

Abstract
Eukaryotes must balance metabolic and cell death actions in mitochondria with control of gene expression and cell fate by chromatin, thereby functionally binding the epigenome and metabolome. This interaction has far reaching implications for chronic disease in humans, the most common of which are those of the cardiovascular system. The most devastating consequence of cardiovascular disease, heart failure, is not a single disease, diagnosis or endpoint. Human and animal studies have revealed that, regardless of etiology and symptoms, heart failure is universally associated with abnormal metabolism and gene expression—to frame as cause or consequence, however, may be to wrongfoot the question. This essay aims to challenge current thinking on metabolic-epigenetic crosstalk in heart failure, presenting hypotheses for how chronic diseases arise, take hold and persist. We unpack assumptions about the order of operations for gene expression and metabolism, exploring recent findings from noncardiac systems linking metabolic intermediates directly to chromatin remodeling. Lastly, we discuss potential mechanisms by which chromatin may serve as a substrate for metabolic memory and how changes in cellular transcriptomes (and hence, cellular behavior) in response to stress correspond to global changes in chromatin accessibility and structure.
Keywords: metabolism, epigenome, chromatin, cardiovascular disease, mitochondria
Introduction
The clinical syndrome of heart failure is characterized by poor oxygen delivery, exhaustion upon minor physical exertion, fluid retention, shortness of breath and impaired function of the heart itself, presenting as depressed diastolic or systolic function [1]. The central cell and organ level observation in heart failure is a reduction in ATP production and decreased phosphocreatine to ATP ratio, indicative of energy starvation and strongly correlating with cardiovascular mortality [2–4]. Heart failure is often commensurate with other pathophysiological changes, such as fatty liver disease in the case of obesity and metabolic syndrome. Over time these maladies precipitate other serious conditions including renal insufficiency, vascular dementia/Alzheimer’s and depression. Management of the heart failure patient relies on modulating fluid balance, vascular tone and cardiomyocyte function, with drugs respectively targeting the renin/angiotensin/aldosterone system to use the kidneys to unload the heart, control of hypertension by modulating vascular tone and improving the work of the heart by β-adrenergic receptor blockade or other modulation of ionotropy. Whereas humans get heart failure from a combination of genetic risk, diet (and accordant BMI), hypertension and atherosclerosis, animal models of heart failure usually arise from a single stress, like high fat diet, infarction, pressure overload or, in many cases, an engineered genetic lesion (i.e. a knockout or transgenic animal). Together these observations present a paradox: the syndrome of heart failure in humans is a spectrum of maladies, unlikely to be treated or cured by a single suite of therapies; yet decades of scientific research has focused on models of heart failure that often times use a set of phenotypes (e.g. myocyte hypertrophy and left ventricular ejection fraction) as a readout that in fact only occur in a portion of the patients suffering from heart failure. One response to this conundrum has been a recent interest in new models of heart failure with preserved ejection fraction, with the ostensible goal of identifying therapies for the ~50% of patients with heart failure that do not exhibit depressed ejection fraction but yet do exhibit diastolic dysfunction and ventricular stiffening.
To understand how chronic diseases develop, we should answer the question of how cells remember what they experience over timescales of seconds, hours or years. A shift in metabolic state can be induced by any number of physiological or experimental factors including substrate availability, physical stress, physiological inputs and energetic demands. These stresses can change gene expression, protein expression, and post-translational modifications, as well as directly alter the substrate pool for metabolic processes, which can themselves store memory in the signaling cascades harboring time delays and feedback loops based on protein and post-translational modification half-lives. Signaling networks and metabolic pathways, due to time delays, post-translational modification and protein lifespans, have the capacity for cellular memory, as do gene expression networks, themselves involving genomic and non-genomic components. But what is the physical substrate of lasting cellular memory and how does this interact with metabolism in a bidirectional manner? We hypothesize that the well characterized cellular bedlam in the failing heart reprograms the epigenome: specifically, it shifts multiple regions of the genome between states of active and inactive conformation—between membraneless suborganelles designed for distinct transcriptional responses—through the actions of chromatin remodelers and transcription factors. In a process of cellular long term memory formation, the entrenching of pathologic metabolism, signaling, calcium handling and myofilament function becomes encoded as a durable substrate via modifications to chromatin structure.
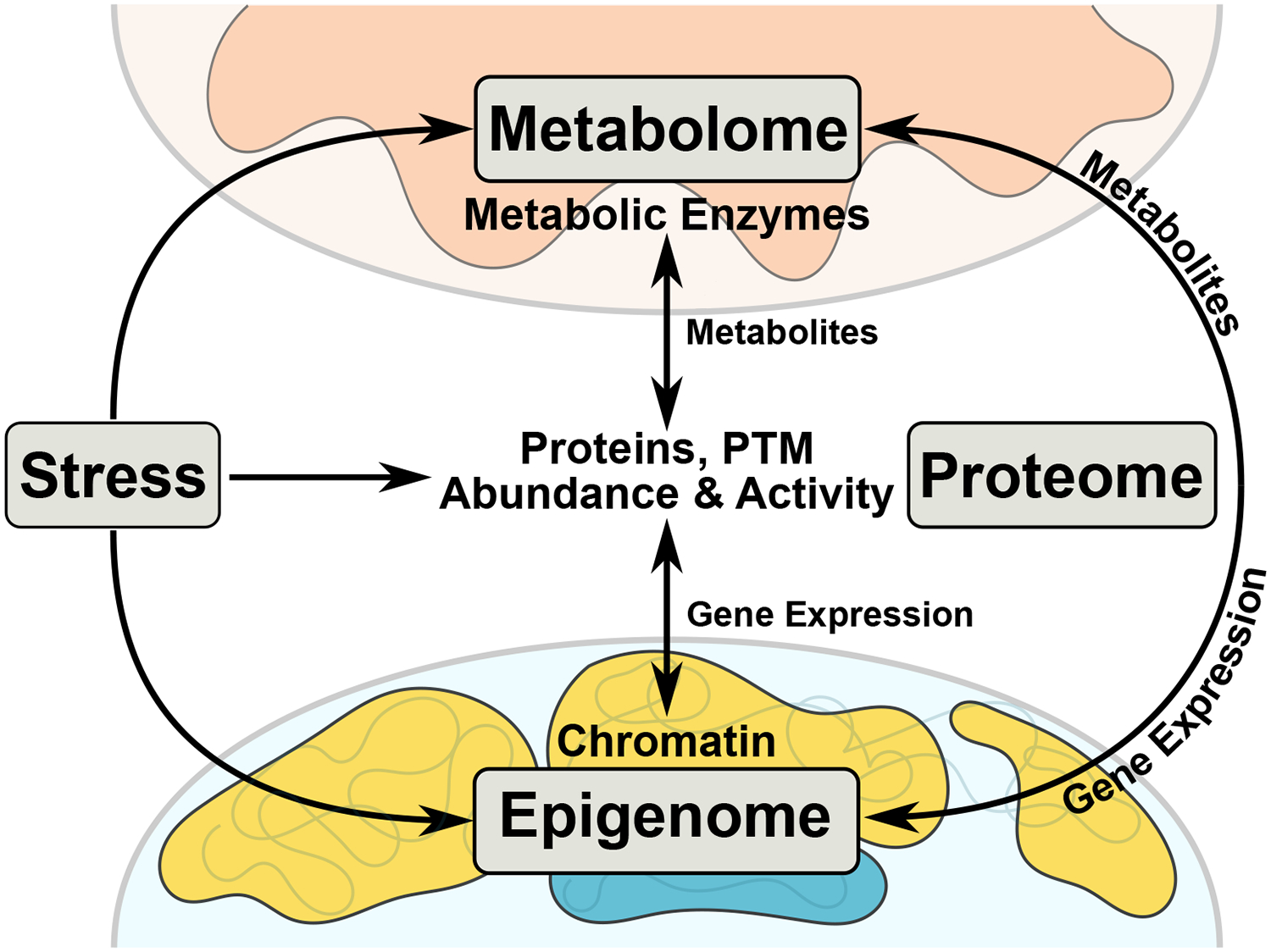
An emerging trend suggests direct coupling of metabolism to chromatin through various molecular means, including substrate availability, enzyme regulation, histone modification, transcription factors and chromatin remodelers (Figure 1) [5, 6]. For metabolic-epigenomic coupling to occur, metabolic precursors must interact with cytoplasmic signaling processes or the nucleus prior to entering the mitochondria or metabolic byproducts of central carbon metabolism (e.g. Kreb’s cycle or oxidative phosphorylation products or intermediates) must leave the mitochondria (or trigger a signaling process that leaves the mitochondria) to impinge upon the nucleus. Let us consider the implications of this relationship in the healthy and failing heart.
Figure 1. Inter-Organelle and Inter-’omic Cooperation in Times of Stress.
Cardiac stress (e.g. ischemia, diet, genetics, hypertension) affects the metabolome, proteome, and epigenome, forcing a coordinated, time-sensitive response. Stress-induced changes in metabolic substrate utilization causes a shift in metabolic enzymatic activity and metabolite pools. These metabolites act on proteins through post-translational modifications (PTMs), changing protein activity or localization. Protein PTM induced nuclear localization directly or indirectly influences epigenetic processes (e.g. histone modifications or DNA methylation), causing changes in chromatin accessibility and gene expression. Metabolites can be shuttled to the nucleus to be utilized as substrates for epigenetic modifications, thereby changing local chromatin accessibility, influencing gene expression and ultimately protein abundance.
Metabolic Flexibility
Being born is stressful, and heralds manifold changes for the heart. Vast glycogen stores are depleted, metabolic machinery for fatty acid utilization is mobilized, mitochondria proliferate, oxygen is suddenly available at much higher concentration, and an inchoate muscle protein scaffold organizes into a rigid, regimented network of fibers engineered for the sole purpose of physical work. Adult cardiac muscle is replete with mitochondria engineered to turn carbohydrates, fats and protein into ATP. Fatty acids are imported to cardiac myocytes from the circulation through fatty acid translocases (CD36 and FAT) and following cytosolic esterification (i.e. conversion to long fatty acyl-CoA by the ATP-dependent acyl-CoA ligase) are directed into the mitochondria through carnitine palmitotransferase isomers (CPT1 and CPT2). These long chain fatty acids are then catabolized by beta oxidation, a multistep process leading to the production of acetyl-CoA, which enters the Kreb’s cycle. This Kreb’s generates FADH2 and NADH, which are utilized by the electron transport chain coupled to oxidative phosphorylation in the inner membrane of the mitochondria. This process is the primary mechanism for generation of large amounts (relative to that from glycolysis) of ATP in cardiac myocytes. In the healthy adult heart, the aforementioned fatty acid metabolic strategy is dominate. Yet carbohydrates are more efficient fuel sources on a per molar oxygen basis compared with fatty acids and, in addition to being the primary fuel source in the fetal heart, carbohydrates are rapidly mobilized in the adult heart during cardiac stresses, such as hypertrophy and failure[7]. Cytosolic glycolysis converts glucose—which can be taken up into cardiac myocytes independent of (GLUT1) or in response to (GLUT4 transporter) insulin, both of which are increased in the failing heart, augmenting the availability of glucose—into pyruvate, which is in turn imported into mitochondria and converted by pyruvate dehydrogenase to acetyl-CoA, entering the Kreb’s cycle. The liver converts fat into ketone bodies, especially during periods of starvation, that navigate to the heart through the circulation and enter cardiac myocytes, presumably via the membrane bound monocarboxylate transporters 1 and 2. The ketone bodies then make their way to the mitochondrial matrix through unknown mechanisms and, after conversion to acetyl-CoA, the rate limiting enzyme for which is succinyl-CoA:3-oxoacid-CoA transferase (helpfully, SCOT), enter the Kreb’s cycle. Proteins, notably branched chain amino acids (BCAAs; leucine, isoleucine and valine—essential amino acids which cannot be synthesized by the body), are catabolized through a series of steps regulated principally by the branched chain alpha keto acid dehydrogenase (BCKDH) to ultimately generate acetyl-CoA and succinyl-CoA (converted from propionyl-CoA), both of which then enter the Kreb’s cycle. Elevation in BCAAs has been linked to metabolic syndrome, a precursor to heart failure, through multi-organ effects reviewed in detail elsewhere[8]. The reader notes that, regardless of the source, the goal of all these metabolic pathways is: get carbon chains into the Kreb’s cycle and on to ETC and oxidative phosphorylation for ATP production.
Metabolic Inflexibility
Although feasible and logical in animal models, separation of hypertension and diabetes as precipitating factors for heart failure does not occur in humans. Lower levels of insulin, greater hemodynamic load, and mitochondrial biogenesis rapidly transforms the anatomy and functionality of the cardiomyocyte after birth. The transcriptional program associated with mitochondrial biogenesis and substrate preference, namely, switching to the preferential metabolism of fatty acids [9], is driven in large part by the nuclear receptor peroxisome proliferator-activated receptor α (PPAR-α) and its coactivator the PPAR-coactivator 1 (PGC-1α). Genomic targets of PGC-1α in cardiac myocytes are unknown, however a recent study from brown adipose tissue demonstrated, unsurprisingly, that genes bound by the transcription factor enrich in pathways involved in fatty acid metabolism[10]. The case for PPAR[11], which is likely also true for other transcription factors whose activation has been witnessed in the diseased heart, is that the actions of the molecule are highly dependent on the nature of the stimulus, the time after the stimulus and the molecular interactions with whichever other DNA binding proteins are activated in the cell.
Heart failure has been termed a state of metabolic inflexibility, an interpretation that comes principally from studies in which the disease state is experimentally induced by a unitary stimulus [12]: experimentally restricting the availability of fuel sources to the heart (either favoring glucose or fat) leads to disease. It has been proposed that metabolic derangements, which are to be found in all forms of heart failure, trigger gene expression changes (implying they also precede the gene expression changes) and may also sustain them [13]. Evidence in support of this observation is: (1) experimental alterations in diet and/or metabolic substrate can induce changes in gene expression; (2) all forms of human heart failure involve metabolic derangement; (3) the gene expression programs initiated by the stresses of heart failure, principally metabolic syndrome itself, hypertension/elevated cardiac pressures, and ischemia—which themselves are often chronic and, especially in humans, rarely perfectly ameliorated—include changes to the transcript and protein levels of metabolic machinery to favor glucose over fatty acid metabolism (e.g. expression of GLUT1/4, pyruvate dehydrogenase kinase 2 and glycogen synthase are all decreased), thereby extending the time window of metabolic alteration. What is the precise molecular chain of events connecting changes in metabolism with gene expression and vice versa? Metabolic syndrome and hypertension/pressure overload can be experimentally separated, wherein studies have revealed the former to be associated with increased fatty acid, and the latter with glucose, oxidation (decreasing and increasing PPARα expression, respectively) [9].
One study showed that visceral adiposity (but neither subcutaneous nor total adiposity) was independently associated with incident hypertension (i.e. the development of high blood pressure following a previously normotensive measurement) in humans[14]. Hypertension—along with high fasting blood glucose, elevated triglycerides, low HDL or waist circumference—is a diagnostic criteria for metabolic syndrome, which in turn is a clinical precedent to obesity and type 2 diabetes. Although high blood pressure and metabolic syndrome may not always be causally linked in a given person, the cooccurrence of these conditions at some stage of the progression of cardiovascular disease in humans is extremely common[1]. Precisely because of the fact that heart failure in humans is not the result of only hypertension, metabolism, ischemia or genetics—but rather, due to an often imperfectly quantified cooccurrence of these factors—we reason that the actual endogenous sensor-regulator of gene expression and metabolic state is chromatin. This process works through shifting regions of accessibility and probably to a lesser degree structure, moving the transcriptome from a more restricted state to a more dynamic one.
Evolution has contrived some aspects of this transition to reverse themselves in response to pathologies to which mammals commonly subject their hearts, although important caveats abound in this characterization. Impairments of glucose uptake and glucose oxidation compounds the diseased myocyte’s ability to metabolize fatty acids; mitochondria go haywire, producing reactive oxygen species as a result of poorly coupled oxidative phosphorylation, losing control of calcium balance, leading to calcium overload (due in part to malfunctioning sarcoplasmic reticulum reuptake during diastole, in turn due to ryanodine receptor leakage), calcium toxicity and catastrophic depolarization (known as permeability transition) and spewing of protein and calcium into the cytosol, causing further havoc, including cell death by apoptosis and necrosis. Ischemia brings with it hypoxia, which further exacerbates reactive oxygen species production, cell death signaling and calcium overload; and the subproteome of contractile molecules is beset by post-translational modification, starved of ATP as a result of the aforementioned mitochondrial impairment. The cardinal observation of a failing heart is a decrease in the high energy reserve compound phosphocreatine, manifest as a decreased phosphocreatine/ATP ratio, resulting from the coincident stresses of increased energy demand (due to higher workload in response to hypertension, e.g.), decreased contractile ability (due to muscle loss or impairment by ischemia, e.g.) and decreased energy supply (due to systemic metabolic crapping out, due to type II diabetes, due to sustained hypercaloric diet, e.g.) and, curiously, both an increase in muscle protein mass and a change in the expression[15, 16] of myosin heavy chain isoforms (See Box 1), sometimes accompanied by gross changes in sarcomeric organization, although without a return to the primitive organization of the contractile apparatus observed in fetal myocardium.
Text Box 1: Isoform Switching in Heart Failure.
A “myosin heavy chain isoform switch”—in which β-MHC (MHC7), which predominates in fetal hearts and is lowly expressed in healthy adults, is upregulated and α-MHC (MHC6), predominating in adult hearts and lowly expressed in the fetus, downregulated—in the hearts of mice with symptoms of hypertrophy or failure has been reported in hundreds of papers, so as to become itself a tacit phenotype of heart failure. Complicating this interpretation are the observations that: (1) in humans the β-MHC isoform predominates throughout life and is upregulated in failing hearts (while the lower expressed α-MHC is further down-regulated); and (2) even in rodent hearts, the cells actually hypertrophying in the wake of pathologic stress are not the same cells with altered MHC isoform expression (i.e., β-MHC expressing cells are smaller, as measured either by antibody-based flow cytometry[15] or mRNA FISH and single cell RNA-seq[16]. The molecular role, thus, of this change in isoform expression in the function of a diseased, hypertrophying myocardium, and as a readout of transcriptional remodeling in heart failure, remains obscured.
Communication Amongst Organelles
The antediluvian pact whereby eukaryotes offload (some) metabolic and cell death decisions to mitochondria in exchange for ceding to the nucleus and cytoplasm substantial control over synthesis of mitochondrial protein machinery, inextricably links chromatin with metabolism. The conceptual framework of the histone code (Figure 2A) in which writers, erasers and readers add, remove and interpret, respectively, nucleosome post-translational modifications to influence genomic function can be helpfully expropriated to discuss post-translational modification of—principally mitochondrial—proteins in the setting of metabolic diseases, an area of research too expansive to do any justice in the present essay but which has been adroitly reviewed elsewhere (for cardiac[17]; for noncardiac and technology [18]). Indeed, research in cancer has revealed multiple direct links between metabolism and gene regulation (see Box 2). Hyperacetylation has been observed [19] on pyruvate dehydrogenase, fatty acid oxidation machinery, Kreb’s cycle enzymes and the electron transport chain proteins and associated with their dysfunction, and attenuate of hyperacetylation, by restoration of the NAD(+)/NADH ratio, is beneficial at the molecular and whole organ level in heart failure [20]. There are many origins of this hyperacetylation, including what might be termed writer-mediated (i.e. changes in the acetyltransferase levels or activity) or eraser-mediated (i.e. changes in the deacetylases); if we stretch the analogy, acetylation machinery also has ink, in the form of the acetyl-CoAs themselves, that are used to write the various types of acetylation modifications that have been observed in mammals and which are present in higher than normal levels during heart failure [21], or in the form of cofactors like NAD+ and the NAD(+)/NADH ratio, which are necessary for sirtuin-mediated deacetylation [19, 20]. Other post-translational modifications, less obviously coupled to metabolism, have been measured in the heart, but a central issue bedevils understanding of their function: stoichiometric and temporal data are sparse, making unknowable the molecular effects of each individual modification. Computational studies have recently been engineered to address this challenge in mass spectrometry experiments[22], but they have yet to be applied in the setting of disease. The metabolic donor pool changes in heart failure, but this is highly dependent on the model and time frame (for example the NAD(+)/NADH ratio has been shown in different studies to increase[23] and decrease [24] after pressure overload). Indeed fatty acid substrates are more depleted in late stage heart failure [25], the time point at which mitochondrial and gross metabolic disturbances are most pronounced [17]. The concept of distinct metabolite pools in different subcompartments of the cell has been established for some time[26]—this hypothesis is receiving renewed interest with the recognition of cross talk between metabolism and various cellular processes, including chromatin accessibility.
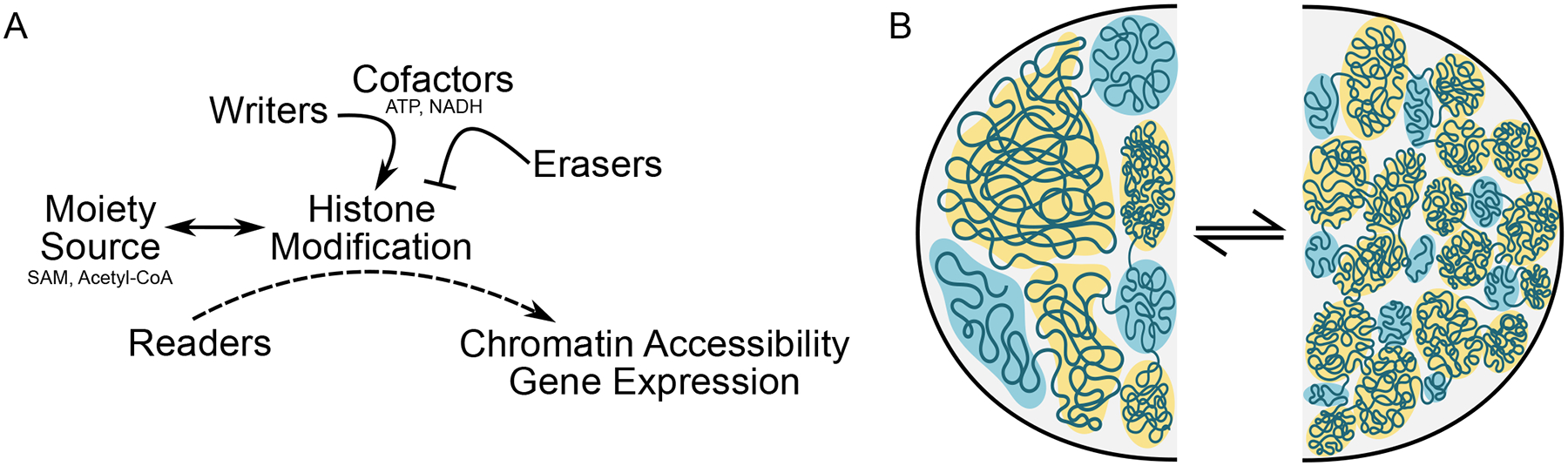
Figure 2. Chromatin Compaction and Histone Modifications Affect Gene Expression.
A. Metabolic cofactors, such as ATP and NADH, work in concert with writers and erasers to orchestrate histone modifications and chromatin condensation. Histone modifications are added by writer, which utilize metabolites as moiety sources (e.g. SAM, Acetyl-CoA and again ATP, and others) to add marks (erasers, accordingly, remove the marks) so that readers, which continuously navigate to and sample histones for proper modifications, can influence gene expression by recruiting transcriptional machinery. B. Rather than a rigid invariant architecture between cells, chromatin organization appears to arise similar to a phase separation, with local regions of accessibility and inaccessibility influenced by protein binding and PTM in response to cell specification cues and environmental stimuli. In this representation, open and active (blue) or closed and inactive (yellow) regions shift in terms of their relative abundance, the features of the domains they occupy and their localization within the nucleus. Chromatin compaction can flow between multiple states, shown here as one with larger more continuous regions of activity or silencing (left) versus smaller more segmented regions of differing accessibility and transcriptional behavior (right).
Text Box 2: Epigenetic-Metabolic Coupling in Cancer.
Cancer research provides insights into how coupling of metabolism and the epigenome can go awry, causing disease. Cytosolic isocitrate dehydrogenase 1 (IDH1) and mitochondrial IDH2 convert isocitrate to α-ketoglutarate (α-KG). Mutations of IDH1 or IDH2, observed most commonly in gliomas, further converts α-KG to 2-hydroxyglutarate (2-HG) [63, 64], and overproduction of this so-called “oncometabolite” inhibits DNA demethylation by binding to ten eleven translocation (TET) family proteins and histone demethylation by Jumonji C (JMJC) family proteins [65]. Hypermethylation occurs as 2-HG competes with α-KG, a hallmark of aggressive tumors [66]. In human IDH mutant gliomas, CTCF and cohesion binding sites are subject to hypermethylation, disrupting TAD domain insulation and resulting in disruption of gene expression, specifically of a constitutive enhancer interacting with the oncogene PDGFRA [67]. Additionally, loss of function mutations to succinate dehydrogenase or fumarase lead to high levels of succinate or fumarate, respectively, to inhibit TET and JMJC demethylation [68].
Post-translational modification of metabolic enzymes alter their enzymatic activity and localization in cancer progression. In the final step of glycolysis, pyruvate kinase (PKM) catalyzes the conversion of phosphoenolpyruvate and ADP to pyruvate and ATP through phosphorylate transfer. Pathological stimulation of EGFR and PDGFR [69] induces increased expression of PKM2 (upregulated in numerous cancer types [70]). Phosphorylation of PKM2 by ERK1 or ERK2 recruits PIN1, inducing isomerization and exposing the nuclear localization sequence [71]. Nuclear translocation of PKM2 enables interaction with phosphorylated β-catenin and recruitment to CCND1 and MYC promoter regions, binding to and phosphorylating histone H3, which displaces HDAC3 for H3K9 acetylation [72]. CCND1 promotes cell division through promoting G1/S transition and MYC induces increased glucose uptake. Additionally, PKM2 drives glucose uptake by binding to hydroxylated HIF1α, inducing increased target gene expression [73].
PPAR nuclear receptors heterodimerize with retinoid receptors to activate transcription in response to excess intracellular fatty acids and genetic disruption of PPARα leads to decreased fatty acid oxidation and increased metabolism of glucose [27]. The PPARγ receptor coactivator, PCG1α, is downregulated in human failing hearts [28] and its deficiency[29], or that of its cousin PGC1β[30], exacerbates heart failure in the setting of pressure overload. Depending on the experimental model of heart failure, the involvement of PPARα signaling can work in opposite directions[9]: pressure overload, mimicking hypertension, is associated with decreased PPARα signaling and increased glucose utilization, whereas mice overexpressing PPARα exhibit a metabolic syndrome like phenotype, increasing utilization of fatty acids (diabetic mouse models also exhibit elevated PPARα). Glucose, more abundant in the failing heart and less efficiently oxidized, may itself directly modify transcription factors, disrupting their function[31]. Myocyte enhancer factor 2A (MEF2A), which plays an essential role in myocyte differentiation, regulates PGC-1α and mitochondrial content and function [32], whereas MEF2A is downregulated in diabetic patients with heart failure [33]. Various histone modifying enzymes have been implicated in heart failure through genetic and pharmacologic means (for review[34–36]) with recent studies implicating cardiac-specific writers in the regulation of normal adult metabolism. The striated muscle-specific histone methyltransferase Smyd1, loss of which induces precipitous heart failure[37] and reduced respiratory capacity of mitochondria[38], is required for normal expression of a panel of metabolic transcription factors including PGC-1α, PPARα, and RXRα through direct regulation of histone H3 lysine 4 trimethylation at gene promoters[38]. Hypoxia, a central component of injury and long term stress in cardiovascular disease, can directly modulate chromatin independent of the hypoxia inducible factor (HIF) transcription factor complex: two studies recently showed Hif-independent modification of locus-selective, but genome-wide, H3K4me3 (KDM5A dependent[39]) and H3K27me3 (KDM6A dependent[40]) modifications, mobilizing gene expression programs in response to hypoxia without transcription factor activation (note: these processes were also found to be independent of the oxygen sensors 2-oxoglutarate (OG)-dependent dioxygenases—implicating the histone modifying enzymes themselves as oxygen sensors). It is also apparent that under certain circumstances, metabolic machinery (e.g. Kreb’s cycle enzymes) can directly localize to the nucleus and regulate cell fate decisions such as zygotic genome activation [41] and cancer progression [42, 43] by coordinating with histone modifications and the binding of other transcriptional regulators, such as histone variants[44]. The frequency of direct genomic actions by metabolic enzymes, and the relative stoichiometry of these proteins in nuclei versus perinuclear mitochondria, are areas of active investigation.
Chromatin as Memory Substrate
Evolution inserted large portions of non-mRNA-encoding DNA into eukaryotic genomes, corresponding with the ability of these genomes to produce dozens and, in higher organisms, hundreds of cell types and transcriptomes from the same genome, ostensibly by varying the structure of the genome at some scale. At present, global chromatin structure-function models are rapidly evolving (i.e. If we wanted to extend the central dogma beyond the codon, what would be the rules?). Mediator complexes, chromatin readers and transcription factors operate within the context of adult myocyte chromatin architecture and a global landscape of accessibility that has been established by writers, erasers, chromatins structural proteins (and perhaps DNA methylation, although this modification seems to follow cues laid down in structure) some of which is cell type and developmental stage specific.
Chromatin conformation capture techniques applied genome-wide (e.g. HiC) initially described topologically associating domains as local regions of higher than average intrachromosomal interactions separated by boundaries, or places at which the interactions abruptly switch from favoring 5’ to 3’ regions (or vice versa). While tempting to view these TADs as structural operons, wherein genes of similar activity are oriented in a similar manner in 3D (rather than along the linear chromosome, as occurs in bacteria), subsequent studies revealed that TADs and their boundaries are largely conserved across adult mammalian cell types, and indeed are specified early in differentiation, thus implying this scale of organization cannot, alone, specify different transcriptomes. Since TADs and their boundaries are conserved, the chromatin structural proteins, like CTCF and cohesins, that preferentially populate these boundaries are unlikely to drive cell type specification, although they have been shown to regulate disease-associated chromatin remodeling and gene expression. During differentiation along the cardiac lineage from embryonic stem cells to neonatal cardiomyocytes, substantial reassignment of the genome to different compartments of chromatin is observed, either as record or driver of changes in the transcriptome [45, 46]. After birth, cardiomyocyte maturation is associated with changes in histone post-translational modifications but minor changes in compartmentalization [45, 46].
The biological relevance of chromatin compartmentalization is not immediately clear however: it may relate to regions within the nucleus of local increased plasticity and accessibility to transcriptional machinery, for example in the center versus the periphery of the nucleus. Chromosomes reproducibly segregate into territories within the interphase nucleus, but within these territories, intra- and inter-chromosomal interaction are dynamic and membraneless suborganelles can form through a process likened to phase separation[47] (Figure 2B). Together, compartmentalization of chromatin (an emergent property of histone isoforms, histone PTMs [and the associated suite of writers, erasers and readers], chromatin structural proteins and remodeling enzymes, themselves regulated by metabolic processes and substrate levels) may combine with chromosome territory cues, subnuclear localization cues (principal among these being tethering of chromatin regions to the nuclear lamina [48]) and phase separation behavior to establish structure-function neighborhoods within the nucleus. Unlike a crystal structure of a nucleosome or the 5–24nm chromatin fibers visible by microscopy, these structural neighborhoods are not perfectly replicated across individual cells, but rather are the lowest energy state organizational feature that segregates active from inactive regions, thereby functionally compartmentalizing the nucleus. Indeed, recent single cell chromatin capture studies indicate a high level of cell-to-cell variability in the absolute structure of TADs (i.e. the exact location of their boundaries)[49]: removal of CTCF or cohesins can disrupt TAD boundaries in bulk HiC experiments[50] (indeed, loss of CTCF from cardiac myocytes is sufficient to induce heart failure and chromatin disorganization [51] [52]), yet similar interventions do not eliminate TADs in single cell experiments—instead, loss of the CTCF-cohesin complex results in greater variability in the formation of TADs [53] suggesting (a) TADs are a basic structural feature of DNA and protein, formed by the physical chemical principles of phase separation (or perhaps due to sequence cues, such as transposons and their transcription [54]), independent of the actions of any single given protein; and (b) the actions of the CTCF-cohesin complex are to entrain/stabilize TADs across different cell types. It has been proposed that long noncoding RNAs may serve as scaffolds for phase separation of chromosomes. Unlike other forms of RNA like mRNA and microRNA, no unifying mechanism of action has been revealed for lncRNAs. Could their role be to seed transcription factories, such that the actual sequence matters less than their positioning along the genome? Nucleosome turnover rate and nucleosome density, along with higher order structure, contribute to accessibility and transcription. Regions with high nucleosome density often have high turnover and low positional stability of nucleosomes, but there are exceptions (e.g. insulators, which are higher density, low turnover, but accessible)[55]. Lastly, histone post-translational modifications, in part responding to metabolic signals, may influence the formation of local microenvironments of activation or silencing, thereby contributing to the formation of membraneless suborganelles (e.g. high transcription of rRNA defining the nucleolus).
Timing
The phenomena of fully reversible ‘physiological’ types of hypertrophy observed in athletes or during pregnancy (neither of which have metabolic syndrome or hypertension, despite increased cardiac mass and chamber volume), which share neither gene expression profiles nor metabolic rearrangements of pathologic hearts [56, 57], support the concept that metabolic syndrome or elevated mean arterial pressure are necessary to maintain the gene expression regime in the diseased state. This gives some window into the stimulus type (elevated MAP and/or metabolic syndrome) and duration (sustained).
How does sensing of metabolic changes occur and what is the order of cellular operations that respond to, adjust and remember these metabolic changes? Changes in gene expression may be unnecessary for short term regulation of metabolism, which can occur through the actions of signaling proteins to modify metabolic enzymes and by the availability of various substrates. However, these processes do not occur in isolation and will simultaneously activate gene expression machinery through the actions of transcription factors and by direct coupling of metabolites to chromatin [39, 40, 58, 59]. Metabolic fluxes (i.e. the time it takes to deplete existing metabolic stores in the absence of input) occur on the timescale of a minute in mammalian cells, whereas protein production takes several minutes. Acute changes in metabolites thus can induce changes in gene and protein expression, but there is a time delay, meaning that some hysteresis will result—at the protein abundance level—if the cell experiences imbalance in metabolite source and utilization. How long does it take to entrain this information in chromatin? ATP-dependent chromatin remodeling complexes regularly physically engage chromatin in nonproductive binding events, waiting for appropriate transcription factor signaling (and perhaps ejection of nucleosomes) to convert this continuous sampling activity into productive remodeling, which can in turn occur on the timescale of seconds to minutes[60, 61]. Furthermore, physical forces alone are sufficient to remodel chromatin, again operating in seconds or minutes[62]. While the specific time constants for these processes in adult cardiac myocytes have not been determined, we can safely estimate that both metabolic gene expression and chromatin remodeling are sensitive to similar cardiac stresses (notably mechanical disruption and metabolic flux) and can respond over similar timescales, making the issue of temporal regulation not a priori obvious and therefore wanting for experimental determination.
Concluding Remarks and Hypotheses
Here is precisely what we think is going on: the abnormal cellular environment in the failing heart entrains—through physical, post-translational and metabolic signaling—chromatin towards a distinct organizational regime that adapts gene expression to resist pathologic stress, thereby producing both the beneficial and detrimental phenotypes observed in cardiac cells. Chromatin is organized based on cues across the genome that draw together regions with similar modifications (to histones and DNA, and due in part to other chromatin binding proteins) to create neighborhoods of transcriptional silencing or activation, similar to phase separation principles operative in oil partitioning from water and driven less by specific enzymatic or transcription factor pathways converging on a handful of target genes, and more by a global shift in the deposition of various modifications controlled by said enzymes as well as by the present and past metabolic environment. The epigenome can serve as an integrator of multiple forms of metabolic stress: if the rules can be uncovered for how it responds in general to stress conditions that cause disease, we may learn how evolution came to utilize chromatin as a memory substrate and cellular decision tree. Therapeutic approaches resulting from our evolving understanding of the coupling of epigenome to metabolome in heart failure may take one of several broad approaches: modify the fuel source (changing diet); modify the metabolic pathways directly (e.g. GLUT transporters, PDH; changing the abundance of enzymes that control flux of metabolic intermediates or utilization of one fuel source over another); modify the molecular intermediates (e.g. acetyl-CoA or NADH; changing the availability of intermediate substrates, thereby influencing processes like post-translational modification of proteins); modifying the crosstalk apparatus that integrates epigenomic and metabolic circuits (changing the transcription factor or chromatin modifying enzymes that regulate metabolism); or targeting chromatin accessibility directly. If chromatin is indeed a substrate of cellular memory, strategies that screen chromatin as a functional readout of new therapies may improve durability of treatment.
Highlights.
Heart failure is a ruinous destination for many afflicted with cardiovascular disease and is not a single condition or single set of diagnostic criteria; rather, it manifests through an intricate series of molecular and systemic malfunctions ranging from the suborganelle level to the multiple organ systems of the body.
Regardless of the etiology, humans with, and animal models of, heart failure are characterized by abnormal metabolism and gene expression, some aspects of which are compensatory responses to the disease and others that promulgate injury.
Close communication between the metabolome and the epigenome sets basal susceptibility to various heart failure symptoms. This communication entrains detrimental conditions in metabolic-epigenetic memory and thus may be a target for novel treatments.
Outstanding Questions.
Protein modification: What is the precise stoichiometry and temporal regulation of post-translational modifications during initiation and progression of heart disease? How do these modifications differ across populations of mitochondria in a cell and across loci in the genome? Which modifications have been evolutionarily selected for to control a specific phenotype? Are there convergent disease-associated modifications that are stimulus independent and yet commonly observed in disease?
Chromatin remodeling: What are the principles for how local accessibility effects global structure of chromatin (and perhaps vice versa)? What are the rules by which DNA sequence targeted transcription factors and chromatin modifying machinery execute locus specific remodeling events?
Metabolic cues: Does heart failure in humans involve the metabolic fuel shifts observed in genetic mouse models (i.e. is metabolic inflexibility a valid target for precision therapeutics of heart failure)? What changes in cardiac metabolism involve changes to chromatin and which changes are independent of gene regulation?
Organelle specification: What are transcriptional implications of metabolic enzymes acting directly on chromatin and what role do these processes play in heart failure? What are the molecular substrates for cellular memory and how is this memory recorded by metabolic cues and physical forces? How different are two nuclei in the same cardiomyocyte and how does chromatin variability over a population of cells confer flexibility to cardiac function in vivo?
Funding:
The Vondriska lab is funded by the National Institutes of Health, the American Heart Association and the David Geffen School of Medicine at UCLA.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts: The authors report no conflicts of interest related to this article.
References
- 1.Benjamin EJ et al. (2019) Heart Disease and Stroke Statistics-2019 Update: A Report From the American Heart Association. Circulation 139 (10), e56–e528. [DOI] [PubMed] [Google Scholar]
- 2.Neubauer S et al. (1997) Myocardial phosphocreatine-to-ATP ratio is a predictor of mortality in patients with dilated cardiomyopathy. Circulation 96 (7), 2190–6. [DOI] [PubMed] [Google Scholar]
- 3.Liao R et al. (1996) Decreased energy reserve in an animal model of dilated cardiomyopathy. Relationship to contractile performance. Circ Res 78 (5), 893–902. [DOI] [PubMed] [Google Scholar]
- 4.Hearse DJ (1979) Oxygen deprivation and early myocardial contractile failure: a reassessment of the possible role of adenosine triphosphate. Am J Cardiol 44 (6), 1115–21. [DOI] [PubMed] [Google Scholar]
- 5.Li X et al. (2018) Regulation of chromatin and gene expression by metabolic enzymes and metabolites. Nat Rev Mol Cell Biol 19 (9), 563–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Keating ST and El-Osta A (2015) Epigenetics and metabolism. Circ Res 116 (4), 715–36. [DOI] [PubMed] [Google Scholar]
- 7.Goodwin GW et al. (1998) Regulation of energy metabolism of the heart during acute increase in heart work. J Biol Chem 273 (45), 29530–9. [DOI] [PubMed] [Google Scholar]
- 8.Arany Z and Neinast M (2018) Branched Chain Amino Acids in Metabolic Disease. Curr Diab Rep 18 (10), 76. [DOI] [PubMed] [Google Scholar]
- 9.Aubert G et al. (2013) Perturbations in the gene regulatory pathways controlling mitochondrial energy production in the failing heart. Biochim Biophys Acta 1833 (4), 840–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chang JS et al. (2018) A map of the PGC-1alpha- and NT-PGC-1alpha-regulated transcriptional network in brown adipose tissue. Sci Rep 8 (1), 7876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Warren JS et al. (2017) Metabolic reprogramming via PPARalpha signaling in cardiac hypertrophy and failure: From metabolomics to epigenetics. Am J Physiol Heart Circ Physiol 313 (3), H584–H596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Karwi QG et al. (2018) Loss of Metabolic Flexibility in the Failing Heart. Front Cardiovasc Med 5, 68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Taegtmeyer H et al. (2010) Return to the fetal gene program: a suggested metabolic link to gene expression in the heart. Ann N Y Acad Sci 1188, 191–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chandra A et al. (2014) The relationship of body mass and fat distribution with incident hypertension: observations from the Dallas Heart Study. J Am Coll Cardiol 64 (10), 997–1002. [DOI] [PubMed] [Google Scholar]
- 15.Lopez JE et al. (2011) {beta}-Myosin Heavy Chain Is Induced by Pressure Overload in a Minor Subpopulation of Smaller Mouse Cardiac Myocytes. Circ Res. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Satoh M et al. (2019) High-throughput single-molecule RNA imaging analysis reveals heterogeneous responses of cardiomyocytes to hemodynamic overload. J Mol Cell Cardiol 128, 77–89. [DOI] [PubMed] [Google Scholar]
- 17.Zhou B and Tian R (2018) Mitochondrial dysfunction in pathophysiology of heart failure. J Clin Invest 128 (9), 3716–3726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Carrico C et al. (2018) The Mitochondrial Acylome Emerges: Proteomics, Regulation by Sirtuins, and Metabolic and Disease Implications. Cell Metab 27 (3), 497–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Horton JL et al. (2016) Mitochondrial protein hyperacetylation in the failing heart. JCI Insight 2 (1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Karamanlidis G et al. (2013) Mitochondrial complex I deficiency increases protein acetylation and accelerates heart failure. Cell Metab 18 (2), 239–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bedi KC Jr. et al. (2016) Evidence for Intramyocardial Disruption of Lipid Metabolism and Increased Myocardial Ketone Utilization in Advanced Human Heart Failure. Circulation 133 (8), 706–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bagwan N et al. (2018) Comprehensive Quantification of the Modified Proteome Reveals Oxidative Heart Damage in Mitochondrial Heteroplasmy. Cell Rep 23 (12), 3685–3697 e4. [DOI] [PubMed] [Google Scholar]
- 23.Oka S et al. (2011) PPARalpha-Sirt1 complex mediates cardiac hypertrophy and failure through suppression of the ERR transcriptional pathway. Cell Metab 14 (5), 598–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee CF et al. (2016) Normalization of NAD+ Redox Balance as a Therapy for Heart Failure. Circulation 134 (12), 883–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sansbury BE et al. (2014) Metabolomic analysis of pressure-overloaded and infarcted mouse hearts. Circ Heart Fail 7 (4), 634–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ingwall JS (2004) Transgenesis and cardiac energetics: new insights into cardiac metabolism. J Mol Cell Cardiol 37 (3), 613–23. [DOI] [PubMed] [Google Scholar]
- 27.Campbell FM et al. (2002) A role for peroxisome proliferator-activated receptor alpha (PPARalpha) in the control of cardiac malonyl-CoA levels: reduced fatty acid oxidation rates and increased glucose oxidation rates in the hearts of mice lacking PPARalpha are associated with higher concentrations of malonyl-CoA and reduced expression of malonyl-CoA decarboxylase. J Biol Chem 277 (6), 4098–103. [DOI] [PubMed] [Google Scholar]
- 28.Sack MN et al. (1996) Fatty acid oxidation enzyme gene expression is downregulated in the failing heart. Circulation 94 (11), 2837–42. [DOI] [PubMed] [Google Scholar]
- 29.Arany Z et al. (2006) Transverse aortic constriction leads to accelerated heart failure in mice lacking PPAR-gamma coactivator 1alpha. Proc Natl Acad Sci U S A 103 (26), 10086–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Riehle C et al. (2011) PGC-1beta deficiency accelerates the transition to heart failure in pressure overload hypertrophy. Circ Res 109 (7), 783–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Young ME et al. (2007) Proposed regulation of gene expression by glucose in rodent heart. Gene Regul Syst Bio 1, 251–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Czubryt MP et al. (2003) Regulation of peroxisome proliferator-activated receptor gamma coactivator 1 alpha (PGC-1 alpha) and mitochondrial function by MEF2 and HDAC5. Proc Natl Acad Sci U S A 100 (4), 1711–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Razeghi P et al. (2002) Downregulation of myocardial myocyte enhancer factor 2C and myocyte enhancer factor 2C-regulated gene expression in diabetic patients with nonischemic heart failure. Circulation 106 (4), 407–11. [DOI] [PubMed] [Google Scholar]
- 34.McKinsey TA and Olson EN (2004) Dual roles of histone deacetylases in the control of cardiac growth. Novartis Found Symp 259, 132–41; discussion 141–5, 163–9. [PubMed] [Google Scholar]
- 35.Rosa-Garrido M et al. (2018) Epigenomes in Cardiovascular Disease. Circ Res 122 (11), 1586–1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gillette TG and Hill JA (2015) Readers, writers, and erasers: chromatin as the whiteboard of heart disease. Circ Res 116 (7), 1245–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Franklin S et al. (2016) The chromatin-binding protein Smyd1 restricts adult mammalian heart growth. Am J Physiol Heart Circ Physiol 311 (5), H1234–H1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Warren JS et al. (2018) Histone methyltransferase Smyd1 regulates mitochondrial energetics in the heart. Proc Natl Acad Sci U S A 115 (33), E7871–E7880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Batie M et al. (2019) Hypoxia induces rapid changes to histone methylation and reprograms chromatin. Science 363 (6432), 1222–1226. [DOI] [PubMed] [Google Scholar]
- 40.Chakraborty AA et al. (2019) Histone demethylase KDM6A directly senses oxygen to control chromatin and cell fate. Science 363 (6432), 1217–1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nagaraj R et al. (2017) Nuclear Localization of Mitochondrial TCA Cycle Enzymes as a Critical Step in Mammalian Zygotic Genome Activation. Cell 168 (1–2), 210–223 e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen J et al. (2018) Compartmentalized activities of the pyruvate dehydrogenase complex sustain lipogenesis in prostate cancer. Nat Genet 50 (2), 219–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang Y et al. (2017) KAT2A coupled with the alpha-KGDH complex acts as a histone H3 succinyltransferase. Nature 552 (7684), 273–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Choi S et al. (2019) Oxoglutarate dehydrogenase and acetyl-CoA acyltransferase 2 selectively associate with H2A.Z-occupied promoters and are required for histone modifications. Biochim Biophys Acta Gene Regul Mech, 194436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nothjunge S et al. (2017) DNA methylation signatures follow preformed chromatin compartments in cardiac myocytes. Nat Commun 8 (1), 1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gilsbach R et al. (2018) Distinct epigenetic programs regulate cardiac myocyte development and disease in the human heart in vivo. Nat Commun 9 (1), 391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Adriaens C et al. (2018) Blank spots on the map: some current questions on nuclear organization and genome architecture. Histochem Cell Biol 150 (6), 579–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Poleshko A et al. (2017) Genome-Nuclear Lamina Interactions Regulate Cardiac Stem Cell Lineage Restriction. Cell 171 (3), 573–587 e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stevens TJ et al. (2017) 3D structures of individual mammalian genomes studied by single-cell Hi-C. Nature 544 (7648), 59–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nora EP et al. (2017) Targeted Degradation of CTCF Decouples Local Insulation of Chromosome Domains from Genomic Compartmentalization. Cell 169 (5), 930–944 e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rosa-Garrido M et al. (2017) High-Resolution Mapping of Chromatin Conformation in Cardiac Myocytes Reveals Structural Remodeling of the Epigenome in Heart Failure. Circulation 136 (17), 1613–1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lee DP et al. (2019) Robust CTCF-Based Chromatin Architecture Underpins Epigenetic Changes in the Heart Failure Stress-Gene Response. Circulation 139 (16), 1937–1956. [DOI] [PubMed] [Google Scholar]
- 53.Bintu B et al. (2018) Super-resolution chromatin tracing reveals domains and cooperative interactions in single cells. Science 362 (6413). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang Y et al. (2019) Transcriptionally active HERV-H retrotransposons demarcate topologically associating domains in human pluripotent stem cells. Nat Genet. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Klemm SL et al. (2019) Chromatin accessibility and the regulatory epigenome. Nat Rev Genet 20 (4), 207–220. [DOI] [PubMed] [Google Scholar]
- 56.Abel ED and Doenst T (2011) Mitochondrial adaptations to physiological vs. pathological cardiac hypertrophy. Cardiovasc Res 90 (2), 234–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gibb AA and Hill BG (2018) Metabolic Coordination of Physiological and Pathological Cardiac Remodeling. Circ Res 123 (1), 107–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Carey BW et al. (2015) Intracellular alpha-ketoglutarate maintains the pluripotency of embryonic stem cells. Nature 518 (7539), 413–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lombardi AA et al. (2019) Mitochondrial calcium exchange links metabolism with the epigenome to control cellular differentiation. Nat Commun 10 (1), 4509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Erdel F et al. (2010) Human ISWI chromatin-remodeling complexes sample nucleosomes via transient binding reactions and become immobilized at active sites. Proc Natl Acad Sci U S A 107 (46), 19873–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Padinhateeri R and Marko JF (2011) Nucleosome positioning in a model of active chromatin remodeling enzymes. Proc Natl Acad Sci U S A 108 (19), 7799–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Iyer KV et al. (2012) Mechanical activation of cells induces chromatin remodeling preceding MKL nuclear transport. Biophys J 103 (7), 1416–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dang L et al. (2009) Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 462 (7274), 739–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ward PS et al. (2010) The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell 17 (3), 225–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Michalak EM et al. (2019) The roles of DNA, RNA and histone methylation in ageing and cancer. Nat Rev Mol Cell Biol 20 (10), 573–589. [DOI] [PubMed] [Google Scholar]
- 66.Janke R et al. (2015) Metabolism and epigenetics. Annu Rev Cell Dev Biol 31, 473–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Flavahan WA et al. (2016) Insulator dysfunction and oncogene activation in IDH mutant gliomas. Nature 529 (7584), 110–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Letouze E et al. (2013) SDH mutations establish a hypermethylator phenotype in paraganglioma. Cancer Cell 23 (6), 739–52. [DOI] [PubMed] [Google Scholar]
- 69.Sigismund S et al. (2018) Emerging functions of the EGFR in cancer. Mol Oncol 12 (1), 3–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yang W et al. (2012) EGFR-induced and PKCepsilon monoubiquitylation-dependent NF-kappaB activation upregulates PKM2 expression and promotes tumorigenesis. Mol Cell 48 (5), 771–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yang W et al. (2012) ERK1/2-dependent phosphorylation and nuclear translocation of PKM2 promotes the Warburg effect. Nat Cell Biol 14 (12), 1295–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yang W et al. (2012) PKM2 phosphorylates histone H3 and promotes gene transcription and tumorigenesis. Cell 150 (4), 685–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Luo W et al. (2011) Pyruvate kinase M2 is a PHD3-stimulated coactivator for hypoxia-inducible factor 1. Cell 145 (5), 732–44. [DOI] [PMC free article] [PubMed] [Google Scholar]