

Abstract
The objective of the current study is to use comparative and functional genomic analysis to help to understand the biological mechanism mediating the effect of single nucleotide polymorphisms (SNPs) on blood pressure. We mapped 26,585 SNPs that are in linkage disequilibrium with 1,071 human blood pressure-associated sentinel SNPs to 9,447 syntenic regions in the mouse genome. 21.8% of the 1,071 linkage disequilibrium regions are located at least 10 kb from any protein-coding gene. Approximately 300 blood pressure-associated SNPs are expression quantitative trait loci (eQTLs) for a few dozen known blood pressure physiology genes in tissues including specific kidney regions. Blood pressure-associated sentinel SNPs are significantly enriched for eQTLs for blood pressure physiology genes compared with randomly selected SNPs (p<0.00023, Fisher’s exact test). Using a newly developed deep learning method and other methods, we identified SNPs that were predicted to influence the conservation of CCCTC-binding factor (CTCF) binding across cell types, transcription factor binding, mRNA splicing or secondary structures of RNA including long noncoding RNA. The SNPs were more likely to be located in CTCF binding regions than what would be expected from the whole genome (p = 4.90 × 10−7, Pearson’s Chi-squared test). One example synonymous SNP rs9337951 was predicted to influence the secondary structure of its host mRNA JCAD and was experimentally validated to influence JCAD protein expression. These findings provide an extensive comparative and functional genomic resource for developing experiments to test the functional significance of human blood pressure-associated SNPs in human cells and animal models.
Keywords: Human blood pressure, Single nucleotide polymorphism, Deep learning, Epigenetics, Comparative genomics
Graphical Abstract
Introduction
Hypertension is the most common identifiable risk factor for disease burden and deaths worldwide (1). Genetic factors play an important role in the development of hypertension (2,3). A powerful approach for identifying common DNA sequence variations associated with phenotypic traits is to perform genome-wide association studies (GWAS). Single nucleotide polymorphisms (SNPs) are substitutions of single nucleotides at specific positions in the genome that occur at significant frequencies, typically >1%, in a population. In a GWAS, phenotypic traits of interest and hundreds of thousands of SNPs are measured in thousands of humans and the association of a SNP with a trait is tested. GWAS for BP-related traits has been highly productive and has provided evidence for significant associations of hundreds of genomic loci with BP (4,5).
The exciting findings of BP GWAS also have revealed one of the most significant challenges for understanding genetic control of BP. That is, only a small fraction of BP-associated SNPs identified by GWAS are nonsynonymous SNPs that could directly influence protein function through known mechanisms (4,5). The vast majority of BP-associated SNPs are either synonymous or are located in noncoding regions of DNA. Protein-coding genes that are located closest to these SNPs in genomic sequence are often reported as names of the GWAS loci. However, strong evidence suggests many SNPs might not regulate their nearest protein-coding genes (6), making it a challenge to predict which gene(s) exhibit altered expression which may ultimately regulate BP. Understanding the molecular function of BP-associated SNPs and linking them with protein-coding genes in physiological pathways that regulate BP are among the most pressing challenges in BP research.
Animal models, including mouse models, are essential tools for understanding molecular and physiological pathways that regulate BP. Identification of mouse genomic regions syntenic with human BP loci is a critical step for developing studies in mouse models to test physiological effects of human BP loci.
The goal of this study was to identify mouse genomic segments that are syntenic with human genomic loci associated with BP and perform a thorough functional annotation for all reported BP-associated SNPs in the human genome and their syntenic regions in the mouse genome. We performed the functional annotation using a newly developed machine learning method along with other analytical tools and data sources that had not been applied to analyze BP SNPs previously. Several specific hypotheses, such as statistically significant enrichment of genomic features in BP-associated SNPs, were tested throughout the study. Proof-of-principle experiments were performed to validate selected results of the functional annotation.
Methods
Detailed methods and data are available in the online supplement.
Human BP-associated SNPs and haplotypes
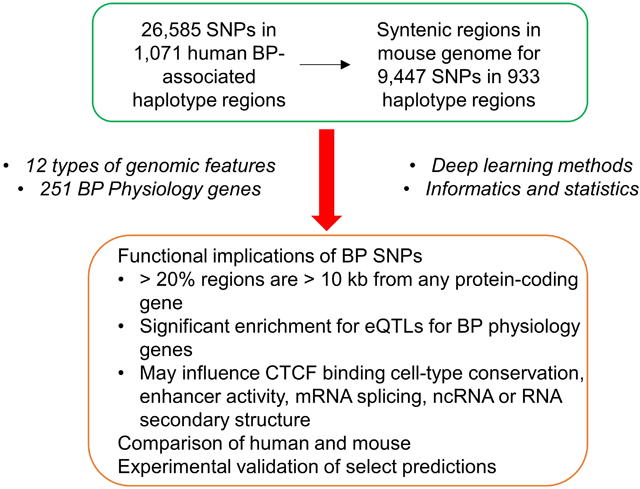
Figure 1 depicts the overall flow of the analysis. Human BP-associated sentinel SNPs were obtained from two recent GWAS reports that included both newly identified and previously reported loci (4,5). The study population involved was manually curated based on the source study. We used haploR (7) to retrieve SNPs in linkage disequilibrium (LD) with each sentinel SNP from HaploReg 4.1 using the threshold of r2 >= 0.8 in the matched population (8). The retrieved SNPs were then combined if more than one population was involved, and the non-redundant SNPs were used to define the haplotype and LD region for the sentinel SNP. Some of the chromosome coordinate information missing in HaploReg was manually filled in based on other sources such as dbSNP (9).
Figure 1. Overall flow of analysis.

See the text for detail.
Identifying syntenic regions in the mouse genome
We converted genomic coordinates of human BP-associated SNPs, including sentinel SNPs and other SNPs in LD, from the hg38 version of the human genome to the hg19 version using R liftOver tools (10). The SNPs in the hg19 version were mapped onto the mm9 mouse genome using the same R liftOver tools to identify syntenic regions. In addition, we identified syntenic regions in a separate analysis using VISTA (11).
Identification of BP “physiology genes”
We defined a protein-coding gene as a BP physiology gene if curated experimental evidence supported a functional role for the gene in BP regulation in humans or experimental models. We retrieved BP physiology genes from Gene Ontology by querying AmiGO 2 (12) with “regulation of blood pressure”, organisms of human/mouse/rat, and limited to “experimental evidence” and “mutant phenotype evidence”. GWAS associations were not part of the evidence that we used to identify BP physiology genes.
Other data sources and tools
Table 1 summarizes the data used for the analysis of long noncoding RNA (lncRNA), microRNA, transcription factor (TF) binding regions, CpG islands, enhancer, reporter assay-based regulatory element activity, expression quantitative trait loci (eQTL), eQTL for kidney regions, topologically associating domains (TAD), CCCTC-binding factor (CTCF) binding regions, mouse CTCF binding regions, mouse enhancer, and mouse eQTL, as well as the sources of these data and the references. We used motifbreakR (13) to examine the effect of SNPs on transcription factor binding. motifbreakR_motif is a collection of motifs from ENCODE, factorBook, hocomoco, and homer (14-17). We used SpliceAI to predict the effect of BP-associated SNPs on mRNA splicing (18).
Table 1.
Data and data sources.
| Feature | Data | Data source | Reference |
|---|---|---|---|
| BP SNPs and LD regions | 26,585 non-duplicated SNPs in 1,071 BP-associated LD regions | GWAS and HaploReg | (4), (5), (8) |
| lncRNA | high-confidence lncRNA including 288,174 lncRNA exons in 107,039 lncRNA transcripts from 49,372 lncRNA genes in hg38 | LNCipedia 5.2 | (41) |
| human microRNA | 1881 precusor microRNAs and 2813 mature microRNAs | miRBase | (42) |
| TF binding sites | predicted TF binding site | HaploReg | (14) |
| CpG islands | 30,477 CpG islands | UCSC genome browser | (10) |
| Enhancer | The human bed broad peak from narrow and broad histone marks | ENCODE | (43) |
| Regulatory element activity | survey of regulatory elements (SuRE) reporter technology assay in K562 and HepG2 cells | (29) | |
| eQTL | eQTL identified in 44 human tissues reported in GTEx pilot analysis v6; other studies | HaploReg | (23) |
| eQTL for kidney regions | 4,221,837 and 4,579,723 eQTLs identified in human glomeruli and tubulointerstitial regions; | (24) | |
| 516 eQTLs from human renal glomerular and 172 from renal tubular compartments | (25) | ||
| TAD | 102,298 and 251,86 TADs from hg38 and hg19, respectively. We merged the data by converting hg19 to hg38. | ENCODE | (43) |
| CTCF binding regions | 109,342 CTCF binding regions identified from 40 cell types | ENCODE (reanalyzed) |
(28) |
| Mouse CTCF binding sites | Based on CTCF ChIP-seq in 19 tissues from mouse | Mouse ENCODE | (44) |
| Mouse enhancer | Based on histone mark ChIP-seq in 19 tissues from mouse | Mouse ENCODE | (44) |
| Mouse eQTL | liver eQTL data from the Hybrid Mouse Diversity Panel | HMDP | (26) |
| mouse microRNA | 1227 primary microRNA transcripts and 2110 mature microRNAs | miRBase | (42) |
A machine learning method for predicting the cell type conservation of CTCF binding
A 1D Convolutional Neural Network (CNN) model was developed to predict the degree of cell type conservation of CTCF binding using a CTCF binding dataset from 40 cell types (19) based on DNA sequences at the loci. The model can be accessed at https://github.com/yliuphys/huCTCF_conservation.
Site-directed mutagenesis, transfection and expression analysis
We performed site-directed mutagenesis and transfection as described previously (19). Reverse transcription and real-time PCR were performed as we described previously (20). Western blot was performed as we descried previously with some modifications (21,22).
Results
BP-associated SNPs and haplotypes and their genomic distribution
Evangelou, et al, reported 984 BP-associated sentinel SNPs, including newly identified and previously reported SNPs (4). Giri, et al, reported another 249 BP-associated sentinel SNPs (5). The total number of LD regions was reduced to 1,071 after duplicated or fully overlapping LD regions were removed. Of the 1,071 LD regions, 701 were identified from European populations (Supplemental Table S1). The 1,071 BP-associated LD regions contain 26,585 non-duplicated SNPs (Supplemental Table S1).
The vast majority of the 26,585 SNPs are intronic or intergenic (Figure 2A). 7,402 SNPs are located at least 10kb from any protein-coding gene based on RefSeq genes (Supplemental Table S1; Figure 2A). 359 SNPs overlap with human CpG islands (Supplemental Table S2). Of the 1,071 LD regions, 932 do not contain any nonsynonymous SNP, of which 290 are completely intergenic and 234 are located at least 10kb from any protein-coding gene (Supplemental Table S1; Figure 2B).
Figure 2. Distribution of human BP-associated SNPs and their mouse syntenic regions.

A. Genomic distribution of human BP-associated SNPs. B. Distribution of human BP-associated linkage disequilibrium (LD) regions. C. Human BP-associated SNPs with or without mouse syntenic regions. D. The majority of human BP-associated SNPs and mouse corresponding sites share the same nearest protein-coding genes. E. Genomic distribution of mouse corresponding sites for human BP-associated SNPs.
Mouse syntenic regions for BP-associated SNPs
We were able to identify syntenic regions in the mouse genome for 9,631 of the 26,585 human BP-associated SNPs using liftOver. 9,447 of them, or more than 98%, were confirmed by VISTA (Supplemental Table S3; Figure 2C). Of the 9,447 sites in human and their corresponding sites in mouse, 6,551 have the same adjacent protein-coding genes in human and mouse (Figure 2D). 5,327 of the 9,447 sites are intronic, 197 and 3 in 3’ and 5’ untranslated regions, respectively, and 2,817 in intergenic regions in both human and mouse (Figure 2E). 2,051 are located at least 10kb from any protein-coding gene.
eQTL
eQTLs are genomic loci with sequence variations that are associated with expression levels of genes across individuals. The association suggests sequence variations at the eQTL may regulate the expression of its associated gene. An eQTL and its associated gene may be up to hundreds of thousands of bp apart (cis eQTL) or located on different chromosomes (trans eQTL). Some eQTLs are shared by multiple tissues, while others are tissue-specific.
The 26,585 BP-associated SNPs include 17,922 SNPs that are eQTLs for 3,801 genes, forming 252,249 eQTL-gene pairs, according to HaploReg, which included data from 44 tissues in several hundred individuals in the GTEx pilot study analysis v6 (23) and other studies. We retrieved 251 BP physiology genes from Gene Ontology (Supplemental Table S4). In 2,626 of the eQTL-gene pairs involving 353 BP-associated SNPs and 26 genes, the genes are BP physiology genes (Supplemental Table S5a, S5b). For example, 16 SNPs are eQTLs of AGT and the SNP rs1230366 is the eQTL of 3 BP physiology genes (ERAP1; ERAP2; LNPEP). 112, 11 and 115 of the 2,626 eQTL-gene pairs were identified in adrenal glands, aorta, and tibial arteries, respectively, which involved 2 genes, ERAP1 and NEDD4L, and 2 haplotypes linked to SNPs rs42398 and rs7235890.
Most of the eQTLs in HaploReg were not identified in the kidney, an essential organ for blood pressure regulation. We examined the eQTLs identified in human microdissected glomeruli and tubulointerstitial regions by Gillies, et al (24). We identified 298 eQTL-gene pairs involving 216 BP-associated SNPs and 20 BP physiology genes in glomeruli and 347 eQTL-gene pairs involving 281 BP-associated SNPs and 22 BP physiology genes in tubulointerstitial regions (Supplemental Table S6a, S6b, S6c, S6d). The eQTLs for ERAP1 and NEDD4L mentioned in the preceding paragraph were also identified in tubulointerstitial regions, and the ERAP1 eQTLs were identified in glomeruli. Several of the eQTL genes are not the nearest protein-coding gene for the eQTL SNPs. For example, several SNPs in the human BP-associated rs9662255 LD region are eQTLs for SLC2A5. The SNPs and the gene are hundreds of kb apart and separated by several other genes (Figure 3A). Several SNPs in the human BP-associated rs12116637 LD region are eQTLs for GUCA2B, while the SNPs are located in introns of other genes (Figure 3B).
Figure 3. Examples of human BP-associated SNPs that are eQTLs for BP physiology genes.

A. Several SNPs in the human BP-associated rs9662255 LD region are eQTLs for SLC2A5. The SNPs and the gene are hundreds of kb apart and separated by several other genes. B. Several SNPs in the human BP-associated rs12116637 LD region are eQTLs for GUCA2B. The SNPs are located in introns of other genes. C. Several SNPs located in and outside of AGT in the human BP-associated rs699 LD region are eQTLs for AGT in human. D. The mouse genomic region syntenic with the human genomic region harboring the SNPs shown in panel C contains an eQTL for mouse Agt. In all panels, eQTL loci are indicated by vertical red lines and eQTL genes by green boxes. Arrows indicate the gene transcription direction.
Of the eQTLs reported by Qiu, et al, in human glomeruli and renal tubular compartments (25), 3 are BP-associated SNPs, and none of the eQTL genes for these 3 SNPs is a BP physiology gene.
Of the 9,447 human BP-associated SNPs that have syntenic regions in the mouse genome, 4,670 are eQTL defined by HaploReg. None of the mouse genomic sites corresponding to these 4,670 SNPs is an eQTL in the mouse liver eQTL dataset that we used (26). liftOver relies on a chain file for genomic coordinate conversion. The chain file includes blocks of aligned regions between human and mouse genomes, which are from whole-genome alignment. We retrieved these aligned regions that liftOver used to convert the 9,447 SNPs from the header of the chain file. We used the aligned regions to query the mouse eQTL dataset. We identified 849 SNPs of which the mouse syntenic regions had the same eQTL genes in mouse as the SNPs in human (Supplemental Table S7a), and 14 of them have eQTL genes that are BP physiology genes (Supplemental Table S7b). For example, several SNPs in the BP-associated rs699 LD region are eQTLs for AGT in human and their syntenic region in mouse contains an eQTL for Agt in mouse (Figure 3C, 3D). A similar analysis using the human kidney regional eQTLs reported by Gillies, et al, identified 4 SNPs that have the same eQTL genes in human and mouse (Supplemental Table S7c).
Enrichment of BP-associated SNPs as eQTLs for BP physiology genes
Of the 1,071 BP-associated sentinel SNPs, 20 are eQTLs for BP physiology genes based on HaploReg. For 1,071 SNPs randomly selected from all SNPs and each representing a different LD block, the number of eQTLs for BP physiology genes ranged from 0 to 3 in 10 times of repeated random selection. BP-associated sentinel SNPs are highly significantly enriched for eQTLs for BP physiology genes compared to randomly selected sentinel SNPs (p < 0.00023, Fisher’s exact test).
TADs
TADs are chromatin domains within which chromatin segments interact frequently and potentially regulate each other (27). We identified a total of 41,178 pairs of BP-associated LD regions and BP physiology genes in the same TADs, involving 787 BP-associated LD regions and 179 BP physiology genes (Supplemental Table S8).
CTCF binding regions
CTCF binding is a key determinant of the boundaries of TADs and sub-domains within TADs and plays an important role in gene transcriptional regulation by enabling or preventing enhancer-promoter interactions. 195 of the 26,585 human BP-associated SNPs are located in CTCF binding regions identified from 40 human cell types (Supplemental Table S9a; Figure 4). The extent of overlap, i.e., 1 of every 136 BP-associated SNPs being located in these CTCF binding regions, is significantly greater than what would be expected from the whole genome where 1 of every 196 base pairs is in these regions (p = 4.90 × 10−7, Pearson’s Chi-squared test). We did not test whether this enrichment was specific to SNPs that are associated with BP.
Figure 4. BP-associated SNPs with large effects on CTCF binding and its conservation across cell types in human and their mouse corresponding sites.

The effect on cell type conservation was predicted using a newly developed machine learning method. CNN, convolutional neural network.
CTCF binding varies between cell types. We developed a deep neural network model to predict the cell type conservation of CTCF binding at a given CTCF binding region. The model achieved 82.82% accuracy in classifying CTCF binding into the most and least conserved CTCF binding regions across 40 human cell types in the testing data. Of the 195 BP-associated SNPs in CTCF binding regions, 5 SNPs had the largest effects on the CTCF binding conservation across cell types (absolute difference of the probability of the CTCF binding to be predicted as the most conserved is >0.2 between major and minor alleles) (Supplemental Table S9b; Figure 4). All 5 SNPs are located in the 35 bp core recognition sequence identified by conserved CTCF binding motif (28).
Of the 195 human BP-associated SNPs that are located in CTCF binding regions, 105 have corresponding sites in the mouse genome. 59 of them are located in at least one CTCF binding region in the mouse (Supplemental Table S9c). Most of the CTCF bindings that are conserved in human across 40 cell types also are conserved across 19 tissues in mouse.
Two human BP-associated SNPs, rs111835543 and rs12481717, that show effects on CTCF binding conservation in 40 human cell types based on deep neural network also have strong effects on CTCF binding according to motifbreakR (Supplemental Table S9d; Figure 4). One of them, rs12481717, has a corresponding site in the mouse genome and is located in a mouse CTCF binding region (Figure 4)
Enhancers
Of the 26,585 human BP-associated SNPs, 16,782 overlap with at least one enhancer mark peak region identified by ENCODE (Supplemental Table S10). A recent study has identified SNPs that influence enhancer or promoter activities based on a high-throughput Survey of Regulatory Element (SuRE) reporter assay performed in K562 and HepG2 cell lines (29). Of the 16,782 BP-associated SNPs that overlap with enhancer mark peak regions, 2,296 showed significantly different enhancer or promoter activities between the two alleles of each SNP in at least one of the two cell lines. For the other 9,803 BP-associated SNPs that do not overlap with any enhancer mark peak region, the number was 1,224, which is significantly less enriched than SNPs overlapping with enhancer mark peak regions (p = 0.003, Fisher’s exact test). It is important to note that substantial cell type specificity was observed in the SuRE analysis (29) and K562 and HepG2 cell lines do not have direct physiological relevance to blood pressure regulation.
Of the 9,447 SNPs that have corresponding sites in the mouse genome, 6,206 overlap with at least one human enhance mark peak region. 2,454 of the 6,206 corresponding sites in mouse also overlap with at least one mouse enhancer mark peak region (Supplemental Table S11a). 446 of them overlap with mouse enhancer mark peak regions that are considered tissue specific (Supplemental Table S11b).
lncRNA
DNA sequence variations may alter the expression or function of lncRNAs, which, in turn, may regulate the expression of protein-coding genes through cis or trans mechanisms (30). We used the “high-confidence” lncRNA collection available in LNCipedia 5.2, which was curated mostly from RNA-seq data. We identified 8,094 SNP-lncRNA gene pairs in which one or more of BP-associated SNPs were located in an lncRNA gene, which involved 6,794 SNPs and 889 lncRNA genes (Supplemental Table S12a). We identified 1,601 SNP-lncRNA exon pairs that involved 892 SNPs and 413 lncRNA exons (Supplemental Table S12b). lncRNA promoter region was defined as −2kb to +200bp relative to the transcriptional start site of an lncRNA transcript. We identified 2,469 pairs of SNPs and lncRNA transcript promoters involving 1,428 SNPs and 1,271 lncRNA transcripts (Supplemental Table S12c).
Of the 9,447 human BP-associated SNPs that have syntenic regions in the mouse genome, 2,044 of the corresponding sites in the mouse genome overlap with 714 mouse lncRNA transcripts, forming 3,678 SNP corresponding site-lncRNA transcript pairs, some of which involve lncRNA exons or promoters (Supplemental Table S12d, S12e, S12f).
745, 53, and 171 SNPs in human and their corresponding sites in mouse are located in lncRNA transcripts, exons, and transcript promoter regions, respectively, in both human and mouse (Supplemental Table S12g).
microRNA
microRNAs are well-established post-transcriptional regulators of protein expression and contribute to the development of hypertension (31). SNP rs2168518, which is in LD with BP-associated rs1378942, and SNP rs2292181, which is in LD with BP-associated rs6792918, are located within human mature miR-4513 and pre-miR-564, respectively (Supplemental Table S13a). Neither of them has a syntenic region in the mouse genome. None of the 9,447 SNP corresponding sites in the mouse genome overlaps with mouse miRNA primary transcripts or mature miRNAs. Approximately 2% of BP-associated SNPs are located less than 10kb from human pre-miRNAs (Supplemental Table S13b), while 0.8% of the mouse corresponding sites are located less than 10kb from mouse pri-miRNAs. 19 SNPs and their mouse corresponding sites are located less than 10kb from a miRNA or pri-miRNA/pre-miRNA in human and mouse (Supplemental Table S13c). 777 SNPs and their mouse corresponding sites share the same nearest microRNA genes (Supplemental Table S13d).
Transcription factor binding sites
23,091 of the 26,585 BP-associated SNPs (or 87%) are located within at least one predicted transcription factor binding region (Supplemental Table S14a). 2,583 and 1,851 of these SNPs are located in gene promoter regions and protein-coding gene promoter regions, respectively. For this analysis, we defined a promoter region as −2kb to +200bp relative to the gene transcriptional start site (Supplemental Table S14b).
An analysis using motifbreakR identified 257,686 pairs of SNPs and TF binding regions in which the SNPs were predicted to have strong effects on TF binding (Supplemental Table S14c). 15,596 of these pairs involve the TF CTCF. 28 of them physically overlapped with CTCF regions identified in our CTCF analysis described earlier in this article (Supplemental Table S14d), which involve 16 unique BP-associated SNPs.
mRNA splicing
SNPs may lead to the gain or loss of splicing donors or acceptors, which may change mRNA splicing patterns. The delta score from SpliceAI represents the probability of the SNP to alter splicing, with scores of 0.2 or above defined as likely pathogenic or pathogenic (18). We identified 19, 9, 22, and 13 SNPs that have a delta value of more than 0.2 for acceptor gain, acceptor loss, donor gain, and donor loss, respectively (Supplemental Table S15). An example is rs12987286, which is located in an intron of ACVR2A. Based on the SpliceAI prediction, the alternative allele of the SNP would result in an extra exon just before the SNP site. Another example is the SNP rs141337782, which might cause a splicing donor loss at 5 bp upstream of the SNP site in ARHGEF25.
Secondary structure of mRNA and lncRNA
The human BP-associated SNP rs9337951 is synonymous and does not have any other SNPs in LD with it. SNP rs9337951 is located in the mRNA encoding JCAD (junctional cadherin 5 associated) protein. Using RNAfold (32), we found modest changes in minimum free energy (+2.9 kcal/mol) and the folding pattern when the rs9337951 sequence in JCAD mRNA was changed from the major allele G to minor allele A (Figure 5A).
Figure 5. BP-associated synonymous SNP rs9337951:G>A in JCAD influences JCAD protein expression.

A. BP-associated synonymous SNP rs9337951:G>A was predicted to have modest effects on the secondary structure of JCAD mRNA. rs9337951 is not in linkage disequilibrium with any other SNP. Red arrows indicate some of the changes of secondary structure. B. BP-associated SNP rs2171690:T>C was predicted to have substantial effects on the secondary structure of lncRNA lnc-LMX1A. rs2171690 is not in linkage disequilibrium with any other SNP. C. Sequence confirmation of plasmid constructs containing JCAD with major or minor allele sequence of rs9337951. D. rs9337951:G>A did not influence JCAD mRNA expression in HEK293 or HK-2 cells. JCAD mRNA abundance was normalized by GAPDH. Real-time PCR was performed using primers specific for cell endogenous and plasmid-encoded (exogenous) JCAD mRNA. E. rs9337951:G>A resulted in significant decreases in JCAD protein abundance in HEK293 cells. F. rs9337951:G>A resulted in significant decreases in JCAD protein abundance in HK-2 cells.
Ten human BP-associated sentinel SNPs and any SNP in LD with them are entirely contained in genomic regions transcribed as lncRNAs. They are rs11222084, rs167479, rs1706003 (with another SNP in LD), rs2171690, rs2854275, rs34517439, rs36006409, rs4984496 (with 3 other SNPs in LD), rs61760904, and rs72914576. The effect of the SNPs on the minimum free energy of the lncRNA transcripts was modest. However, the SNPs are predicted to cause substantial changes in the folding pattern of several of the lncRNAs, which were much more pronounced than what we observed above in JCAD mRNA. An example is shown in Figure 5B.
Experimental testing of the effect of a synonymous SNP on protein expression
We performed a proof of concept experiment to test whether a synonymous BP-associated SNP that is predicted to influence mRNA secondary structure would affect protein expression, focusing on rs9337951:G>A in JCAD (Figure 5A). We obtained plasmids containing the coding sequence for JCAD with G at rs9337951 and used site-directed mutagenesis to generate plasmids containing A at rs9337951 (Figure 5C). Transfection in HEK293 and HK-2 cells with these plasmids resulted in robust overexpression of JCAD mRNA. The allelic sequence at rs9337951 did not influence JCAD mRNA abundance significantly in either cell type (Figure 5D). However, transfection with plasmids containing the minor allele (A) at rs9337951 resulted in significantly lower expression of JCAD protein compared with plasmids containing the major allele (G) in both HEK293 and HK-2 cells (Figure 5E, 5F).
Discussion
The majority of BP-associated haplotypes that have been identified by GWAS do not contain any nonsynonymous, damaging SNP. This suggests that the majority of BP-associated SNPs might influence BP by regulating gene expression, although the effect of some of these SNPs on BP might be explained by nonsynonymous sequence variants that occur at frequencies lower than 1%. SNPs may regulate gene expression through a variety of mechanisms, such as altering the binding of transcriptional factors, enhancer function, chromatin conformation, mRNA splicing or the function or expression of regulatory RNA (33).
In this study we identified specific BP-associated SNPs that could influence each of these mechanisms. This was accomplished by a thorough analysis of the most comprehensive set of BP-associated SNPs to date. Our analytical procedure used newly developed methods and approaches as well as traditional approaches to synthesize the SNP data with a broad range of data sources including recently reported data sources.
For example, the neural network model that we developed has high accuracy in predicting the conservation of CTCF binding across cell types, which allowed us to identify BP-associated SNPs located in such CTCF binding regions. Similar to any other deep neural network model, our model would benefit from further testing with additional datasets. Our model only uses 151bp DNA sequence regions as the basis for the prediction. Incorporating other factors such as chromatin conformation might further improve the prediction. Experimental analysis will be required to understand the biological basis of the identified conservation and the effect of BP-associated SNPs on such conservation.
To connect SNPs with physiological pathways known to be important in BP regulation, we developed a list of 251 “BP physiology genes”. Given the current state of gene annotation, the list is unlikely to include all genes that are physiologically important in BP regulation and it may include genes of which physiological significance in BP regulation is debatable. Nevertheless, this list is a powerful tool for prioritizing SNPs and regulatory mechanisms for future studies. Fewer than 20 of these BP physiology genes have been reported as BP GWAS genes, which, again, highlights the gap between BP GWAS findings and physiological understanding of BP regulation.
eQTL mapping is an excellent approach for identifying genes that may be regulated by a SNP. An important consideration when using eQTL data for this purpose is that eQTLs are often tissue-specific (6). Several specific tissues, such as renal microvasculature and tubules and peripheral resistance arterioles, have well-established physiological importance in BP regulation. Data from broad, multi-tissue eQTL studies such as GTEx are highly valuable but do not provide tissue resolution at the level of renal microvasculature and tubules (6). In this study, we used recently reported kidney region-specific eQTL to complement the data from GTEx and other studies, which allowed us to identify BP-associated SNPs that are eQTLs in tissue regions in an organ that is physiologically essential for BP regulation.
A major new result of this study is the annotation of mouse syntenic regions for human BP-associated SNPs. BP is an in vivo trait that cannot be modeled in cell culture. Studies performed in animal models, such as dogs, rats and mice, are a major basis for our understanding of physiological mechanisms that regulate BP. We have identified mouse syntenic regions for a large fraction of human BP-associated SNPs. Most of these mouse syntenic regions share genomic or functional genomic features with human genomic regions harboring BP-associated SNPs. These findings provide a strong foundation for developing mouse experiments to help understand physiological mechanisms mediating the effect of BP-associated SNPs. It is important to recognize that BP regulatory mechanisms in mice may not be identical to those in humans. This point is highlighted in our study by the fact that mouse syntenic regions cannot be identified for all human BP GWAS SNPs. Moreover, one should be cautious in extrapolating mechanisms identified in mice to humans even if the identification of the mechanism is driven by GWAS SNPs and genomic synteny.
While synonymous SNPs are traditionally considered inconsequential, as they do not alter amino acid sequence, increasing evidence suggests synonymous SNPs could influence protein abundance and function via several mechanisms (34,35). One of such mechanisms involves alterations of RNA secondary structure that in turn influence RNA stability and translational activity as our proof-of-principle experiment on rs9337951 in JCAD would suggest. It remains to be determined whether the change in JCAD protein expression results from the changes in mRNA secondary structure. We observed prominent effects of several BP-associated SNPs on the secondary structures of lncRNAs. lncRNAs might be under less evolutionary pressure than mRNA in general (36), allowing SNPs to have more pronounced effects on their secondary structures.
The effect of rs9337951 on JCAD protein expression adds to emerging studies supporting the notion that BP-associated regulatory SNPs could have substantial effects on gene expression or cellular function via unsuspected mechanisms (37-39). The substantial effects of SNPs reported in these studies were observed in specific genomic contexts in well-controlled experiments. This raises the possibility that the effect of some of the BP-associated SNPs on BP might be larger in specific sub-populations or under specific clinical conditions than that reported in the original GWAS.
The most important future studies that the findings of the current study call for and will facilitate are experimental validations, as illustrated by our proof-of-principle study of the synonymous SNP in JCAD. The findings of this study also highlight the value of other future studies. For example, we used r2 >= 0.8 as the threshold and data from the 1000 Genomes Project as the basis for defining LD regions. Using a different threshold or haplotype information from other whole genome sequencing projects may change the list of SNPs in LD with BP-associated sentinel SNPs, the analysis of which may lead to additional insights into the genetic underpinning of BP regulation. We compared SNPs associated with BP and other features such as eQTLs and identified overlaps. Overlaps also may be identified through a formal statistical testing using the original data (40). We mapped human BP-associated SNPs to mouse. Similar mapping in other animal models commonly used for BP research, such as rats or dogs, would be valuable but is currently limited by the scarcity of functional genomic data available for these species. Even the data available from mice would benefit from further expansion. It is important to note though that it is inherently difficult to generate directly comparable data for some human data in animal models. An example is eQTL data, which rely on naturally present sequence variations in human populations that are difficult to model in animals.
Perspectives
The result of the current study provides a systematic basis for linking BP-associated SNPs with biological and physiological regulatory mechanisms. The finding may facilitate the effort to utilize BP GWAS discoveries to better understand BP regulation and develop new or improved approaches for predicting, preventing and managing BP abnormalities.
Supplementary Material
Novelty and Significance.
What is new?
We mapped 26,585 human blood pressure (BP)-associated single nucleotide polymorphisms (SNPs) in 1,071 linkage disequilibrium regions to 9,447 syntenic regions in the mouse genome.
We systematically identified genomic features associated with these SNPs and regions using approaches that include newly developed data resources and machine learning methods and experimental testing.
What is relevant?
The findings provide novel insights into the potential functional significance of human BP-associated SNPs and an extensive resource for developing experiments to test such significance in human cells and animal models.
Summary.
More than 20% of the 1,071 BP-associated linkage disequilibrium regions are located at least 10 kb from any protein-coding gene.
BP-associated sentinel SNPs are significantly enriched for eQTLs for BP physiology genes.
BP SNPs may influence the conservation of CTCF binding across cell types, transcription factor binding, mRNA splicing, or secondary structures of RNA.
Acknowledgments
Sources of Funding
This work was supported by the U.S. National Institutes of Health [HL121233, HL125409, GM066730, HL116264], American Heart Association [15SFRN23910002], and the Advancing a Healthier Wisconsin Endowment.
Footnotes
Disclosures
None.
References
- 1.Collaborators GBDRF. Global, regional, and national comparative risk assessment of 79 behavioural, environmental and occupational, and metabolic risks or clusters of risks, 1990-2015: a systematic analysis for the Global Burden of Disease Study 2015. Lancet. 2016; 388, 1659–1724. doi: 10.1016/S0140-6736(16)31679-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cowley AW Jr. The genetic dissection of essential hypertension. Nat Rev Genet. 2006, 7, 829–840. doi: 10.1038/nrg1967 [DOI] [PubMed] [Google Scholar]
- 3.Padmanabhan S, Joe B. Towards Precision Medicine for Hypertension: A Review of Genomic, Epigenomic, and Microbiomic Effects on Blood Pressure in Experimental Rat Models and Humans. Physiol Rev. 2017; 97, 1469–1528. doi: 10.1152/physrev.00035.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Evangelou E, Warren HR, Mosen-Ansorena D, Mifsud B, Pazoki R, Gao H, Ntritsos G, Dimou N, Cabrera CP, Karaman I, Ng FL, Evangelou M, Witkowska K, Tzanis E, Hellwege JN, Giri A, Velez Edwards DR, Sun YV, Cho K, Gaziano JM, Wilson PWF, Tsao PS, Kovesdy CP, Esko T, Magi R, Milani L, Almgren P, Boutin T, Debette S, Ding J, Giulianini F, Holliday EG, Jackson AU, Li-Gao R, Lin WY, Luan J, Mangino M, Oldmeadow C, Prins BP, Qian Y, Sargurupremraj M, Shah N, Surendran P, Theriault S, Verweij N, Willems SM, Zhao JH, Amouyel P, Connell J, de Mutsert R, Doney ASF, Farrall M, Menni C, Morris AD, Noordam R, Pare G, Poulter NR, Shields DC, Stanton A, Thom S, Abecasis G, Amin N, Arking DE, Ayers KL, Barbieri CM, Batini C, Bis JC, Blake T, Bochud M, Boehnke M, Boerwinkle E, Boomsma DI, Bottinger EP, Braund PS, Brumat M, Campbell A, Campbell H, Chakravarti A, Chambers JC, Chauhan G, Ciullo M, Cocca M, Collins F, Cordell HJ, Davies G, de Borst MH, de Geus EJ, Deary IJ, Deelen J, Del Greco MF, Demirkale CY, Dorr M, Ehret GB, Elosua R, Enroth S, Erzurumluoglu AM, Ferreira T, Franberg M, Franco OH, Gandin I, Gasparini P, Giedraitis V, Gieger C, Girotto G, Goel A, Gow AJ, Gudnason V, Guo X, Gyllensten U, Hamsten A, Harris TB, Harris SE, Hartman CA, Havulinna AS, Hicks AA, Hofer E, Hofman A, Hottenga JJ, Huffman JE, Hwang SJ, Ingelsson E, James A, Jansen R, Jarvelin MR, Joehanes R, Johansson A, Johnson AD, Joshi PK, Jousilahti P, Jukema JW, Jula A, Kahonen M, Kathiresan S, Keavney BD, Khaw KT, Knekt P, Knight J, Kolcic I, Kooner JS, Koskinen S, Kristiansson K, Kutalik Z, Laan M, Larson M, Launer LJ, Lehne B, Lehtimaki T, Liewald DCM, Lin L, Lind L, Lindgren CM, Liu Y, Loos RJF, Lopez LM, Lu Y, Lyytikainen LP, Mahajan A, Mamasoula C, Marrugat J, Marten J, Milaneschi Y, Morgan A, Morris AP, Morrison AC, Munson PJ, Nalls MA, Nandakumar P, Nelson CP, Niiranen T, Nolte IM, Nutile T, Oldehinkel AJ, Oostra BA, O’Reilly PF, Org E, Padmanabhan S, Palmas W, Palotie A, Pattie A, Penninx B, Perola M, Peters A, Polasek O, Pramstaller PP, Nguyen QT, Raitakari OT, Ren M, Rettig R, Rice K, Ridker PM, Ried JS, Riese H, Ripatti S, Robino A, Rose LM, Rotter JI, Rudan I, Ruggiero D, Saba Y, Sala CF, Salomaa V, Samani NJ, Sarin AP, Schmidt R, Schmidt H, Shrine N, Siscovick D, Smith AV, Snieder H, Sober S, Sorice R, Starr JM, Stott DJ, Strachan DP, Strawbridge RJ, Sundstrom J, Swertz MA, Taylor KD, Teumer A, Tobin MD, Tomaszewski M, Toniolo D, Traglia M, Trompet S, Tuomilehto J, Tzourio C, Uitterlinden AG, Vaez A, van der Most PJ, van Duijn CM, Vergnaud AC, Verwoert GC, Vitart V, Volker U, Vollenweider P, Vuckovic D, Watkins H, Wild SH, Willemsen G, Wilson JF, Wright AF, Yao J, Zemunik T, Zhang W, Attia JR, Butterworth AS, Chasman DI, Conen D, Cucca F, Danesh J, Hayward C, Howson JMM, Laakso M, Lakatta EG, Langenberg C, Melander O, Mook-Kanamori DO, Palmer CNA, Risch L, Scott RA, Scott RJ, Sever P, Spector TD, van der Harst P, Wareham NJ, Zeggini E, Levy D, Munroe PB, Newton-Cheh C, Brown MJ, Metspalu A, Hung AM, O’Donnell CJ, Edwards TL, Psaty BM, Tzoulaki I, Barnes MR, Wain LV, Elliott P, Caulfield MJ, Million Veteran P. Genetic analysis of over 1 million people identifies 535 new loci associated with blood pressure traits. Nat Genet. 2018; 50, 1412–1425. doi: 10.1038/s41588-018-0205-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Giri A, Hellwege JN, Keaton JM, Park J, Qiu C, Warren HR, Torstenson ES, Kovesdy CP, Sun YV, Wilson OD, Robinson-Cohen C, Roumie CL, Chung CP, Birdwell KA, Damrauer SM, DuVall SL, Klarin D, Cho K, Wang Y, Evangelou E, Cabrera CP, Wain LV, Shrestha R, Mautz BS, Akwo EA, Sargurupremraj M, Debette S, Boehnke M, Scott LJ, Luan J, Zhao JH, Willems SM, Theriault S, Shah N, Oldmeadow C, Almgren P, Li-Gao R, Verweij N, Boutin TS, Mangino M, Ntalla I, Feofanova E, Surendran P, Cook JP, Karthikeyan S, Lahrouchi N, Liu C, Sepulveda N, Richardson TG, Kraja A, Amouyel P, Farrall M, Poulter NR, Understanding Society Scientific G, International Consortium for Blood P, Blood Pressure-International Consortium of Exome Chip S, Laakso M, Zeggini E, Sever P, Scott RA, Langenberg C, Wareham NJ, Conen D, Palmer CNA, Attia J, Chasman DI, Ridker PM, Melander O, Mook-Kanamori DO, Harst PV, Cucca F, Schlessinger D, Hayward C, Spector TD, Jarvelin MR, Hennig BJ, Timpson NJ, Wei WQ, Smith JC, Xu Y, Matheny ME, Siew EE, Lindgren C, Herzig KH, Dedoussis G, Denny JC, Psaty BM, Howson JMM, Munroe PB, Newton-Cheh C, Caulfield MJ, Elliott P, Gaziano JM, Concato J, Wilson PWF, Tsao PS, Velez Edwards DR, Susztak K, Million Veteran P, O’Donnell CJ, Hung AM, Edwards TL. Trans-ethnic association study of blood pressure determinants in over 750,000 individuals. Nat Genet. 2019; 51, 51–62. doi: 10.1038/s41588-018-0303-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Consortium GT, Laboratory DA, Coordinating Center -Analysis Working G, Statistical Methods groups-Analysis Working G, Enhancing Gg, Fund NIHC, Nih/Nci, Nih/Nhgri, Nih/Nimh, Nih/Nida, Biospecimen Collection Source Site N, Biospecimen Collection Source Site R, Biospecimen Core Resource V, Brain Bank Repository-University of Miami Brain Endowment B, Leidos Biomedical-Project M, Study E, Genome Browser Data I, Visualization EBI, Genome Browser Data I, Visualization-Ucsc Genomics Institute UoCSC, Lead a, Laboratory DA, Coordinating C, management NIHp, Biospecimen c, Pathology, e QTLmwg, Battle A, Brown CD, Engelhardt BE, Montgomery SB. Genetic effects on gene expression across human tissues. Nature. 2017; 550, 204–213. doi: 10.1038/nature24277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhbannikov IY, Arbeev K, Ukraintseva S, Yashin AI. haploR: an R package for querying web-based annotation tools. F1000Res. 2017; 6, 97. doi: 10.12688/f1000research.10742.2. eCollection 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ward LD, Kellis M. HaploReg: a resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucleic Acids Res. 2012; 40, D930–934. doi: 10.1093/nar/gkr917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sherry ST, Ward MH, Kholodov M, Baker J, Phan L, Smigielski EM, Sirotkin K. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 2001; 29, 308–311. doi: 10.1093/nar/29.1.308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Haeussler M, Zweig AS, Tyner C, Speir ML, Rosenbloom KR, Raney BJ, Lee CM, Lee BT, Hinrichs AS, Gonzalez JN, Gibson D, Diekhans M, Clawson H, Casper J, Barber GP, Haussler D, Kuhn RM, Kent WJ. The UCSC Genome Browser database: 2019 update Nucleic Acids Res. 2019; 47, D853–D858. doi: 10.1093/nar/gky1095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Frazer KA, Pachter L, Poliakov A, Rubin EM, Dubchak I. VISTA: computational tools for comparative genomics. Nucleic Acids Res. 2004; 32, W273–279. doi: 10.1093/nar/gkh458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.The Gene Ontology C. The Gene Ontology Resource: 20 years and still GOing strong. Nucleic Acids Res. 2019; 47, D330–d338. doi: 10.1093/nar/gky1055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Coetzee SG, Coetzee GA, Hazelett DJ. motifbreakR: an R/Bioconductor package for predicting variant effects at transcription factor binding sites. Bioinformatics. 2015; 31, 3847–3849. doi: 10.1093/bioinformatics/btv470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kheradpour P, Kellis M. Systematic discovery and characterization of regulatory motifs in ENCODE TF binding experiments. Nucleic Acids Res. 2014; 42, 2976–2987. doi: 10.1093/nar/gkt1249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang J, Zhuang J, Iyer S, Lin X, Whitfield TW, Greven MC, Pierce BG, Dong X, Kundaje A, Cheng Y, Rando OJ, Birney E, Myers RM, Noble WS, Snyder M, Weng Z. Sequence features and chromatin structure around the genomic regions bound by 119 human transcription factors. Genome Res. 2012; 22, 1798–1812. doi: 10.1101/gr.139105.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kulakovskiy IV, Medvedeva YA, Schaefer U, Kasianov AS, Vorontsov IE, Bajic VB, Makeev VJ. HOCOMOCO: a comprehensive collection of human transcription factor binding sites models. Nucleic Acids Res. 2013; 41, D195–202. doi: 10.1093/nar/gks1089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, Glass CK. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell. 2010; 38, 576–589. doi: 10.1016/j.molcel.2010.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jaganathan K, Kyriazopoulou Panagiotopoulou S, McRae JF, Darbandi SF, Knowles D, Li YI, Kosmicki JA, Arbelaez J, Cui W, Schwartz GB, Chow ED, Kanterakis E, Gao H, Kia A, Batzoglou S, Sanders SJ, Farh KK. Predicting Splicing from Primary Sequence with Deep Learning. Cell. 2019; 176, 535–548. doi: 10.1016/j.cell.2018.12.015 [DOI] [PubMed] [Google Scholar]
- 19.Liu Y, Mladinov D, Pietrusz JL, Usa K, Liang M. Glucocorticoid response elements and 11 beta-hydroxysteroid dehydrogenases in the regulation of endothelial nitric oxide synthase expression. Cardiovasc Res. 2009; 81, 140–147. doi: 10.1093/cvr/cvn231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Widlansky ME, Jensen DM, Wang J, Liu Y, Geurts AM, Kriegel AJ, Liu P, Ying R, Zhang G, Casati M, Chu C, Malik M, Branum A, Tanner MJ, Tyagi S, Usa K, Liang M. miR-29 contributes to normal endothelial function and can restore it in cardiometabolic disorders. EMBO Mol Med. 2018; 10. doi: 10.15252/emmm.201708046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liang M, Pietrusz JL. Thiol-related genes in diabetic complications: a novel protective role for endogenous thioredoxin 2. Arterioscler Thromb Vasc Biol. 2007; 27, 77–83. doi: 10.1161/01.ATV.0000251006.54632.bb [DOI] [PubMed] [Google Scholar]
- 22.Tian Z, Greene AS, Usa K, Matus IR, Bauwens J, Pietrusz JL, Cowley AW Jr., Liang M. Renal regional proteomes in young Dahl salt-sensitive rats. Hypertension. 2008; 51, 899–904. doi: 10.1161/HYPERTENSIONAHA.107.109173 [DOI] [PubMed] [Google Scholar]
- 23.Ward LD, Kellis M. HaploReg v4: systematic mining of putative causal variants, cell types, regulators and target genes for human complex traits and disease. Nucleic Acids Res. 2016; D877–D881. doi: 10.1093/nar/gkv1340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gillies CE, Putler R, Menon R, Otto E, Yasutake K, Nair V, Hoover P, Lieb D, Li S, Eddy S, Fermin D, McNulty MT, Nephrotic Syndrome Study N, Hacohen N, Kiryluk K, Kretzler M, Wen X, Sampson MG. An eQTL Landscape of Kidney Tissue in Human Nephrotic Syndrome. Am J Hum Genet. 2018; 103, 232–244. doi: 10.1016/j.ajhg.2018.07.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Qiu C, Huang S, Park J, Park Y, Ko YA, Seasock MJ, Bryer JS, Xu XX, Song WC, Palmer M, Hill J, Guarnieri P, Hawkins J, Boustany-Kari CM, Pullen SS, Brown CD, Susztak K. Renal compartment-sepecific genetic variation analyses identify new pathways in chronic kidney disease. Nat Med. 2018; 24, 1721–1731. doi: 10.1038/s41591-018-0194-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bennett BJ, Farber CR, Orozco L, Kang HM, Ghazalpour A, Siemers N, Neubauer M, Neuhaus I, Yordanova R, Guan B, Truong A, Yang WP, He A, Kayne P, Gargalovic P, Kirchgessner T, Pan C, Castellani LW, Kostem E, Furlotte N, Drake TA, Eskin E, Lusis AJ. A high-resolution association mapping panel for the dissection of complex traits in mice. Genome Res. 2010; 20, 281–290. doi: 10.1101/gr.099234.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dekker J, Mirny L. The 3D Genome as Moderator of Chromosomal Communication. Cell. 2016; 164, 1110–1121. doi: 10.1016/j.cell.2016.02.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Maurano MT, Wang H, John S, Shafer A, Canfield T, Lee K, Stamatoyannopoulos JA. Role of DNA Methylation in Modulating Transcription Factor Occupancy. Cell Rep. 2015; 12, 1184–1195. doi: 10.1016/j.celrep.2015.07.024 [DOI] [PubMed] [Google Scholar]
- 29.van Arensbergen J, Pagie L, FitzPatrick VD, de Haas M, Baltissen MP, Comoglio F, van der Weide RH, Teunissen H, Vosa U, Franke L, de Wit E, Vermeulen M, Bussemaker HJ, van Steensel B. High-throughput identification of human SNPs affecting regulatory element activity. Nat Genet. 2019; 51, 1160–1169. doi: 10.1038/s41588-019-0455-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kong Y, Lu Z, Liu P, Liu Y, Wang F, Liang EY, Hou FF, Liang M. Long Noncoding RNA: Genomics and Relevance to Physiology. Compr Physiol. 2019; 9, 933–946. doi: 10.1002/cphy.c180032 [DOI] [PubMed] [Google Scholar]
- 31.Liu Y, Usa K, Wang F, Liu P, Geurts AM, Li J, Williams AM, Regner KR, Kong Y, Liu H, Nie J, Liang M. MicroRNA-214-3p in the Kidney Contributes to the Development of Hypertension. J Am Soc Nephrol. 2018; 29, 2518–2528. doi: 10.1681/ASN.2018020117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hofacker IL. Vienna RNA secondary structure server. Nucleic Acids Res. 2003; 31, 3429–3431. doi: 10.1093/nar/gkg599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liang M Epigenetic Mechanisms and Hypertension. Hypertension. 2018; 72, 1244–1254. doi: 10.1161/HYPERTENSIONAHA.118.11171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sauna ZE, Kimchi-Sarfaty C. Understanding the contribution of synonymous mutations to human disease. Nat Rev. Genet. 2011; 12, 683–691. doi: 10.1038/nrg3051 [DOI] [PubMed] [Google Scholar]
- 35.Polfus LM, Khajuria RK, Schick UM, Pankratz N, Pazoki R, Brody JA, Chen MH, Auer PL, Floyd JS, Huang J, Lange L, van Rooij FJA, Gibbs RA, Metcalf G, Muzny D, Veeraraghavan N, Walter K, Chen L, Yanek L, Becker LC, Peloso GM, Wakabayashi A, Kals M, Metspalu A, Esko T, Fox K, Wallace R, Franceschini N, Matijevic N, Rice KM, Bartz TM, Lyytikainen LP, Kahonen M, Lehtimaki T, Raitakari OT, Li-Gao R, Mook-Kanamori DO, Lettre G, van Duijn CM, Franco OH, Rich SS, Rivadeneira F, Hofman A, Uitterlinden AG, Wilson JG, Psaty BM, Soranzo N, Dehghan A, Boerwinkle E, Zhang X, Johnson AD, O’Donnell CJ, Johnsen JM, Reiner AP, Ganesh SK, Sankaran VG. Whole-Exome Sequencing Identifies Loci Associated with Blood Cell Traits and Reveals a Role for Alternative GFI1B Splice Variants in Human Hematopoiesis. Am J Hum Genet. 2016; 99, 481–488. doi: 10.1016/j.ajhg.2016.06.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Necsulea A, Soumillon M, Warnefors M, Liechti A, Daish T, Zeller U, Baker JC, Grutzner F, Kaessmann H. The evolution of lncRNA repertoires and expression patterns in tetrapods. Nature. 2014; 505, 635–640. doi: 10.1038/nature12943 [DOI] [PubMed] [Google Scholar]
- 37.Gupta RM, Hadaya J, Trehan A, Zekavat SM, Roselli C, Klarin D, Emdin CA, Hilvering CRE, Bianchi V, Mueller C, Khera AV, Ryan RJH, Engreitz JM, Issner R, Shoresh N, Epstein CB, de Laat W, Brown JD, Schnabel RB, Bernstein BE, Kathiresan S. A Genetic Variant Associated with Five Vascular Diseases Is a Distal Regulator of Endothelin-1 Gene Expression. Cell. 2017; 170, 522–533.e15. doi: 10.1016/j.cell.2017.06.049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lo Sardo V, Chubukov P, Ferguson W, Kumar A, Teng EL, Duran M, Zhang L, Cost G, Engler AJ, Urnov F, Topol EJ, Torkamani A, Baldwin KK. Unveiling the Role of the Most Impactful Cardiovascular Risk Locus through Haplotype Editing. Cell. 2018; 175, 1796–1810.e1720. doi: 10.1016/j.cell.2018.11.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bai X, Mangum KD, Dee RA, Stouffer GA, Lee CR, Oni-Orisan A, Patterson C, Schisler JC, Viera AJ, Taylor JM, Mack CP. Blood pressure-associated polymorphism controls ARHGAP42 expression via serum response factor DNA binding. J Clin Invest. 2017; 127, 670–680. doi: 10.1172/JCI88899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Giambartolomei C, Vukcevic D, Schadt EE, Franke L, Hingorani AD, Wallace C, Plagnol V. Bayesian test for colocalisation between pairs of genetic association studies using summary statistics. PLoS Genet. 2014; 10, e1004383. doi: 10.1371/journal.pgen.1004383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Volders PJ, Anckaert J, Verheggen K, Nuytens J, Martens L, Mestdagh P, Vandesompele J. LNCipedia 5: towards a reference set of human long noncoding RNAs. Nucleic Rcids Aes. 2019; 47, D135–d139. doi: 10.1093/nar/gky1031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kozomara A, Birgaoanu M, Griffiths-Jones S. miRBase: from microRNA sequences to function. Nucleic Acids Res. 2019; 47, D155–d162. doi: 10.1093/nar/gky1141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Consortium EP An integrated encyclopedia of DNA elements in the human genome. Nature. 2012; 489, 57–74. doi: 10.1038/nature11247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shen Y, Yue F, McCleary DF, Ye Z, Edsall L, Kuan S, Wagner U, Dixon J, Lee L, Lobanenkov VV, Ren B. A map of the cis-regulatory sequences in the mouse genome. Nature. 2012; 488, 116–120. doi: 10.1038/nature11243 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.