

Abstract
Rationale:
TRIC-A and TRIC-B are distributed to endoplasmic reticulum (ER)/sarcoplasmic reticulum (SR) intracellular Ca2+ stores. The crystal structure of TRIC has been determined, confirming the homo-trimeric structure of a potassium channel. While the pore architectures of TRIC-A and TRIC-B are conserved, the carboxyl-terminal tail domains (CTT) of TRIC-A and TRIC-B are different from each other. Aside from its recognized role as a counter-ion channel that participates in excitation-contraction coupling of striated muscles, the physiological function of TRIC-A in heart physiology and disease has remained largely unexplored.
Objective:
In cardiomyocytes, spontaneous Ca2+ waves, triggered by store overload-induced Ca2+ release (SOICR) mediated by the type 2 ryanodine receptor (RyR2), develop extra-systolic contractions often associated with arrhythmic events. Here we test the hypothesis that TRIC-A is a physiologic component of RyR2-mediated Ca2+ release machinery that directly modulates SOICR activity via CTT.
Methods and Results:
We show that cardiomyocytes derived from the TRIC-A−/− mice display dysregulated Ca2+ movement across SR. Biochemical studies demonstrate a direct interaction between CTT-A and RyR2. Modeling and docking studies reveal potential sites on RyR2 that show differential interactions with CTT-A and CTT-B. In HEK293 cells with stable expression of RyR2, transient expression of TRIC-A, but not TRIC-B, leads to apparent suppression of spontaneous Ca2+ oscillations. Ca2+ measurements using the cytosolic indicator Fura-2 and the ER luminal store indicator D1ER suggest that TRIC-A enhances Ca2+ leak across the ER by directly targeting RyR2 to modulate SOICR. Moreover, synthetic CTT-A peptide facilitates RyR2 activity in lipid bilayer reconstitution system, enhances Ca2+ sparks in permeabilized TRIC-A−/− cardiomyocytes, and induces intracellular Ca2+ release after microinjection into isolated cardiomyocytes, whereas such effects were not observed with the CTT-B peptide. In response to isoproterenol stimulation, the TRIC-A−/− mice display irregular electrocardiogram and develop more fibrosis than the WT littermates.
Conclusions:
In addition to the ion-conducting function, TRIC-A functions as an accessory protein of RyR2 to modulate SR Ca2+ handling in cardiac muscle.
Keywords: RYR2 gene, calcium signaling, excitation-contraction coupling, SOICR, TMEM38a
Subject Terms: Basic Science Research, Calcium Cycling/Excitation-Contraction Coupling, Myocardial Biology
Graphical Abstract
TRIC channels were discovered in 2007. Aside from its recognized role as a counter-ion channel, the physiological function of TRIC-A in the heart has remained largely unexplored. Even with the recent resolution of the crystal structure of the TRIC channels, the interacting partners for TRIC have yet to be defined. This study uncovered a novel function for TRIC-A as an accessory protein that interacts with the RyR to modulate intracellular Ca2+ release in cardiomyocytes. The CTT domain of TRIC-A, an important portion of the channel that is left out of the crystal structure determination, constitutes an active motif that interacts with RyR. The vulnerability of the TRIC-A knockout mouse heart to handling of isoproterenol stimulation further support the important physiological function of TRIC-A in heart physiology and disease.
INTRODUCTION
In cardiac muscle, sarcolemma depolarization triggers the release of Ca2+ from the sarcoplasmic reticulum (SR) via Ca2+-induced Ca2+ release (CICR)1–3. During this process, Ca2+ influx through the voltage-dependent Ca2+ channels on the sarcolemma activates the type 2 ryanodine receptor (RyR2) channel located on the SR. Under certain pathological conditions, spontaneous Ca2+ release from the SR can take place in the absence of membrane excitation due to store overload induced Ca2+ release (SOICR)4–10. SOICR may evoke propagating Ca2+ waves that can further result in delayed after-depolarizations, which may cause arrhythmias in heart failure.
The SR Ca2+ store of cardiomyocytes is maintained by uptake and release processes, both of which are electrogenic events. The release of Ca2+ through the RyR2 channels will lead to the development of a negative potential inside the SR lumen, and this would further limit Ca2+ release from the SR if uncompensated. Likewise, SERCA-mediated uptake of Ca2+ into the SR would lead to the accumulation of a positive potential within the SR lumen, and that would tend to inhibit Ca2+-pumping function. Thus coordinated counter-ion movements are required to balance the SR membrane potential in order to maintain efficient Ca2+ release and uptake in cardiomyocytes.
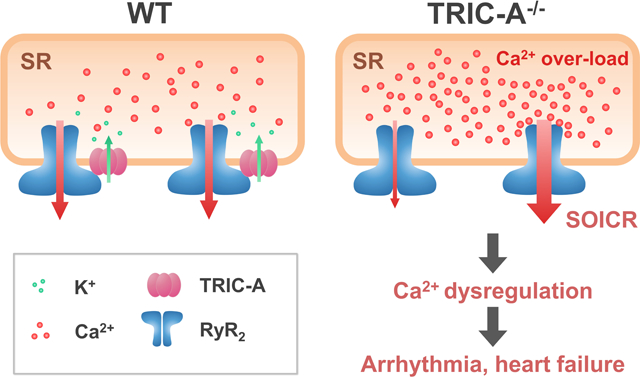
In 2007, Takeshima and colleagues identified trimeric intracellular cation (TRIC) channels located on the SR and endoplasmic reticulum (ER) of multiple cell types11. In human and mouse genomes, two isoforms of TRIC genes were identified: TRIC-A, a subtype predominantly expressed in SR of excitable cells, and TRIC-B, a ubiquitous subtype expressed in ER of all tissues11–21. TRIC-A and TRIC-B appear to have different functions in Ca2+ signaling in excitable and non-excitable cells, since knocking out TRIC-B affects IP3 receptor mediated Ca2+ release in airway epithelial cells, which results in respiratory defects and neonatal death with TRIC-B−/− pups21. The TRIC-A−/− mice, in addition to dysfunction of skeletal muscle20, develop hypertension that is linked to defective Ca2+ sparks and spontaneous transient outward currents in arterial smooth muscle19. A common feature with the TRIC-A−/− and TRIC-B−/− mice is the development of Ca2+ overload inside the SR/ER of multiple tissues11, 19–21. This Ca2+ overload can impact the function of SOICR, causing instability of Ca2+ movement across the SR membrane in muscle cells, which could further contribute to tissue dysfunctions associated with ablation of the TRIC genes.
While researchers over the past ten years have established that genetic ablations or mutations of TRIC channels are associated with hypertension, muscle dysfunction, respiratory defects, and brittle bone disease11–21, the function of TRIC channels in heart physiology and disease has yet to be established. El-Ajouz et al22 reported dampened activity of RyR channels in skeletal muscle lacking TRIC-A, suggesting TRIC-A could play a modulatory role in control of RyR-mediated intracellular Ca2+ release. Recently, the crystal structure of TRIC has been determined, confirming the homo-trimeric structure of a potassium channel23–26. While the pore architectures of TRIC-A and TRIC-B appear to be conserved, the carboxyl-terminal tail (CTT) domains of TRIC-A and TRIC-B are different from each other. These CTT domains show the flexible structure and potentially interfere with crystal formation, thus all available structural determinations of the TRIC channels were obtained without the CTT domain.
Here we report that cardiomyocytes derived from the TRIC-A−/− mice show dysregulated Ca2+ movement across the SR membrane. Our biochemical and immunohistochemical studies show that TRIC-A can physically associate with RyR2 via the CTT domain. Reconstitution studies in the lipid bilayer membrane, Ca2+ measurements in adult cardiomyocytes and heterologous cells demonstrate that TRIC-A, in addition to serving as a counter-ion channel, can interact with RyR2 to modulate the intracellular Ca2+ homeostasis and consequently the operation of SOICR.
METHODS
The data, analytical methods, and other study materials will be provided upon reasonable request to the corresponding author.
Cardiomyocyte isolation from adult mice.
TRIC-A knock out (TRIC-A−/−) mice used in this study were reported in our previous publication11. Handling of mice followed IACUC guidelines from The Ohio State University. Isolated hearts from adult TRIC-A−/− and wild type (WT) littermate mice (10–12 weeks of both genders) were perfused with a Langendorff apparatus at 37°C. The enzyme digestion step consisted of perfusing Tyrode’s solution containing 1 mg/ml collagenase (Type II, 300 U/mg; Worthington) and 0.1 mg/ml protease (Type XIV) for 6 min. Cardiomyocytes were dissociated from digested ventricles by gentle mechanical dissociation and used within 3 hours. The Tyrode’s solution contained (in mM) 136 NaCl, 5.4 KCl, 0.33 NaH2PO4, 1.0 MgCl2, 10 glucose and 10 HEPES (pH 7.4).
Mice and isoproterenol-treatment protocol.
Alzet mini-osmotic pump (Alza Corp.) containing either isoproterenol (ISO), phosphate-buffered saline (PBS) as a control were surgically inserted subcutaneously in mice under isoflurane anesthesia. The mini-osmotic pump provides controlled delivery of ISO (60 mg/kg/day). In separate study, 80 mg/kg ISO was injected once intraperitoneally in mice under isoflurane anesthesia. 1 hour later, electrocardiogram (ECG) was measured using Animal Bio Amp and PowerLab 8/30, and analyzed by LabChart 7.0 (ADInstruments).
Hearts from the TRIC-A−/− and WT mice were collected at the indicated times after ISO treatment, fixed in 10% formalin-containing PBS, and embedded in paraffin. Serial 4-μm heart sections were cut and stained with H & E and Masson’s trichrome. Blinding was not performed because the genotype of mice was already known before the experiments were conducted.
Ca2+ sparks and waves measurement in cardiomyocytes.
Intracellular Ca2+ sparks of intact cardiomyocytes isolated from adult mouse (4–8 months age) were recorded using a Zeiss780 confocal microscope with a 40× 1.42 NA oil immersion objective. Cardiomyocytes were loaded with Fluo-4-AM (2 μM) and then stimulated with a field stimulation of 0.5 Hz for 20 seconds in a normal Tyrode’s solution containing 1.8 mM Ca2+. Spontaneous Ca2+ spark activities were measured afterward. Experiments were conducted at room temperature (24–26 °C).
Isolated cardiomyocytes were loaded with 5 μM Fluo-4 AM (Thermo Fisher Scientific) in 1.8 mM Ca2+ physiological salt solution (PSS) (in mM): 130 NaCl, 5.6 KCl, 1 MgCl2, 11 glucose, and 10 Hepes, pH 7.4 for 30 min at room temperature. Line scan images of Fluo-4 fluorescence intensity were acquired at a sampling rate of 2 ms per line using the Galvano scan mode of a Nikon A1R confocal microscope. During the 1 min line scan imaging, cardiomyocytes were subjected to an electric field stimulation of 0.2 Hz, 10 ms duration pulses of 6 V.
To examine the impact of store-overload induced intracellular Ca2+ release in the isolated cardiomyocytes, extracellular Ca2+ was increased from 1.8 mM to 10 mM in PSS containing 50 μM BTS to mitigate myocyte contraction. Serial x-y images of cardiomyocytes in 10 mM Ca2+ PSS were acquired at 130 ms per frame using the resonant scan mode of Nikon A1R confocal microscope. All image data were analyzed by Fiji-ImageJ.
Separate studies were performed with Ca2+ spark measurement in saponin-permeabilized cardiomyocytes, following the protocol of Lukyanenko and Gyorke27 and Guo et al28. Specifically, cardiomyocytes were permeabilized with saponin (0.01% for ~45 s). The MaxChelator program (URL: maxchelator.stanford.edu) developed by Bers et al29 was used to prepare the solution for precise control of free cytosolic Ca2+. The cytosolic solution contained (mM): 120 potassium aspartate, 5 MgATP, 10 phosphocreatine, 0.03 Fluo-4 pentapotassium salt, 10 Hepes (pH 7.25) and 5 U/ml creatine phosphokinase, with 0.4 EGTA and 0.087 CaCl2 which buffers free Ca2+ at 100 nM. The Fluo-4 dye was excited with the 488 nm line of a LED laser (0.30% of maximum power, Nikon-A1R), pinhole 2.0 (equivalent to 39.6 μm). The use of low laser-power avoids laser-induced damage to cardiomyocytes during line-scan imaging of Ca2+ sparks using the Nikon-A1R imaging system. During Ca2+ spark measurement, 0.001% saponin was continuously present in the cytosolic solution to avoid resealing of the sarcolemmal membrane. Quantification and characterization of Ca2+ sparks were performed using SparkMaster ImageJ program30.
Plasmid construction.
The mouse TRIC-A or TRIC-B cDNA was cloned into pCMS-mRFP vector (Clontech). The pCMS-mRFP plasmid contained a red fluorescent protein (RFP) reporter gene driven under a separate SV40 promoter, allowing for selection of cells transfected with TRIC-A or TRIC-B using red fluorescence. For the investigation of the carboxyl-terminal tail (CTT) domain of TRIC on Ca2+ signaling, two chimeras were constructed, TRIC-AB and TRIC-BA. TRIC-AB contains a.a. 1–230 from TRIC-A plus a.a. 229–291 from TRIC-B. TRIC-BA contains a.a. 1–228 from TRIC-B plus a.a. 231–299 from TRIC-A. The cDNA coding sequences were chemically synthesized (Quintara Biosciences, CA) and cloned into the pCMS-mRFP vector to express the chimeric TRIC proteins in mammalian cells. All plasmid identities were confirmed by gene sequencing.
Baculovirus production and its infection to HEK293 cells.
Baculovirus carrying vesicular stomatitis virus G-protein (VSVG) on the virus envelope that effectively infects mammalian cells was produced as described previously31. cDNAs for TRIC-A and TRIC-B were cloned into the modified pFastBac1 vector (Invitrogen). Baculovirus was produced in Sf9 cells according to the manufacturer’s instructions (Invitrogen). P2 virus was used for the experiments. HEK293 cells inducibly expressing RyR2 which were kindly provided by Dr. Nagomi Kurebayashi or control HEK293 cells were cultured in DMEM, 5% FBS, 1% penicillin, and streptomycin at 37°C, 5% CO2. P2 virus solution was supplemented to the culture media to infect the virus and cells were observed at 36 hours post infection.
Ca2 + imaging in HEK293 cells.
HEK293 cells with inducible expression of RyR2 were provided by Dr. Wayne Chen and cultured in DMEM, 5% FBS, 1% penicillin, and streptomycin at 37°C, 5% CO25. pCMS-mRFP plasmids containing the various TRIC-A and TRIC-B cDNAs were transfected into HEK293 cells using lipofectamine reagent following manufacture’s instruction. 24 hours later, tetracycline (0.1 μg/ml) was added to the culture medium to induce RyR2 expression. Measurements of cytosolic Ca2 + were conducted at 16–18 hours post tetracycline induction. Cells were loaded with Fura-2-AM (5 μM) or Fluo-4 AM (2 μM) in 0 Ca2 + Krebs–Ringer Hepes (KRH) buffer containing (mM): 125 NaCl, 5 KCl, 25 HEPES, 6 glucose, and 1.2 MgCl2, pH 7.4 for 40 min at room temperature. All solutions were prepared with double-distilled H2O (18.2 MΩ cm resistance). Cells were then continuously perfused with KRH solution containing different concentrations of CaCl2 (0–2.0 mM) at room temperature. Fura-2 fluorescence was captured by a dual-wavelength excitation spectrofluorometer (Photon Technology International, Monmouth Junction, NJ) with fast switching of 340 nm and 380nm excitations. Fluorescence of Fluo-4 was excited at 488 nm and detected by a Zeiss 780 confocal microscope.
In separate studies, HEK-293 cells with inducible expression of RyR2 were co-transfected with TRIC-A and D1ER cDNA, or TRIC-B and D1ER to determine the impact of TRIC-A/TRIC-B on the ER Ca2+ stores. Cells were perfused continuously at room temperature with KRH containing 2 mM CaCl2. Tetracaine (2 mM) was used to block RyR2 activity, and caffeine (10 mM) was used to activate RyR2 to deplete the ER Ca2+ store. Cells were excited at 458 nm, and emissions of yellow fluorescent protein (YFP) and cyan fluorescent protein (CFP) from D1ER were captured every 0.5 s. The amount of fluorescence resonance energy transfer (FRET) was determined by the ratio of the emissions at 535 and 480 nm5, 32.
Immunoblotting.
HEK293 cells expressing RyR2 and TRIC-A were harvested and lysed in RIPA buffer (50 mM Tris-HCl pH 7.4, 150 mM NaCl, 1% Triton x-100, 1% sodium deoxycholate, 0.1% SDS, 1 mM EDTA) supplemented with a protease inhibitor cocktail (PIC, Sigma) for 10 min at 4°C. PIC contains (final concentration) AEBSF (1 mM), aprotinin (0.8 μM), bestatin (40 μM), E-64 (14 μM), leupeptin (20 μM) and pepstatin A (15 μM). Heart lysates derived from wild type mouse were used for positive control of TRIC-A and RyR2. Mouse hearts were minced and re-suspended in PIC containing RIPA buffer. Heart protein extraction was done by three-cycle of freeze-thawing on dry ice and extracted with pestle homogenizer with disposable polypropylene pestles (Thermo Fisher Scientific). Cells or tissue lysates were centrifuged (20,000 × g, 15 min. at 4°C) and protein containing supernatant was collected. Protein concentration was determined using a Bio-Rad Protein Assay kit. 10 μg of protein were loaded on 12% or 5% SDS-PAGE and transferred onto a polyvinylidene difluoride membrane for detection of TRIC-A or RyR2 expressions, respectively. The expression levels of TRIC-A or RyR2 were visualized using specific antibodies anti-RyR2 (34C) (1:3000) (Abcam, ab2868) and anti-TRIC-A (1:2000) (Proteintech Group, 19920–1-AP).
Immunofluorescence staining.
Isolated mouse cardiomyocytes were washed by phosphate buffered saline (PBS) and fixed in 4% phosphate-buffered paraformaldehyde (PFA) for 20 min at room temperature, and then permeabilized for 15 min with 0.2% Triton X-100 in PBS. Cells were blocked by 3% BSA for 40 min and then incubated with TRIC-A (Proteintech Group, 19920–1-AP) and RyR2 antibodies (1:200, Abcam, ab2868) at 4°C overnight. Alexa 647 labeled goat anti-rabbit (Thermo Fischer A21244) and Alexa 546 labeled goat anti-mouse (Thermo Fisher A11003) secondary antibodies were applied and incubated for 1 h at 37 °C. After a brief wash, DAPI was used to stain the nucleus for 5 min and then washed by PBST 5 times. TRIC-A−/− mice heart was used as negative control for TRIC-A antibody and IgG only staining was used for RyR2 antibody staining that confirmed the specificity of both antibodies. Images were captured by Zeiss LSM 780 confocal microscope and analyzed by ImageJ.
GST-CTT-A protein expression and beads binding assay.
cDNA coding for the CTT domain of TRIC-A (a.a. 231–299) was cloned into the pGEX4T-1 bacteria expression plasmid. The pGEX-CTT-A plasmid was transformed into E. coli (DH5α), and expression of GST-CTT-A protein was induced with 0.3 mM isopropyl-β-D-thiogalactopyranoside (IPTG, Sigma) for 3.5 h at 30°C. Harvested bacterial pellets were re-suspended in a buffer containing 20 mM Tris-HCl, 200 mM NaCl, and 0.2 mM PMSF (pH 7.5) supplemented with 0.5% protease inhibitor cocktails (PIC, Sigma), 1 mg/ml lysozyme, and 1 mg/ml MgCl2, and sonicated (Branson Ultrasonics) for 15 s for 5 cycles at 30% power output. The lysates were centrifuged twice at 15,000 rpm for 30 min at 4°C. The cleared supernatants were diluted 1:4 into the suspension buffer, mixed with Glutathione Sepharose Beads (Amersham) and incubated at 4°C overnight on an orbital shaker. The mixture was then loaded into a poly-prep chromatography column (Bio-Rad), allowed to precipitate by gravity, and then washed with 50 ml buffer containing 20 mM Tris-HCl, 200 mM NaCl, 0.2 mM PMSF and PIC, pH 7.5. C1148 cells with stable expression of RyR1 were described previously33, 34. Microsomal vesicles derived from C1148 cells or SR vesicles derived from rat heart (kindly provided by Dr. Michael Fill, Rush University, Chicago) were lysed in RIPA buffer supplemented with PIC, and loaded to either GST or GST-CTT-A bound beads and incubated at 4°C on an orbital shaker for 3 hours. The bound proteins were washed 4 times with buffer containing 0.5% NP-40, 20 mM Tris (pH 7.4) supplemented with PIC, eluted with sample loading buffer, and loaded onto SDS-PAGE gel and western blotted with anti-RyR (1:3000, Abcam, ab2868).
Channel activity assay on planar bilayer system.
SR vesicles isolated from rat hearts were used for lipid bilayer reconstitution studies of single RyR2 channels35, 36. The SR vesicles were kindly provided by Dr. Michael Fill at Rush Medical University28. Planar lipid bilayers were formed with a lipid composition of phosphatidylethanolamine and phosphatidylserine (Avanti Polar Lipids, Birmingham, AL) (1:1 ratio, dissolved in decane, 50 mg/ml) across a 200 μm diameter hole in a polystyrene partition. CTT-A and CTT-B peptides were synthesized by GL Biochem (Shanghai, China) with >95% purity. SR vesicles and CTT-A or CTT-B peptides were added to one chamber using cis design, composed of 250 mM CsCl, 0.22 mM CaCl2, 1 mM EGTA, 10 mM HEPES, pH 7.4. The other chamber, designated as trans, contained 50 mM CsCl, 10 mM HEPES, pH 7.4. Single channel data acquisition and analysis were conducted with pClamp software.
Microinjection of CTT-A and CTT-B peptide.
CTT-A and CTT-B peptides were dissolved in PBS solution at a concentration of 10 μM. Freshly isolated wild type mouse cardiomyocytes were loaded with 5 μM Fluo-4 AM. Microinjection pipettes made of borosilicate glass (Sutter Instrument Co.) were prepared using a micropipette puller (Model P-97, Sutter Instrument Co.). The microfilament inside the pipette ensures that the solution reaches the tip for microinjection. The tip pore of the pipette was adjusted to allow for delivery of 10–20 pico liter of peptide solution in 60 ms using a picospritzer (Parker Instrumentation) coupled to PatchMan micromanipulator (Eppendorf). Changes in intracellular [Ca]i were monitored under a BioRad confocal microscope.
Modeling and Docking of CTT-A and CTT-B with RyR2.
The atomic structures of CTT-A and CTT-B, were modeled using the I-TASSER server37. The atomic structure of human RyR2 (hRyR2) has not been solved. However, the cryo-EM structures of porcine RyR2 (pRyR2) are available in the Protein Data Bank38 (PDB ID: 5goa, open state; PDB ID: 5go9, closed state)39. Due to the large size of RyR2, traditional homology modeling cannot be used to model human RyR2 using pRyR2 structures. Instead, mutations were introduced in the structures of pRyR2 (5goa and 5go9, respectively) following the hRyR2 sequence, using UCSF Chimera40. The structures of CTT-A and CTT-B were respectively docked onto the homology model of hRyR2 using our in-house docking software, MDockPP41, 42. The P1 domain, P2 domain, and channel domain were blocked from docking to reduce the enormous search space on the huge RyR2 protein, as the peptides unlikely interact with these domains. Four potential binding sites of CTT-A/CTT-B were identified, and the representative binding mode in each site was optimized using the minimization tool of UCSF Chimera to remove intermolecular side chain clashes. The differential binding modes in the binding sites were further compared between the open and closed state of the hRyR2 channel.
Data handling and statistical analysis.
D’Agostino & Pearson normality test was used to evaluate normality of the data. If normal, we used unpaired t-test for single comparison and Tukey’s test for multiple comparisons. If not normal, we used non-parametric test (Mann-Whitney test for single comparison or Dunn’s test for multiple comparisons) to derive the p-value. Before doing a Tuckey’s or Dunn’s test for multiple comparisons, ANOVA or Kruskal-Wallis (if not passed normality test) test was conducted for the whole group.
Data are represented as mean ± standard deviation. A value of P<0.05 was considered significant. All data were analyzed using Excel and Prism 8 software.
RESULTS
Abnormal Ca2+ signaling in cardiomyocytes derived from the TRIC-A−/− mice.
We have generated knockout mice carrying the deletion of either TRIC-A or TRIC-B. While TRIC-A−/− mice survived past their adolescent age, homozygous ablation of TRIC-B proved lethal as the TRIC-B−/− mice died at the neonatal stage due to respiratory dysfunction21. Moreover, the aggravated embryonic fatality was observed with the TRIC-A−/−TRIC-B−/− mice, demonstrating the essential role of TRIC in development11, 17. Using cardiomyocytes derived from the TRIC-A−/− adult mice and littermate wild type (WT) controls (10–12 weeks age), we examined Ca2+ spark signaling properties following electrical pacing. As shown in Fig. 1A, following a 0.5 Hz electrical field stimulation for 20 s, spontaneous Ca2+ sparks were observed in the isolated cardiomyocytes upon the termination of the electric stimulation. Compared with the WT cardiomyocytes, the TRIC-A−/− cardiomyocytes showed less frequent Ca2+ sparks (Fig. 1B, left). Interestingly, the amplitudes of individual Ca2+ sparks appear to be significantly higher in TRIC-A−/− cardiomyocytes than those in WT cardiomyocytes (Fig. 1B, middle).
Fig. 1. Compromised Ca2+ signaling in cardiomyocytes derived from TRIC-A−/− mice.

A. Confocal line scan images of Ca2+ sparks in isolated cardiomyocytes from WT and TRIC-A−/− mice, after pacing by 0.5 Hz of electric field stimulation for 20 s. B. Ca2+ spark amplitude was higher (p=0.0091, 2-tailed t-test) and spark frequency was lower (p=0.0098, 2-tailed t-test) in TRIC-A−/− cardiomyocytes, compared with WT cardiomyocytes. Caffeine-induced Ca2+ release was significantly higher (p=0.0487, 2-tailed t-test) in TRIC-A−/− cardiomyocytes. C. Line-scan imaging of electric pacing induced intracellular Ca2+ transients, demonstrating the different patterns in WT and TRIC-A−/− cardiomyocytes. Data from individual cardiomyocytes were plotted. Rising time (D) (P=0.0090, 2-tailed t-test), full-duration-half-maximum (FDHM) (E) (P= <0.0001, Mann-Whitney test), and peak amplitude of electric pacing induced Ca2+ transients (F) (P=0.7473, Mann-Whitney test) are plotted for TRIC-A−/− (red) and WT (black) cardiomyocytes. G. Different propagating patterns of store-overload induced Ca2+ waves in WT and TRIC-A−/− cardiomyocytes. Speed of Ca2+ wave propagation (H) (P=0.0001, 2-tailed t-test), percentage of oscillating cells (I) (p=0.0205, 2-tailed t-test), and interval between Ca2+ waves (J) (n=165 for WT, n=89 for TRIC-A−/−, p=0.0047, Mann-Whitney test) were significantly larger in TRIC-A−/− cardiomyocytes. * P<0.05, ** P<0.01, **** P<0.0001.
We also compared caffeine-induced Ca2+ release from the WT and TRIC-A−/− cardiomyocytes. As illustrated in (Fig. 1B, right), the total caffeine-releasable Ca2+ pool from the SR was significantly higher in TRIC-A−/− than in WT cardiomyocytes. This observation is consistent with our published data with skeletal muscle20 and epithelial cells21 where the absence of TRIC-A or TRIC-B led to elevated Ca2+ storage inside the SR or ER.
Confocal line scan imaging of Fluo-4 fluorescence revealed different patterns of intracellular Ca2+ transients in WT and TRIC-A−/− cardiomyocytes following electric field stimulation (Fig. 1C). There appeared to be a delayed onset of intracellular Ca2+ transients and a prolonged decaying phase of Ca2+ transients in the TRIC-A−/− cardiomyocytes compared with those in the WT cardiomyocytes. On average, the rising time of Ca2+ transients was significantly longer in TRIC-A−/− cardiomyocytes when compared with WT cardiomyocytes (Fig. 1D). The full-duration-half-maximal (FDHM) of Ca2+ transients was significantly longer in the TRIC-A−/− cardiomyocytes (Fig 1E), whereas the amplitude of Ca2+ transients showed no significant difference (Fig. 1F) between the WT and TRIC-A−/− cardiomyocytes. Studies from other investigators demonstrated that reduced RyR2 channel activity could contribute to the delayed time-to-peak and prolonged decaying phase of electric pacing-induced Ca2+ release from cardiomyocytes43–45. This suggests the possibility that ablation of TRIC-A in cardiomyocytes could lead to reduced function of the RyR2 channel.
Using the resonant scan mode of the Nikon A1R confocal microscope, we performed continuous x-y imaging of spontaneous Ca2+ waves (at a rate of 130 ms per frame) in the isolated cardiomyocytes upon elevation of the extracellular Ca2+ from 1.8 mM to 10 mM. Such fast time-lapse imaging enabled us to examine the impact of TRIC-A ablation on the speed of Ca2+ wave propagation. As shown in Fig. 1G, the speed of Ca2+ wave propagation appeared to be faster in WT than in TRIC-A−/− cardiomyocytes (see Online Movie I and II). Measurements from multiple experiments support the statistical difference in the speed of Ca2+ wave propagation between WT and TRIC-A−/− cardiomyocytes (Fig. 1H). Moreover, the percentage of cells that displayed Ca2+-overload induced spontaneous Ca2+ waves was significantly lower in TRIC-A−/− cardiomyocytes than that in WT cardiomyocytes (Fig. 1I). In Online Movie III and IV, one could see the different behavior of Ca2+ waves in WT and TRIC-A−/− cardiomyocytes.
Isoproterenol treatment induces irregular ECG and cardiac fibrosis in TRIC-A−/− mice.
Under basal condition, there was no clear difference in electrocardiograms (ECG) recorded in the TRIC-A−/− mice and WT littermates (Fig. 2A). However, following a bolus intraperitoneal injection of isoproterenol (ISO, 80 mg/kg), irregular ECG was observed with the TRIC-A−/− mice at 1 hour post ISO injection, whereas the WT mice displayed regular patterns of ECG. Analysis of the R-R intervals between the individual ECGs demonstrated large variations, confirming the irregularity of ISO-induced ECG with the TRIC-A−/− mice (Fig. 2B).
Fig. 2. Irregular ECG and increased cardiac fibrosis in TRIC-A−/− mice upon stimulation with isoproterenol.

A. Representative traces of ECG in WT and TRIC-A−/− mice before and after treatment of ISO. B. Distribution of ECG R-R intervals at 1 hr after 80 mg/kg ISO injection. (P<0.0001, Mann-Whitney test) C. Trichrome stain of WT and TRIC-A−/− heart at 2 weeks after treatment of PBS or ISO (60 mg/kg/day). D. Summary data with percentage of fibrosis area in WT and TRIC-A−/− heart stained by Mason’s trichrome (P<0.001 across the groups, ANOVA test; WT+ISO vs TRIC-A−/−+ISO: P<0.0001, Tukey’s test).
We next tested if ablation of TRIC-A could alter the response of the mice to chronic treatment with ISO. With chronic treatment of ISO (60 mg/kg/day for 2 weeks, delivered using an Alzet mini-osmotic pump), the TRIC-A−/− heart would develop extensive fibrosis than the WT heart (Fig. 2C). Mason’s trichrome staining revealed significant increase in fibrosis in the heart tissue derived from the TRIC-A−/− mice at 2 weeks post ISO treatment, compared with the WT mice (Fig. 2D).
Thus, chronic exposure to ISO likely caused increased death of cardiomyocytes and fibrotic remodeling, which may be linked to the overload of SR Ca2+ associated with ablation of TRIC-A. In response to ISO, SR Ca2+ overload may cause mitochondria Ca2+ toxicity and apoptosis in the TRIC-A−/− cardiomyocytes. Further dissecting the role of TRIC-A in modulating the Ca2+ signaling crosstalk from SR to mitochondria will require dedicated effort in future studies.
Co-expression of TRIC-A and RyR2 in HEK293 cells modulates SOICR.
We used HEK293 cells with tetracycline-inducible expression of RyR2 to investigate the impact of TRIC-A and TRIC-B on RyR2-mediated intracellular Ca2+ signaling5, 6 (Fig. 3A). In this model, the elevation of extracellular [Ca]o lead to increased Ca2+ content inside the ER, which triggered the opening of the RyR2 channel via its luminal Ca2+ sensing mechanism, leading to the appearance of SOICR. Previously, we showed that co-expression of RyR1 and MG29 in Chinese hamster ovary (CHO) cells caused depletion of the ER Ca2+ store and consequently apoptotic cell death46. Interestingly, we found that transient co-expression of RyR2 and TRIC-A, but not TRIC-B, in HEK293 cells led to changes in cell morphology, and a large portion of the cells expressing TRIC-A and RyR2 would die within 36 hours (Fig. 3B), suggesting cytotoxicity by potential perturbation of ER Ca2+ homeostasis.
Fig. 3. Effect of TRIC-A and TRIC-B on RyR2-mediated SOICR in HEK293 cells.

A. Western blot of RyR2 and TRIC-A in HEK293 cells with tetracycline-inducible expression of RyR2. Mouse heart lysate was used as positive control of RyR2 and TRIC-A expression. B. Representative images of HEK293 cells without RyR2 expression (top) and with RyR2 expression (bottom). Following baculovirus infection, only cells co-expressing RyR2 and TRIC-A became round and detached from the plate (middle-bottom panel). Co-expression of RyR2 and TRIC-B had no effect on cell viability. Scale bar, 200 μm. C. Line-scan image of Ca2+ oscillation, representing SOICR, in HEK293 cells expressing RyR2 alone (non-red cell) and lack of Ca2+ oscillation in HEK293 cells co-expressing RyR2 and TRIC-A (red cell). D. Representative Fura-2 Ca2+ measurement in HEK293 cells with stable expression of RyR2 (blue trace), co-expression of TRIC-A and RyR2 (red trace), and co-expression of TRIC-B and RyR2 (green trace). E. Summary data with percentage of cells showing spontaneous Ca2+ oscillations in 0.5mM Ca2+. (P=0.0003 across groups, 2-way ANOVA; TRIC-A vs NT: P<0.0001, Sidak’s test). F. Co-expression of TRIC-A and RyR2 alters the dependence of SOICR on extracellular Ca2+ concentration. Co-expression of TRIC-B and RyR2 produced similar response of SOICR with changes of [Ca]o, as cells transfected with pCMS-RFP vector control. Data from 4 individual experiments were used for the statistical analyses. (P<0.0001 across groups, 2-way ANOVA; RyR2+TRIC-A vs RyR2+TRIC-B: P<0.0001, Sidak’s test) ****P<0.0001.
For assaying the effect of TRIC-A in HEK293 cells, we used a dual-reporter plasmid with mRFP-expression cassette driven by a separate promoter. This allows selection of transfected vs non-transfected cells in the same dish (Fig. 3C). From the line-scan measurements, it is clear that cells co-expressing RyR2 and TRIC-A (red labeled) did not show oscillating patterns of Ca2+ signaling, unlike those cells expressing RyR2 alone, which often display Ca2+ oscillations. A representative trace of Ca2+ measurement in a HEK293 cell with co-expression of TRIC-A and RyR2 (labelled red, Fig. 3D), illustrates the apparent absence of spontaneous Ca2+ oscillations. After treatment of tetracaine, a RyR2 blocker, SOICR was diminished and cytosolic Ca2+ level was decreased. At the end of the recording, caffeine was introduced to the dish to test the release of Ca2+ via RyR2. Ca2+ release from TRIC-A transfected cells triggered by caffeine confirmed the apparent suppression of SOICR is not due to the dysfunction of RyR2 (Fig. 3D, top). This effect appears to be specific to TRIC-A, as co-expression with TRIC-B did not affect RyR2-mediated SOICR in HEK293 cells (Fig. 3D, bottom). Summary data from multiple experiments is shown in Fig. 3E, indicating that TRIC-A, not TRIC-B, could modulate SOICR in HEK293 cells.
Studies from Chen and colleagues demonstrated that the frequency of RyR2-mediated SOICR in HEK293 cells was dependent on the concentration of Ca2+ in the extracellular solution5. Here we tested if co-expression of TRIC-A and RyR2 could modulate SOICR in a Ca2+-dependent manner. The data shown in Fig. 3F clearly suggest that the presence of TRIC-A could alter [Ca]o-dependent activation of SOICR. Moreover, this effect is TRIC-A specific, since cells with co-expression of TRIC-B and RyR2 showed similar response as those transfected with vector alone (as control).
To elucidate the mechanism of TRIC-A mediated modulation of SOICR, we used D1ER, a Ca2+ biosensor targeted to ER, to directly measure ER luminal Ca2+ level5, 32, 47. We followed the approach developed by Bers and colleagues to assay the ER Ca2+ content using tetracaine to inhibit RyR2 activity48, 49. This will allow for testing to what extent co-expression of TRIC-A alters the Ca2+ leak rate across the ER. As shown in Fig. 4A, HEK293 cells co-expressing RyR2 and TRIC-A displayed reduced ER Ca2+ content (measured with the D1ER probe), when compared with cells expressing RyR2 alone. While cells expressing RyR2 alone show oscillating patterns of ER Ca2+ content (Fig. 4A, blue trace) that mirrors the SOICR signal (Fig. 4B), co-expression with TRIC-A led to apparent suppression of the ER Ca2+ content below the oscillating threshold levels (Fig. 4A, red trace). In addition, we observed less Ca2+ release by direct caffeine treatment in TRIC-A expressing cells (Fig. 4B). This data further supports that TRIC-A reduced the ER Ca2+ content in the RYR2 expressing HEK293 cells.
Fig. 4. Effect of TRIC-A on changes in ER Ca2+ load require interaction with RyR2.

A. ER luminal Ca2+ measured by D1ER in cells expressing RyR2 (blue), or RyR2 + TRIC-A (red). 2 mM tetracaine inhibited ER Ca2+ oscillations and increased ER Ca2+ store and 10 mM caffeine caused depletion of ER Ca2+ store. (P=0.0042, 2-tailed t-test) B. Caffeine induced Ca2+ release from the ER store in cells expressing RyR2 alone (blue) and in cells with co-expression of TRIC-A and RyR2 (red) (p=0.0001, 2-tailed t-test) . C. Cytosolic Ca2+ release by ATP in HEK293 cells without RyR2 expression showed similar response with or without TRIC-A expression. (P=0.2667, Mann-Whitney test) D. D1ER measurement of CPA (SERCA inhibitor) treatment showed that TRIC-A overexpression did not affect the total ER Ca2+ and the ER Ca2+ uptake in HEK293 cells (2 mM Ca2+: P=0.2627; 2 mM Ca2++CPA: P=0.2695, 2-way ANOVA, Sidak’s test).
The effect of TRIC-A on ER Ca2+ handling likely require the participation of the RyR2 channel, since transfection of TRIC-A in HEK293 cells (in the absence of RyR2) did not affect the ER Ca2+ content as measured by ATP-induced Ca2+ release through IP3R (Fig. 4C) nor cytotoxicity (Fig. 3B). Moreover, when blocking ER Ca2+ uptake by cyclopiazonic acid (CPA), the decline of luminal Ca2+ is not affected by TRIC-A (Fig. 4D). These results support that altered ER Ca2+ signaling by TRIC-A is RyR2 dependent.
TRIC-A interact with RyR2 through its carboxyl-terminal tail (CTT) domain.
We performed immunohistochemical (IHC) staining of TRIC-A and RyR2 in cardiomyocytes derived from the WT mice. The clear overlap between TRIC-A and RyR2 could be observed (Fig. 5A). Images were analyzed by Pearson and Manders coefficients and showed significant co-localization (Fig. 5B).
Fig. 5. Co-localization of TRIC-A and RyR2 in mouse cardiomyocytes and CTT-A interaction with RyR.

A. Fluorescent staining shows TRIC-A and RyR2 are highly co-localized together in mouse isolated cardiomyocytes. B. Images were analyzed by scatter plot, Pearson coefficient (R) and Mander’s coefficient (M1 and M2). Bright yellow color of the scatterplot across the line indicates stronger co-localization. A value of 1 for R, M1/M2 indicates perfect co-localization and 0 indicates no co-localization; a threshold of 0.5 usually indicates good co-localization. M1 represents the degree of green fluorescence (TRIC-A) overlay with red fluorescence (RyR2); M2 represents the degree of red fluorescence overlay with green fluorescence. C. CTT-A and CTT-B diverge from each other. CTT-A contains a histidine-rich motif (orang) and a polylysine domain (purple), which are flanked by a hydrophobic domain (blue). Such structure is similar to the intracellular-loop joining repeats II and III of the L-type Ca2+ channel. Only the polylysine domain is present in CTT-B. D. GST-CTT-A can pull down RyR1 from C1148 cells. E. GST-CTT-A can pull down RyR2 from SR vesicle isolated from rat heart, and three other candidate proteins (labeled as a, b and c.). Western blot confirmed the pull-down of RyR2 from heart SR vesicle. GST alone does not pull down RyR2. F. TRIC-A could be immuno-precipitated with RyR2 in HEK293 cells co-expressing RyR2 and TRIC-A.
Topology analysis revealed that both TRIC-A and TRIC-B contained large CTT domains that reside in the cytosol (Fig. 5C). This CTT domain likely represents a flexible structure that interfered with crystallization and thus was not included in the structural determination of TRIC channels by Liu and colleagues50. The CTTs of TRIC-A and TRIC-B diverge from each other. CTT-A contains a histidine-rich motif and a polylysine domain that are flanked by a hydrophobic domain. This structure is similar to the intracellular loop joining repeats II and III of the L-type Ca2+ channel, which has been shown to be a critical domain that regulates the activity of the SR Ca2+ release in muscle cells51–53. Only the polylysine domain appears in the CTT-B domain.
To test if the divergent CTT-A constitutes a site for interaction with the RyR channel, we generated a GST-fusion peptide containing the CTT-A (a.a. 231–299) and CTT-B (a.a. 229–291). These recombinant peptides were purified from E. coli as GST-fusion proteins. Using a protein pull-down assay, we observed that CTT-A could interact with RyR1 stably expressed in CHO cells54, 55 (Fig. 5D), and RyR2 from rat heavy SR vesicles (Fig. 5E). Using antibodies against RyR2, we could co-immunoprecipitate TRIC-A from HEK293 cells that co-express RyR2 and TRIC-A (Fig. 5F). These interactions appear to be specific for CTT-A, as GST peptide alone could not pull down either RyR1 or RyR2. In addition to RyR2, several other candidate proteins were pulled-down by CTT-A as marked by a, b and c in Fig. 5E. MALDI-MS identification revealed a-band contains SERCA, MPP1, and C2CD2; b-band contains TRIC-A, destrin, Rtn2, and proteolytic fragment derived from RyR2; and the c-band contains mostly proteolytic fragment for RyR2 and SERCA.
TRIC-A carboxyl-terminal tail peptide increases RyR2 channel activity.
Based on these findings, we generated chimeric constructs of TRIC-AB or TRIC-BA. TRIC-AB contains a.a. 1–230 from TRIC-A plus a.a. 229–291 from TRIC-B. TRIC-BA contains a.a. 1–228 from TRIC-B plus a.a. 231–299 from TRIC-A. These constructs were co-expressed with RyR2 in HEK293 cells. We found that replacement of the CTT domain in TRIC-A with CTT-B could alleviate the impact on SOICR (Fig. 6). Consistent with the data shown in Fig. 3, co-expression of TRIC-A with RyR2 could reduce the spontaneous Ca2+ oscillation in HEK293 cells (Fig. 6A, red color), whereas co-expression of TRIC-B with RyR2 had no effect on SOICR activity (Fig. 6A, green color). Co-expression of TRIC-AB with RyR2 resulted in the elimination of the inhibitory effect of TRIC-A on SOICR (Fig. 6A, orange color). This finding supports the important function of CTT-A on RyR2-mediated SOICR in HEK293 cells. Interestingly, co-expression of TRIC-BA with RyR2 also had minimal effect on RyR2-mediated SOICR (Fig. 6B, blue color), suggesting the possibility that CTT-A alone in the context of TRIC-B might lose its capability to interact with the RyR2 channel.
Fig 6. CTT-A directly modulates RyR2 channel function.

A. TRIC-A, TRIC-B, TRIC-AB and TRIC-BA were transiently transfected into HEK293 cells with stable expression of RyR2. Represent traces of spontaneous Ca2+ oscillations in 2 mM Ca2+ were shown. B. Percentage of cells with SOICR was quantified. (P<0.0001 across groups, ANOVA; Control vs TRIC-A: P<0.0001; TRIC-A vs TRIC-AB: P=0.0009, Tukey’s test) C. Caffeine-induced Ca2+ release was used to quantify the impact of the various TRIC-A and TRIC-B constructs on ER Ca2+ stores. (P<0.001 for Kruskal-Wallis test for whole group; TRIC-A vs TRIC-AB: P<0.0001, TRIC-A vs Control: P<0.0001, Dunn’s test). D. Reconstitution of RyR2 channels in lipid bilayer. Single channel activity was measured with 250 mM Cs (cis)/50 mM Cs (trans) and 1 μM free Ca2+ in the cis solution. Holding potential was −60 mV. Open probability of individual channels was calculated from a continuous recording of 5 min, and data from multiple measurements were averaged. Addition of 1 μM CTT-A to cis solution led to significant increase in the bursting pattern of RyR2 channel, whereas addition of 10 μM TRIC-B-tail did not affect RyR2 activity. E. Confocal line scan images of Ca2+ events measured by Fluo-4 after microinjection of CTT-A or CTT-B into isolated cardiomyocytes. F. Statistical analysis of RyR2 channel open probabilities. (P<0.0001 across groups, ANOVA; TRIC-A vs Control: P<0.0001, TRIC-A vs TRIC-B: P<0.0001, Tukey’s test) G. CTT-A induced significantly more Ca2+ release events in cardiomyocytes than CTT-B and PBS does. (CTT-A vs PBS: P=0.0174; CTT-A vs CTT-B: P=0.0301, chi-square test)
We used 10 mM caffeine to induce activation of the RyR2 channel as the assessment of the total ER Ca2+ load (Fig. 6C). Clearly, only TRIC-A had a significant impact on the caffeine response, whereas TRIC-B, TRIC-AB and RIC-BA had a negligible effect on ER Ca2+ content in HEK293 cells expressing RyR2.
For direct evaluation of the CTT-A and CTT-B peptides on RyR2 channel function, we reconstituted the RyR2 channels from rat cardiac muscle into the lipid bilayer membrane. As shown in Fig. 6D, the addition of CTT-A to the cis-cytoplasmic solution significantly enhanced the RyR2 channel activity (n=5). The effect appears to be specific for CTT-A, as the addition of CTT-B peptide did not result in significant changes in RyR2 channel activity (n=4). Data from multiple experiments are summarized in Fig. 6F.
To further evaluate the functional effect of CTT-A and CTT-B peptides on Ca2+ signaling in cardiomyocytes, we performed microinjection of CTT-A or CTT-B peptides into isolated mouse cardiomyocytes (Fig. 6E). We found that microinjection of CTT-A peptide elicited significantly more intracellular Ca2+ events as compared to those cardiomyocytes injected with CTT-B peptide (Fig. 6G).
CTT-A modulates Ca2+ spark activity in cardiomyocytes derived from TRIC-A−/− cardiomyocytes.
We followed the protocol of Lukyanenko and Gyorke27 to characterize the Ca2+ spark activity in saponin-permeabilized cardiomyocytes derived from wild type and TRIC-A−/− mice (Fig. 7A). Saponin permeabilization of the sarcolemma allows access of CTT-A and CTT-B to the intracellular organelles of the cardiomyocytes. Starting from a basal condition of 100 nM free Ca2+ in the cytosolic solution, we observed the significantly lower frequency of spontaneous Ca2+ sparks in the TRIC-A−/− cardiomyocytes compared with the wild type cardiomyocytes (Fig. 7B). For testing the effect of CTT-A or CTT-B on Ca2+ spark signaling, the free cytosolic Ca2+ was reduced to 50 nM, which led to lower activity of Ca2+ spark events in the TRIC-A−/− cardiomyocytes (Fig. 7C). When CTT-A (10 μM) was added to the cytosolic solution, transient elevation of Ca2+ spark activity was observed in the early phase (within 3 min after CTT-A addition), which is followed by a decline of Ca2+ spark activity during the later phase (starting from ~3 min following CTT-A addition) (Fig. 7C, left). In the presence of CTT-A, we frequently observed Ca2+ spark events that occur in a consecutive streaming pattern of opening-closings, reflecting increased RyR2 channel activity. Such streaming patterns of Ca2+ spark events were rarely observed under control conditions and did not appear to change with either early or late treatment of CTT-B (10 μM) (Fig. 7C, right). Diary plot of Ca2+ spark activity over the 10 min time period remained steady under the control condition of 50 nM free Ca2+ in the cytosol (Fig. 7D, left). Clearly, the addition of CTT-A led to transient stimulation of the spontaneous Ca2+ spark events (Fig 7D, middle). In contrast, CTT-B did not have a measurable impact on Ca2+ spark activity (Fig 7D, right). Data from multiple experiments were summarized in Fig. 7E, showing a significant increase in CTT-A-induced transient Ca2+ spark activity which did not occur with CTT-B. Consistent with the streaming pattern of Ca2+ spark events observed with CTT-A treatment that may cause depletion of the SR Ca2+ store (Fig. 7C, left), quantitative analysis revealed significant reduction of Ca2+ spark amplitude in the presence of CTT-A, but not CTT-B (Fig. 7F). The effect of CTT-A on Ca2+ sparks measured from the saponin-permeabilized cardiomyocytes derived from in the TRIC-A−/− mice is reversible, as washout of CTT-A led to the recovery of Ca2+ spark activities to the basal level (Supplemental Online Figure I).
Fig. 7. CTT-A enhances Ca2+ spark activities in permeabilized TRIC-A−/− cardiomyocytes.

A. Line-scan images of Ca2+ sparks in saponin-permeabilized wild type (top) and TRIC-A−/− cardiomyocytes (bottom). B. Dot plot of spontaneous Ca2+ spark activities in wild type and TRIC-A cardiomyocytes at 100 nM free cytosolic Ca2+. (P =0.0147, 2-tailed t-test) . C. Left panel shows distinct streaming-pattern of Ca2+ sparks observed in TRIC-A−/− cardiomyocytes following 1 min application of 10 μM CTT-A (early), with apparent suppression of Ca2+ sparks at 5 min after CTT-A treatment (late). Treatment with CTT-B (10 μM) did not induce measurable changes in Ca2+ spark activity (right panels). All Ca2+ sparks were measured with a cytosolic solution containing 50 nM free Ca2+. D. Diary plots of Ca2+ spark activities under control condition of 50 nM free Ca2+ (left), with addition of CTT-A (middle) and CTT-B (right). E. Scattered plots of Ca2+ spark events from individual cardiomyocytes. Two separate groups of experiments were performed with CTT-A and CTT-B, all with cytosolic free Ca2+ buffered at 50 nM. Early – indicates 0–3 min following CTT-A/B treatment; late – indicates 3–6 min following CTT-A/B treatment. (P<0.0001 across groups, ANOVA; 50nM Ca2+ vs early: P=0.0130, early vs late: P=0.0002, Tukey’s test) F. Averaged value of Ca2+ spark amplitudes measured at 50 nM free cytosolic Ca2+ (Control, blue, n=3939), with early addition of CTT-A (red, n=3434) or CTT-B (green, n=2126). (P<0.0001 for ANOVA test; P<0.0001, CTT-A vs Control, CTT-A vs CTT-B, Tukey’s test) **P<0.001, ****P<0.0001
CTT-A and CTT-B dock to different motifs on RyR2.
To gain insights into the interaction of CTT-A or CTT-B with the RyR2 channel, we performed modeling and docking studies using our established molecular dynamic simulation method37. We first compared the 3-D structure of CTT-A with CTT-B and found divergent structural patterns between the two peptides (Fig. 8A). Interestingly, the close similarity between CTT-A and the II-III loop peptide of the dihydropyridine receptor (DHPR) was observed (Fig. 8B).
Fig. 8. Docking of CTT-A and CTT-B with open and closed-state of the RyR2 channel.

A. Structural comparison between CTT-A (cyan) and CTT-B (pink) in ribbon representation. The histidine-rich motif in CTT-A is colored red. B. Structural comparison of CTT-A, CTT-B and the II-III loop of DHPR. C. Four potential binding sites of CTT-A interaction with hRyR2 were predicted from molecular docking. Both hRyR2 (grey) and CTT-A are plotted in ribbon representation. CTT-A is colored differently at the four sites. D. CTT-A and CTT-B bind to hRyR2 distinctively on Site 3 and Site 4. Left - binding to the closed structure of hRyR2; right - binding to the open structure of hRyR2. CTT-A (blue), CTT-B (pink) and hRyR2 (grey) are plotted in ribbon representation.
Molecular docking of CTT-A and CTT-B onto the human RyR2 (hRyR2) showed four potential binding sites, which were numbered according to their rankings by the energy scoring function in MDockPP56. Representative binding modes of CTT-A at these four sites on the closed structure of hRyR2 are plotted in Fig. 8C. Among them, Site 1 and Site 2 exist in both open and closed structures of hRyR2 for both CTT-A and CTT-B. Site 1, the best ranked site, is the hinge domain of hRyR239, which has been shown to play a critical role in the activation of the RyR2 channel. Site 2 consists of binding domain with FKBP12 (see PDB ID: 3J8H57), which has demonstrated function in control of RyR channel opening. The zoom-in picture of CCT-A interacting with the closed structure of hRyR2 is shown in Supplemental Online Fig. II.
Interestingly, CTT-A and CTT-B bind distinctively on Site 3 and Site 4 (Fig. 8D). Site 3 locates on the N-terminal domain (NTD), and Site 4 locates on the SPRY domain. CTT-A binds to Site 3 for both the open and closed state of hRyR2, whereas CTT-B binds to Site 3 only for the closed state of hRyR2. CTT-A binds to Site 4 only in the closed state, whereas CTT-B binds to Site 4 only in the open state. Previous studies by Tae et al showed that the SPRY domain of RyR potentially contributes to binding to DHPR for control of RyR channel activity58. While our modeling study suggested conformation-dependent interaction of CTT-A with the SPRY domain, further biochemical and structure-function studies will be required to test if the modeled CTT-A/SPRY interaction or CTT-A’s binding to other domains of RyR2 indeed play a role in the control of Ca2+ release from the SR.
DISCUSSION
The present study uncovered a novel function for TRIC-A as an accessory protein that interacts with RyR2 to modulate intracellular Ca2+ signaling. Aside from its recognized role as a counter-ion channel that participates in excitation-contraction coupling of striated muscles, the physiological function of TRIC-A in heart physiology and disease has remained largely unexplored. Even with the recent resolution of the crystal structure of the TRIC channels, the interacting partners for TRIC have yet to be defined. We showed that CTT of TRIC-A, an important portion of the channel that is left out of the crystal structure determination, constitutes an active motif that interacts with RyR2 to control SOICR function. We found that TRIC-B is ineffective in control of RyR2-mediated SOICR in cultured cells, suggesting the specificity of TRIC-A interaction with the RyR2 channel. Using reconstitution studies and Ca2+ spark measurements, we demonstrated that the synthetic CTT-A peptide could directly enhance the RyR2 channel activity and stimulate intracellular Ca2+ release in isolated cardiomyocytes.
While many studies have suggested that altered function of SOICR from the SR in cardiomyocytes may contribute to the development of cardiac arrhythmias32, 47, 59–61, searching for accessory proteins that modulate RyR2 channel function and SR Ca2+ homeostasis should yield important clues to the function of SOICR in physiological and pathophysiological settings. We showed that TRIC-A ablation led to SR Ca2+ overload, yet the spontaneous Ca2+ sparks and store-overload induced Ca2+ waves were less frequent in cardiomyocytes derived from the TRIC-A−/− mice compared with those from the WT mice. Moreover, such SR Ca2+ overload did not trigger SOICR in the TRIC-A−/− cardiomyocytes under basal conditions. Through the determination of the ER Ca2+ content, we showed that overexpression of TRIC-A could increase Ca2+ leak across the ER in HEK293 cells. The TRIC-A mediated increase of ER Ca2+ leak reflects direct activation of the RyR2 channel, for expression of TRIC-A alone in the absence of RyR2 did not affect ER Ca2+ storage or passive Ca2+ leak across the ER. From these studies, we conclude that TRIC-A constitutes a physiological component of the SR Ca2+ release machinery, and TRIC-A deficiency could render RyR2 channels less sensitive to physiological activation of Ca2+ signaling in cardiac muscle. The vulnerability of the TRIC-A knockout mouse heart to handling of isoproterenol stimulation further support the important physiological function of TRIC-A in heart physiology and disease.
It is known that changes in RyR2 activity can impact the frequency, as well as the speed of stress-induced Ca2+ waves in cardiomyocytes. Studies from other investigators have shown that experimental manipulations or mutagenesis of the RyR2 channel that caused reduction of the RyR2 channel activity led to slower onset and prolonged decaying phase of voltage-induced Ca2+ transients and reduced speed of Ca2+ wave propagation43–45; and conversely mutations of RyR2 (e.g. R4496C) that enhance RyR2 channel activity had the opposite effect on intracellular Ca2+ transients62. Moreover, Ca2+ wave velocity increased with treatment of caffeine and decreased with treatment of tetracaine or flecainide62. Ullrich et al63 reported that cardiomyocytes expressing RyR2-S2808A displayed slower Ca2+ wave velocity. Our data demonstrated that cardiomyocytes derived from the TRIC-A−/− mice displayed reduced frequency of Ca2+ sparks and reduced speed of Ca2+ wave propagation, likely reflecting reduced RyR2 activity. When cardiomyocytes are exposed to a high Ca2+ (10 mM) extracellular solution, less frequent store-overload induced Ca2+ waves were observed in the TRIC-A−/− cardiomyocytes than those in the WT cardiomyocytes. Moreover, the frequency of stress-induced Ca2+ waves was less in the TRIC-A−/− cardiomyocytes than that in the WT cardiomyocytes. Following electric pacing, there was a delayed onset of intracellular Ca2+ transients and prolonged decaying phase of Ca2+ transients in the TRIC-A−/− cardiomyocytes compared with those in the WT cardiomyocytes. Similar observations were also made by other investigators who reported extended decay of Ca2+ transients in cardiomyocytes derived from the A4860G mice (with reduced RyR2 activity)45 and from the RyR2-Ex3-del+/− mice44, indicating that alteration of RyR2 activity alone would impact the decaying phase of intracellular Ca2+ transient. However, the apparent delayed phase of intracellular Ca2+ release from the TRIC-A−/− cardiomyocytes may involve additional actions on other components of Ca2+ uptake and release across the SR, e.g. counter-ion movement or SERCA activity. While our data support the notion that alteration of RyR2 activity associated with genetic ablation of TRIC-A could be a main contributing factor for the dysfunctional intracellular Ca2+ signaling in cardiomyocytes, we could not rule out the possibility that changes in SERCA activity could also contribute to the altered Ca2+ handling in the TRIC-A−/− cardiomyocytes.
Smith and O’Neill64 described the effect of low ATP and tetracaine on spontaneous Ca2+ waves in permeabilized cardiomyocytes. Lower ATP was shown to cause reduced frequency and slower Ca2+ wave propagation, and the changes in Ca2+ wave characteristics in 0.5 mM ATP were similar to those observed during perfusion with 50 μM tetracaine. Smith and O’Neill64 also reported that the SR Ca2+ content increased as ATP was reduced or tetracaine was increased. Both tetracaine and lowering ATP are known to cause reduction of the RyR2 channel activity. Many features of the changes in Ca2+ sparks and Ca2+ waves reported by Smith and O’Neill64 are very similar to what we observed with the TRIC-A−/− cardiomyocytes. This further support the notion that ablation of TRIC-A leads to reduced RyR2 activity in cardiomyocytes.
A recent study from Sitsapesan and colleagues found that in skeletal muscle, the RyR1 channel from the TRIC-A−/− mice displays increased sensitivity to Mg2+ inhibition, defective response to protein kinase A phosphorylation, and physiological activator such as ATP is less effective in activating the individual RyR1 channels reconstituted into the lipid bilayer membrane22. They also reported that the Ca2+-dependent control of RyR1 channel was not altered in the absence of TRIC-A, suggesting that the modulatory effect of TRIC-A may involve other factors. These findings are consistent with a potential role of TRIC-A as an enhancer of RyR channels, such that the absence of TRIC-A leads to reduced RyR channel function under stress conditions.
Crystal structures of several TRIC channels from invertebrate and prokaryotic TRIC channels have been reported23–26. All of these TRIC proteins share very similar folding and trimeric organizations. Interestingly, all of these crystal structures lack the highly flexible CTT domain, which investigators claim may decrease the stability of the crystal23–26. Our findings that TRIC-A directly modulates RyR2-mediated SOICR may have wide implications in cardiovascular research. Potential therapeutic interventions can be used to target the functional interaction between TRIC-A and RyR2 to restore defective Ca2+ signaling in cardiovascular diseases.
In our studies testing CTT-A on Ca2+ sparks measured in permeabilized cardiomyocytes, we found an interesting phenomenon. The stimulatory effect of CTT-A on Ca2+ sparks was transient, and there appeared to be a delayed inhibitory effect on Ca2+ sparks with prolonged incubation of CTT-A. The transient increase in Ca2+ sparks induced by CTT-A may reduce the SR Ca2+ content that would reduce the frequency of Ca2+ sparks after the CTT-A stimulation, or alternatively the inhibitory effect of CTT-A likely reflected other components of Ca2+ movement across the SR membrane. Since the functional effect on intracellular Ca2+ release was only observed with CTT-A, not CTT-B, differential interaction between RyR2 and CTT-A would be required. Using the available information about the 3-D structure of the RyR2 channel39, we performed modeling and docking studies and discovered potential sites of interaction between RyR2 and CTT-A/CTT-B. Of the four sites plotted in Fig. 8C (see also Supplemental Online Fig. II), one of them is particularly interesting. The SPRY domain of RyR2 constitutes a potential binding site (site 4) for interaction with CTT-A and CTT-B. CTT-A preferentially accesses the SPRY domain when the channel is in the closed state, whereas CTT-B appears to recognize the SPRY domain when the channel is in the open state. Several studies have demonstrated that the SPRY domain of RyR plays an important role in modulation of the Ca2+ release channel activity58, 65. It should be emphasized that our modeling studies only provide a hint of possible domain-domain interaction between CTT-A/B and RyR. Further mutagenesis studies will be required to ascertain the functional impact of such interaction in control of intracellular Ca2+ release in muscle cells.
Previous studies from Chen and colleagues showed that reconstitution of RyR2 gain-of-function mutants in HEK293 cells could lead to enhanced SOICR as reflected by increased Ca2+ oscillations and/or reduced threshold of luminal Ca2+ to trigger Ca2+ oscillations5, 6, 59, 66. Interestingly, our study revealed that co-expression of TRIC-A and RyR2 led to an apparent reduction of Ca2+ oscillation in HEK293 cells, which seems unexpected based on the role of TRIC-A as an enhancer of RyR2 function. In fact, when TRIC-A was introduced into HEK293 cells, increased Ca2+ leak across the ER was observed that may reflect direct activation of the RyR2 channel. It is possible that the TRIC-A/RyR2-mediated ER Ca2+ leak may take place in the form of small Ca2+ release events which are un-synchronized and therefore do not appear as Ca2+ oscillations. One would ask why TRIC-A would lead to over activation of RyR2 in HEK293 cells but not in native cardiomyocytes. It is known that coordinated activation and inactivation mechanisms must exist in cardiomyocytes for the proper function of Ca2+ signaling in the heart, and many of the stabilizers or inhibitory factors of RyR2-mediated Ca2+ release are likely absent in HEK293 cells. One avenue of future study may take advantage of HEK293 cells to reconstitute the potential functional interaction of TRIC-A with other regulatory components of the intracellular Ca2+ release machinery. Our data support the dual function of TRIC-A as a counter-ion channel and an activator of RyR2, and both functions would enhance RyR2-mediated Ca2+ release in cardiac muscle. A direct interaction between TRIC-A and RyR2 constitutes an important physiologic component of intracellular Ca2+ signaling in the heart. Studies from Fill and colleagues suggested that due to the non-selective nature of the Ca2+ release channels, RyR channels can provide a certain extent of counter currents by themselves28, 67, 68. Quantitative simulations suggested that TRIC channels could contribute to the network of SR membrane potential to support Ca2+ release and reuptake68. Thus, in addition to regulating the acute phase of Ca2+ release from the SR/ER store, TRIC-mediated movement of counter current flow could also play a role in balancing the electronegative influence of Ca2+ release on other aspects of Ca2+ homeostasis inside the SR/ER.
TRIC-A and TRIC-B have different functions in regulating SR and ER Ca2+ homeostasis in excitable and non-excitable cells, respectively11, 16–21. TRIC-B is present in both excitable and non-excitable cells, whereas TR-A is predominantly expressed in excitable tissues such as striated muscles and brain tissues. We showed that the divergent CTT domains of TRIC-A and TRIC-B have different functions in modulation of RyR2 channel activity. Previously we have demonstrated that epithelial cells derived from the TRIC-B knockout mice display abnormal function of IP3 receptor (IP3R) mediated Ca2+ release from the ER store21, and skeletal muscle derived from the TRIC-A knockout mice display abnormal function of RyR1-mediated Ca2+ release from the SR store20. The functional crosstalk between IP3R and RyR-mediated Ca2+ signaling has been implicated in muscle and heart cells under physiologic and pathologic conditions69–75. Dissecting the function of TRIC-A and TRIC-B in modulating RyR/IP3R cross-talk for control of Ca2+ signaling in health and disease will be an important task of future research.
Supplementary Material
NOVELTY AND SIGNIFICANCE.
What Is Known?
Dysfunction of ryanodine receptor (RyR)-mediated Ca2+ signaling contributes to arrhythmia and heart failure.
TRIC channels provide counter-current movement during Ca2+ release from intracellular stores.
Crystal structure of TRIC lacking the carboxyl-terminal tail (CTT) domain has been solved.
What New Information Does This Article Contribute?
TRIC-A knockout mouse heart shows abnormal response to isoproterenol stimulation due to dysfunctional Ca2+ signaling.
TRIC-A interacts with RyR to directly control Ca2+ release in cardiomyocytes.
ACKNOWLEDGMENTS
We thank Dr. S.R Wayne Chen for critical reading of this manuscript. We also thank Dr. Michael Fill for providing the cardiac SR vesicles for the biochemical and lipid-bilayer reconstitution studies, and Dr. Lianbo Yu for help with the statistical analysis.
SOURCES OF FUNDING
This work is supported by NIH grants to J. Ma, J. Zhou and JSPS Core-to-Core Program, JSPS Bilateral Open Partnership Joint Research Projects to HT.
Nonstandard Abbreviations and Acronyms:
- SR
sarcoplasmic reticulum
- CICR
Calcium induced Calcium release
- SOICR
store overload induced calcium release
- ECG
electrocardiogram
- RyR
ryanodine receptor
- TRIC
trimeric intracellular cation channels
- CTT
carboxyl-terminal tail
- CPA
cyclopiazonic acid
Footnotes
DISCLOSURES
None.
REFERENCES
- 1.Bers DM. Cardiac excitation-contraction coupling. Nature. 2002;415:198–205. [DOI] [PubMed] [Google Scholar]
- 2.Cannell MB and Kong CH. Local control in cardiac E-C coupling. J Mol Cell Cardiol. 2012;52:298–303. [DOI] [PubMed] [Google Scholar]
- 3.Bround MJ, Asghari P, Wambolt RB, Bohunek L, Smits C, Philit M, Kieffer TJ, Lakatta EG, Boheler KR, Moore ED, Allard MF and Johnson JD. Cardiac ryanodine receptors control heart rate and rhythmicity in adult mice. Cardiovasc Res. 2012;96:372–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Priori SG and Chen SR. Inherited dysfunction of sarcoplasmic reticulum Ca2+ handling and arrhythmogenesis. Circ Res. 2011;108:871–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jiang D, Xiao B, Yang D, Wang R, Choi P, Zhang L, Cheng H and Chen SR. RyR2 mutations linked to ventricular tachycardia and sudden death reduce the threshold for store-overload-induced Ca2+ release (SOICR). Proc Natl Acad Sci U S A. 2004;101:13062–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jiang D, Wang R, Xiao B, Kong H, Hunt DJ, Choi P, Zhang L and Chen SR. Enhanced store overload-induced Ca2+ release and channel sensitivity to luminal Ca2+ activation are common defects of RyR2 mutations linked to ventricular tachycardia and sudden death. Circ Res. 2005;97:1173–81. [DOI] [PubMed] [Google Scholar]
- 7.Fabiato A Two kinds of calcium-induced release of calcium from the sarcoplasmic reticulum of skinned cardiac cells. Adv Exp Med Biol. 1992;311:245–62. [DOI] [PubMed] [Google Scholar]
- 8.Lakatta EG. Functional implications of spontaneous sarcoplasmic reticulum Ca2+ release in the heart. Cardiovasc Res. 1992;26:193–214. [DOI] [PubMed] [Google Scholar]
- 9.Marban E, Robinson SW and Wier WG. Mechanisms of arrhythmogenic delayed and early afterdepolarizations in ferret ventricular muscle. J Clin Invest. 1986;78:1185–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Orchard CH, Eisner DA and Allen DG. Oscillations of intracellular Ca2+ in mammalian cardiac muscle. Nature. 1983;304:735–8. [DOI] [PubMed] [Google Scholar]
- 11.Yazawa M, Ferrante C, Feng J, Mio K, Ogura T, Zhang M, Lin PH, Pan Z, Komazaki S, Kato K, Nishi M, Zhao X, Weisleder N, Sato C, Ma J and Takeshima H. TRIC channels are essential for Ca2+ handling in intracellular stores. Nature. 2007;448:78–82. [DOI] [PubMed] [Google Scholar]
- 12.Yamazaki D, Yamazaki T and Takeshima H. New molecular components supporting ryanodine receptor-mediated Ca(2+) release: roles of junctophilin and TRIC channel in embryonic cardiomyocytes. Pharmacol Ther. 2009;121:265–72. [DOI] [PubMed] [Google Scholar]
- 13.Zhao X, Yamazaki D, Kakizawa S, Pan Z, Takeshima H and Ma J. Molecular architecture of Ca2+ signaling control in muscle and heart cells. Channels (Austin). 2011;5:391–6. [DOI] [PubMed] [Google Scholar]
- 14.Weisleder N, Takeshima H and Ma J. Immuno-proteomic approach to excitation--contraction coupling in skeletal and cardiac muscle: molecular insights revealed by the mitsugumins. Cell Calcium. 2008;43:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pitt SJ, Park KH, Nishi M, Urashima T, Aoki S, Yamazaki D, Ma J, Takeshima H and Sitsapesan R. Charade of the SR K+-channel: two ion-channels, TRIC-A and TRIC-B, masquerade as a single K+-channel. Biophys J. 2010;99:417–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhao C, Ichimura A, Qian N, Iida T, Yamazaki D, Noma N, Asagiri M, Yamamoto K, Komazaki S, Sato C, Aoyama F, Sawaguchi A, Kakizawa S, Nishi M and Takeshima H. Mice lacking the intracellular cation channel TRIC-B have compromised collagen production and impaired bone mineralization. Sci Signal. 2016;9:ra49. [DOI] [PubMed] [Google Scholar]
- 17.Zhou X, Lin P, Yamazaki D, Park KH, Komazaki S, Chen SR, Takeshima H and Ma J. Trimeric intracellular cation channels and sarcoplasmic/endoplasmic reticulum calcium homeostasis. Circ Res. 2014;114:706–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tao S, Yamazaki D, Komazaki S, Zhao C, Iida T, Kakizawa S, Imaizumi Y and Takeshima H. Facilitated hyperpolarization signaling in vascular smooth muscle-overexpressing TRIC-A channels. J Biol Chem. 2013;288:15581–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yamazaki D, Tabara Y, Kita S, Hanada H, Komazaki S, Naitou D, Mishima A, Nishi M, Yamamura H, Yamamoto S, Kakizawa S, Miyachi H, Yamamoto S, Miyata T, Kawano Y, Kamide K, Ogihara T, Hata A, Umemura S, Soma M, Takahashi N, Imaizumi Y, Miki T, Iwamoto T and Takeshima H. TRIC-A channels in vascular smooth muscle contribute to blood pressure maintenance. Cell Metab. 2011;14:231–41. [DOI] [PubMed] [Google Scholar]
- 20.Zhao X, Yamazaki D, Park KH, Komazaki S, Tjondrokoesoemo A, Nishi M, Lin P, Hirata Y, Brotto M, Takeshima H and Ma J. Ca2+ overload and sarcoplasmic reticulum instability in tric-a null skeletal muscle. J Biol Chem. 2010;285:37370–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yamazaki D, Komazaki S, Nakanishi H, Mishima A, Nishi M, Yazawa M, Yamazaki T, Taguchi R and Takeshima H. Essential role of the TRIC-B channel in Ca2+ handling of alveolar epithelial cells and in perinatal lung maturation. Development. 2009;136:2355–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.El-Ajouz S, Venturi E, Witschas K, Beech M, Wilson AD, Lindsay C, Eberhardt D, O’Brien F, Iida T, Nishi M, Takeshima H and Sitsapesan R. Dampened activity of ryanodine receptor channels in mutant skeletal muscle lacking TRIC-A. J Physiol. 2017;595:4769–4784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Su M, Gao F, Yuan Q, Mao Y, Li DL, Guo Y, Yang C, Wang XH, Bruni R, Kloss B, Zhao H, Zeng Y, Zhang FB, Marks AR, Hendrickson WA and Chen YH. Structural basis for conductance through TRIC cation channels. Nat Commun. 2017;8:15103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ou X, Guo J, Wang L, Yang H, Liu X, Sun J and Liu Z. Ion- and water-binding sites inside an occluded hourglass pore of a trimeric intracellular cation (TRIC) channel. BMC Biol. 2017;15:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang H, Hu M, Guo J, Ou X, Cai T and Liu Z. Pore architecture of TRIC channels and insights into their gating mechanism. Nature. 2016;538:537–541. [DOI] [PubMed] [Google Scholar]
- 26.Kasuya G, Hiraizumi M, Maturana AD, Kumazaki K, Fujiwara Y, Liu K, Nakada-Nakura Y, Iwata S, Tsukada K, Komori T, Uemura S, Goto Y, Nakane T, Takemoto M, Kato HE, Yamashita K, Wada M, Ito K, Ishitani R, Hattori M and Nureki O. Crystal structures of the TRIC trimeric intracellular cation channel orthologues. Cell Res. 2016;26:1288–1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lukyanenko V and Gyorke S. Ca2+ sparks and Ca2+ waves in saponin-permeabilized rat ventricular myocytes. J Physiol. 1999;521 Pt 3:575–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Guo T, Nani A, Shonts S, Perryman M, Chen H, Shannon T, Gillespie D and Fill M. Sarcoplasmic reticulum K(+) (TRIC) channel does not carry essential countercurrent during Ca(2+) release. Biophys J. 2013;105:1151–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bers DM, Patton CW and Nuccitelli R. A practical guide to the preparation of Ca(2+) buffers. Methods Cell Biol. 2010;99:1–26. [DOI] [PubMed] [Google Scholar]
- 30.Picht E, Zima AV, Blatter LA and Bers DM. SparkMaster: automated calcium spark analysis with ImageJ. Am J Physiol Cell Physiol. 2007;293:C1073–81. [DOI] [PubMed] [Google Scholar]
- 31.Uehara A, Murayama T, Yasukochi M, Fill M, Horie M, Okamoto T, Matsuura Y, Uehara K, Fujimoto T, Sakurai T and Kurebayashi N. Extensive Ca2+ leak through K4750Q cardiac ryanodine receptors caused by cytosolic and luminal Ca2+ hypersensitivity. J Gen Physiol. 2017;149:199–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tang Y, Tian X, Wang R, Fill M and Chen SR. Abnormal termination of Ca2+ release is a common defect of RyR2 mutations associated with cardiomyopathies. Circ Res. 2012;110:968–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Penner R, Neher E, Takeshima H, Nishimura S and Numa S. Functional expression of the calcium release channel from skeletal muscle ryanodine receptor cDNA. FEBS Lett. 1989;259:217–21. [DOI] [PubMed] [Google Scholar]
- 34.Takekura H, Takeshima H, Nishimura S, Takahashi M, Tanabe T, Flockerzi V, Hofmann F and Franzini-Armstrong C. Co-expression in CHO cells of two muscle proteins involved in excitation-contraction coupling. J Muscle Res Cell Motil. 1995;16:465–80. [DOI] [PubMed] [Google Scholar]
- 35.Ma J, Fill M, Knudson CM, Campbell KP and Coronado R. Ryanodine receptor of skeletal muscle is a gap junction-type channel. Science. 1988;242:99–102. [DOI] [PubMed] [Google Scholar]
- 36.Fill M, Coronado R, Mickelson JR, Vilven J, Ma J, Jacobson B and Louis CF. Abnormal ryanodine receptor channels in malignant hyperthermia. Biophysical Journal. 1990;57:471–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang Y I-TASSER server for protein 3D structure prediction. BMC Bioinformatics. 2008;9:40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN and Bourne PE. The Protein Data Bank. Nucleic acids research. 2000;28:235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Peng W, Shen H, Wu J, Guo W, Pan X, Wang R, Chen SR and Yan N. Structural basis for the gating mechanism of the type 2 ryanodine receptor RyR2. Science. 2016;354. [DOI] [PubMed] [Google Scholar]
- 40.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC and Ferrin TE. UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–12. [DOI] [PubMed] [Google Scholar]
- 41.Huang SY, Yan C, Grinter SZ, Chang S, Jiang L and Zou X. Inclusion of the orientational entropic effect and low-resolution experimental information for protein-protein docking in Critical Assessment of PRedicted Interactions (CAPRI). Proteins. 2013;81:2183–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Huang SY and Zou X. MDockPP: A hierarchical approach for protein-protein docking and its application to CAPRI rounds 15–19. Proteins. 2010;78:3096–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kolstad TR, van den Brink J, MacQuaide N, Lunde PK, Frisk M, Aronsen JM, Norden ES, Cataliotti A, Sjaastad I, Sejersted OM, Edwards AG, Lines GT and Louch WE. Ryanodine receptor dispersion disrupts Ca(2+) release in failing cardiac myocytes. Elife. 2018;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu Y, Wang R, Sun B, Mi T, Zhang J, Mu Y, Chen J, Bround MJ, Johnson JD, Gillis AM and Chen SR. Generation and characterization of a mouse model harboring the exon-3 deletion in the cardiac ryanodine receptor. PLoS One. 2014;9:e95615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhao YT, Valdivia CR, Gurrola GB, Powers PP, Willis BC, Moss RL, Jalife J and Valdivia HH. Arrhythmogenesis in a catecholaminergic polymorphic ventricular tachycardia mutation that depresses ryanodine receptor function. Proc Natl Acad Sci U S A. 2015;112:E1669–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pan Z, Hirata Y, Nagaraj RY, Zhao J, Nishi M, Hayek SM, Bhat MB, Takeshima H and Ma J. Co-expression of MG29 and ryanodine receptor leads to apoptotic cell death: effect mediated by intracellular Ca2+ release. J Biol Chem. 2004;279:19387–90. [DOI] [PubMed] [Google Scholar]
- 47.Jones PP, Jiang D, Bolstad J, Hunt DJ, Zhang L, Demaurex N and Chen SR. Endoplasmic reticulum Ca2+ measurements reveal that the cardiac ryanodine receptor mutations linked to cardiac arrhythmia and sudden death alter the threshold for store-overload-induced Ca2+ release. Biochem J. 2008;412:171–8. [DOI] [PubMed] [Google Scholar]
- 48.Curran J, Hinton MJ, Rios E, Bers DM and Shannon TR. Beta-adrenergic enhancement of sarcoplasmic reticulum calcium leak in cardiac myocytes is mediated by calcium/calmodulin-dependent protein kinase. Circ Res. 2007;100:391–8. [DOI] [PubMed] [Google Scholar]
- 49.Shannon TR, Ginsburg KS and Bers DM. Quantitative assessment of the SR Ca2+ leak-load relationship. Circ Res. 2002;91:594–600. [DOI] [PubMed] [Google Scholar]
- 50.Yang H, Hu M, Guo J, Ou X, Cai T and Liu Z. Pore architecture of TRIC channels and insights into their gating mechanism. Nature. 2016. [DOI] [PubMed] [Google Scholar]
- 51.Ogawa Y Role of ryanodine receptors. Crit Rev Biochem Mol Biol. 1994;29:229–74. [DOI] [PubMed] [Google Scholar]
- 52.Tanabe T, Beam KG, Adams BA, Niidome T and Numa S. Regions of the skeletal muscle dihydropyridine receptor critical for excitation-contraction coupling. Nature. 1990;346:567–9. [DOI] [PubMed] [Google Scholar]
- 53.Ogawa Y, Kurebayashi N and Murayama T. Ryanodine receptor isoforms in excitation-contraction coupling. Adv Biophys. 1999;36:27–64. [DOI] [PubMed] [Google Scholar]
- 54.Bhat MB, Zhao J, Takeshima H and Ma J. Functional calcium release channel formed by the carboxyl-terminal portion of ryanodine receptor. Biophys J. 1997;73:1329–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bhat MB, Zhao J, Zang W, Balke CW, Takeshima H, Wier WG and Ma J. Caffeine-induced release of intracellular Ca2+ from Chinese hamster ovary cells expressing skeletal muscle ryanodine receptor. Effects on full-length and carboxyl-terminal portion of Ca2+ release channels. J Gen Physiol. 1997;110:749–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Huang SY and Zou X. An iterative knowledge-based scoring function for protein-protein recognition. Proteins. 2008;72:557–79. [DOI] [PubMed] [Google Scholar]
- 57.Yan Z, Bai X-c, Yan C, Wu J, Li Z, Xie T, Peng W, Yin C-c, Li X, Scheres SHW, Shi Y and Yan N. Structure of the rabbit ryanodine receptor RyR1 at near-atomic resolution. Nature. 2014;517:50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tae H, Wei L, Willemse H, Mirza S, Gallant EM, Board PG, Dirksen RT, Casarotto MG and Dulhunty A. The elusive role of the SPRY2 domain in RyR1. Channels (Austin). 2011;5:148–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Liu Y, Wei J, Wong King Yuen SM, Sun B, Tang Y, Wang R, Van Petegem F and Chen SRW. CPVT-associated cardiac ryanodine receptor mutation G357S with reduced penetrance impairs Ca2+ release termination and diminishes protein expression. PLoS One. 2017;12:e0184177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zvaritch E, Depreux F, Kraeva N, Loy RE, Goonasekera SA, Boncompagni S, Kraev A, Gramolini AO, Dirksen RT, Franzini-Armstrong C, Seidman CE, Seidman JG and Maclennan DH. An Ryr1I4895T mutation abolishes Ca2+ release channel function and delays development in homozygous offspring of a mutant mouse line. Proc Natl Acad Sci U S A. 2007;104:18537–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Thomas NL, Lai FA and George CH. Differential Ca2+ sensitivity of RyR2 mutations reveals distinct mechanisms of channel dysfunction in sudden cardiac death. Biochem Biophys Res Commun. 2005;331:231–8. [DOI] [PubMed] [Google Scholar]
- 62.Savio-Galimberti E and Knollmann BC. Channel Activity of Cardiac Ryanodine Receptors (RyR2) Determines Potency and Efficacy of Flecainide and R-Propafenone against Arrhythmogenic Calcium Waves in Ventricular Cardiomyocytes. PLoS One. 2015;10:e0131179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ullrich ND, Valdivia HH and Niggli E. PKA phosphorylation of cardiac ryanodine receptor modulates SR luminal Ca2+ sensitivity. J Mol Cell Cardiol. 2012;53:33–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Smith GL and O’Neill SC. A comparison of the effects of ATP and tetracaine on spontaneous Ca(2+) release from rat permeabilised cardiac myocytes. J Physiol. 2001;534:37–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Peralvarez-Marin A, Tae H, Board PG, Casarotto MG, Dulhunty AF and Samso M. 3D Mapping of the SPRY2 domain of ryanodine receptor 1 by single-particle cryo-EM. PLoS One. 2011;6:e25813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.MacLennan DH and Chen SR. Store overload-induced Ca2+ release as a triggering mechanism for CPVT and MH episodes caused by mutations in RYR and CASQ genes. J Physiol. 2009;587:3113–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gillespie D and Fill M. Intracellular calcium release channels mediate their own countercurrent: the ryanodine receptor case study. Biophys J. 2008;95:3706–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zsolnay V, Fill M and Gillespie D. Sarcoplasmic Reticulum Ca(2+) Release Uses a Cascading Network of Intra-SR and Channel Countercurrents. Biophys J. 2018;114:462–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zima AV and Blatter LA. Inositol-1,4,5-trisphosphate-dependent Ca(2+) signalling in cat atrial excitation-contraction coupling and arrhythmias. J Physiol. 2004;555:607–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Domeier TL, Zima AV, Maxwell JT, Huke S, Mignery GA and Blatter LA. IP3 receptor-dependent Ca2+ release modulates excitation-contraction coupling in rabbit ventricular myocytes. Am J Physiol Heart Circ Physiol. 2008;294:H596–604. [DOI] [PubMed] [Google Scholar]
- 71.Kockskamper J, Zima AV, Roderick HL, Pieske B, Blatter LA and Bootman MD. Emerging roles of inositol 1,4,5-trisphosphate signaling in cardiac myocytes. J Mol Cell Cardiol. 2008;45:128–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Li X, Zima AV, Sheikh F, Blatter LA and Chen J. Endothelin-1-induced arrhythmogenic Ca2+ signaling is abolished in atrial myocytes of inositol-1,4,5-trisphosphate(IP3)-receptor type 2-deficient mice. Circ Res. 2005;96:1274–81. [DOI] [PubMed] [Google Scholar]
- 73.Zhang WM, Lin MJ and Sham JS. Endothelin-1 and IP3 induced Ca2+ sparks in pulmonary arterial smooth muscle cells. J Cardiovasc Pharmacol. 2004;44 Suppl 1:S121–4. [DOI] [PubMed] [Google Scholar]
- 74.Zhang WM, Yip KP, Lin MJ, Shimoda LA, Li WH and Sham JS. ET-1 activates Ca2+ sparks in PASMC: local Ca2+ signaling between inositol trisphosphate and ryanodine receptors. Am J Physiol Lung Cell Mol Physiol. 2003;285:L680–90. [DOI] [PubMed] [Google Scholar]
- 75.Tjondrokoesoemo A, Li N, Lin PH, Pan Z, Ferrante CJ, Shirokova N, Brotto M, Weisleder N and Ma J. Type 1 inositol (1,4,5)-trisphosphate receptor activates ryanodine receptor 1 to mediate calcium spark signaling in adult mammalian skeletal muscle. J Biol Chem. 2013;288:2103–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.