

Abstract
Rationale:
Bone marrow kinase on the X chromosome (BMX) is highly expressed in the arterial endothelium from the embryonic stage to the adult stage in mice. It is also expressed in microvessels and the lymphatics in response to pathological stimuli. However, its role in endothelial permeability and sepsis remains unknown.
Objective:
We aimed to delineate the function of BMX in thrombin-mediated endothelial permeability and the vascular leakage that occurs with sepsis in cecal ligation and puncture (CLP) models
Methods and Results:
The CLP model was applied to wild-type and BMX knockout mice to induce sepsis. Meanwhile, the electric cell-substrate impedance sensing assay was used to detect trans-endothelial electrical resistance in vitro and the modified Miles assay was used to evaluate vascular leakage in vivo. We showed that BMX loss caused lung injury and inflammation in early CLP-induced sepsis. Disruption of BMX increased thrombin-mediated permeability in mice and cultured endothelial cells (ECs) by 2–3-fold. The expression of BMX in macrophages, neutrophils, platelets and lung epithelial cells was undetectable compared with that in ECs, indicating that endothelium dysfunction, rather than leukocyte and platelet dysfunction, was involved in vascular permeability and sepsis. Mechanistically, biochemical and cellular analyses demonstrated that BMX specifically repressed thrombin-protease-activated receptor 1 (PAR1) signaling in ECs by directly phosphorylating PAR1 and promoting its internalization and deactivation. Importantly, pre-treatment with the selective PAR1 antagonist SCH79797 rescued BMX loss-mediated endothelial permeability and pulmonary leakage in early CLP-induced sepsis.
Conclusion:
Acting as a negative regulator of PAR1, BMX promotes PAR1 internalization and signal inactivation through PAR1 phosphorylation. Moreover, BMX-mediated PAR1 internalization attenuates endothelial permeability to protect vascular leakage during early sepsis.
Keywords: BMX, thrombin, PAR1, vascular permeability, sepsis, vascular endothelium
Subject Terms: Basic Science Research, Inflammation, Pathophysiology, Vascular Biology
Graphical Abstract
Sepsis has high mortality and is pathologically characterized by diffuse alveolar damage caused by increased alveolar capillary permeability. However, the pathogenesis of alveolar capillary leak during sepsis remains unknown and no current therapy is available that directly prevents or reverses this underlying capillary leak. The thrombin-PAR1 signaling play a crucial role in the pathogenesis of sepsis by mediating endothelial permeability and alveolar edema. In the present study, we demonstrate that in a CLP model BMX functions as an endogenous antagonist of PAR1 in the endothelium to regulate vascular homeostasis during sepsis. Mechanistically, we show that BMX directly phosphorylates PAR1 and promotes its internalization and signal inactivation to repress thrombin-PAR1-mediated endothelial permeability. The identification of BMX as a novel antagonist of PAR1 in the endothelium provides a new therapeutic target for PAR1-mediated vascular disease.
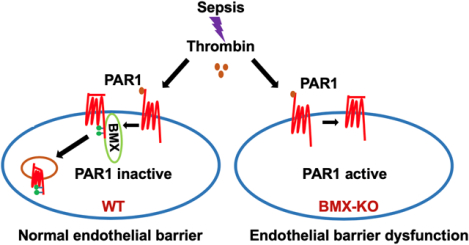
INTRODUCTION
Sepsis is a common cause of hospital admission and has high mortality. Sepsis is characterized by dysregulated inflammation and can rapidly lead to multiorgan failure affecting most commonly of the cardiovascular and respiratory systems.1–3 Lung injury in sepsis is initially characterized by transudation of fluid and proteins across the alveolar wall, described clinically as acute respiratory distress syndrome (ARDS) and pathologically as diffuse alveolar damage.4 The increased alveolar capillary permeability causes the paracellular leak of fluid, solutes and proteins across the endothelium.5–7 However, the pathogenesis of alveolar capillary leak during sepsis remains unknown and no current therapy is available that directly prevents or reverses this underlying capillary leak.
One hallmark of acute sepsis is microvessel dysfunction, in which increased endothelial permeability and deposited thrombosis appear to play pivotal roles.8 Accompanied by activation of the coagulation system, sepsis typically leads to deposition of thrombin in the microvasculature.8–11 Thrombin plays a crucial role in regulating vascular homeostasis, including vascular permeability, thrombosis, and vascular tone, primarily through its receptor protease-activated receptor (PAR1).12–16 PAR1, which is primarily expressed in the endothelium and platelets in the vasculature,12, 17 is proteolytically activated by thrombin at the N-terminal extracellular domain, generating a new N-terminal site that functions as a tethered ligation by binding to the second loop of PAR1.18 In sepsis, thrombin-cleaved PAR1 activation causes platelet activation and aggregation; meanwhile, thrombin-activated PAR1 in ECs causes increased endothelial permeability and promotes vWF secretion from the endothelium to recruit platelets and then initiate thrombosis.12–14 PAR1 signaling is regulated by multiple independent mechanisms at different temporal and spatial levels. Among the multiple regulatory mechanisms, PAR1 internalization has been proven to be crucial for the regulation of PAR1 signaling. It has been reported that a tyrosine-based motif, YXXL, is involved in PAR1 internalization,19–21 but the mechanism is unclear.
Bone marrow kinase on the X chromosome (BMX), which is expressed mainly in the arterial endothelium and cardiac endothelium, is a non-receptor tyrosine kinase belonging to the Tec family that plays pivotal roles in cardiovascular pathology.22, 23 Various proteins, including tyrosine receptors and G-protein-coupled receptors, have been shown to interact with specific domains of BMX to facilitate intracellular signaling pathways that control cellular processes such as migration, proliferation, survival, and differentiation.24–28 The association between BMX and PAR1 has been reported in tumor cells. PAR1 activation induces BMX, with subsequent Shc binding to its C-tail to form a complex. BMX via its PH domain binds to a region of seven residues containing the YVY motif at the PAR1 C-tail. Functionally, this binding is required for PAR1-mediated tumor migration and invasion.29 It has also been reported that PAR1-induced tumor-promoting processes depend on the presence of PAR2 by forming PAR1-PAR2 complex.30 Interestingly, BMX-deficient mice do not have an obvious phenotype in the steady state and BMX appears to be dispensable for embryonic and postnatal development in mice.31 However, BMX plays important roles in inflammatory responses. We first reported that BMX in the endothelium and bone marrow cells are required for ischemia/inflammation-induced angiogenesis.32–34 BMX-deficient mice exhibit reduced cardiac hypertrophy induced by Angiotensin II or transverse aortic constriction.35, 36 Furthermore, BMX can be upregulated in blood capillaries and LYVE-1+ lymphatic vessels during endothelial remodeling.32, 37, 38 Importantly, BMX contributes to vascular remodeling in these models. However, the role of BMX in endothelial permeability remains unknown.
In this study, using a mouse model of severe sepsis with ARDS and the “gold standard” technique of CLP,39–41 we demonstrated that BMX regulates endothelial integrity and vascular permeability in early CLP-induced sepsis and thrombin-mediated permeability in ECs and in mice in vivo. Mechanically, BMX represses PAR1 signaling in ECs by promoting PAR1 phosphorylation and internalization. Furthermore, BMX loss-mediated endothelial barrier dysfunction and pulmonary leakage in early CLP-induced sepsis is PAR1 dependent. Thus, BMX functions as an endogenous antagonist of PAR1 in the endothelium to regulate vascular homeostasis and influences the vascular leakage that occurs with sepsis.
METHODS
The authors declare that all supporting data are available within the article or in the online-only Data Supplement or from the corresponding author on request.
Sepsis Model.
BMX-deficient mice were from Dr. Kari Alitalo (University of Helsinki, Finland) under MTA31 and were subsequently bred 6 generations in the C57BL/6 (Jackson Laboratory) background. Mice were cared for in accordance with National Institutes of Health guidelines, and all procedures were approved by the Yale University Animal Care and Use Committee. Both male and female mice that were 8–10 weeks of age were used to select age- and sex-matched WT and BMX-KO mice for experiments. Sepsis was induced in mice by CLP with 19-gauge needle as described39–41. Briefly, age- and weight-matched WT and BMX-KO mice were anesthetized with ketamine (80–100 mg/kg body weight). The cecum was ligated at the designated position which depends on the desired severity grade, and then the ligated cecum was subjected to a single “through and through” puncture with 19-gauge needle. Sham-operated mice underwent the same procedure except for ligation and perforation of the cecum. Paraffin-embedded sections 5 μm in thickness prepared from the lungs were used for hematoxylin and eosin staining (HE), immunohistochemical staining (IHC) and immunofluorescence (IF). Inflammatory cells in the BALF were collected and measured by Complete Blood Count (CBC). Cytokines in the BALF were measured by enzyme-linked immunosorbent assay (ELISA).
Trans-endothelial electrical resistance (TEER) measurement by electric cell-substrate impedance sensing (ECIS) assay.
The barrier function in human umbilical vein ECs (HUVECs) and human pulmonary microvascular ECs (HPMVECs) were assayed by measuring the resistance of cell-covered electrode by using an ECIS instrument (Applied BioPhysics). An 8W10E plate was incubated for 15 min with L-cysteine (10 mM) solution, followed by gelatin 0.1% for 30 min. Cells were plated on the electrode at 7×104 cells per well. After 72 h incubation, ECs were exposed to thrombin, and the resistance was monitored.
Study Design and Statistical Analysis.
Group sizes were determined by an a priori power analysis for a two-tailed, two-sample t-test with an α of 0.05 and power of 0.8, in order to detect a 10% difference in lesion size at the endpoint. Animal were grouped with no blinding but randomized during the experiments. Male and female animals were used in equal numbers for all experiments. No samples or animals were excluded from analysis. All quantifications (survival, permeability, junctional integrity, histology analyses, confocal imaging) were performed in a blind fashion. All figures are representative of at least three experiments unless otherwise noted. Representative figures/images reflected the average level of each experiment. All graphs report mean ± SEM values of biological replicates. Normality of the data (using Shapiro-Wilk test) and the equality of group variance (using Brown-Forsythe test) were performed on all data using SigmaPlot 14. Comparisons between two groups were performed by unpaired, two tailed t-test, between more than two groups by one-way ANOVA or by two-way ANOVA followed by Bonferroni’s post-hoc using Prism 6.0 software (GraphPad). Experiment-wide multiple test correction was not applied. P values were two-tailed and values < 0.05 were considered to indicate statistical significance.
Expanded materials and methods are provided in the Supplemental Materials and include details about the mouse CLP sepsis model, the modified Miles assay, CRISPR/Cas9 knockdown and lentivirus packaging, cell surface, biotinylation and internalization assays, cell resistance measurement, cell culture, in vitro kinase assays, immunoprecipitation and western blot analysis, immunofluorescence microscopy, and statistical analyses.
RESULTS
BMX ablation causes lung injury and inflammation in early sepsis.
To define the role of BMX in sepsis, age- and weight-matched male C57BL/6 (WT) and BMX global knockout (BMX-KO) mice were subjected to the CLP-mediated sepsis model. Assessments of the survival rate indicated that 100% of BMX-KO mice died within 40 h after CLP surgery, whereas only 33% of WT mice died within the same time period and 100% died 52 h post-CLP (Figure 1A). Moreover, we observed edema and hemorrhage in the lungs of WT mice, as indicated by fresh tissue images and the increased lung wet weight at 6 and 12 h post-CLP (Figure 1B–C). BMX-KO mice exhibited accelerated CLP-induced lung injury, as indicated by more severe lung edema and increased lung weight at 6 h post-CLP (Figure 1B–C). We further measured lung vascular permeability using a modified Miles assay and Evans blue dye.42 As shown in Figure 1D with quantifications in 1E, ablation of BMX augmented CLP-induced pulmonary vascular permeability by 3-fold at 6 h post-CLP. We also observed increased edema and permeability as measured by Evans blue dye in the brain, kidney, spleen and liver tissues of BMX-KO mice at 12 h post-CLP compared to WT mice. However, overall responses in these tissues of both WT and BMX-KO were 3-fold (kidney, spleen and liver) and 10-fold (brain) less than the lung tissues (Online Figure I A–E). Similar augmented CLP-induced lung injury and tissue edema were observed in female BMX-mice.
Figure 1. BMX ablation augments lung injury and inflammation during early sepsis.
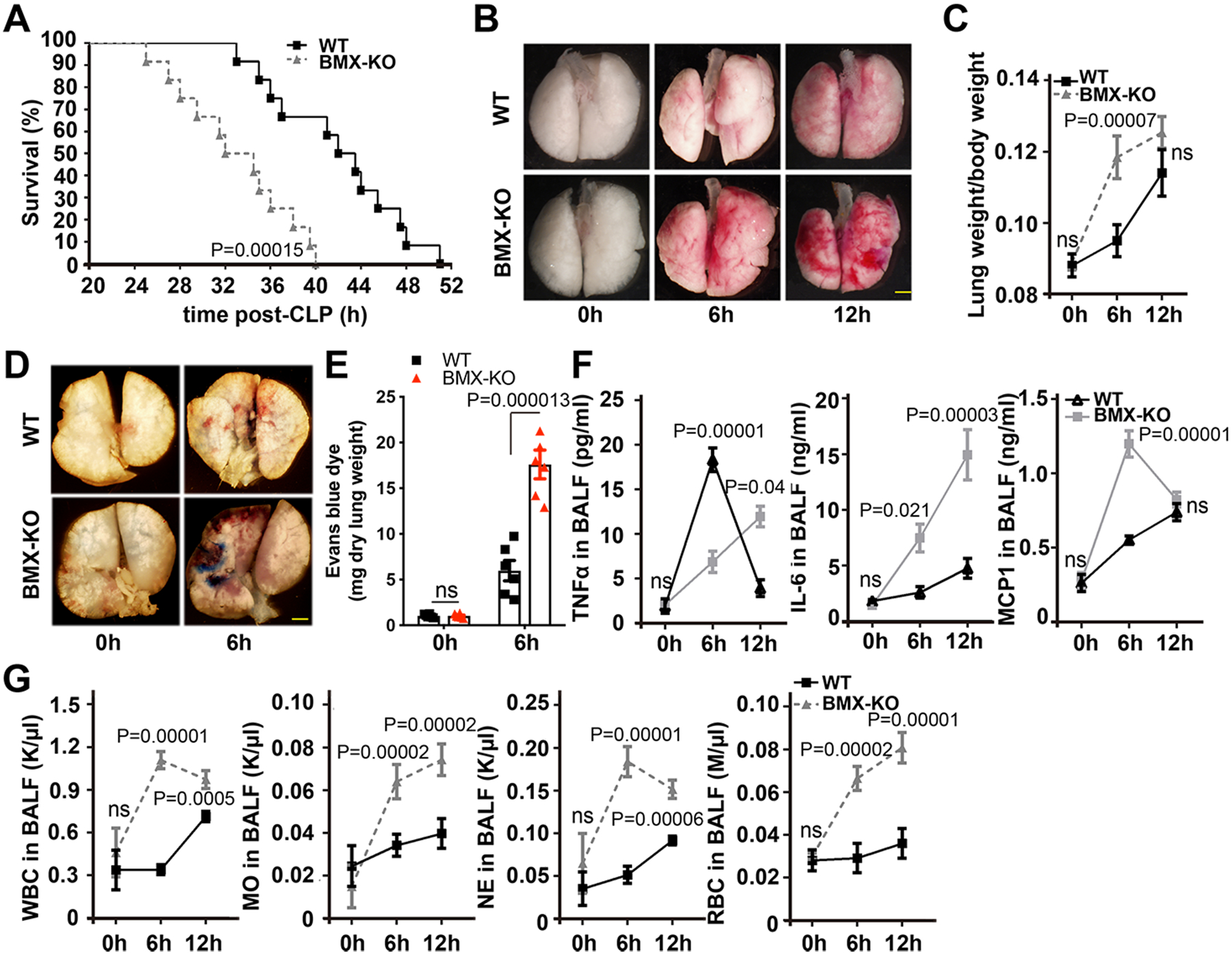
(A–G) WT and BMX-KO mice were subjected to CLP-induced sepsis surgery. (A) Survival analysis post-CLP, with 12 mice in each group. P<0.001, comparison of survival curves between WT and BMX-KO mice using the log-rank (Mantel-Cox) test. (B) Gross morphology of lung tissue. (C) Lung weight per body weight at the indicated times post-CLP. (D) Evans blue dye (30 mg/ml) was injected into mice at 6 h post-CLP, and then lung tissue was harvested for imaging. (E) The data were quantified as the relative fold of Evans blue dye per mg of the dry tissue weight. (F) ELISA for IL-6, TNFα and MCP1 in BALF at the indicated times post-CLP. (G) CBC test for WBCs, neutrophils, monocytes, and RBCs in BALF at the indicated times post-CLP. (B–G) Six mice in each group were quantified. Error bars represent the mean±SEM. ns: non significance; P < 0.05 were considered to indicate statistical significance between WT and BMX-KO mice at various time points using two-way ANOVA and Bonferroni post-hoc multiple comparisons (C, E, F, G). Scale bar: 2 mm (B,D).
CLP typically induces ARDS, as indicated by pulmonary fluid collections. The transudate, which may be sampled by bronchoalveolar lavage fluid (BALF), is initially cell poor but becomes enriched in infiltrated neutrophils and monocytes at later times.43–45 Increased levels of proinflammatory cytokines in local lung tissues are tightly associated with ARDS in sepsis.46 We measured the cytokine levels in BALF by enzyme-linked immunosorbent assay (ELISA). Tumor necrosis factor (TNF)α was detected at a low level in the BALF of WT and BMX-KO mice (15–20 pg/ml) at 6–12 h post-CLP (Figure 1F). However, the levels of monocyte chemoattractant protein-1 (MCP1; also known as chemokine C-C motif ligand 2 CCL2) and interleukin-6 (IL-6) were detected at high levels in BALF of WT after CLP (Figure 1F). Deletion of BMX caused 3-fold increase in MCP-1 at 6 h, followed by a decline at 12 h, and a steady increase of IL-6 at 6–12 h post-CLP (Figure 1F). This finding is consistent with that in clinical samples where the IL-6 level correlates with sepsis symptoms.47 Complete blood count (CBC) analyses indicated increased numbers of infiltrating white blood cells (WBCs), monocytes, neutrophils and red blood cells (RBCs) in the BALF of BMX-KO mice at 6–12 h post-CLP compared with those of WT mice (Figure 1G). Additionally, the numbers of infiltrating WBCs, monocytes and neutrophils in blood were increased by 2-fold in BMX-KO mice at 6 h compared with that in WT mice (Online Figure II). Together, these experiments indicated that BMX loss promotes lung injury and inflammatory response in early sepsis.
Depletion of BMX increases pulmonary vascular permeability during early sepsis.
CLP typically induces a marked increase in alveolar capillary permeability and increased lung weight, and infiltrated cells in the BALF of BMX-KO mice were likely due to an augmented disruption of the alveolar vascular integrity. It is known that paracellular leak through the endothelial adhesion junction and tight junction disruption, as well as dissociations of EC-pericyte interactions, contribute to vascular barrier dysfunction.48 We examined CLP-induced damage to the alveolar vasculature in the early phases of sepsis. Hematoxylin and eosin (H&E) staining indicated that CLP induced ARDS-like changes in the alveolar wall of WT mice, as evident by an enlarged alveolar space and infiltrated cells; these changes were more dramatic in BMX-KO lung tissue (Figure 2A). Sepsis is well known to induce expression of inducible nitric oxide synthase (iNOS) and large amount of peroxynitrite from iNOS can disrupt endothelial barrier function49. We detected 4-fold increases in iNOS expression and peroxynitrite-mediated protein modification nitrotyrosine by immunohistochemistry (IHC) in BMX-KO lung tissues (Figure 2B–E). Next, we examined endothelium integrity by immunostaining to detect the EC adhesion junctional protein vascular endothelial (VE)-cadherin, tight junctional protein Claudin-5 (CLDN5) and pericyte marker NG2. CLP induced disruptions of the continuity of VE-cadherin, CLDN5 and NG2 in alveolar capillaries (Figure 2F, H, J) with a 65% reduction in VE-cadherin, CLDN5 and NG2-coverages (Figure 2G, I, K). Again, these changes were more drastic in BMX-KO mice. These results suggest that BMX loss augments CLP-induced disruptions of the alveolar vascular barrier, leading to enhanced CLP-induced vascular permeability and a sepsis phenotype.
Figure 2. BMX ablation exacerbates pulmonary vascular permeability in early sepsis.
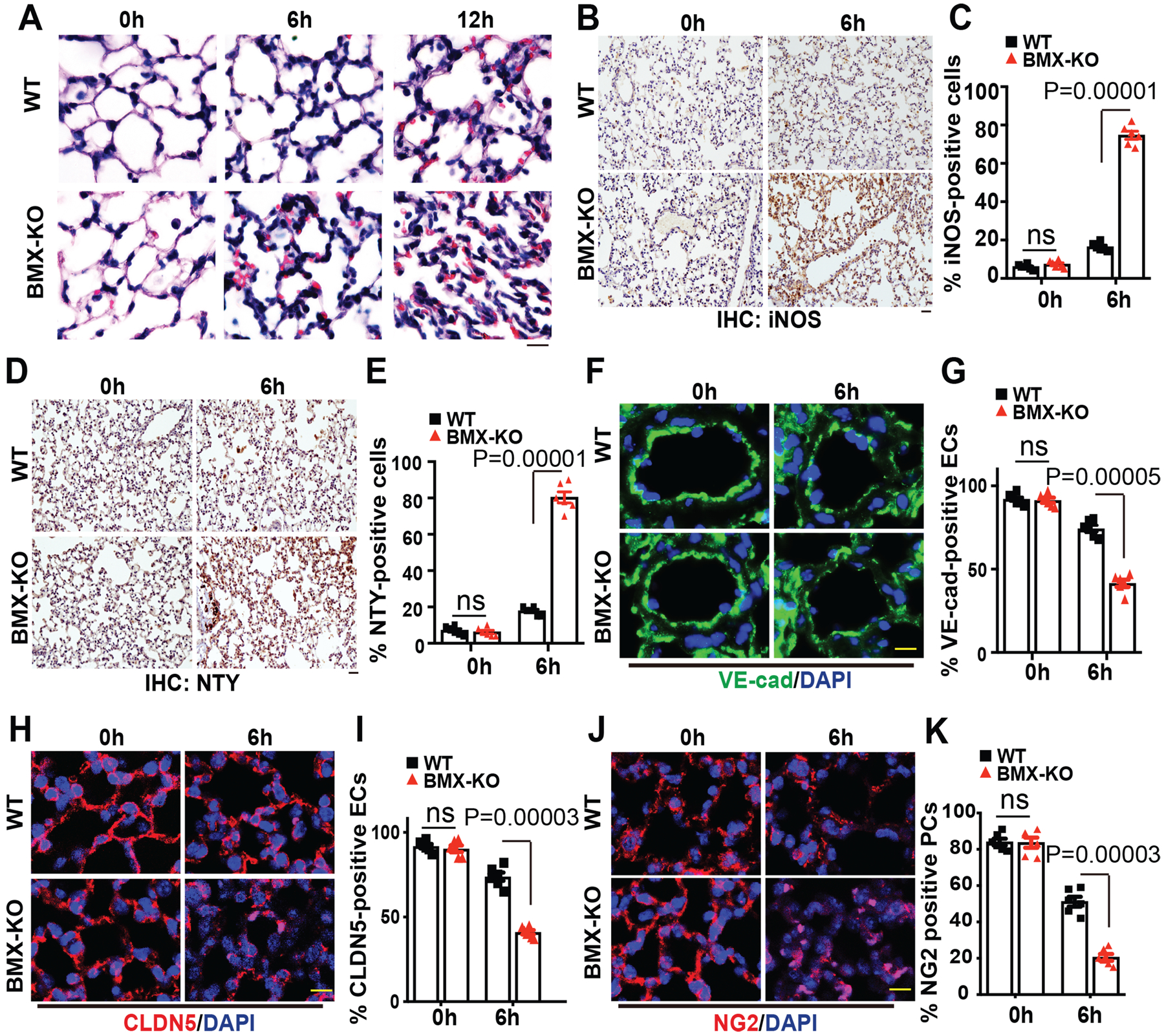
WT and BMX-KO mice were subjected to CLP-induced sepsis surgery. (A) H&E staining of lung tissue after CLP at the indicated times. (B-E) Immunohistochemistry (IHC) for iNOS (B) and nitrotyrosine (D) with quantifications of % positive cells in (C) and (E). (F-K) Immunofluorescence analysis of the EC marker VE-cadherin (F), endothelial adherens junction marker CLDN5 (H) and the pericyte marker NG2 (J) in lung tissue sections after CLP at the indicated times with quantifications of % positive cells in (G), (I) and (J), respectively. Six mice in each group were used and quantified. Error bars represent the mean±SEM. ns: non significance; P < 0.05 were considered to indicate statistical significance between WT and BMX-KO mice at various time points using two-way ANOVA and Bonferroni post-hoc multiple comparisons. Scale bar: 20 μm (A,B,D,F,H,J).
BMX specifically block augments thrombin-mediated permeability both in vivo and in vitro.
To determine whether the dysfunction of inflammatory cells mainly contributes to BMX-mediated lung injury in CLP-induced sepsis, we first tested the expression of BMX in different cell types. BMX is highly expressed in mouse lung microvascular ECs but not in neutrophils, macrophages or platelets (Online Figure III A). Interestingly, we observed that blood clotting was increased in BMX-KO mice after CLP. Consistently, the bleeding time was markedly decreased to 1.6 min in BMX-KO mice compared to 4 min in WT mice at 12 h post-CLP in the tail-bleeding assay (Online Figure III B). The blood platelet count was higher in BMX-KO mice than in WT mice after CLP (Online Figure IIIC). Similarly, the pulmonary deposition of CD41-positive platelets was significantly increased in BMX-KO mice at 6 h post-CLP (Online Figure III D–F). However, we detected no change at the basal and in fact slightly decreased upon CLP in the total levels of prothrombin and thrombin in the serum of BMX-KO compared to WT mice (Online Figure III F). In summary, these data suggest that BMX-KO mice may have augmented vascular permeability during sepsis through endothelial barrier dysfunction rather than leukocyte or platelet dysfunction. To test the idea that BMX potentially regulates thrombin-mediated endothelial permeability, we first determined the effect of BMX on thrombin-mediated vascular leakage in vivo using the modified Miles assay. After the intradermal injection of thrombin, the extravasation of Evans blue dye, as an index of protein leakage into tissue, was significantly increased (>4-fold) in BMX-KO mice compare with that in WT mice (Figure 3A–3B). To determine whether BMX directly regulates thrombin-mediated endothelial permeability, we measured the thrombin-induced change in the trans-endothelial monolayer electrical resistance (TEER) of endothelial cells by the electric cell-substrate impedance sensing (ECIS) assay. As shown in Figure 3C, thrombin treatment in human umbilical vein endothelial cells (HUVECs) caused a sharp reduction in the trans-endothelial monolayer electrical resistance of ECs by about 35% in 15 min, and then this resistance returned to nearly the normal level by 2 h. By contrast, knockdown of BMX by short interfering RNA (siRNA) decreased this electrical resistance by >50% in 15 min and this decreased resistance only recovered to about 70% after 2 h (Figure 3C–D). The delayed recovery in electrical resistance in BMX-siRNA-transfected HUVECs was correlated with a slower recovery in VE-cadherin junctional expression without increasing cell death compared to Scr-siRNA-transfected cells (Figure 3E; Online Figure IV A–B). Phospho-MLC2 functions as a thrombin downstream effector to regulate thrombin-mediated endothelial permeability.12, 13 To address the effect of BMX on thrombin signaling in ECs in vitro, we silenced BMX expression in HUVECs using BMX-siRNA. As shown in Figure 3F, knockdown of BMX markedly increased thrombin-induced phosphor-MLC2 (4-fold at 2 min compared to 2.8-fold at 30 min), indicating that BMX negatively regulates thrombin signaling in vitro. Furthermore, we found that the knockdown of BMX in HUVECs significantly promoted thrombin-mediated secretion of Angiopoietin-2 (Angpt2) and vWF (Figure 3G).50 In contrast to the thrombin response, BMX knockdown had no effects on TNF-induced EC barrier dysfunction while BMX knockdown actually attenuated LPS and VEGF-induced EC permeability by 2-fold, consistent with its effects on the TNF, LPS and VEGF signals (Online Figure V A–D).
Figure 3. BMX ablation augments thrombin-mediated permeability in mice and in cultured ECs.
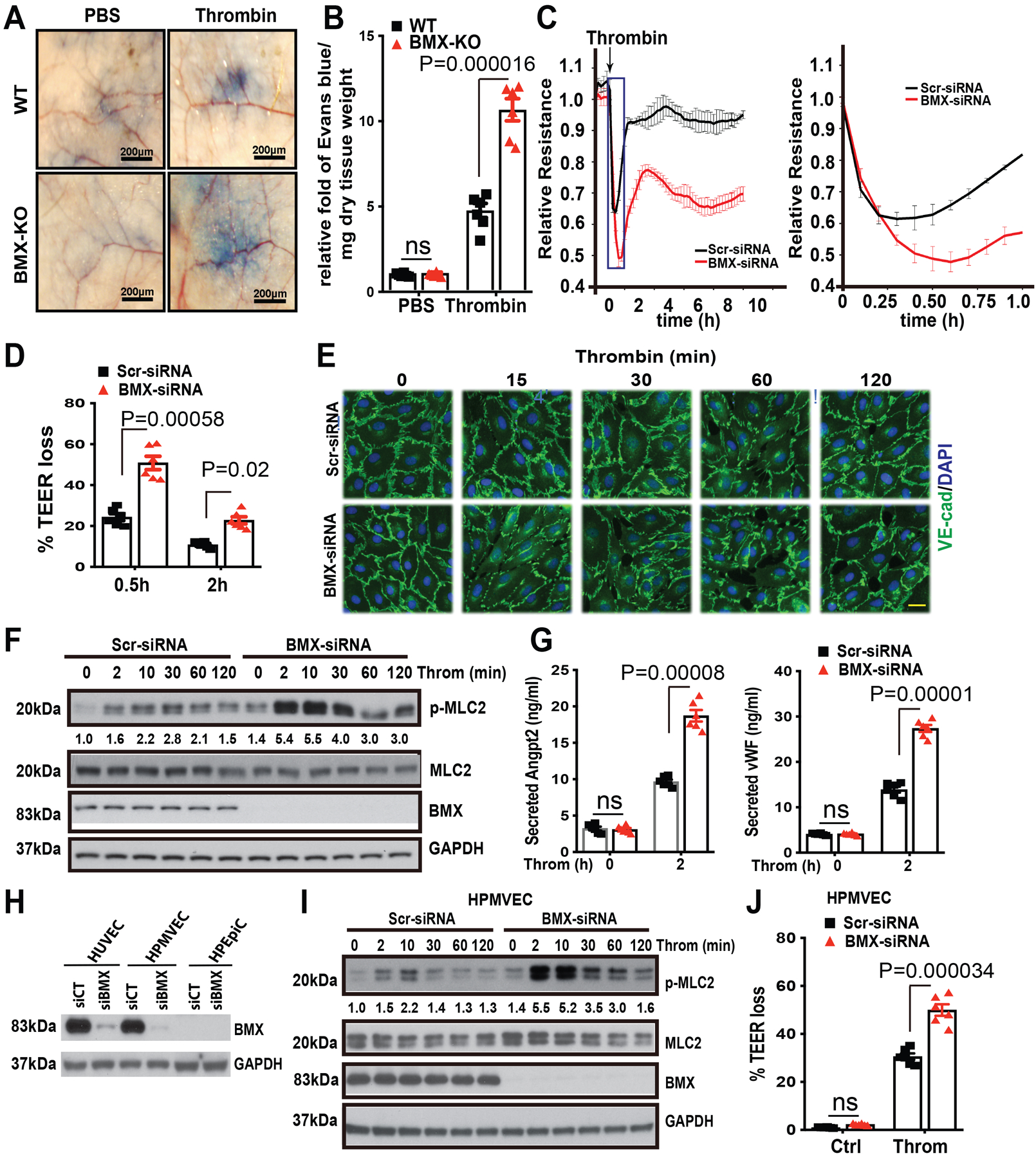
(A) The mouse tail vein was injected with Evans blue dye (30 mg/ml), and then thrombin (20 ng/ml) or phosphate-buffered saline was immediately injected into the ventral skin. (B) The relative fold of Evans blue dye per mg of dry tissue weight was quantified, n=6. (C) Post-confluent HUVECs were treated with thrombin (1.5 U/ml) as shown by the arrow, and then TEER was immediately measured for the indicated times. An expanded 0–1 h window is shown on the right. (D) % TEER losses at indicated times are presented. (E-F) HUVECs were transfected with Scr-siRNA or BMX-siRNA. About 72 h after transfection, HUVECs were starved overnight and then were treated with thrombin (1.5 U/ml) at the indicated times, followed by immunofluorescence analysis of VE-Cadherin (E) or by Western blot for thrombin signaling as indicated (F). (G) After starvation overnight, Scr-siRNA-transfected or BMX-deficient HUVECs were treated with thrombin (1.5 U/ml) for 2 h, and then the medium was collected for ELISA to test secreted Ang2 and vWF levels. (H) HUVECs, HPMVECs and HPEpiC were transfected with Scr-siRNA or BMX-siRNA. BMX and PAR1 protein levels were determined by Western blot with specific antibodies. (I) HPMVECs were transfected with Scr-siRNA or BMX-siRNA. About 72 h after transfection, HPMVECs were starved overnight and then were treated with thrombin (1.5 U/ml) at the indicated times, followed by Western blot for thrombin signaling as indicated. (J) Post-confluent HPMVECs were treated with thrombin (1.5 U/ml) and then TEER was measured. % TEER losses at 0.5 h are presented. All experiments were repeated three times with biological replicates. Error bars indicate SEM. ns: non significance; P < 0.05 were considered to indicate statistical significance between WT and BMX-KO mice (B) or between Scr-siRNA and BMX-siRNA groups (D,G,J) using two-way ANOVA and Bonferroni post-hoc multiple comparisons. Scale bar: 200 μm (A); 20 μm (E).
Since permeability changes in lung epithelial cells may also contribute to alveoli dysfunction, we examined if BMX was expressed in human lung epithelial cells (HPEpiC). We did not detected any BMX protein expression in HPEpiC (Figure 3H). We next verified expression and function of BMX in lung microvascular ECs, a cell type more close to sepsis. We found human pulmonary microvascular ECs (HPMVECs) and HUVECs expressed similar levels of BMX (Figure 3H). Knockdown of BMX also augmented thrombin-induced MLC activation in HPMVEC (4-fold at 2 min compared to 2.2-fold at 10 min; Figure 3I). More important, similar to the observations in HUVECs, BMX deletion sensitized thrombin-induced barrier dysfunction in HPMVECs (Online Figure V E; Figure 3J). These data suggested that the disruption of BMX specifically augments the pro-permeability effect of thrombin in vivo and in vitro potentially by up-regulating thrombin-mediated PAR1 signaling in ECs.
BMX directly phosphorylates PAR1 at the C-terminal Y381/Y381 motif.
PAR1 is the main receptor for thrombin-mediated signaling.12 Our data showed that BMX is essential for thrombin-mediated permeability in ECs. Here, we decided to test whether PAR1 was involved. Because BMX acts as a crucial kinase in many cellular processes,23, 28 we first tested whether BMX phosphorylated PAR1. To this end, we created the BMX-knockout (KO) HUVECs by CRISPR/Cas9 and thrombin-induced PAR1 phosphorylation was determined in control and BMX-KO HUVECs. As shown in Figure 4A–B, thrombin increased PAR1 phosphorylation while BMX deletion abolished it. Conversely, overexpression of BMX-WT by lentivirus in HUVECs increased PAR1 phosphorylation by 4-fold while BMX-KR (kinase active site mutation) attenuated PAR1 phosphorylation by 2-fold compared to vector control (Figure 4C–D), consistent with BMX-KR as a dominant negative form. Importantly, we detected reduced tyrosine phosphorylation of PAR1 in BMX-KO lung tissues in response to CLP injury (Online Figure VI).
Figure 4. BMX directly phosphorylates at the Y381/383 sites.
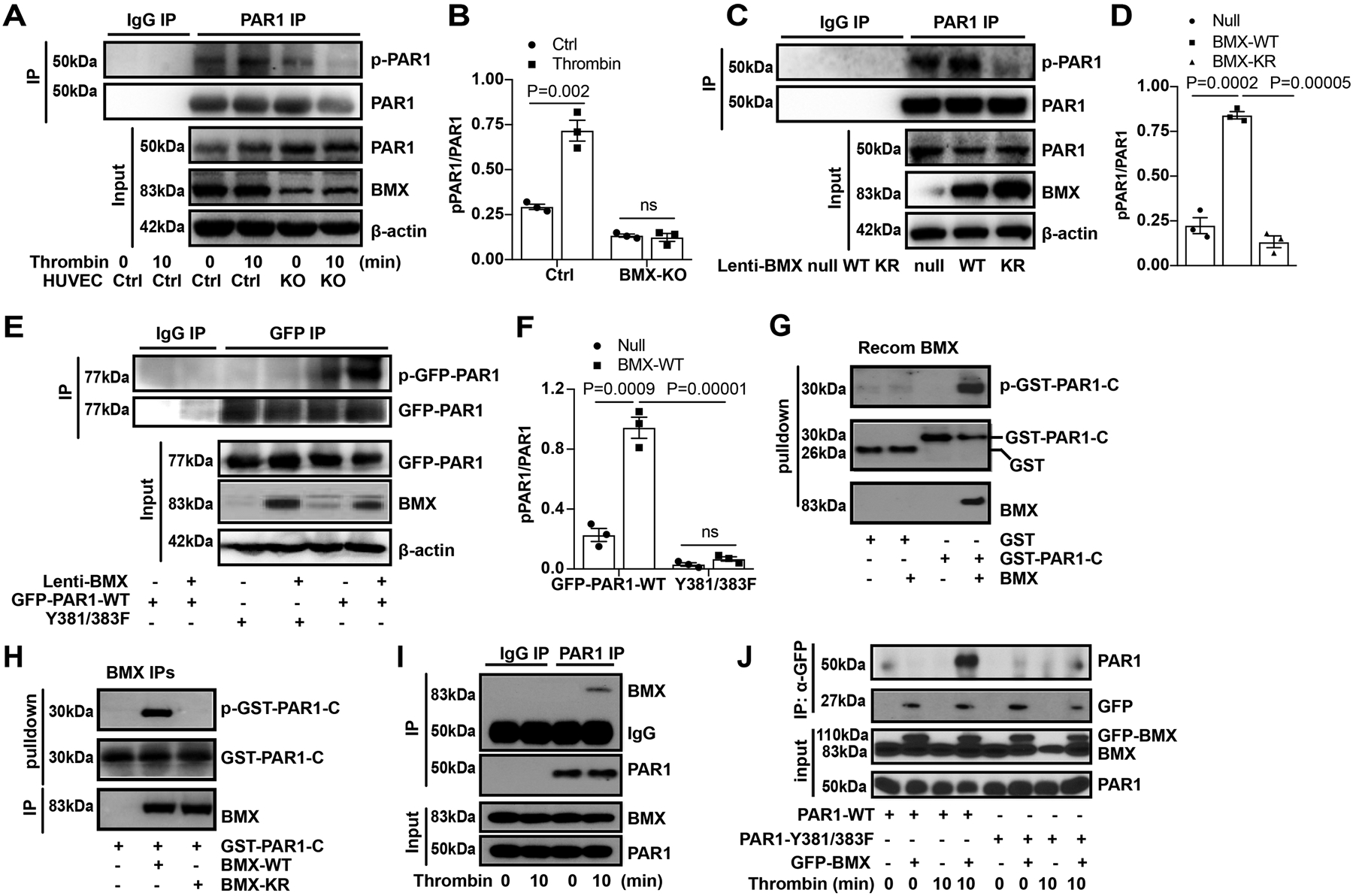
(A-D). Phosphorylation of endogenous PAR1. The endogenous BMX was knocked down by CRISPR/Cas9 in HUVECs. The cells were treated with thrombin (1.5 U/ml) for the indicated times. PAR1 was isolated by immunoprecipitation using PAR1 antibody, and PAR1 phosphorylation was detected by western blot using a p-Y antibody (A with quantification of p-PAR1 in B). BMX-WT or BMX-KR was overexpressed by lentivirus in ECs, and PAR1 phosphorylation was detected (C with quantification of p-PAR1 in D). (E-F). Phosphorylation of PAR1 at Y381/383 sites. Co-expression of BMX-WT with GFP-PAR1-WT or GFP-PAR1-Y381/383F in HUVECs by lentivirus. Exogenous PAR1 was isolated by immunoprecipitation using a GFP antibody, and PAR1 phosphorylation was detected by Western blot using a p-Y antibody (E with quantification in F). (G-H). In vitro kinase assay. Recombinant BMX (G), immunoprecipitates containing BMX-WT or BMX-KR (H) was incubated with GST-PAR1-C in a kinase reaction. The p-PAR1-C, GST-PAR1 and BMX proteins were detected by immunoblotting with specific antibodies. (I). Interactions of endogenous BMX and PAR1 in HUVECs. The HUVECs were starved overnight and then were treated with thrombin (1.5 U/ml) for 10 min. Association of PAR1 with BMX was determined by co-IP with anti-PAR1 followed by Western blot with anti-BMX. (J). The Y381/383 sites on PAR1 is critical for the BMX binding. GFP-BMX and PAR-WT/PAR1-Y381/383F constructs were transfected into 293T cells. At 48 h after transfection, the cells were starved overnight, followed by thrombin treatment (1.5 U/ml). The cells were harvested for IP with a GFP antibody followed by immunoblotting with respective antibodies. All experiments were repeated at least three times. Error bars indicate SEM. ns: non significance; P < 0.05 were considered to indicate statistical significance between indicated groups using one-way ANOVA (D) or two-way ANOVA and Bonferroni post-hoc multiple comparisons (B,F).
To determine the phosphorylation sites on PAR1, we created the PAR1 Y381FY383F mutant because these two sites were predicted phosphorylation sites of BMX on PAR1. As shown in Figure 4E–F, BMX overexpression increased PAR1-WT phosphorylation but had no effect on PAR1 Y381FY383F mutant, indicating that BMX phosphorylated PAR1 at Y381 and Y383. To determine BMX directly phosphorylates PAR1, we performed in vitro kinase assays using recombinant BMX or immunoprecipitated BMX or with purified GST fused to the PAR1 C-terminal tail (375–425 aa) as a substrate. Recombinant BMX phosphorylated GST-PAR1-C but not GST (Figure 4G). Similar to the observations in HUVECs, GST-PAR1-C was phosphorylated by BMX-WT but not by BMX-KR (Figure 4H). Interestingly, we detected GST-PAR1-C, not GST, pulled down BMX (see Figure 4G), suggesting that BMX could bind to the PAR1 at the C-terminal motif. To further confirm the interactions between BMX and PAR1 in cells, endogenous co-immunoprecipitation was performed to determine the interaction between BMX and PAR1 in HUVECs. As shown in Figure 4I, BMX could associate with PAR1 in the presence of thrombin treatment but not at the basal level. Furthermore, domain mapping showed that the mutation of Y381FY383F in the C-terminus of PAR1 markedly decreased its interaction with BMX in vitro (Figure 4J), indicating that the Y381VY383S motif in the C-terminus of PAR1 is critical for PAR1 binding to BMX. In summary, these data showed that BMX binds to the C-terminal domain of PAR1 and directly phosphorylates PAR1 on Y381 and Y381 sites.
BMX promotes PAR1 internalization via phosphorylation of PAR1 without significant effects on PAR1 protein stability.
Given that BMX attenuates thrombin-induced PAR1 signaling, we proposed that BMX could regulate thrombin-mediated PAR1 internalization in ECs. To test the hypothesis, we first measured the internalized and surface PAR1 levels in the presence of thrombin treatment in a biotin assay. BMX knockdown by siRNA decreased the internalized PAR1 protein level by 3-fold and retained the surface PAR1 protein level in HUVECs (Figure 5A and 5B), indicating that BMX regulates thrombin-mediated PAR1 internalization. We determined the internalization of surface PAR1 in the presence of thrombin treatment by immunostaining. As shown in Figure 5C, thrombin treatment caused PAR1 endosome localization in Scr-siRNA-transfected HUVECs, whereas BMX-siRNA retained part of PAR1 on the membrane surface in response to thrombin treatment. Consistent with this finding, pre-treatment with the BMX inhibitor AG879, but not with dimethyl sulfoxide, the ERK inhibitor PD98059 or the Src inhibitor PP2, caused part of PAR1 to localize on the membrane surface in response to thrombin treatment (Figure 5D).
Figure 5. PAR1 phosphorylation by BMX promotes PAR1 internalization.
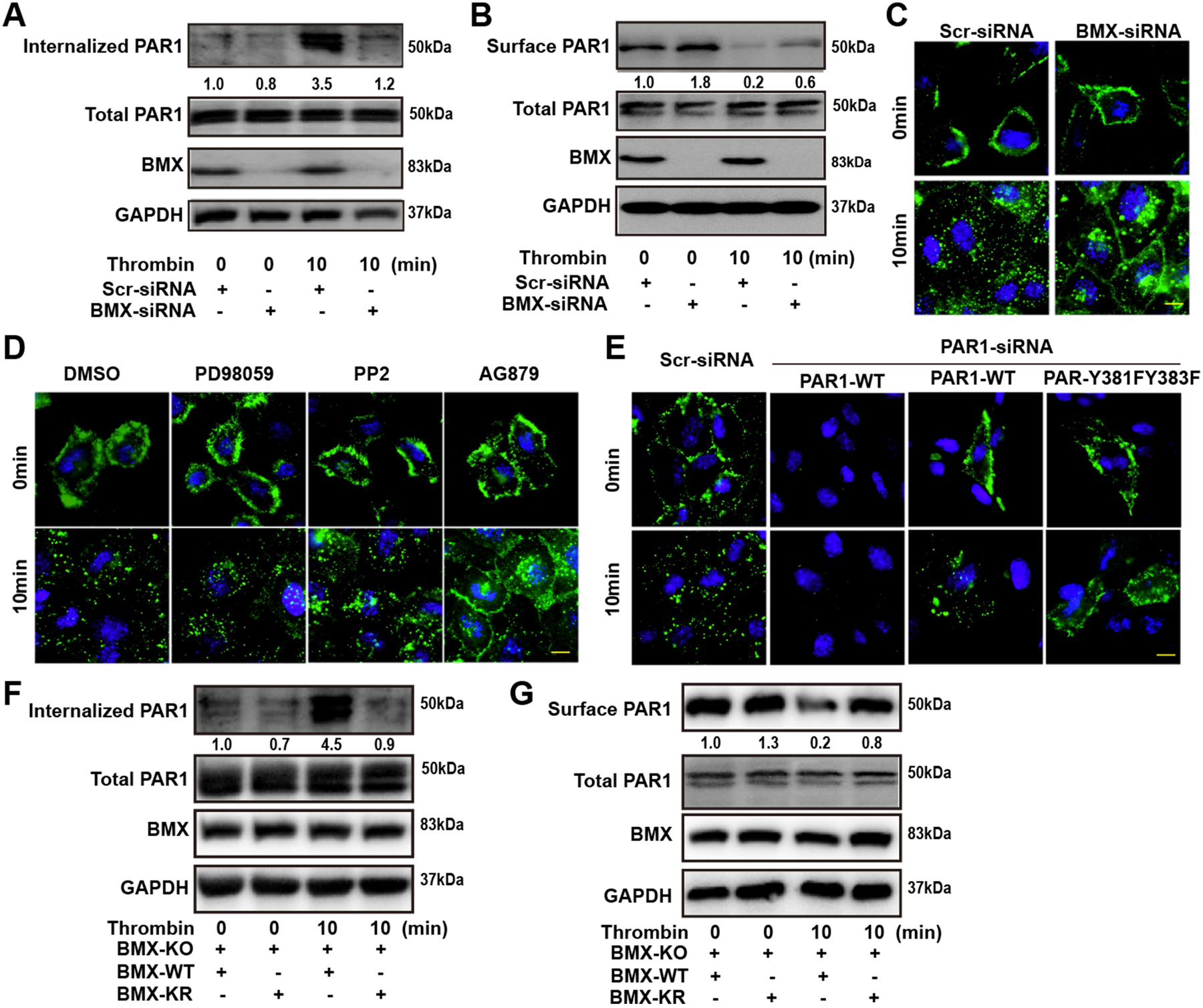
(A) Scr-siRNA-transfected or BMX-deficient HUVECs were biotinylated with sulfo-NHS-SS-biotin at 4°C to label cell surface protein for 1 h. The cells were then treated with thrombin (1.5 U/ml) at 37°C for the indicated times. After internalization, the cells were immediately chilled to stop endocytosis and residual biotin on the cell surface was stripped with a reducing agent. The cells were collected for IP with streptavidin beads. (B) HUVECs transfected by Scr-siRNA or BMX-siRNA were starved overnight, followed by thrombin (1.5 U/ml) treatment at the indicated times. Next, the cells were biotinylated with sulfo-NHS-SS-biotin at 4°C to label cell surface protein for 1 h. After washing, the cells were collected for IP with streptavidin beads. (C) Scr-siRNA-transfected or BMX-deficient HUVECs were starved overnight. (D) HUVECs were starved overnight, followed by treatment with PD98059, PP2 or AG879. (E) HUVECs were transfected with PAR1-siRNA against PAR1–3UTR and then were co-transfected with the PAR-WT or PAR1-Y381FY383F construct for 48 h. After starvation, HUVECs were incubated with the PAR1 antibody at 4°C for 1h, followed by thrombin treatment (1.5 U/ml) at 37°C for the indicated times. Immunofluorescence was performed to analyze antibody-labeled PAR1 (Green) distribution (C–E). The internalized PAR1 (F) and surface PAR1 (G) in BMX-WT or BMX-KR overexpressed BMX-KO HUVECs under thrombin treatment were detected by the biotin label method as described previously. All experiments were repeated at least three times. Scale bar: 5 μm (C,D,E).
To determine if the Y381VY383S motif in the C-terminus of PAR1 is critical for PAR1 internalization, siRNA against the PAR1 3’- untranslated region was transfected into HUVECs, followed by transient transfection with WT-PAR1 or PAR1-Y381FY383F constructs. After starvation, the HUVECs were treated with thrombin for immunofluorescence staining. The PAR1 mutant, but not WT-PAR1, maintained membrane surface localization in response to thrombin treatment (Figure 5E). To further prove that BMX-phosphorylated PAR1 participates in PAR1 internalization, we overexpressed BMX-WT and BMX-KR in BMX-KO ECs. As shown in Figure 5F and 5G, thrombin treatment increased the levels of internalized PAR1 but decreased the levels of surface PAR1 protein in BMX-WT-overexpressed ECs by 4-fold compared with those in BMX-KR-overexpressed ECs, indicating that BMX phosphorylation activity is essential for thrombin-mediated PAR1 internalization. Taken together, these data indicated that BMX promotes PAR1 internalization via phosphorylation.
As we shown previously,51 increased receptor endocytosis is associated with increased protein degradation. To study whether BMX regulated PAR1 protein stability, BMX-KO ECs and control ECs were treated with cycloheximide (CHX) to block translation at the indicated time. Western blot analysis showed no significant difference in the PAR1 protein level between BMX-KO ECs and control ECs (Online Figure VII A–B). We also found that the PAR1 protein level was comparable in BMX-WT- and BMX-KR-overexpressed ECs (Online Figure VII C–D), indicating that BMX-phosphorylated PAR1 was not involved in PAR1 protein stability. Despite inducing PAR1 endocytosis and signaling, thrombin did not significantly affect the PAR1 protein level in control and BMX-KO ECs (Online Figure VII E–F). Taken together, these data suggest that BMX-promoted PAR1 internalization is not associated with its PAR1 protein stability.
Pre-treatment with SCH79797 rescues BMX loss-mediated lung injury during early sepsis.
To determine whether BMX loss-mediated lung injury in early CLP-induced sepsis could be PAR1 dependent, WT and BMX-KO mice were pretreated with the PAR1-selective antagonist SCH79797 every 12 h over 3 days, followed by CLP-mediated sepsis. As shown in Figure 6A, pre-treatment with SCH79797 extended the survival rate of BMX-KO mice from 28 h to 44.5 h post-CLP. We also found that pre-treatment with SCH79797 significantly alleviated BMX-mediated lung injury at 6 h post-CLP (Figure 6B and Online Figure VIII). Moreover, pre-treatment with SCH79797 markedly decreased the numbers of infiltrating WBCs, monocytes, neutrophils, and RBCs within BALF in BMX-KO mice at 6 h post-CLP (Figure 6C). Furthermore, pre-treatment with the PAR1 antagonist SCH79797 significantly protected against the deposition of CD41-positive platelets in the BMX-KO lung tissue at 6 h post-CLP (Figure 6D). Consistent with these data, we also found that the BMX loss-mediated disruption of endothelial integrity was markedly alleviated at 6 h post-CLP under SCH79797 treatment (Figure 6E). Moreover, the number of NG2-positive pericytes at 6 h post-CLP could largely be rescued by pre-treatment with SCH79797 (Figure 6F). To further confirm these results in vitro, we blocked PAR1 by SCH 79797 (Figure 6G–6H) or PAR1 siRNA (Figure 6I–6J) in BMX-KO HUVECs or control HUVECs. The results showed that BMX loss disrupted endothelial integrity, as indicated by immunofluorescence analysis of VE-cadherin (Figure 6G, 6I) and CLDN5 (Figure 6H, 6J). However, blocking PAR1 significantly ameliorated this effect (Figure 6G–6J). Together, these data indicated that pre-treatment with PAR1 antagonist SCH79797 can rescue BMX loss-mediated lung injury in early CLP-induced sepsis.
Figure 6. The PAR inhibitor SCH79797 rescues BMX loss-mediated lung injury during early sepsis.
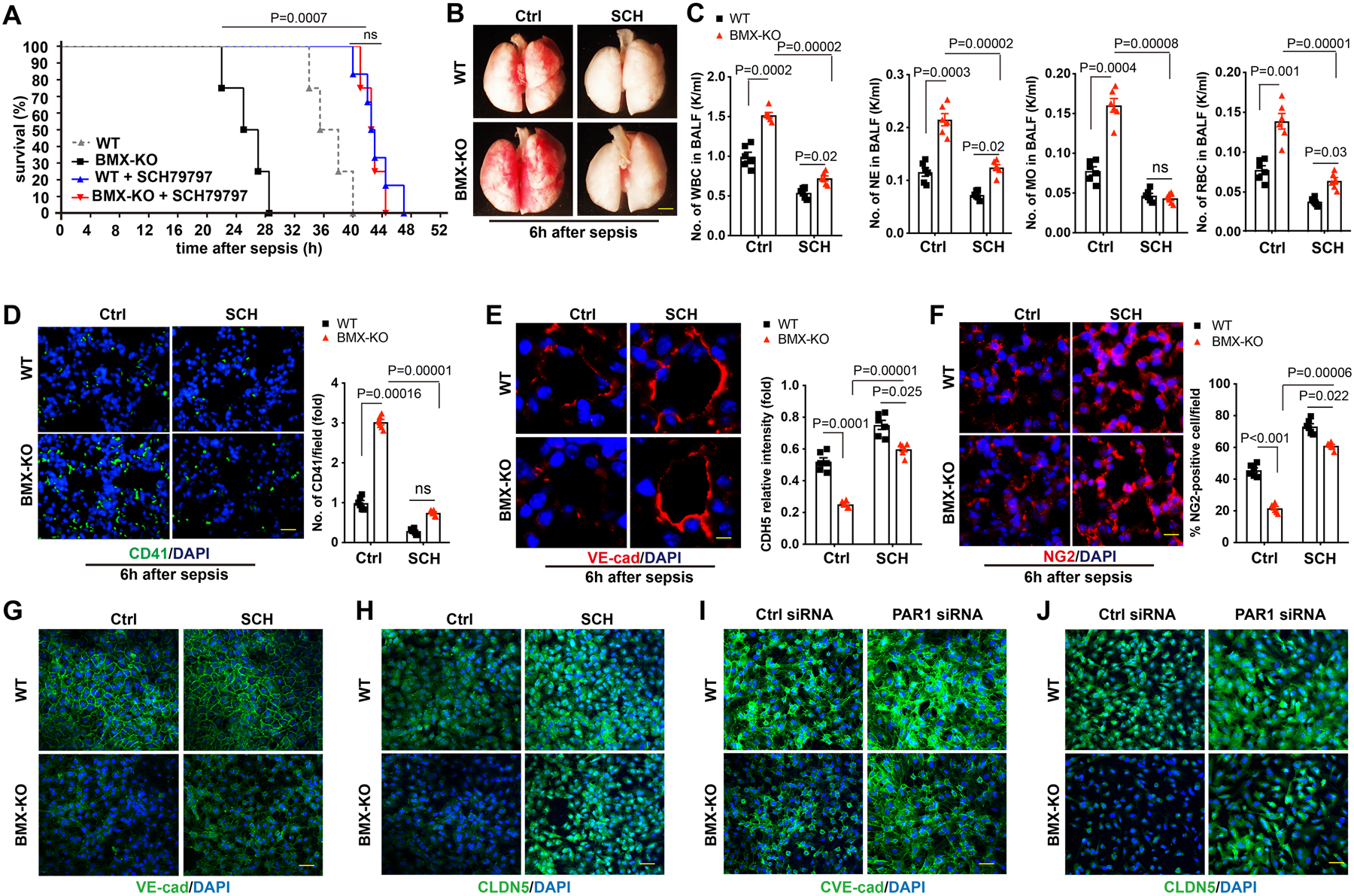
WT and BMX-KO mice were intraperitoneally injected with 100 μM SCH79797 or saline every 12 h over 3 days, followed by sepsis surgery, with six mice in each group. (A) Survival analysis post-CLP. P<0.001 comparison of survival curves between untreated and SCH79797-treated BMX-KO mice whereas ns (P>0.05) between SCH79797-treated WT and SCH79797-treated BMX-KO mice, using the log-rank (Mantel-Cox) test. (B) Gross morphology of lung tissue. (C) CBC test for WBCs, neutrophils, monocytes and RBCs in BALF at 6 h post-CLP. (D) Platelet deposition in the lung tissue at 6 h post-CLP. (E) Quantification of deposited platelets in lung tissue. (F) Immunofluorescence analysis of the endothelial adherens junction marker VE-cadherin in the lung tissue at 6 h post-CLP. (G) Quantification of the VE-cadherin relative intensity per vessel. (H) Immunofluorescence analysis of the pericyte marker NG2 in lung tissue sections at 6 h post-CLP. (I) Quantification of the percent of the NG2-positive pericyte number per field. (G–J) BMX was knocked down by CRISPR/CAS9 in HUVECs. The cells were then treated with SCH79797 (150 nM) (G–H) or were transfected with PAR1 siRNA (20 nM) to block PAR1 for 48 h. Immunofluorescence analysis of VE-cadherin (G, I) and CLDN5 (H, J). All experiments were repeated at least three times. Error bars represent the mean±SEM. Data were analyzed between WT and BMX-KO, and between untreated and SCH79797-treated BMX-KO mice, using 2-way ANOVA and Bonferroni post-hoc multiple comparisons (C,D,E,F). ns, non-significant (P>0.05). Scale bar: 2 mm (B); 20 μm (D,E,F); 50 μm (G,H,I,J).
DISCUSSION
Thrombin generation, thrombin-mediated immunothrombosis, and thrombin-mediated endothelial permeability and alveolar edema play a crucial role in the pathogenesis of sepsis.52 Our data present compelling evidence that BMX protects mice from sepsis-induced lung injury in early CLP-mediated sepsis through suppressing the thrombin pathway. BMX loss in vivo markedly accelerated CLP-induced lung injury, and increased disruption of vascular integrity can be observed in BMX-KO mice in early CLP-induced sepsis. Furthermore, BMX loss caused increased levels of inflammatory cytokines (IL-6 and MCP1) and increased numbers of infiltrating cells from BALF in lung tissue in CLP-induced sepsis. Consequently, we found that sepsis-induced BMX-KO mice died earlier than WT mice after post-CLP.
BMX is not only highly expressed in arteries but also expressed in capillaries and shows inducible expression in the lymphatics as well as in epithelial cells.37, 53 Previously, we have found that BMX-KO mice have reduced inflammatory angiogenesis by regulating and vascular endothelial growth factor receptor signaling.32, 37 Although TNFα plays a crucial role in sepsis,54, 55 our evidence demonstrated that TNFα did not contribute to BMX-mediated endothelial permeability lung injury in early sepsis. First, the TNFα, level from BMX-KO mice was lower than that in WT mice at 6 h post-CLP. Second, knockdown of BMX did not increase TNFα-induced phosphor-nuclear factor (NF)-κB activity and TNFα induced the ICAM-1 protein level. BMX has been found involving in VEGF signaling and acting as both upstream activator and downstream effector of VEGFR2.37 Given VEGFR2 playing crucial role in endothelial permeability in response VEGF stimulation,56, 57 BMX potentially promoted endothelial permeability in response to VEGF. However, BMX knockdown had no effects on TNF-induced EC barrier dysfunction while BMX knockdown actually attenuated LPS and VEGF-induced EC permeability. In this study we proved that BMX loss increased thrombin-mediated permeability in vitro in ECs and in vivo in mice during early sepsis. Moreover, inhibition PAR1 signaling rescued BMX loss-induced endothelial permeability. Further investigation will be need to delineated the function of BMX in endothelial permeability under different stimulations and pathological conditions.
PAR1 is highly expressed in ECs and platelets.12, 17 However, BMX expression is predominantly detectable in ECs but not in platelets, indicating that endothelial BMX specifically regulates PAR1-mediated endothelial barrier dysfunction. To support this idea, we found that the disruption of BMX significantly promotes thrombin-mediated permeability in ECs in vitro and in mice in vivo. Furthermore, BMX-deficient ECs showed a marked increase in the thrombin-mediated p-MLC2 level and thrombin-mediated vWF and Angpt2 secretion from ECs. Consistent with this data, thrombin-activated PAR1 in ECs activated MLC2 signal, and caused an increase in vWF secretion from endothelial cells.12, 13, 17, 58
PAR1 activation on ECs promotes the conversion of these cells into a proinflammatory phenotype, causing increased vascular permeability, increased exposure/secretion of proteins and increased levels of cytokines mediating the local accumulation of platelets and leukocytes.12, 13, 17 These effects contribute to the vascular consequences of sepsis and diseases such as acute lung injury and ARDS.59 Thrombin-activated PAR1 can transactivate PAR2 through the donation of its tethered ligand domain. Interestingly, this transactivation in ECs during the late stage of sepsis switches thrombin signaling from barrier disruptive to barrier protective.60 Here, several lines of evidence strongly support that BMX-mediated lung injury in early sepsis is mainly caused by PAR1 dysfunction. First, BMX depletion markedly increased platelet deposition in lung tissue. Second, pre-treatment with a PAR1 selective antagonist rescued BMX loss-mediated vascular barrier dysfunction and lung injury in early sepsis.
Receptor internalization plays a crucial role in the regulation of PAR1 signaling, which is clathrin and dynamin dependent.19, 61, 62 Here, our data supported that BMX negatively regulates PAR1 signaling in ECs through directly promoting PAR1 phosphorylation and internalization in response to thrombin treatment. First, an endogenous interaction between BMX and PAR1 can be detected in response to thrombin treatment. Second, thrombin treatment increases PAR1 phosphorylation through BMX activity. Interestingly, both thrombin and BMX have no effect on PAR1 protein stability and degradation. Although the Y381VY383SIL tyrosine-based motif in the C-terminus of PAR1 has been reported to regulate thrombin-triggered PAR1 internalization,63 the mechanism is not clear. Here, we first discovered that BMX phosphorylated PAR1 at Y381 and Y383 through binding to this Y381VY383SIL tyrosine-based motif of the PAR1 C-tail. Given that the PAR1 C-tail plays a pivotal role in its internalization,19 we supposed that BMX negatively regulated the PAR1 signal by PAR1 phosphorylation and internalization. Knockdown of BMX decreased thrombin-mediated PAR1 internalization. Consistent with this finding, treatment with the BMX/ETK inhibitor AG879 treatment retained partial PAR1 surface localization in response to thrombin treatment. Furthermore, BMX-mediated PAR1 internalization could occur in connection with the Y381VY383SIL tyrosine-based motif of PAR1. The PAR1- Y381FY383F mutant mimicked BMX loss-mediated thrombin-induced PAR1 internalization. Finally, overexpression of BMX-WT, rather than overexpression of BMX-KR, in BMX-deficient ECs promoted thrombin-mediated PAR1 internalization. Consistent with these data, the suggested mechanism constitutes a novel regulator of BMX for PAR1 phosphorylation and internalization that depends on the Y381VY383SIL tyrosine-based motif in the PAR1 C-tail and that suppresses the PAR1 signal, endothelial permeability, and lung injury in early sepsis (Figure 7: A model for the role of BMX in thrombin-PAR1 signaling). Although our in vitro data suggest that BMX-promoted PAR1 internalization is not associated with its PAR1 protein stability, we notice that PAR1 is degraded in lung tissues in response to CLP injury (see Online Figure VI). It needs further investigation to determine if increased proinflammatory cytokines during sepsis induce PAR1 degradation.
Figure 7. A model for the role of BMX as a novel negative regulator in thrombin-PAR1-mediated vascular permeability and sepsis.
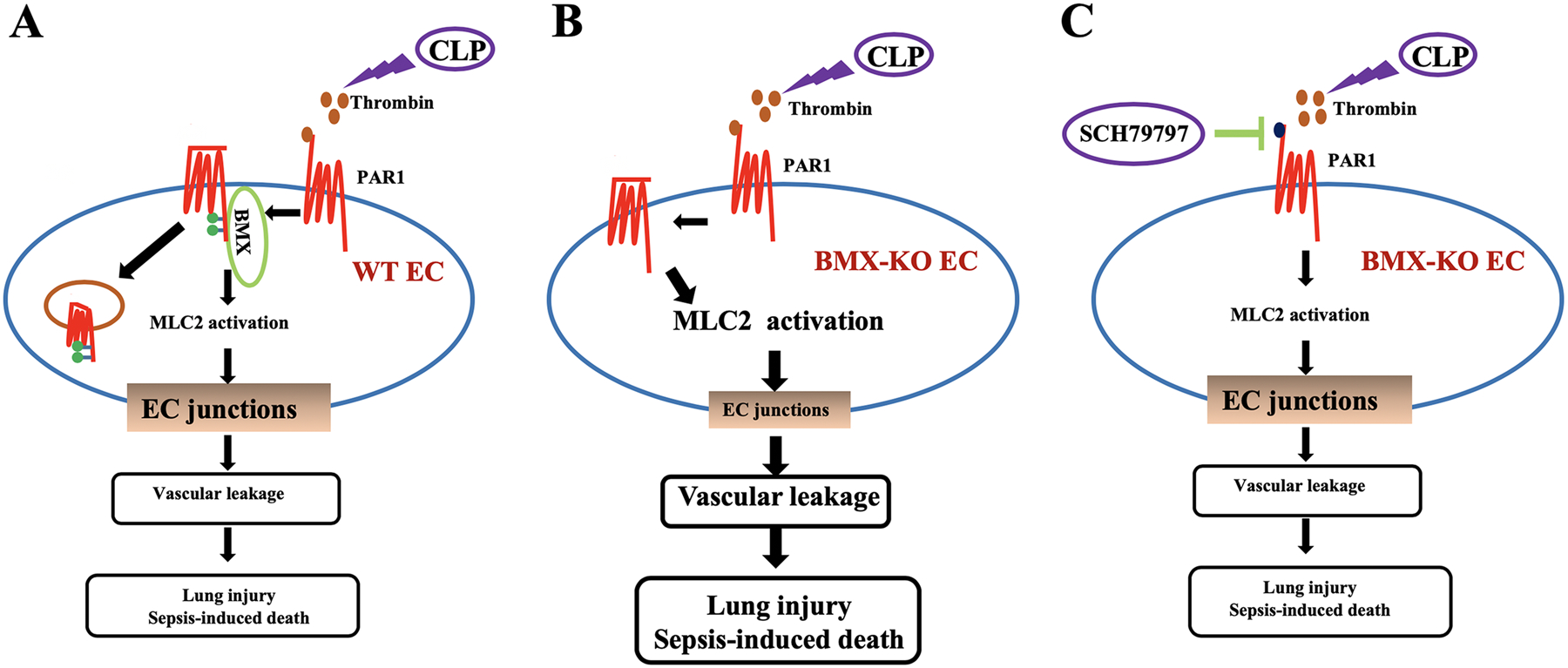
CLP causes thrombin deposition in lung vasculature, and, in turn, PAR1 is activated on the endothelial membrane and promotes vascular permeability during sepsis. (A) Endothelial BMX increases PAR1 phosphorylation and internalization to attenuate PAR1 signaling. (B) BMX loss exacerbates CLP-induced endothelial permeability, lung injury and sepsis. (C) However, the PAR1 selective antagonist SCH79797 ameliorates these effects, providing a therapeutic approach for the treatment of sepsis.
In summary, this work illustrates that BMX represses lung injury in CLP-induced sepsis in a PAR1-dependent manner and that BMX antagonizes thrombin-mediated permeability in ECs in vitro and in mice in vivo through regulating PAR1 phosphorylation and internalization. The identification of BMX as a novel antagonist of PAR1 in the endothelium provides a new therapeutic target for PAR1-mediated vascular disease.
Supplementary Material
NOVELTY AND SIGNIFICANCE.
What Is Known?
In sepsis, thrombin-cleaved PAR1 activation increases endothelial cell (EC) permeability in lung, causing diffuse alveolar damage and associated acute respiratory distress syndrome (ARDS).
Among the multiple regulatory mechanisms, PAR1 internalization is crucial for the regulation of PAR1 signaling.
BMX is highly expressed in the arterial endothelium and is also expressed in microvessels in response to pathological stimuli. However, its role in endothelial permeability and sepsis is unknown.
What New Information Does This Article Contribute?
Using a mouse cecal ligation and puncture (CLP) model of severe sepsis with ARDS, we demonstrated that BMX regulates endothelial integrity and vascular permeability in early CLP-induced sepsis.
BMX loss-mediated endothelial barrier dysfunction and pulmonary leakage in early CLP-induced sepsis is PAR1 dependent.
BMX represses PAR1 signaling in ECs by promoting PAR1 phosphorylation and internalization followed by signal inactivation.
SOURCES OF FUNDING
This work was partly supported by NIH grants HL109420 and HL115148 (WM), National Career Development Award from American Heart Association 19CDA34760284 (HJZ). This work was also supported by National Key Research and Development Program of China (2016YFC1300600), National Natural Science Foundation of China U1601219, 8197042 and 81900234, the postdoctoral innovative talent support program BX20180393, and the China Postdoctoral Science Foundation grant 2018M640859 (Zhao Li).
Nonstandard Abbreviations and Acronyms:
- Angpt2
angiopoietin-2
- ARDS
acute respiratory distress syndrome
- BALF
bronchoalveolar lavage fluid
- BMX
Bone marrow kinase on the X chromosome
- CBC
Complete blood count
- CLDN5
Claudin-5
- CLP
cecal ligation and puncture
- CHX
cycloheximide
- ELISA
enzyme-linked immunosorbent assay
- EC
endothelial cell
- ECIS
electric cell-substrate impedance sensing assay
- HE
hematoxylin and eosin staining
- HUVEC
human umbilical vein endothelial cells
- ICAM-1
intercellular adhesion molecule-1
- IHC
immunohistochemistry
- iNOS
inducible nitric oxide synthase
- PAR1
protease-activated receptor
- TEER
trans-endothelial monolayer electrical resistance
Footnotes
DISCLOSURES
The authors confirm that there are no conflicts of interest.
REFERENCES
- 1.Rhee C, Dantes R, Epstein L, Murphy DJ, Seymour CW, Iwashyna TJ, Kadri SS, Angus DC, Danner RL, Fiore AE, Jernigan JA, Martin GS, Septimus E, Warren DK, Karcz A, Chan C, Menchaca JT, Wang R, Gruber S, Klompas M and Program CDCPE. Incidence and Trends of Sepsis in US Hospitals Using Clinical vs Claims Data, 2009–2014. JAMA. 2017;318:1241–1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hershey TB and Kahn JM. State Sepsis Mandates - A New Era for Regulation of Hospital Quality. The New England journal of medicine. 2017;376:2311–2313. [DOI] [PubMed] [Google Scholar]
- 3.Hotchkiss RS, Moldawer LL, Opal SM, Reinhart K, Turnbull IR and Vincent JL. Sepsis and septic shock. Nature reviews Disease primers. 2016;2:16045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ranieri VM, Rubenfeld GD, Thompson BT, Ferguson ND, Caldwell E, Fan E, Camporota L and Slutsky AS. Acute respiratory distress syndrome: the Berlin Definition. Jama. 2012;307:2526–33. [DOI] [PubMed] [Google Scholar]
- 5.Zhang Z, Wu Z, Xu Y, Lu D and Zhang S. Vascular endothelial growth factor increased the permeability of respiratory barrier in acute respiratory distress syndrome model in mice. Biomedicine & pharmacotherapy = Biomedecine & pharmacotherapie. 2019;109:2434–2440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ince C, Mayeux PR, Nguyen T, Gomez H, Kellum JA, Ospina-Tascón GA, Hernandez G, Murray P, De Backer D and Workgroup AX. THE ENDOTHELIUM IN SEPSIS. Shock. 2016;45:259–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Herrero R, Sanchez G and Lorente JA. New insights into the mechanisms of pulmonary edema in acute lung injury. Ann Transl Med. 2018;6:32–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schouten M, Wiersinga WJ, Levi M and van der Poll T. Inflammation, endothelium, and coagulation in sepsis. Journal of leukocyte biology. 2008;83:536–45. [DOI] [PubMed] [Google Scholar]
- 9.Vincent JL. Microvascular endothelial dysfunction: a renewed appreciation of sepsis pathophysiology. Crit Care. 2001;5:S1–S5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hoppensteadt D, Tsuruta K, Cunanan J, Hirman J, Kaul I, Osawa Y and Fareed J. Thrombin generation mediators and markers in sepsis-associated coagulopathy and their modulation by recombinant thrombomodulin. Clin Appl Thromb Hemost. 2014;20:129–35. [DOI] [PubMed] [Google Scholar]
- 11.Petros S, Kliem P, Siegemund T and Siegemund R. Thrombin generation in severe sepsis. Thromb Res. 2012;129:797–800. [DOI] [PubMed] [Google Scholar]
- 12.Coughlin SR. Thrombin signalling and protease-activated receptors. Nature. 2000;407:258–64. [DOI] [PubMed] [Google Scholar]
- 13.Coughlin SR. Protease-activated receptors in hemostasis, thrombosis and vascular biology. Journal of thrombosis and haemostasis: JTH. 2005;3:1800–14. [DOI] [PubMed] [Google Scholar]
- 14.Malik AB and Fenton JW 2nd. Thrombin-mediated increase in vascular endothelial permeability. Seminars in thrombosis and hemostasis. 1992;18:193–9. [DOI] [PubMed] [Google Scholar]
- 15.De Mey JG, Claeys M and Vanhoutte PM. Endothelium-dependent inhibitory effects of acetylcholine, adenosine triphosphate, thrombin and arachidonic acid in the canine femoral artery. The Journal of pharmacology and experimental therapeutics. 1982;222:166–73. [PubMed] [Google Scholar]
- 16.Cleator JH and Vaughan DE. Clinical implications of the contrasting effects of in vivo thrombin receptor activation (protease-activated receptor type 1) on the human vasculature. Journal of the American College of Cardiology. 2008;51:1757–9. [DOI] [PubMed] [Google Scholar]
- 17.Kataoka H, Hamilton JR, McKemy DD, Camerer E, Zheng YW, Cheng A, Griffin C and Coughlin SR. Protease-activated receptors 1 and 4 mediate thrombin signaling in endothelial cells. Blood. 2003;102:3224–31. [DOI] [PubMed] [Google Scholar]
- 18.Vu TK, Hung DT, Wheaton VI and Coughlin SR. Molecular cloning of a functional thrombin receptor reveals a novel proteolytic mechanism of receptor activation. Cell. 1991;64:1057–68. [DOI] [PubMed] [Google Scholar]
- 19.Paing MM, Johnston CA, Siderovski DP and Trejo J. Clathrin adaptor AP2 regulates thrombin receptor constitutive internalization and endothelial cell resensitization. Mol Cell Biol. 2006;26:3231–3242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen B, Dores MR, Grimsey N, Canto I, Barker BL and Trejo J. Adaptor protein complex-2 (AP-2) and epsin-1 mediate protease-activated receptor-1 internalization via phosphorylation- and ubiquitination-dependent sorting signals. The Journal of biological chemistry. 2011;286:40760–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen CH, Paing MM and Trejo J. Termination of protease-activated receptor-1 signaling by beta-arrestins is independent of receptor phosphorylation. The Journal of biological chemistry. 2004;279:10020–31. [DOI] [PubMed] [Google Scholar]
- 22.Ekman N, Lymboussaki A, Vastrik I, Sarvas K, Kaipainen A and Alitalo K. Bmx tyrosine kinase is specifically expressed in the endocardium and the endothelium of large arteries. Circulation. 1997;96:1729–32. [DOI] [PubMed] [Google Scholar]
- 23.Cenni B, Gutmann S and Gottar-Guillier M. BMX and its role in inflammation, cardiovascular disease, and cancer. International reviews of immunology. 2012;31:166–73. [DOI] [PubMed] [Google Scholar]
- 24.Pan S, An P, Zhang R, He X, Yin G and Min W. Etk/Bmx as a tumor necrosis factor receptor type 2-specific kinase: role in endothelial cell migration and angiogenesis. Mol Cell Biol. 2002;22:7512–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang R, Xu Y, Ekman N, Wu Z, Wu J, Alitalo K and Min W. Etk/Bmx transactivates vascular endothelial growth factor 2 and recruits phosphatidylinositol 3-kinase to mediate the tumor necrosis factor-induced angiogenic pathway. J Biol Chem. 2003;278:51267–76. [DOI] [PubMed] [Google Scholar]
- 26.Chau CH, Chen KY, Deng HT, Kim KJ, Hosoya K, Terasaki T, Shih HM and Ann DK. Coordinating Etk/Bmx activation and VEGF upregulation to promote cell survival and proliferation. Oncogene. 2002;21:8817–29. [DOI] [PubMed] [Google Scholar]
- 27.Tamagnone L, Lahtinen I, Mustonen T, Virtaneva K, Francis F, Muscatelli F, Alitalo R, Smith CI, Larsson C and Alitalo K. BMX, a novel nonreceptor tyrosine kinase gene of the BTK/ITK/TEC/TXK family located in chromosome Xp22.2. Oncogene. 1994;9:3683–8. [PubMed] [Google Scholar]
- 28.Qiu Y and Kung HJ. Signaling network of the Btk family kinases. Oncogene. 2000;19:5651–61. [DOI] [PubMed] [Google Scholar]
- 29.Cohen I, Maoz M, Turm H, Grisaru-Granovsky S, Maly B, Uziely B, Weiss E, Abramovitch R, Gross E, Barzilay O, Qiu Y and Bar-Shavit R. Etk/Bmx regulates proteinase-activated-receptor1 (PAR1) in breast cancer invasion: signaling partners, hierarchy and physiological significance. PloS one. 2010;5:e11135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jaber M, Maoz M, Kancharla A, Agranovich D, Peretz T, Grisaru-Granovsky S, Uziely B and Bar-Shavit R. Protease-activated-receptor-2 affects protease-activated-receptor-1-driven breast cancer. Cellular and molecular life sciences: CMLS. 2014;71:2517–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rajantie I, Ekman N, Iljin K, Arighi E, Gunji Y, Kaukonen J, Palotie A, Dewerchin M, Carmeliet P and Alitalo K. Bmx tyrosine kinase has a redundant function downstream of angiopoietin and vascular endothelial growth factor receptors in arterial endothelium. Mol Cell Biol. 2001;21:4647–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.He Y, Luo Y, Tang S, Rajantie I, Salven P, Heil M, Zhang R, Luo D, Li X, Chi H, Yu J, Carmeliet P, Schaper W, Sinusas AJ, Sessa WC, Alitalo K and Min W. Critical function of Bmx/Etk in ischemia-mediated arteriogenesis and angiogenesis. J Clin Invest. 2006;116:2344–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Luo D, Luo Y, He Y, Zhang H, Zhang R, Li X, Dobrucki WL, Sinusas AJ, Sessa WC and Min W. Differential functions of tumor necrosis factor receptor 1 and 2 signaling in ischemia-mediated arteriogenesis and angiogenesis. Am J Pathol. 2006;169:1886–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Luo Y, Xu Z, Wan T, He Y, Jones D, Zhang H and Min W. Endothelial-specific transgenesis of TNFR2 promotes adaptive arteriogenesis and angiogenesis. Arteriosclerosis, thrombosis, and vascular biology. 2010;30:1307–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Holopainen T, Räsänen M, Anisimov A, Tuomainen T, Zheng W, Tvorogov D, Hulmi JJ, Andersson LC, Cenni B, Tavi P, Mervaala E, Kivelä R and Alitalo K. Endothelial Bmx tyrosine kinase activity is essential for myocardial hypertrophy and remodeling. Proc Natl Acad Sci U S A. 2015;112:13063–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mitchell-Jordan SA, Holopainen T, Ren S, Wang S, Warburton S, Zhang MJ, Alitalo K, Wang Y and Vondriska TM. Loss of Bmx nonreceptor tyrosine kinase prevents pressure overload-induced cardiac hypertrophy. Circulation research. 2008;103:1359–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jones D, Xu Z, Zhang H, He Y, Kluger MS, Chen H and Min W. Functional analyses of the bone marrow kinase in the X chromosome in vascular endothelial growth factor-induced lymphangiogenesis. Arteriosclerosis, thrombosis, and vascular biology. 2010;30:2553–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Holopainen T, Lopez-Alpuche V, Zheng W, Heljasvaara R, Jones D, He Y, Tvorogov D, D’Amico G, Wiener Z, Andersson LC, Pihlajaniemi T, Min W and Alitalo K. Deletion of the endothelial Bmx tyrosine kinase decreases tumor angiogenesis and growth. Cancer Res. 2012;72:3512–21. [DOI] [PubMed] [Google Scholar]
- 39.Dejager L, Pinheiro I, Dejonckheere E and Libert C. Cecal ligation and puncture: the gold standard model for polymicrobial sepsis? Trends in microbiology. 2011;19:198–208. [DOI] [PubMed] [Google Scholar]
- 40.Remick DG, Newcomb DE, Bolgos GL and Call DR. Comparison of the mortality and inflammatory response of two models of sepsis: lipopolysaccharide vs. cecal ligation and puncture. Shock. 2000;13:110–6. [DOI] [PubMed] [Google Scholar]
- 41.Rittirsch D, Huber-Lang MS, Flierl MA and Ward PA. Immunodesign of experimental sepsis by cecal ligation and puncture. Nature protocols. 2009;4:31–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Radu M and Chernoff J. An in vivo assay to test blood vessel permeability. Journal of visualized experiments: JoVE. 2013:e50062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.de Torre C, Ying SX, Munson PJ, Meduri GU and Suffredini AF. Proteomic analysis of inflammatory biomarkers in bronchoalveolar lavage. Proteomics. 2006;6:3949–57. [DOI] [PubMed] [Google Scholar]
- 44.Czermak BJ, Breckwoldt M, Ravage ZB, Huber-Lang M, Schmal H, Bless NM, Friedl HP and Ward PA. Mechanisms of enhanced lung injury during sepsis. The American journal of pathology. 1999;154:1057–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.DiStasi MR and Ley K. Opening the flood-gates: how neutrophil-endothelial interactions regulate permeability. Trends in immunology. 2009;30:547–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Englert JA, Bobba C and Baron RM. Integrating molecular pathogenesis and clinical translation in sepsis-induced acute respiratory distress syndrome. JCI insight. 2019;4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Damas P, Ledoux D, Nys M, Vrindts Y, De Groote D, Franchimont P and Lamy M. Cytokine serum level during severe sepsis in human IL-6 as a marker of severity. Annals of surgery. 1992;215:356–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cerutti C and Ridley AJ. Endothelial cell-cell adhesion and signaling. Exp Cell Res. 2017;358:31–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Filep JG, Delalandre A and Beauchamp M. Dual role for nitric oxide in the regulation of plasma volume and albumin escape during endotoxin shock in conscious rats. Circ Res. 1997;81:840–7. [DOI] [PubMed] [Google Scholar]
- 50.Tsangaris I, Tsantes A, Vrigkou E, Kopterides P, Pelekanou A, Zerva K, Antonakos G, Konstantonis D, Mavrou I, Tsaknis G, Kyriazopoulou E, Mouktaroudi M, Kokori S, Orfanos SE, Giamarellos-Bourboulis EJ and Armaganidis A. Angiopoietin-2 Levels as Predictors of Outcome in Mechanically Ventilated Patients with Acute Respiratory Distress Syndrome. Dis Markers. 2017;2017:6758721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pasula S, Cai X, Dong Y, Messa M, McManus J, Chang B, Liu X, Zhu H, Mansat RS, Yoon SJ, Hahn S, Keeling J, Saunders D, Ko G, Knight J, Newton G, Luscinskas F, Sun X, Towner R, Lupu F, Xia L, Cremona O, De Camilli P, Min W and Chen H. Endothelial epsin deficiency decreases tumor growth by enhancing VEGF signaling. J Clin Invest. 2012;122:4424–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Frantzeskaki F, Armaganidis A and Orfanos SE. Immunothrombosis in Acute Respiratory Distress Syndrome: Cross Talks between Inflammation and Coagulation. Respiration. 2017;93:212–225. [DOI] [PubMed] [Google Scholar]
- 53.Xu XF, Wang JJ, Ding L, Ye JS, Huang LJ, Tao L, Gao F and Ji Y. Suppression of BMX-ARHGAP fusion gene inhibits epithelial-mesenchymal transition in gastric cancer cells via RhoA-mediated blockade of JAK/STAT axis. Journal of cellular biochemistry. 2019;120:439–451. [DOI] [PubMed] [Google Scholar]
- 54.Lv S, Han M, Yi R, Kwon S, Dai C and Wang R. Anti-TNF-alpha therapy for patients with sepsis: a systematic meta-analysis. International journal of clinical practice. 2014;68:520–8. [DOI] [PubMed] [Google Scholar]
- 55.Gordon AC, Lagan AL, Aganna E, Cheung L, Peters CJ, McDermott MF, Millo JL, Welsh KI, Holloway P, Hitman GA, Piper RD, Garrard CS and Hinds CJ. TNF and TNFR polymorphisms in severe sepsis and septic shock: a prospective multicentre study. Genes and immunity. 2004;5:631–40. [DOI] [PubMed] [Google Scholar]
- 56.Weis S, Shintani S, Weber A, Kirchmair R, Wood M, Cravens A, McSharry H, Iwakura A, Yoon YS, Himes N, Burstein D, Doukas J, Soll R, Losordo D and Cheresh D. Src blockade stabilizes a Flk/cadherin complex, reducing edema and tissue injury following myocardial infarction. J Clin Invest. 2004;113:885–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Weis SM and Cheresh DA. Pathophysiological consequences of VEGF-induced vascular permeability. Nature. 2005;437:497–504. [DOI] [PubMed] [Google Scholar]
- 58.Grimsey NJ and Trejo J. Integration of endothelial protease-activated receptor-1 inflammatory signaling by ubiquitin. Curr Opin Hematol. 2016;23:274–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Moussa MD, Santonocito C, Fagnoul D, Donadello K, Pradier O, Gaussem P, De Backer D and Vincent JL. Evaluation of endothelial damage in sepsis-related ARDS using circulating endothelial cells. Intensive care medicine. 2015;41:231–8. [DOI] [PubMed] [Google Scholar]
- 60.Kaneider NC, Leger AJ, Agarwal A, Nguyen N, Perides G, Derian C, Covic L and Kuliopulos A. ‘Role reversal’ for the receptor PAR1 in sepsis-induced vascular damage. Nature immunology. 2007;8:1303–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Trejo J, Altschuler Y, Fu HW, Mostov KE and Coughlin SR. Protease-activated receptor-1 down-regulation: a mutant HeLa cell line suggests novel requirements for PAR1 phosphorylation and recruitment to clathrin-coated pits. The Journal of biological chemistry. 2000;275:31255–65. [DOI] [PubMed] [Google Scholar]
- 62.Paing MM, Stutts AB, Kohout TA, Lefkowitz RJ and Trejo J. beta -Arrestins regulate protease-activated receptor-1 desensitization but not internalization or Down-regulation. The Journal of biological chemistry. 2002;277:1292–300. [DOI] [PubMed] [Google Scholar]
- 63.Paing MM, Temple BR and Trejo J. A tyrosine-based sorting signal regulates intracellular trafficking of protease-activated receptor-1: multiple regulatory mechanisms for agonist-induced G protein-coupled receptor internalization. The Journal of biological chemistry. 2004;279:21938–47. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.