVC configuration gains intracellular access also by passing a negative electrode current to cause membrane permeabilization. This electrode current is set up indirectly via Ve which we directly apply, and the pVC configuration also measures this current (Fig. 2c): as we set Ve in the range of −0.6 V to −0.7 V, a negative bias current Ie ~ −900 pA flows into the electrode. The magnitude of Ie at the onset of intracellular access is similar to that found for the pCC (see Supplementary Fig. 13 for the determination of these voltage/current values) resulting in a similar small portion (1 ~ 10%) of the Ie entering the neuron through Rjm. Unlike the pCC with a high input impedance (Zeq in Fig. 2b), the low input impedance of the pVC (Fig. 2c) prevents spontaneous neuron APs during the permeabilization process. Stimulation is therefore used to activate the neuron’s ion-channels (Fig. 2f). Negative Na+ spikes and positive K+ repolarization currents are observed during these stimulations, as these ion channel currents Im cause the change ΔIe = γ·ΔIm in the electrode current from the bias value. Application of the ion-channel drugs tetrodotoxin (TTX, Na+ channel blocker) and tetraethylammonium (TEA, K+ channel blocker) confirm these signal origins (Fig. 2g). Determination of the pVC attenuation (γ) is more difficult than the pCC attenuation (α) because Im is related to the surface area of the neuron’s cell membrane; Na+ spikes ranging in magnitude from 100 pA to more than 1 nA (e.g. Supplementary Fig. 13a) have been measured. This pVC configuration will be particularly useful for high throughput ion-channel drug screening applications where planar patch-clamp arrays14–16 are currently a dominant tool for such applications but are limited to non-neuronal, artificial cell lines.
Network-wide intracellular recording and stimulation
As demonstrated, an individual pixel of our CNEI performs a stable, controlled intracellular recording of subthreshold PSPs as well as ion-channel currents, similarly to the patch clamp electrode. The key trait that separates the CNEI from the patch-clamp electrode is its scalability: the CNEI parallelizes the high-fidelity intracellular recording with its 4,096 densely populated channels (Fig. 3a–b). We demonstrate this with each pixel configured in the pCC mode: with a negative current applied to each pixel electrode (Ie = -1.1 nA), we measure intracellular membrane potentials across a network of rat cortical neurons cultured on top. For example, in the data presented in Fig. 3a (see also Supplementary Videos 1–2), we observe bursts of synchronized firings across the CNEI, and during these bursts, we measure from up to 1,837 pixels in parallel, achieving a coupling rate of ~44 %. The median amplitude of APs during the network bursts in Fig. 3a is ~200 μV but individual amplitudes can be as large ~10 mV.
Fig. 3 |. Network-wide intracellular measurements of dissociated rat neurons in the pCC configuration.
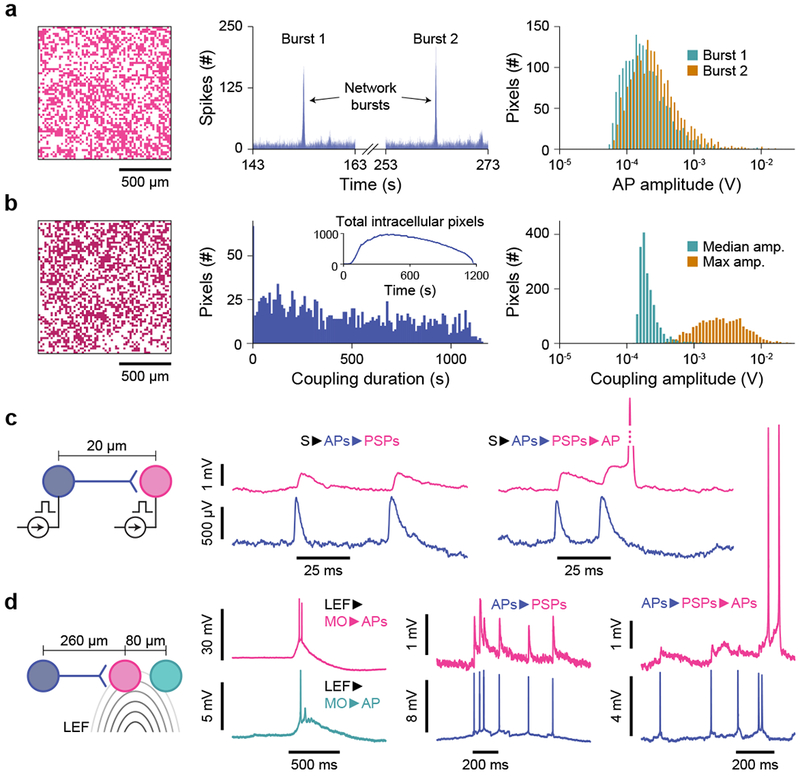
a, Intracellular recordings across the array using Ie = -1.1 nA show synchronized firings: large network bursts involving 1,837 pixels (left, Burst 1) and 1,882 pixels (Burst 2), show correlated spikes (middle, histogram bins of 10 ms) at 153 s and 263 s of the experiment, respectively. The AP spike amplitudes are extracted from both events (right), with an array median of 172 μV and 229 μV, respectively. b, Another array-wide intracellular recording, with continual stimulation and a total of 1,728 pixels (left) intracellularly coupled throughout the 19-minute recording. The coupling duration (middle, median of 495 s) and AP spike amplitude (right) are characterized. The total number of simultaneously intracellularly coupled pixels as a function of time is plotted in the middle inset, with a maximum of 982 pixels being simultaneously intracellularly coupled at time 396 s. c, Array-wide stimulation increases the overall synaptic network activity: stimulated APs of the presynaptic neuron (blue: S►APs) induce excitatory PSPs in the postsynaptic neuron (magenta, APs►PSPs). When the PSPs are close enough to summate and exceed threshold, an AP fires (PSPs►APs). d, Spontaneous (non-stimulated) membrane potentials of the middle neuron (magenta) can be related to the left neuron via their synaptic connection (blue) and the local electrochemical field (LFF) that also excites the right neuron (green). APs from the blue neuron induce excitatory PSPs that in turn induce APs. The LEF induces membrane oscillations (MOs); large MOs cause the neurons (magenta and green) to exceed their thresholds to fire APs (MO►APs).
In another array-wide recording with a duration of 19 min, we perform periodic stimulation (6 s every 1 min) to excite the network and induce more neuronal activity (Fig. 3b and Supplementary Video 3). In the 19-min span, we record intracellular signals from 1,728 pixels, with a maximum of 982 pixels intracellularly coupled simultaneously at ~6 min after the start of the current injection. The inset of Fig. 3b, middle, also shows how the number of the simultaneously intracellularly coupled pixels varies with time. The coupling duration of a pixel, defined as the time difference between the first and last recorded intracellular APs, ranges from <10 s to the full extent of the recording, 19 min (limited by the data acquisition rate of 4 GBytes/min), with a median of ~8 min (495 s). Figure 3b, middle, is the histogram that shows the distribution of the count of pixels (out of 1,728 intracellularly coupled pixels) over the range of the coupling duration up to 19 min: 569 pixels remain intracellularly coupled over 10 minutes. The median AP amplitude of each pixel is similar to that of the spontaneous burst of Fig. 3a, but because the typical signal magnitude increases over time (Supplementary Fig. 10, Supplementary Fig. 11, and Supplementary Discussion 2), the maximum AP amplitude measured at a typical pixel in the experiment of Fig. 3b with the increased activity is much higher, with the median value of 2.54 mV.
Network-wide intracellular recording and stimulation
With the unique ability to measure subthreshold membrane potentials from many neurons simultaneously, the CNEI in the pCC configuration can locate and characterize synaptic connections and other signal pathways across neuronal networks with the fluency unmatched by patch clamp electrodes. Figure 3c–d shows example recordings that illustrate this point (extended data for these examples are located in Supplementary Fig. 14). Network-wide stimulation is first used to find an example of a pair of connected neurons (blue and magenta, Fig. 3c): as we apply a current pulse to every pixel, APs fire in both neurons, but the APs of the presynaptic neuron (blue) also cause closely spaced PSPs in the postsynaptic neuron (magenta), which can summate to reach threshold to fire a new AP ~11.8 ms after the second EPSP (selective stimulation of just a presynaptic neuron is shown in Supplementary Fig. 14b). In Fig. 3d, unstimulated APs in the presynaptic neuron (blue) cause EPSPs in the postsynaptic neuron (magenta) ~260 μm away, where closely spaced EPSPs summate to trigger eventual APs. Separately, in Fig. 3d, two neurons (magenta and green) ~80 μm apart with no direct synaptic connection slowly oscillate together, and when these slow oscillations exceed threshold, the neurons fire temporarily correlated APs often with multiple APs per oscillation (see also Supplementary Fig. 4c). These spatially localized slow oscillations are caused by a local electrochemical field (LEF): this is confirmed by the addition of isoguvacine (GABA agonist) that increases the frequency of such oscillations (Supplementary Fig. 15 and Supplementary Video 4).
The summation of PSPs into an AP seen in Fig. 3c–d is a characteristic typical of chemical synapses between neurons. A further analysis of measured PSPs reveals another characteristic essential to chemical synapses: the quantization of the PSP amplitudes. For example, consider an excitatory synaptic connection found from a network-wide recording (Fig. 4a and Supplementary Fig. 14d). In this connection between two neurons separated by ~398 μm, each AP in the presynaptic neuron (blue) induces an EPSP in the postsynaptic neuron (magenta) with an average delay of 1.7 ms (this time is on par with patch recordings of synaptic delays in in vitro networks42), with a varying amplitude of the EPSP (Fig. 4a, left). These variable EPSP amplitudes are clearly quantized as multiples of 45 μV (in comparison to the AP amplitude of 5.48 mV) (Fig. 4a, right), which is due to the quantal nature of the chemical synapse. The PSP amplitude quantization has thus far been able to be resolved only with patch clamp recordings43,44. So the study here not only confirms that the PSPs we measure are indeed from chemical synaptic connections, but also highlights the high sensitivity of the CNEI’s intracellular recording (these essential characteristics of PSPs have not been apparent in other electrode-based works27,30,41 that have claimed to measure PSPs). A control experiment (Fig. 4b) shows the reduction of measured EPSP and IPSP activity with the application of synaptic blockers, which further confirms the chemical origin of the synapses.
Fig. 4 |. Measurement of chemical synapse characteristics and network-wide mapping of synaptic connectivity with the pCC configuration.
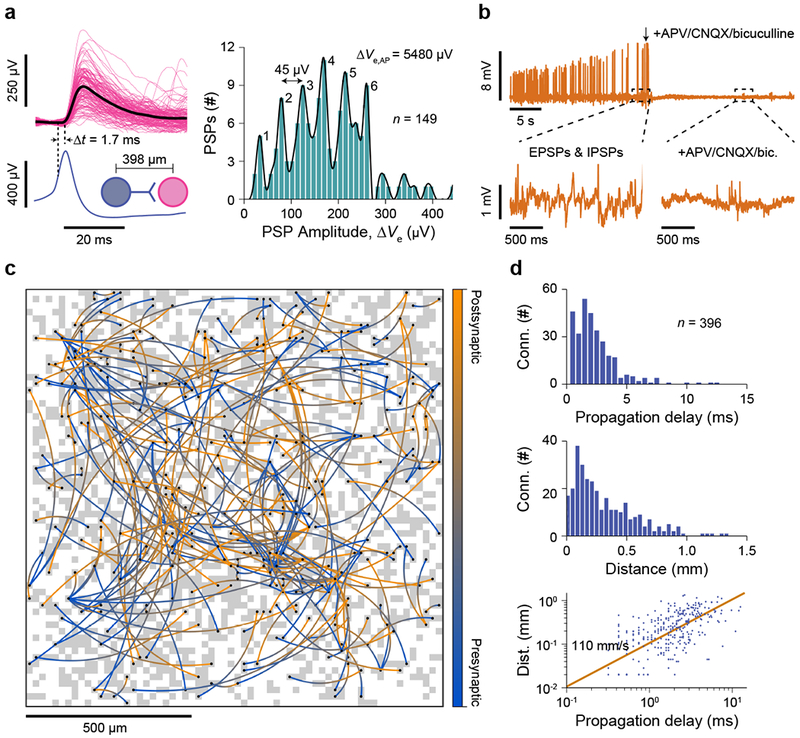
a, A connection from the presynaptic neuron (blue) to postsynaptic neuron (magenta) displays varying PSP amplitudes (left) with their average shown in black. The synaptic propagation delay, Δt = 1.7 ms, from the presynaptic neuron AP (average in blue) is estimated. Amplitude histogram of 149 PSPs (right) shows a distinct quantal amplitude of 45 μV (in comparison to an AP amplitude of 5480 μV). Six quantized amplitudes are clearly observed, the amplitude of ~180 μV being the most frequent. b, Addition of APV (NMDA antagonist), CNQX (AMPA antagonist), and bicuculline (GABA antagonist) to a neuron which exhibits both EPSPs and IPSPs (t1, upper right), quickly reduces the frequency of PSPs and further AP activity (t2, lower right). c, Connections between presynaptic and postsynaptic neurons are mapped across a neuronal network using spike-triggered-averaging. Pixels recording intracellular membrane potentials during the experiment are indicated in gray (the same neuronal network and coupling as Fig. 3b). A total of 304 synaptic connections between 396 neurons are mapped and indicated with an arced line from the presynaptic (blue) to postsynaptic (orange) neuron’s pixels. d, Propagation delay (top) and propagation distance (middle) extracted from the network in (c). Bin sizes are 0.5 ms and 40 μm. Correlation between propagation distance and the propagation delay (bottom), with a best linear speed fit of 110 mm/s.
The highlight demonstration of the unprecedented parallelism of PSP-level recording is the explicit mapping of the synaptic connectivity among thousands of neurons across the entire array (Fig. 4c–d). The basic program for this mapping is to identify and correlate presynaptic APs and resulting PSPs—as we have shown a few examples in Fig. 3c–d and Fig. 4a—across the array. In particular, we use APs of any given pixel (“origin” pixel) as a trigger for averaging the other 4,095 pixels’ signals and identify the “target” pixels exhibiting clearly time-correlated PSPs. The detection of these PSPs indicates synaptic connections between the neurons on the “origin” and “target” pixels (Supplementary Fig. 16–17, Methods). By applying this process to the array-wide 19-min long intracellular recording data of Fig. 3b, we map 304 synaptic connections between 396 neurons (Fig. 4c) amongst 1,728 neurons that are intracellularly coupled.
Most of these synaptic connections have propagation delays less than 5 ms with a median of ~2 ms (Fig. 4d, top), suggesting that these connections are monosynaptic, or without intervening neuronal connections. The median pixel distance between pre- and post-synaptic neurons (Fig. 4d, middle) is 228 μm with preponderance of shorter synaptic connections: this observation provides a rough measure of the distribution of neurite routing lengths in our array. Moreover, the plot of the propagation delay and distance indicates that the signal propagation speed is ~100 mm/s (Fig. 4d, bottom), a value consistent with other measured propagation speeds ex vivo45. The large variance in the delay-distance correlation is likely due to the random distribution of synaptic cleft propagation delays (as short as 300 μs and up to ~5 ms42,46) and the actual neurite routing being deviant from the pre-to-post synaptic pixel distance.
Discussion
The CNEI’s combination of subthreshold sensitivity and parallelism is crucial for accurate and efficient synaptic connectivity mapping. For instance, the analysis of the data in Fig. 3b using AP spike cross correlations (one of the common methods employed in MEA studies32,47: Supplementary Fig. 18) identifies only 63 (21%) out of the 304 connections detected in Fig. 4c. This accentuates the importance of the CNEI’s ability to measure subthreshold PSP signals45. Other synaptic mapping methods, notably the ones that rely upon multiple patch clamps6 or patch clamp in conjunction with Ca2+ imaging1, optogenetic stimulation2,3, or MEAs4, are limited by the number of simultaneous patch recordings (less than ~10 vs. 4,096 in our CNEI), resulting in prohibitively low throughput for network-wide mapping. Our mapping of the ~300 synaptic connections amongst the intracellularly coupled ~1,700 neurons with only the 19 min recording time highlights the experimental throughput.
High-fidelity, simultaneous recording of a large number of connected neurons has long been an important target milestone in electrophysiology. Our CNEI accomplishes this feat at least for in vitro mammalian neuronal networks. Since in vitro cultures are frequently used in the electrophysiological screening of pharmaceutical candidates for neurological disorders, the capability of the CNEI should be readily applicable to, and improve, such pharmaceutical screening applications. Furthermore, its demonstrated compatibility with iPSC-derived neurons (Supplementary Fig. 7c) shows its flexibility of cell-type for these applications. The patch clamp has been traditionally the method of choice for measuring electrical responses of neurons to chemical inputs, but is inherently low throughput and incapable of measuring network dynamics and connections efficiently; in contrast, the CNEI can examine the effects of chemical inputs with much higher throughput and at a network level. Further major improvements in the passive electrode design and the active circuit functions can expand the device capability to ex vivo and in vivo samples, which could benefit the synaptic connectivity mapping of the brain.
One immediate problem to tackle to further the device capability is to elongate the intracellular coupling. Intracellular recording of neurons is known for trading sensitivity with invasiveness, so it is difficult to repeat from the same neuron. Case in point, patch-clamp recording results in cell death, and likewise the CNEI intracellular recording could not be repeated from the same neuronal network. On the other hand, prolonging the intracellular coupling from the aforementioned median duration of 8 min—which is already on a par with patch recording—may be more easily achievable by perfecting the leakage compensation. For example, in the pCC configuration, the DC level of the membrane potential could be directly measured and fed back to modify Ie so as to accurately maintain the DC membrane potential at a proper value, thus lengthening intracellular recording. This feedback control would be particularly well suited to semiconductor electronics.
Methods
Electrode fabrication and packaging.
We designed the CMOS IC and outsourced its fabrication in 0.18 μm technology to the United Microelectronics Corporation. Subsequently, we post-fabricated the Pt electrodes on the surface Al pads of the CMOS IC in house.
The PtB vertical nanoneedle with pad edge electrodes were fabricated with the following steps (Supplementary Fig. 1a): (1) The original foundry passivation was removed via dry etching, and a thick metal layer (20 nm Ti, 200 nm Pt) was deposited on the Al pads. (2) A 1-μm thick amorphous Si layer was deposited via chemical vapor deposition. (3) Electron-beam lithography was used to define vertical nanoneedles. (4) Dry etching was performed to etch the amorphous Si layer into ~100-nm diameter vertical nanoneedles. (5) A thin layer of metal (5 nm Ti, 10 nm Pt) was sputtered to make connection to the pad. A 20-nm thick SiO2 passivation layer was then deposited via atomic layer deposition. (6) A thin layer of photoresist was spun on the device to cover the nanoneedle base and flat portions surrounding the pad. (7) Wet etching was used to remove the oxide only at the tip of the nanoneedles and at the edge of the pad.
The PtB planar hole electrodes (Supplementary Fig. 1c) were fabricated with the following steps: (1) Photolithography was used to define a 2 μm hole in the foundry passivation layer. (2) The foundry passivation was removed to expose the Al pad via dry etching. (3) A thick layer of metal (20 nm Ti and 200 nm Pt) was deposited to form Pt electrodes.
After the electrode post-fabrication, the CMOS ICs were wire-bonded to chip carriers (Spectrum Semiconductor Materials, San Jose, CA) with Au wedge bonding. A glass inner ring and outer ring (Friedrich & Dimmock, Millville, NJ) were glued to the chip and chip carrier, respectively, using polydimethylsiloxane (PDMS). PDMS was then poured into the moat between these two glass rings to encapsulate the wire bonds. The well formed by inner ring is where the neurons are cultured after the device is completed with the PtB electrodeposition, as explained next.
PtB electrodeposition.
PtB deposition on the Pt electrodes was performed with packaged CMOS ICs, using the same experimental setup as in the electrophysiology experiments (Supplementary Fig. 1f). A solution of 0.5 mM H2PtCl6 (Sigma-Aldrich, Atlanta, GA) and 25 mM NaNO3 dissolved in doubly ionized water26 was poured onto the Pt electrode array. Electrodeposition was then performed by cycling the electrodes’ voltages from 0 V to −1.2 V versus a Pt reference electrode at a scan rate of 50 mV/s. AC impedance measurements before and after deposition were performed by applying a 10 mV, 5 kHz sine wave to each electrode in the array sequentially and measuring the resultant current through the Pt reference electrode. To generate uniform impedance across the array, the AC impedance was measured periodically throughout the cyclic electrodeposition, and an individual pixel pad’s PtB deposition was stopped when its overall impedance reached down to ~300 kΩ (~95 pF). Typically 10 ~ 20 cycles were needed to achieve the desired impedance. The devices were re-deposited for ~5 cycles after each neuron experiment to re-gain the low electrode impedance.
Neuron culture and drug application.
E18 combined rat neurons from the cortex, hippocampus and ventricular zones were purchased (Brainbits llc, Springfield, Il) and cultured according to the recommended company protocols. Before each neuron plating, the devices were plasma treated at 18 W for 5 min, soaked in ethanol for ~5 min while inside a bio-hood, rinsed 5+ times in sterile DI water, and air dried in ambient conditions. The devices were then soaked in 0.1% poly-d-lysine solution dissolved in filtered water (Sigma-Aldrich, Atlanta, GA) at 4°C overnight, and then washed 5 times to form a cell-adhesive coating. The neurons were plated on the same day upon delivery: neural tissues were dissociated in company recommended dissociation media and adjusted to 1 Million/mL concentration. 150k cells (150 μL of media) were seeded per device and replenished with 4 mL of media 30 min after seeding. Half media swaps with NBactive4 (Brainbits llc, Springfield, Il) were performed on the second day of plating and every 3 days afterwards to maintain cell health.
All intracellular rat neuron experiments were performed on 10 to 14 day in vitro (DIV) cultures; the extracellular experiments of Supplementary Fig. 6 were performed on 26 to 33 DIV cultures. The iPSC neurons measurements of Supplementary Fig. 7c were performed on 20 DIV iCell GlutaNeurons (FUJIFILM Cellular Dynamics, Madison, WI) cultured according to the recommended company protocols. The electrical measurements were performed in neuron culture media with a Pt reference electrode. All measurements were performed in the dark to minimize the light sensitivity of the CMOS electronics. The temperature of the CMOS IC was set to 34–37°C for experiments using the integrated temperature sensors and heater (Supplementary Fig. 3a).
For the drug experiment of Supplementary Fig. 6b, a small amount of drug was added to form a final concentration of APV/CNQX/bicuculline (25 μM/10 μM/50 μM); 8× half media exchanges were then performed before the final measurement. For the drug experiment of Supplementary Fig. 6c, small amounts of CNQX were added to form the concentrations stated; 8× half media exchanges were then performed before the final measurement. For the drug experiments of Fig. 2g, the electrode voltage was adjusted to gain intracellular access and voltage stimulations of the same amplitude were repeated every 5 s. Either 2 ml of 100 nM tetrodotoxin (TTX) dissolved in media was added to form a final concentration of 33 nM or a small amount (200 μL) of high concentration tetraethylammonium (TEA) was added to form a final concentration of 1 mM. For the drug experiment of Fig. 4b, a small amount (200 μL) of high concentration APV/CNQX/bicuculline (500 μM/200 μM/1 mM), was added to form a final concentration of APV/CNQX/bicuculline (25 μM/10 μM/50 μM). For the drug experiment of Supplementary Fig. 15, a small amount of high concentration isoguvacine (10 mM) was added to form a final concentration of 10 μM.
The devices were cleaned with trypsin, soap water and DI water after each neuron cultures and were reused after re-deposition of PtB. A typical device could be used for a total of ~100 DIV showing that the PtB electrodes are capable of passing the required Faradaic current without formation of neurotoxic deposits. 39 successful pCC intracellular experiments used 19 CNEI devices (Supplementary Table 1) and 13 rat neuron orders (multiple devices were plated per order). All of the pCC data in the manuscript and Supplementary Information are presented from 13 of these experiments. Fewer pVC intracellular experiments were performed and focused on single neuron measurements: the data in the manuscript and Supplementary Information are presented from 4 of these experiments from an additional 3 rat neuron orders. Specifically, Fig. 2d, Supplementary Fig. 8 was recorded from the PtB vertical nanoneedle and pad edge electrode device NE1 and neuron culture C1; Figs. 2e, 3c, 4a, and Supplementary Figs. 14a,d from device NE2 and C2; Fig. 2f, Supplementary Fig. 13a from PtB vertical nanoneedle (without edge electrode) device N1 and C10; Fig. 2g, left from N2 and C11; Fig. 2g, right from N1 and C12; Figs. 3a, 3d, Supplementary Figs. 7b, 10, 14c, 16–17 Supplementary Videos 1, 2 from NE3 and C3; Figs. 3b, 4c–d, Supplementary Figs. 9d, 18, Supplementary Video 3 from NE4 and C2; Fig. 4b from NE6 and C5; Supplementary Fig. 3a–c from NE2 and C9; Supplementary Fig. 4, top from NE4 and C6; Supplementary Fig. 5, bottom from PtB-coated hole electrode device H1 and C7; Supplementary Fig. 5, middle from PtB vertical nanoneedles (without edge electrode) device N1 and C8; Supplementary Fig. 7c from NE8 and iPSC derived neuron culture I1; Supplementary Fig. 9b from NE7 and C4; Supplementary Figs. 9c and 11 from NE3 and C6; Supplementary Fig. 13a–b from NE4 and C10; Supplementary Fig. 13a, bottom from N1 and C10; Supplementary Fig. 14b from NE5 and C4; Supplementary Fig. 15, Supplementary Video 4 from NE4 and C3. The extracellular measurements of Supplementary Fig. 6b–d were recorded from two separate neuron orders/cultures using NE6 and NE9.
CMOS IC design and measurement.
The architecture and performance of the CMOS IC is a significant improvement from our previous work8, not only in the array scale and pixel density but more fundamentally in the pixel configurations combining the current injector and the voltage amplifier, and noise reduction in the voltage amplifier. The amplifier is a variable gain bandpass topology, and intracellular experiments were typically performed using a passband gain of ~30 V/V (Supplementary Fig. 3c–d); extracellular experiments were performed with a passband gain of ~300 V/V. The 4,096 amplifier outputs were sampled using 32 integrated 128:1 analog output multiplexers8 operating with a 1.20512 MHz clock, resulting in an effective pixel sampling rate of 9.415 kHz. Two National Instruments PXIe-6358 DAQ cards were used for analog to digital conversion.
We characterized the pixel amplifier’s voltage gain by applying a sine wave (1 mV amplitude and variable frequency ranging from 0.1 Hz to 100 kHz for Supplementary Fig. 3c; 1 mV amplitude and 100 Hz for Supplementary Fig. 3d) to the amplifier input, with the amplifier output (Vamp, with reference to Supplementary Fig. 6a, Fig. 2b–c, and Supplementary Fig. 3b), sampled at 10× the applied frequency. The input referred noise at the Ve node (Supplementary Fig. 3e–f) was determined by measuring the output noise with a DC input signal applied to Ve and dividing it by the measured passband gain (the noise and gain here are measured without solution, as we calculate the input referred noise at the Ve node). The current injector (Supplementary Fig. 3g–h) of each pixel was characterized by an external transimpedance amplifier. We measured and calculated the parasitic routing capacitance (Supplementary Fig. 3i–j), by first measuring the gain from Vs,1 to Vamp with Ve set to a DC voltage and then measuring it again with Ve floating. We then used these two gain values and the designed value of C1 (3.5 pF) and C2 (110 fF) to calculate Cp,routing. We measure the amplifier’s transimpedance gain in the pVC configuration (Supplementary Fig. 3k–m) by applying voltage ramps through C1 connected to negative terminal of the amplifier to generate current pulses of varying magnitude. The input current noise (Supplementary Fig. 3n) was determined by measuring the output voltage noise with no voltage ramp applied to C1 and dividing by the measured transimpedance gain (the noise and gain here are measured without solution).
Signal filtering.
Data was acquired using LabVIEW software and post-processed using MATLAB. Long (>10 s) trace signals were filtered in the time domain using a single-pole (frequency ranging from 1–100 Hz) high pass filter. Long traces with stimulations (except the pVC data of Fig. 2f) were first high pass filtered with the remaining edge of the electrode response zeroed or omitted from the trace (approximately 10 ms to 1 s of data omitted). Short signal traces (<10 s) were not high pass filtered after acquisition except for Fig. 2g: to eliminate the lower frequency electrode responses (resulting from the electrode’s Faradaic resistance, Rs, and Rjm in conjunction with the electrode’s double layer capacitance) to compare ion-channel currents before and after drug application, we applied a high-pass filter of ~50 Hz. A rolling window average of 3–10 data points was used to lower discretization noise of the analog-to-digital conversion for slower signals, including Fig. 3c, Fig. 3d, and Fig. 4b. The short signals of Fig. 2d and Fig. 2f were unfiltered. As mentioned in the main text, all recorded voltages are referred to the Ve node. Scale bars are omitted for synaptic connection data in Supplementary Figs. 16–17 due to the variability of the recording attenuation from pixel to pixel and the variation of the coupling amplitude over time.
Spike detection was used to generate the Supplementary Videos 1, 2, and 3, for the PSTH of Supplementary Fig. 6b–d, for characterization in Fig. 3a–b, for the quantal PSP amplitude analysis of Fig. 4a, and for the spike-triggered-averaging (STA) of Fig. 4c. Data was first high pass filtered using a frequency of 94 Hz, and then stimulation artifacts were omitted if applicable. Positive spikes larger than 5 standard deviations were calculated, with each spike time adjusted to the spike’s maximum. For the intracellular coupling characterization in Fig. 4b, only pixels that showed clear intracellular signals were included.
Peristimulus time histogram (PSTH), CNQX titration, and STDP stimulation.
For the PSTHs in Supplementary Fig. 6b–d, repeated extracellular stimulations were applied to stimulate pixels and spike detection was performed on recording pixels with the time difference between the spike and stimulation binned (histogram bins of 1/9.415 kHz). The extracellular recordings were performed using the transimpedance configuration of the amplifier with Ve adjusted such that Ie,DC = 0 A. For the titration of Supplementary Fig. 6c, small amounts of high concentration CNQX were added to the culture media immediately after the PSTH stimulations to form the final concentrations stated, the PSTH stimulation were started at intervals of 10 min. The STDP stimulation of Supplementary Fig. 6d was performed by repeatedly stimulating the stimulation group 1 pixels 10 ms before the stimulation group 2 pixels every 100 ms for 3000 total stimulations.
Quantal PSP amplitude analysis.
For the quantal PSP amplitude analysis of Fig. 4a, spike detection was performed to detect the presynaptic neuron’s APs during a 93 s window, totaling 475 spikes. Time windows of 50 ms (10 ms before and 40 ms after the spikes) were then extracted for the postsynaptic neuron’s PSPs. Only time windows with a stable baseline for 10 ms before the AP spikes, calculated as ΔVe < 33 μV, and time windows without APs were used to allow for an accurate PSP amplitude measurement: a total of 149 PSP windows met this criteria. Data was smoothed with a rolling window average of 33 data points (3.5 ms) and the difference between maximum to the average of the 10 ms before the AP spikes was used as the PSP amplitude.
Spike-triggered-averaging.
For STA, time windows of 75 ms (25 ms before and 50 ms after the spike time) were used to align and average other pixel’s recordings. Only pixels with >150 spike times (~1500 pixels) were used for the STA of Fig. 4c and only the largest 500 spikes were used for pixels with >500 spikes. Windows with abnormally large signals (e.g. action potentials) in comparison to the other windows were omitted from the averaging to prevent false positives. Averaged signals were sorted by the ratio of the maximum amplitude in the data window to the standard deviation of the outer most 25 ms and then manually inspected. To calculate the propagation delays of Fig. 4d, the averaged data was high pass filtered using a 100 Hz filter frequency, and the time difference was calculated between the maximum of the presynaptic neuron’s signal to the maximum of the postsynaptic neuron’s signal. The spike cross correlation calculation of Supplementary Fig. 18 was performed using a bin width of 1 ms47.
Reporting summary.
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
The authors declare that all data supporting the findings of this study are available within the paper and its Supplementary Information.
Supplementary Material
Acknowledgements
Post-fabrication and characterization were performed, in part, at the Center for Nanoscale Systems at Harvard University. The authors are grateful for the support of this research by Samsung Advanced Institute of Technology, Samsung Electronics, Suwon, Republic of Korea (A37734 to H.P. and D.H.), Catalyst Foundation, Valhalla, NY (J.A., H.P., and D.H.), the Army Research Office (W911NF-15-1-0565 to D.H.), the Army Research Office (W911NF-17-1-0425 to D.H.), the National Science Foundation Graduate Research Fellowship Program (DGE1745303 to K.K.), the National Institutes of Health (1-U01-MH105960-01 to H.P.), the Gordon and Betty Moore Foundation (to H.P.), and the U. S. Army Research Laboratory and the U. S. Army Research Office (W911NF1510548 to H.P.).
Footnotes
Competing interests
The authors declare no competing interests.
Supplementary information is available for this paper at https://doi.org/10.1038/s41551-01X-XXXX-X.
References
- 1.Sasaki T, Minamisawa G, Takahashi N, Matsuki N & Ikegaya Y Reverse optical trawling for synaptic connections in situ. J. Neurophysiol. 102, 636–643 (2009). [DOI] [PubMed] [Google Scholar]
- 2.Petreanu L, Huber D, Sobczyk A & Svoboda K Channelrhodopsin-2-assisted circuit mapping of long-range callosal projections. Nat. Neurosci. 10, 663–668 (2007). [DOI] [PubMed] [Google Scholar]
- 3.Shemesh OA et al. Temporally precise single-cell-resolution optogenetics. Nat. Neurosci. 20, 1796–1806 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jäckel D et al. Combination of High-density Microelectrode Array and Patch Clamp Recordings to Enable Studies of Multisynaptic Integration. Sci. Rep. 7, 978 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Spira ME & Hai A Multi-electrode array technologies for neuroscience and cardiology. Nature Nanotech. 8, 83–94 (2013). [DOI] [PubMed] [Google Scholar]
- 6.Perin R, Berger TK & Markram H A synaptic organizing principle for cortical neuronal groups. Proc. Natl. Acad. Sci. U. S. A. 108, 5419–5424 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Robinson JT et al. Vertical nanowire electrode arrays as a scalable platform for intracellular interfacing to neuronal circuits. Nature Nanotech. 7, 180–184 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Abbott J et al. CMOS nanoelectrode array for all-electrical intracellular electrophysiological imaging. Nature Nanotech. 12, 460–466 (2017). [DOI] [PubMed] [Google Scholar]
- 9.Eversmann B et al. A 128 × 128 CMOS Biosensor Array for Extracellular Recording of Neural Activity. IEEE J. Solid-State Circuits 38, 2306–2317 (2003). [Google Scholar]
- 10.Berdondini L et al. Active pixel sensor array for high spatio-temporal resolution electrophysiological recordings from single cell to large scale neuronal networks. Lab Chip 9, 2644–51 (2009). [DOI] [PubMed] [Google Scholar]
- 11.Frey U et al. Switch-matrix-based high-density microelectrode array in CMOS technology. IEEE J. Solid-State Circuits 45, 467–482 (2010). [Google Scholar]
- 12.Tsai D, Sawyer D, Bradd A, Yuste R & Shepard KL A very large-scale microelectrode array for cellular-resolution electrophysiology. Nat. Commun. 8, 1802 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lopez CM et al. A 16384-electrode 1024-channel multimodal CMOS MEA for high-throughput intracellular action potential measurements and impedance spectroscopy in drug-screening applications. in IEEE International Solid-State Circuits Conference 61, 464–466 (IEEE, 2018). [Google Scholar]
- 14.Fertig N, Blick RH & Behrends JC Whole cell patch clamp recording performed on a planar glass chip. Biophys. J. 82, 3056–62 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lau AY, Hung PJ, Wu AR & Lee LP Open-access microfluidic patch-clamp array with raised lateral cell trapping sites. Lab Chip 6, 1510–1515 (2006). [DOI] [PubMed] [Google Scholar]
- 16.Dunlop J, Bowlby M, Peri R, Vasilyev D & Arias R High-throughput electrophysiology: an emerging paradigm for ion-channel screening and physiology. Nat. Rev. Drug Discov. 7, 358–368 (2008). [DOI] [PubMed] [Google Scholar]
- 17.Hochbaum DR et al. All-optical electrophysiology in mammalian neurons using engineered microbial rhodopsins. Nat. Methods 11, 825–833 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim CK, Adhikari A & Deisseroth K Integration of optogenetics with complementary methodologies in systems neuroscience. Nat. Rev. Neurosci. 18, 222–235 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ikegaya Y et al. Synfire Chains and Cortical Songs: Temporal Modules of Cortical Activity. Science 304, 559–564 (2004). [DOI] [PubMed] [Google Scholar]
- 20.Stetter O, Battaglia D, Soriano J & Geisel T Model-Free Reconstruction of Excitatory Neuronal Connectivity from Calcium Imaging Signals. PLoS Comput. Biol. 8, e1002653 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ahrens MB et al. Brain-wide neuronal dynamics during motor adaptation in zebrafish. Nature 485, 471–477 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Akerboom J et al. Optimization of a GCaMP Calcium Indicator for Neural Activity Imaging. J. Neurosci. 32, 13819–13840 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gong Y et al. High-speed recording of neural spikes in awake mice and flies with a fluorescent voltage sensor. Science 350, 1361–1366 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Woodford CR et al. Improved PeT molecules for optically sensing voltage in neurons. J. Am. Chem. Soc. 137, 1817–1824 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lou S et al. Genetically Targeted All-Optical Electrophysiology with a Transgenic Cre-Dependent Optopatch Mouse. J. Neurosci. 36, 11059–11073 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li L-L et al. Morphological control of platinum nanostructures for highly efficient dye-sensitized solar cells. J. Mater. Chem. 22, 6267 (2012). [Google Scholar]
- 27.Dipalo M et al. Intracellular and Extracellular Recording of Spontaneous Action Potentials in Mammalian Neurons and Cardiac Cells with 3D Plasmonic Nanoelectrodes. Nano Lett. 17, 3932–3939 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lin ZC, Xie C, Osakada Y, Cui Y & Cui B Iridium oxide nanotube electrodes for sensitive and prolonged intracellular measurement of action potentials. Nat. Commun. 5, 3206 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hai A, Shappir J & Spira ME In-cell recordings by extracellular microelectrodes. Nat. Methods 7, 200–2 (2010). [DOI] [PubMed] [Google Scholar]
- 30.Liu R et al. High Density Individually Addressable Nanowire Arrays Record Intracellular Activity from Primary Rodent and Human Stem Cell Derived Neurons. Nano Lett. 17, 2757–2764 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fromherz P Self-Gating of Ion Channels in Cell Adhesion. Phys. Rev. Lett. 78, 4131–4134 (1997). [Google Scholar]
- 32.Obien MEJ, Deligkaris K, Bullmann T, Bakkum DJ & Frey U Revealing neuronal function through microelectrode array recordings. Front. Neurosci. 9, 423 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Massobrio P, Tessadori J, Chiappalone M & Ghirardi M In vitro studies of neuronal networks and synaptic plasticity in invertebrates and in mammals using multielectrode arrays. Neural Plast. 2015, 1–18 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Froemke RC, Debanne D & Bi GQ Temporal modulation of spike-timing-dependent plasticity. Front. Synaptic Neurosci. 1–16 (2010). doi: 10.3389/fnsyn.2010.00019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hai A & Spira ME On-chip electroporation, membrane repair dynamics and transient in-cell recordings by arrays of gold mushroom-shaped microelectrodes. Lab Chip 12, 2865–2873 (2012). [DOI] [PubMed] [Google Scholar]
- 36.Xie C, Lin Z, Hanson L, Cui Y & Cui B Intracellular recording of action potentials by nanopillar electroporation. Nature Nanotech. 7, 185–190 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Duan X et al. Intracellular recordings of action potentials by an extracellular nanoscale field-effect transistor. Nat Nanotechnol 7, 174–179 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lee K-Y et al. Vertical nanowire probes for intracellular signaling of living cells. Nanoscale Res. Lett. 9, 56 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tian B et al. Three-Dimensional, Flexible Nanoscale Field-Effect Transistors as Localized Bioprobes. Science 329, 830–834 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hai A, Shappir J & Spira ME Long-term, multisite, parallel, in-cell recording and stimulation by an array of extracellular microelectrodes. J. Neurophysiol. 104, 559–68 (2010). [DOI] [PubMed] [Google Scholar]
- 41.Shmoel N et al. Multisite electrophysiological recordings by self-assembled loose-patch-like junctions between cultured hippocampal neurons and mushroom-shaped microelectrodes. Sci. Rep. 6, 27110 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fitzsimonds RM, Song HJ & Poo MM Propagation of activity-dependent synaptic depression in simple neural networks. Nature 388, 439–448 (1997). [DOI] [PubMed] [Google Scholar]
- 43.Kullmann DM & Nicoll RA Long-term potentiation is associated with increases in quantal content and quantal amplitude. Nature 357, 240–244 (1992). [DOI] [PubMed] [Google Scholar]
- 44.Hardingham NR et al. Quantal Analysis Reveals a Functional Correlation between Presynaptic and Postsynaptic Efficacy in Excitatory Connections from Rat Neocortex. J. Neurosci. 30, 1441–1451 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shein-Idelson M, Pammer L, Hemberger M & Laurent G Large-scale mapping of cortical synaptic projections with extracellular electrode arrays. Nat. Methods 14, 882–890 (2017). [DOI] [PubMed] [Google Scholar]
- 46.Sabatini BL & Regehr WG Timing of neurotransmission at fast synapses in the mammalian brain. Nature 384, 170–172 (1996). [DOI] [PubMed] [Google Scholar]
- 47.Barthó P et al. Characterization of Neocortical Principal Cells and Interneurons by Network Interactions and Extracellular Features. J. Neurophysiol. 92, 600–608 (2004). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The authors declare that all data supporting the findings of this study are available within the paper and its Supplementary Information.