

Abstract
Background
Removal of the Bax gene from mice completely protects the somas of retinal ganglion cells (RGCs) from apoptosis following optic nerve injury. This makes BAX a promising therapeutic target to prevent neurodegeneration. In this study, Bax+/− mice were used to test the hypothesis that lowering the quantity of BAX in RGCs would delay apoptosis following optic nerve injury.
Methods
RGCs were damaged by performing optic nerve crush (ONC) and then immunostaining for phospho-cJUN and quantitative PCR were used to monitor the status of the BAX activation mechanism in the months following injury. The apoptotic susceptibility of injured cells was directly tested by virally introducing GFP-BAX into Bax−/− RGCs after injury. The competency of quiescent RGCs to reactivate their BAX activation mechanism was tested by intravitreal injection of the JNK pathway agonist, anisomycin.
Results
24 weeks after ONC, Bax+/− mice had significantly less cell loss in their RGC layer than Bax+/+ mice 3 weeks after ONC. Bax+/− and Bax+/+ RGCs exhibited similar patterns of nuclear phospho-cJUN accumulation immediately after ONC, which persisted in Bax+/− RGCs for up to 7 weeks before abating. The transcriptional activation of BAX activating genes was similar in Bax+/− and Bax+/+ RGCs following ONC. Intriguingly, cells deactivated their BAX activation mechanism between 7 and 12 weeks after crush. Introduction of GFP-BAX into Bax−/− cells at 4 weeks after ONC showed that these cells had a nearly normal capacity to activate this protein, but this capacity was lost 8 weeks after crush. Collectively, these data suggest that 8–12 weeks after crush, damaged cells no longer displayed increased susceptibility to BAX activation relative to their naïve counterparts. In this same timeframe, retinal glial activation and the signaling of the pro-apoptotic JNK pathway also abated. Quiescent RGCs did not show a timely reactivation of their JNK pathway following intravitreal injection with anisomycin.
Conclusions
These findings demonstrate that lowering the quantity of BAX in RGCs is neuroprotective after acute injury. Damaged RGCs enter a quiescent state months after injury and are no longer responsive to an apoptotic stimulus. Quiescent RGCs will require rejuvenation to reacquire functionality.
Keywords: BAX, Retinal Ganglion Cells, Optic Nerve Crush, intrinsic apoptosis, cJun, Glia, Neuroprotection
Background
Retinal ganglion cells (RGCs) are long-projection neurons of the central nervous system whose axons carry visual signals from the retina to the brain. RGC axons converge at the optic nerve head, which is the principal site of axonal injury in optic neuropathies like glaucoma [1], and project through the optic nerve. RGC axons can be synchronously damaged using optic nerve crush (ONC), which is a widely used acute model of optic nerve injury that mimics many of the molecular hallmarks of glaucomatous degeneration [2].
Neuronal apoptosis is governed by the BCL2 gene family, which consists of three groups of proteins; the anti-apoptotic proteins, the BH3-only proteins and the effector proteins, that work in concert to arbitrate the cellular commitment to apoptosis [3]. When activated, the effector proteins undergo a conformational shift that causes them to insert into and oligomerize on the mitochondrial outer membrane (MOM), forming pores that cause mitochondrial outer membrane permeabilization (MOMP), which leads to caspase activation and subsequent apoptosis. The anti-apoptotic BCL2 family proteins work to prevent apoptosis by preventing activation of the effector proteins and retrotranslocating them from the MOM [4]. In RGCs, the principal anti-apoptotic Bcl2 family gene is BclX [5,6]. The third group of Bcl2 family genes are the BH3-only genes, so named because they share only the third BCL2-homology (BH3) domain with the other members of the family. These proteins promote apoptosis by inhibiting anti-apoptotic protein function and also by directly causing effector activation.
The BCL2-associated X protein (BAX) is a member of the effector group of BCL2 family genes. Due to an alternative splicing event that affects another effector protein, BAK, BAX is the lone arbiter of MOMP and intrinsic apoptosis in neurons [7]. This fact is illustrated in Bax−/− mice whose RGC somas survive indefinitely following acute or chronic optic nerve injury [8–10]. By contrast, performing ONC on a Bax+/+ mouse causes near complete loss of RGCs within 3 weeks after injury [10]. This profound preservation of RGC somas in Bax−/− mice makes BAX a promising target for a neuroprotective therapy.
Crosstalk between the BCL2 gene family ultimately governs cellular fate. The balance between the three groups of proteins is controlled by several upstream pathways, which shift the balance between BH3only proteins and anti-apoptotic proteins in response to environmental stimuli [11]. Here, pathways that are activated in response to a damaging stimulus that contribute to BAX activation are collectively referred to as the BAX activation mechanism (BAM). In RGCs, a predominant BAM pathway is the Jun N-terminal Kinase (JNK) pathway [12]. Following axonal injury, Dual Leucine Zipper Kinase (DLK) is phosphorylated in the axon and then transported to the soma where successive phosphorylation events of MKK4/7, JNK2/3 and cJUN contribute to apoptosis through their interactions with the Bcl2 gene family and by driving the transcription of BH3-only genes [13–16]. Importantly, the redundancy of the BAM pathways is not yet fully understood, since even disruption of two critical mediators of the BAM does not result in the same level of protection as genetic deletion of Bax [17].
In order to assess if targeting BAX activation is beneficial for preventing neurodegeneration, it is necessary to consider pragmatic treatment paradigms. Complete genetic deletion of the Bax gene in glaucoma patients is technically challenging and inadvisable since BAX functions as a tumor suppressor gene and has an emerging role in the maintenance of the mitochondrial network under normal conditions [18–20]. A more viable therapeutic strategy is to lower the effective quantity of BAX in RGCs, but the effect of lowered BAX dosage on RGC survival after injury has not been studied in depth. Therefore, we tested the hypothesis that Bax+/− RGCs would be resistant to apoptosis following ONC.
This hypothesis was tested by performing ONC on Bax+/− mice, which produce half as much retinal Bax mRNA and protein as a wild type mouse [10]. Thus, these mice effectively model a therapy that lowers the quantity of BAX in RGCs by half, without impairing developmental pruning of redundant RGCs, which is a hallmark of Bax−/− mice [21].
This study demonstrates a profound effect of lowered BAX dosage on long term RGC survival following crush injury and demonstrates that these RGCs deactivate their BAM several months after ONC and become quiescent. These findings have important implications for the development of a neuroprotective strategy targeted at preventing neuronal apoptosis.
Methods
Animals
Adult mice (3–6 months of age) were used for this study and handled according to the Association for Research in Vision and Ophthalmology statement for the use of animals in research. Every protocol described here was approved by the Animal Care and Use Committee at the University of Wisconsin-Madison. Bax+/+, Bax+/− and Bax−/− mice were obtained by breeding Bax+/− mice [22]. All mice were on a C57BL/6J background and an equal proportion of sexes were included in each cohort. Mice were housed in microisolator cages, fed a 4% fat diet and kept in a facility that uses a 12-hour light/dark cycle. Mice were removed from the study if any ocular abnormalities, such as persistent cataract, were observed.
Surgical protocols, ONC, intravitreal injections
For all surgical procedures, mice were anesthetized with a mixture of ketamine (16 mg/mL) and xylazine (1.5 mg/mL). To alleviate post-operative discomfort, anesthetized mice also received a subcutaneous injection of buprenorphine (0.03 mg/mL). Each eye to be operated on was anesthetized using a drop of proparacaine hydrochloride.
ONC was performed as previously described [2]. The left eye of each mouse underwent the procedure. The optic nerve was exposed by cutting the conjunctiva at the limbal junction. N7 curved, self-closing forceps (Fine Science Tools, Foster City, CA or Roboz Surgical Instrument Co, Gaithersburg, MD) were used to pinch the optic nerve for 5 seconds. After surgery, the eye was covered in antibiotic ointment to prevent infection and the mice were allowed to recover in their cages.
Both anisomycin and viral vectors were injected intravitreally as follows. First, a 30 gauge needle was used to puncture the conjunctiva and the sclera of the eye near the limbus. Then a 35 gauge NanoFil needle attached to a 10 μL NanoFil syringe (World Precision Instruments, Sarasota, FL) was used to inject 1 μL of solution into the vitreal chamber, which was accessed through the hole created using the 30 gauge needle. Following injection, the NanoFil needle was held in place for 10 seconds to prevent backflow of solution through the hole made by the needle. AAV2/2-Pgk-GFP-BAX viral preparations were packaged by the University of North Carolina – Chapel Hill Vector Core (Chapel Hill, NC) with a viral titer of at least 1012 viral particles per mL. For experiments involving anisomycin, 1 μL of 10 μM anisomycin in dimethyl sulfoxide (Alfa Aesar, Haverhill, MA) was injected intravitreally. For vehicle injections, 1 μL of dimethyl sulfoxide was injected intravitreally. Mice were collected 24 hours later, processed and sectioned as described below.
Whole-mounting, frozen sectioning and immunofluorescence
Following euthanasia with 0.1 mL of Euthasol (Virbac AH, Fort Worth, TX), eyes were enucleated, punctured with a 30 gauge needle and fixed in 4% paraformaldehyde in phosphate buffered saline (PBS) for 50 minutes at room temperature, then washed once in PBS before being immediately used in downstream applications.
Retinal whole-mounts were prepared by removing the anterior chamber of the eye, dissecting the retina out of the posterior chamber and laying it flat on a slide by making 4 relaxing cuts. Nuclei were labeled by staining whole-mounted retinas with 4′,6-diamidino-2-phenylindole (DAPI) at a concentration of 500 ng/mL for 15 minutes, followed by three washes in PBS. Immumount (Thermo Fisher Scientific, Waltham, Massachusetts) was used to adhere the coverslip to the whole-mount on the slide.
DAPI-stained, whole-mounted retinas were imaged on an Andor Revolution XD Spinning-Disk Confocal microscope (Andor, Belfast, United Kingdom). Four images were taken using a 100X objective in each quadrant of the mid-peripheral retina and the number of total neuronal cells (defined by nuclear size and morphology) in the RGC layer were counted and compared between the injured and the contralateral control eyes. A minimum of 4 mice were used per group.
In preparation for sectioning, fixed eye cups were infiltrated with 30% Sucrose in PBS overnight at 4°C. Tissues were submerged in Optimal Cutting Temperature media and frozen on dry ice. 10 μM longitudinal sections, which contained the optic nerve head, were cut so that the entire retina could be viewed. Every other section was discarded to avoid counting the same cell twice.
Sections were rehydrated in PBS for 10 minutes and then blocked in 1% Horse Serum (Lonza, Basel, Switzerland) in PBS for one hour at room temperature. Sections were incubated with mouse anti-BRN3A monoclonal antibody (Millipore, Temecula, CA, MAB1585, 1:50 dilution) and rabbit anti-Phospho-cJUN (Ser73) (D47G9) monoclonal antibody (Cell Signaling Technology, Danvers, MA, #3270, 1:400 dilution) diluted in PBS containing 2% Bovine Serum Albumin (W/V) and 0.3% Triton-X 100 (V/V) overnight at 4°C. Sections were then washed briefly three times in PBS, before being incubated with Texas Red conjugated goat anti-rabbit (Jackson Immunoresearch, West Grove, PA, 1:500 dilution) and Alexa Fluor 488-conjugated goat anti-mouse IgG (Jackson Immunoresearch, 1:500 dilution) for 1 hour at room temperature. Sections were then incubated in 500 ng/mL DAPI for 10 minutes before being washed briefly three times in PBS and then cover slipped using Immumount (Thermo Fisher Scientific).
For TOM20 staining, eye cups containing GFP-BAX transduced retinas were blocked using PBS containing 2% Bovine Serum Albumin (W/V) and 0.3% Triton-X 100 (V/V) overnight at 4°C. Eye cups were then incubated with 10 μg/mL moue monoclonal anti-TOM20 (4F3) (Sigma-Aldrich, Saint Louis, MO) in 2% BSA and 5% donkey serum in PBS for 2 days at 4°C. The samples were then washed 3 times in PBS before being incubated in Texas Red conjugated goat anti-mouse IgG (Jackson Immunoresearch, 1:500 dilution) in 2% BSA, 0.3% Triton-X 100 overnight at 4°C, before being washed 3 times in PBS, stained with 500 ng/mL DAPI and whole-mounted for imaging.
For each group, a minimum of 3 mice were used and a minimum of 5 sections per eye were counted. Sections were imaged on a Zeiss Axioimager Z2 upright microscope (Carl Zeiss, Oberkochen, Germany). For each section, images were taken using a 20X objective of the entire RGC layer from one end of the retina to the other so that the entire RGC layer of each section was counted. Cell counting was performed using Zen Blue image analysis software (v2.3, Carl Zeiss). Counts of cells that were stained positive for BRN3A and/or phospho-cJUN were expressed as a percentage of the DAPI positive cells in the RGC layer.
Viral introduction of exogenous BAX to test the activation status of the BAM
Following intravitreal injection of AAV2/2-Pgk-GFP-BAX, four weeks were given before analysis to allow for adequate expression of the transgene [23]. Some groups of mice were transduced prior to ONC and some were transduced at varying times after ONC. To quantify GFP-BAX expression and activation, retinas were fixed and whole-mounted as described above.
Images were taken on a Zeiss Axioimager Z2 upright microscope (Carl Zeiss). Images were taken with a 20X objective in each quadrant of each whole-mounted retina and no fewer than 200 GFP-BAX transduced cells were counted in each retina. Images were analyzed using Zen Blue image analysis software to mark and count each GFP-BAX positive cell in each image.
The quantification of cells with punctate BAX is reported as a percentage of the total number of cells labeled with GFP-BAX. A one tailed T-test was used to assess significance between groups. A minimum of 5 mice per group were used for each group that was transduced with virus prior to ONC and 7 mice per group were used for groups that were transduced with virus after ONC.
Quantitative PCR
Total RNA was isolated from flash frozen retinas using the IBI DNA/RNA/Protein Extraction Kit (IBI Scientific, Dubuque, Iowa). Contaminating genomic DNA was digested using an IBI in-column DNase I kit (IBI).
To synthesize cDNA, 2 μg of total RNA from each sample were combined with 500 ng of random hexamers (Promega, Madison, Wisconsin, USA) and heated to 70°C, quenched on ice and then incubated with 10 mM dNTPs and M-MLV Reverse Transcriptase (Promega) for 90 minutes at 37°C.
The resulting cDNA was diluted 1:10 and transcript abundance for each sample was analyzed using customized TaqMan Array Cards (Thermo Fisher Scientific). Cards were customized to contain triplicate primers for the Bcl2 family genes Bcl2l1 (BclXL), Bax, Bim, Bbc3, Hrk, Bad, Bmf and Noxa. The transcript for Bid was excluded because this BH3-only protein is activated by cleavage of a latent protein, and there is little evidence that transcriptional upregulation plays a role in its activation. The array also contained primers for the RGC markers Nrn1 and Sncg, the glial markers Gfap and Aif1, and the phospho-cJUN transcription target genes Ecel1, Gal and Atf3. The transcript for 18S rRNA, was used as an internal reference for the amount of cDNA loaded in each sample. Quantitative PCR (qPCR) was performed on a QuantStudio 7 Flex system (Thermo Fisher Scientific) using a PCR procedure of 40 cycles of 15 seconds at 95°C and 60 seconds at 60°C. Transcript abundances were measured 1, 7 and 20 weeks after ONC for Bax+/− mice and 1, 3, and 7 days after ONC for Bax+/+ mice. Each time point contained 4 replicate groups. Whole retinas from 3 mice were pooled for each group. Changes in the quantity of mRNA of each gene of interest between samples were quantified using the ΔΔCt method [24], using the 18S rRNA abundance for each sample to normalize the total amount of cDNA and then calculating fold change between the experimental and contralateral control retinas for each group. Significance of observed fold changes for genes, relative to the control sample, was calculated using a 1 tailed, paired t-test, assuming equal variance. The significance of differences in fold change between timepoints was assessed using a 2 tailed, unpaired t-test, assuming equal variance between groups.
Results
Bax+/− mice experience delayed and diminished loss of RGCs after ONC
In wild type mice, ONC produces substantial loss of cells in the ganglion cell layer by 3 weeks after damage. Previously, we reported that Bax+/− mice exhibited complete resistance to ONC at this time point, but that some cell loss was evident after 8 weeks [10]. To investigate the time course of cell loss in Bax+/− mice in greater detail, ONC was performed on Bax+/− mice and the pattern of cell loss in the ganglion cell layer was examined at times up to 24 weeks after optic nerve damage (Figure 1). RGCs constitute roughly half of the cells in the ganglion cell layer [25], so the maximal cell loss expected in this experiment was 50% of the total neuronal population in this layer. In Bax+/+ mice, 3 weeks after ONC there was a 27.4%±2.4% decrease in cell density in the crushed eye compared to the contralateral control eye. Conversely, Bax+/− mice did not lose a significant percentage of the cells in their ganglion cell layer until 12 weeks (11% cell loss ± 5%, p = 0.009) after ONC and at 24 weeks they had only lost 13% of the cells in their RGC layer (Figure 1). Importantly, there was no significant cell loss observed between the 12 and 24 week post-ONC groups, implying that the protective effect of BAX depletion was sustained for months after acute optic nerve injury. Thus, BAX depletion provides sustained protection to RGC somas following acute optic nerve injury.
Figure 1: Bax deficiency attenuates the loss of cells in the RGC layer following optic nerve crush (ONC).

The change in the density of neuronal cells in the retinal ganglion cell layer of an eye that had undergone ONC versus the contralateral control eye of the same mouse. The mean ± standard deviation is graphed for both Bax+/− (blue bars) and Bax+/+ (orange bars) mice. P values were calculated comparing groups to the 0 week timepoint by using a one sided t-test that assumed equal variance between groups. Cell loss appears to be slowly progressive in Bax+/− mice, but only reaches significance by 12 weeks post ONC (pONC). No further cell loss is evident even 24 weeks after crush. At no time point did cell loss in Bax+/− mice reach the level observed in wild type animals after 3 weeks. *P<0.05, ***P<0.001, n.s. = not significant. N = 5 mice per group for cohorts collected 3–12 weeks and N=3 for the cohort collected 24 weeks post ONC.
Apoptotic susceptibility of RGCs is determined by BAX dosage
To further demonstrate the effect of BAX dosage on apoptotic susceptibility, the extent of BAX translocation to the MOM after ONC was compared between Bax+/+ and Bax+/− mice that had been transduced with AAV2/2-Pgk-GFP-Bax. Virally transduced BRN3A positive RGCs expressed the GFP-BAX transgene 4 weeks after intravitreal injection (Figure 2A-D). Furthermore, induction of apoptosis via ONC caused GFP-BAX to aggregate on the mitochondria, as demonstrated by colocalization of GFP-BAX and TOM20 (Figure 2E–J).
Figure 2: AAV2/2-Pgk-GFP-Bax transduction of RGCs to monitor BAX activity in vivo.

(A) Tiled image of a retina that has been transduced with AAV2/2-Pgk-GFP-Bax. Size bar = 75 μm. (B-D) The GFP-BAX transgene is expressed in cells in the RGC layer, the majority of which are positive for the RGC marker BRN3A (asterisks indicate examples of co-localization). GFP-BAX expressing cells that are not BRN3A positive (arrows) may represent displaced amacrines or some of the 15% of RGCs that do not express this marker [55]. Size bar = 10 μm. (E-G) Immunostained sections of a retina from a control eye. In healthy RGCs, GFP-BAX is localized throughout the cytosol (E). TOM20 staining shows the distribution of mitochondria in these cells. (H-J) Following optic nerve crush, GFP-BAX translocates to the mitochondrial outer membrane, as evidenced by colocalization with TOM20, and exhibits a punctate pattern. Size bars in G and J = 5 μm.
Apoptotic susceptibility was quantified by counting the percentage of GFP-BAX expressing cells with a punctate localization of GFP-BAX (Figure 3). Both groups of mice had a significantly greater number of punctate cells in the damaged retina, compared to unoperated eyes (p = 3E-7 for Bax+/+, p = 0.0001 for Bax+/−) (Figure 3E). There were significantly more punctate labeled cells in the injured eyes of Bax+/+ mice than in the injured eyes of Bax+/− mice (p = 0.018). Thus, overall concentration of latent BAX protein in RGCs influences their apoptotic susceptibility, which is consistent with the attenuated loss of RGCs in Bax+/− mice after optic nerve injury.
Figure 3: The apoptotic susceptibility of RGCs is determined by BAX dosage.

(A,C) Representative images of AAV2/2-Pgk-GFP-Bax transduced cells in the contralateral (Con) eye of a Bax+/+ and Bax+/− mouse, respectively. Images are of DAPI stained, whole-mounted retinas (B,D) Representative images of transduced cells in the eye 7 days post optic nerve crush (pONC) in a Bax+/+ and Bax+/− mouse, respectively. Arrows point to cells with punctate GFP-BAX localization. Size bar = 20 μm. (E) Box and whisker plots illustrating the percentage of GFP-BAX labeled cells that exhibit punctate localization of GFP-BAX in Bax+/+ and Bax+/− genotypes after ONC. P values were calculated using a one sided t test, assuming equal variance between groups. Both genotypes exhibited significantly more cells with punctate GFP-BAX in the eyes that received ONC (relative to Con eyes), while significantly more cells were punctate in Bax+/+ mice compared to Bax+/− mice. *P<0.05, ***P<0.001. N = 10 mice for Bax+/+ and 5 mice for Bax+/−. An average of 226 and 280 GFP-BAX transduced cells were counted for each Bax+/+ and Bax+/− retina, respectively.
The BAM remains active for months after ONC
Since Bax+/− mice exhibited reduced BAX translocation relative to their wild type counterparts, we tested the ability of these cells to activate their BAM. At various times from 3 days to 20 weeks after ONC, sections from Bax+/+ and Bax+/− animals were stained for the ganglion cell marker BRN3A, a marker of BAM activation, phospho-cJUN, and for DAPI (Figure 4A–G). Within 3 days after ONC, phospho-cJUN accumulated in the nuclei of a majority of BRN3A positive cells in the ganglion cell layer in both Bax+/+ and Bax+/− mice, suggesting that Bax+/− cells had a functional BAM and were actively attempting to undergo apoptosis (Figure 4H). Both BRN3A and phospho-cJUN levels decreased over 2 weeks in wild type mice, reflective of the loss of RGCs in these animals (Figure 4I,4K). Surprisingly, Bax+/− mice exhibited elevated levels of phospho-cJUN labelling up to and including 7 weeks after ONC (Figure 4L). Between 7 and 12 weeks after ONC, however, there was a decrease in the percentage of cells labeled with phospho-cJUN that was not accompanied by a decrease in the percentage of cells labeled with BRN3A (Compare Figure 4L and Figure 4J). These results implied that Bax+/− RGCs were deactivating their BAM after failing to undergoing apoptosis.
Figure 4: The BAX activation mechanism is rapidly activated and then deactivated months after optic nerve crush.

(A-G) Representative images of the RGC layer at various times post optic nerve crush (pONC) in Bax+/− mice. The control retina depicts the contralateral eye. BRN3A (green) and phospho-cJUN (red) have been labeled using immunofluorescence and the nuclei have been labeled via DAPI staining (blue). Size bar = 30 μm. (H) Quantification of the percentage of nuclei in the RGC layer that are positively labeled for BRN3A (green bar), phospho-cJUN (red bar), or both (yellow). The total height of the bar at each time point represents the total percentage of cells in the RGC layer that were labeled with either immunofluorescent marker. Error bars represent the standard deviation from the mean for each of the three labelling patterns. Bax+/+ mice exhibit rapid accumulation of phospho-cJUN after ONC, which declines by 2 weeks. This decline is accompanied by a similar decline in BRN3A staining cells, consistent with cell loss in these animals. Bax+/− mice exhibit a similar rapid accumulation of phospho-cJUN. These animals also exhibit a temporal decrease in phospho-cJUN labeling, but this is not accompanied by a dramatic decrease in BRN3A staining. (I,J) Individual graphs of the data shown in (H) showing the percentage of cells labeled for BRN3A at various times after ONC in Bax+/+ and Bax+/− retinas. (K,L) Individual graphs of the data shown in (H) showing the percentage of cells labeled for phospho-cJUN at various times after ONC in Bax+/+ and Bax+/− retinas. Error bars represent the standard deviation from the mean. P values were calculated using a one sided t test, assuming equal variance between groups. *P<0.05, **P<0.01, ***P<0.001. N = minimum of 4 mice per group, except 1 week group for which N = 3 mice.
The BAM transcriptional response is normal in Bax+/− RGCs
To further characterize the BAM in Bax+/− mice, qPCR was used to compare the transcriptional responses to ONC between Bax+/+ and Bax+/− mice. Because RGC apoptosis becomes an increasingly confounding factor in Bax+/+ mice by 1 week after ONC, we compared the transcriptional response in these mice 3 days after injury with Bax+/− mice 1 week after injury. Three days also marks the peak of transcript abundance in wild type animals for a majority of the BAM-related genes being assayed with the microfluidic array cards (Supplemental Figure 1).
The BH3-only transcripts Bim, Hrk, Bbc3, and Noxa, were all significantly more abundant in the Bax+/− retinas that underwent ONC than the contralateral control eyes and there was no significant difference in the fold changes between wild type and Bax+/− retinas, except for Noxa, which was more potently upregulated in Bax+/− retinas after ONC (p = 0.01) (Figure 5A). Furthermore, markers of glial activation, Aif1 and Gfap, and markers of activated JUN transcription, Ecel1, Gal and Atf3, showed similar increases in abundance in both genotypes, indicating similar glial responses and JUN transcriptional responses between genotypes (Figure 5B,C). Ecel1 was slightly, but significantly, more upregulated in Bax+/− than wild types (p = 0.004). The depletion of the neuronal transcripts Nrn1 and Sncg was not different between genotypes, implying that RGC specific gene expression loss was not protected by Bax depletion (Figure 5D). These data demonstrate that Bax+/− RGCs have a competent BAM and that BAX depletion confers their resistance to apoptosis.
Figure 5: Bax+/− and Bax+/+ retinas exhibit a similar transcriptional response of BAM related genes after optic nerve crush.

The transcriptional responses of Bax+/+ retinas 3 days after ONC and Bax+/− retinas 1 week post optic nerve crush (pONC). The 3 day time point represents the time when a majority of BAM-related genes showed maximal changes in Bax+/+ mice (see Figure S1). A comparison of the changes in transcript abundance of BH3-only genes (A), transcripts expressed by activated glia (B), RGC specific transcripts (C) and transcripts expressed as a result of JUN directed transcription (D). Data is expressed as the mean, with error bars representing standard deviation. Transcript abundance changes were the same, or slightly lower, in Bax+/+ mice compared to Bax+/− mice for all 4 groups of assessed genes. Bax+/+ data consists of the pooled average from 2 groups where 3 retinas were pooled for each group. Bax+/− data consists of the pooled average from 4 groups where 3 retinas were pooled for each group. Error bars represent standard deviation from the mean. P values were calculated using a one sided t test, assuming equal variance. *P<0.05.
The relative abundances of Bcl2-family transcripts is similar between Bax+/+ and Bax+/− retinas
Another way to compare the BAMs between Bax+/+ and Bax +/− is to examine their relative abundance of Bcl2-family transcripts (Figure 6). We compared Bax+/+ transcript abundances 3 days after ONC to Bax+/− transcript abundances 1 week after ONC. The overall relative distribution of molecules appeared very similar between the two genotypes. In both genotypes, Bcl2l1 (BclX) was the most abundant transcript, followed by Bax, Bad, and Bim. Conversely, Noxa and Hrk transcripts represented only 1–2% of the mRNAs of all the Bcl2-family genes, and only 2–7% of the BH3-only genes, interrogated in this study. As expected, Bax transcripts were less abundant in Bax+/− retinas than wild type retinas (26.6% and 14.4% of Bcl2 family transcripts in Bax+/+ and Bax+/− retinas, respectively) (Figure 6), and levels were between 3–5 fold less abundant than Bcl2l1 transcripts, depending on genotype. The relative quantities of Bcl2 family transcripts are similar between Bax+/+ and Bax+/− mice.
Figure 6: The relative abundance of Bcl2-family transcripts in whole retina after optic nerve crush.

(A) The relative abundance of each Bcl2-gene family transcripts 3 days after ONC in Bax+/+ retinas and 1 week after ONC in Bax+/− retinas. Values were normalized to the least abundant transcript (Noxa in both genotypes, which was assigned a value of 1%). Both genotypes exhibit a similar relative abundance of transcripts for Bcl2-family genes, with the exception of Bax, which is reduced by approximately 45% in Bax+/− retinas. (B) The relative abundance of BH3-only gene transcripts from the same groups (delineated by a line in the graphs in (A)).
The BAM is deactivated in RGCs when they become quiescent
The decrease in the percentage of phospho-cJUN labeled RGCs between 7 and 12 weeks after ONC suggests that the BAM is turned off in these cells. This phenomenon was quantified further by comparing changes in transcript abundance for BAM-relevant genes in Bax+/− retinas at 1, 7 and 20 weeks after ONC. The transcripts for Bax and BclX were not significantly more abundant in the injured eyes than in the contralateral eyes at any of the three timepoints (data not shown). We also quantified the relative change in transcript levels for 6 BH3-only genes, (Bim, Hrk, Bbc3, Bad, Bmf, and Noxa), many of which have been reported to be active in RGC death [13,26,27]. The abundances of 4 BH3-only transcripts (Bim, Hrk, Bbc3, and Noxa) were initially increased, but then decreased progressively throughout the experiment (Figure 7A), while neither Bad or Bmf showed any change compared to fellow control retinas, possibly because their activation can occur via post-translational modification of latent proteins similar to BID [28,29]. Only Hrk and Noxa mRNAs were significantly more abundant in the injured eyes than in the contralateral control eyes 20 weeks post-ONC. This may be due to the fact that some RGCs remain positive for phospho-cJUN (Figure 4), and therefore may still have an active BAM.
Figure 7: The transcriptional activation in the retina associated with optic nerve crush gradually subsides in the months after injury.

A comparison of the changes in transcript abundance of BH3-only genes (A), transcripts expressed by activated glia (B), RGC specific transcripts (C) and transcripts expressed as a result of JUN directed transcription (D) in Bax+/− retinas 1, 7, and 20 weeks post optic nerve crush (pONC). Data is expressed as the mean, with error bars representing standard deviation. Each bar combines data from 4 groups where each group contains cDNA derived from 3 pooled retinas. P values were calculated using a one sided t test, assuming equal variance. *P<0.05, **P<0.01, ***P<0.001.
The same trend of initial accumulation followed by decrease in mRNA abundance was also observed in the markers of glial activation (Gfap and Aif1) and in the markers of JUN dependent transcription (Ecel1, Gal, and Atf3) (Figure 7B and 7C, respectively). Therefore, the transcriptional arm of the BAM, particularly with respect to JUN-mediated gene expression, tapers off by 20 weeks post-ONC and pathologic glial activation, a contributor to RGC death [30], tapers off by 7 weeks post-ONC. Importantly, the abundance of RGC-specific transcripts showed a marked decrease between 1 and 7 weeks, implying that damaged RGCs continue to atrophy in the months after injury (Figure 7D).
Characterization of the BAM in damaged RGCs in Bax−/−mice
Because of redundancy in the pathways that constitute the BAM, it was necessary to directly test if the BAM was sufficiently activated to cause BAX translocation at various times following ONC. To do this, ONC was performed on Bax−/− mice which were then transduced with AAV2/2-Pgk-GFP-Bax 4 or 8 weeks after ONC and then collected 4 weeks after transduction, allowing time for transduction and expression of GFP-BAX (Figure 3A). Previously, we reported that RGCs from Bax−/− mice could be efficiently transduced, and express a transgene from the Pgk promoter, several months after ONC [31].
We found that there were significantly more punctate cells in the damaged eyes of mice transduced with virus 4 weeks after ONC (p = 0.0414, relative to the undamaged contralateral control eye) (Figure 8). Additionally, the percentage of punctate cells at this time point was statistically similar to the number of punctate cells induced by ONC in Bax−/− mice that were transduced with virus prior to ONC (p = 0.0868). Thus, RGCs transduced with GFP-BAX virus 4 weeks after ONC are still susceptible to BAX translocation due to an activated BAM.
Figure 8: Damaged RGCs exhibit a diminishing capacity to activate exogenous BAX after optic nerve crush.

(A-F) Representative images of AAV2/2-Pgk-GFP-Bax transduced retinas from Bax−/− mice. Contralateral control (Con) retinas principally contain cells with diffuse GFP-BAX (A,C,E), while retinas transduced either before (B) optic nerve crush (ONC) or 4 weeks after (D), exhibit an increase in cells with punctate GFP-BAX (arrows). Retinas transduced 8 weeks after ONC, however, show no increase in the presence of punctate cells (F). Size bar = 30 μm. (G) Quantification of the percentage of GFP-BAX labeled cells that display a punctate localization of GFP-BAX. The X axis denotes when the mice were transduced with AAV2/2-Pgk-GFP-Bax relative to ONC. For mice that were transduced before ONC (Pre-Crush), ONC was performed 4 weeks after viral injection and the mice were analyzed 7 days after ONC. For mice transduced at the times indicated post-ONC (pONC), the data were analyzed after 4 additional weeks to allow for expression of the GFP-BAX transgene. Therefore mice transduced at 4 weeks pONC were analyzed at 8 weeks pONC, and mice transduced at 8 weeks pONC were analyzed at 12 weeks. ONC induced a significant increase in punctate GFP-BAX distribution in retinas transduced pre-crush and retinas transduced 4 weeks after ONC. Retinas transduced 8 weeks pONC showed no increase in punctate GFP-BAX labeling over background, suggesting the crush-induced BAX activation mechanism was no longer functioning. Error bars represent standard deviation from the mean. P values were calculated using a one sided t test, assuming equal variance between groups. n.s. = not significant, *P<0.05, ***P<0.001. N = 9 mice for the Pre-crush group, 7 mice for the 4 week group and 7 mice for the 8 week group. An average of 434, 305, and 320 cells were counted in each transduced retina for the Pre-Crush, 4 week post-crush and 8 week post-crush groups, respectively. There was no significant difference in the average number of cells labeled per image between groups (Supplemental Figure 2).
In the cohort of mice transduced with virus 8 weeks after ONC, there was no statistical difference in the percentage of punctate cells between the experimental and control eyes (p = 0.3788). This indicates that the ONC injury is no longer contributing the BAX translocation in these eyes. Importantly, there was no difference in the number of labeled cells per field between the experimental and contralateral control eyes of mice injected 8 weeks after ONC (p = 0.33), which rules out apoptosis of cells prior to fixation, or reduced transduction efficiency, as reasons for the decreased percentage of punctate labeled cells (Supplemental Figure 2).
The reduced susceptibility of Bax−/− RGCs injected 8 weeks after ONC is consistent with the data showing a reduction of phospho-cJUN staining, and reduced levels of transcript abundance for genes involved in the BAM. Taken together, these results suggest that the BAX activation mechanism is deactivated in surviving RGCs months after ONC. Since these RGCs have decreased expression of RGC specific transcripts and have deactivated their apoptotic machinery, we classify them as quiescent RGCs (qRGCs).
The BAM is not restimulated by anisomycin treatment in qRGCs
Our results indicate that the BAM is deactivated in surviving RGCs after ONC. We then asked if this was a permanent or transient phenomenon. Our experimental paradigm involved acute damage to the optic nerve, which precluded a second ONC surgery to induce the BAM in these RGCs. Therefore, we applied a secondary stimulus via intravitreal injection of anisomycin, an activator of the JNK pathway [32]. Stimulation of the JNK pathway was confirmed by the accumulation of phospho-cJUN in the nuclei of anisomycin injected retinas (Figure 9A, 9C). In naïve eyes, anisomycin injection resulted in the majority of nuclei in the RGC layer and inner nuclear layer to stain positively for phospho-cJUN 1 day after injection (Figure 9A), including 90% of cells that were also positive for BRN3A (Figure 9D). Next, we attempted to reactivate JNK pathway in qRGCs by injecting anisomycin into eyes 20 weeks after ONC. Anisomycin injection at this time caused phospho-cJUN accumulation in the nuclei of the inner nuclear layer (Figure 9C). Among, cells in the RGC layer, anisomycin injection failed to increase the percentage of cells labeled for both BRN3A and phospho-cJUN (Figure 9D). This indicates that qRGCs are deficient at responding to external stimulation by reactivating the BAM.
Figure 9: qRGCs do not respond to anisomycin treatment 20 weeks after optic nerve crush.

(A-C) Images of the RGC layer (GCL) and inner nuclear layer (INL) of retinas from Bax+/− mice, where BRN3A (green) and phospho-cJUN (red) have been labeled using immunofluorescence and the nuclei have been labeled via DAPI staining (blue). Anisomycin stimulates wide-spread accumulation of phospho-cJUN in nuclei of cells in both layers within 1 day after injection (A). (B) Retina from a mouse 20 weeks post optic nerve crush (pONC) and 1 day after injection of vehicle showing minimal labeling for phospho-cJUN in either layer. (C) A retina, 20 weeks pONC, 1 day after anisomycin injection. Minimal phospho-cJUN labeling is detected in the GCL, but extensive labeling is observed in the INL. Size bar = 30 μm. (D) Quantification of the percentage of BRN3A positive cells that are also positive for phosphor-cJUN. The retinas of eyes injected with anisomycin at 20 weeks pONC showed no significant increase in phospho-cJUN accumulation in RGCs compared to the vehicle injected 20 week pONC group. Error bars represent standard deviation from the mean. *P<0.05, **P<0.01, n.s. = not significant. 4 mice were used for the naïve cohort that received an anisomycin injection. The 20 week post-ONC groups contained 10 mice for each group.
Discussion
BAX depletion protects RGCs from apoptosis following acute injury
Bax+/− RGCs were robustly protected against apoptosis following acute optic nerve injury. This protection was not due to a deficient BAM in Bax+/− RGCs or because of a dampened glial response. These findings demonstrate that lowering the effective quantity of BAX in a neuron is an effective strategy for preventing apoptosis.
A small but significant minority of Bax+/− RGCs did undergo apoptosis following ONC, indicating a spectrum of susceptibilities among the population of RGCs. This finding may reflect recent studies, which have demonstrated subtype specific susceptibility of RGCs to apoptosis following optic nerve injury. Although these studies show conflicting sensitivity of αRGCs, they are in agreement that intrinsically photosensitive RGCs are most resilient [33,34]. It is possible that the cells exhibiting continued activation of phospho-cJUN represent a sub-population of RGCs that are most sensitive to optic nerve damage. Similarly, more resistant sub-types of RGCs may represent cells that have constitutively lower BAX levels. It is important to note that studies monitoring different susceptibility of RGC subtypes have only successfully examined a small fraction of the 30 or so different subtypes described in the mouse retina. Further study is needed to determine if BAX dosage is different among different subtypes of RGCs or if the extent of BAM activation is different between RGC subtypes.
What is the mechanism for the neuroprotective effect of BAX depletion? The mechanism of BAX-mediated MOMP has at least two distinct steps, translocation of BAX to the MOM and BAX dimerization and oligomerization leading to MOMP [35]. The translocation process is only partially understood. A recent study postulated that latent globular BAX molecules likely exist as dimers in an ensemble of different structures. These dimers need to be disassembled in order for BAX to translocate, a process that is presumed to occur by the interaction with active BH3-only proteins [36]. BH3-only proteins play an integral part in this early activation process, because they must also interact and antagonize the function of anti-apoptotic proteins such as BCL21L (BCL-X). The stoichiometry of all the different players in this process may be critically important, but not necessarily from the standpoint of BAX concentration. Semi-quantitative western blotting and quantitative PCR of mouse retina samples indicate that the concentration of anti-apoptotic molecules exceeds the concentration of BAX by several fold [36,37, this study]. Additionally, the transcript levels of Bcl2l1 are neither increased nor decreased in Bax+/− mouse retinas (this study). As a consequence, cell death requires the expression of sufficient BH3-only proteins to accommodate both functions of anti-apoptotic protein antagonism and BAX activation, which may explain the wide variety and redundancy of BH3-only proteins expressed by dying cells. How then could the depletion of BAX affect the activation process? One explanation may reside in studies that show that only some BH3-only proteins (BIM, PUMA, and NOXA) have the capacity to bind to BAX, and these actually have greater binding affinity for anti-apoptotic BCL2 family members [39]. It is possible that the reduction of latent BAX substrate, combined with the poor binding affinity of critical BH3-only proteins, increases the activation energy of this reaction. Importantly, the studies reported here demonstrate that Bax+/− mice express the expected complement of BH3-only proteins.
The second component of MOMP is the formation of activated BAX dimers, and then large oligomers, in the MOM. A critical element of this step is that once activated, the BH3 domain of BAX becomes exposed. Evidence suggests that BAX itself may function to activate more latent BAX molecules to amplify the recruitment of BAX to the MOM [37,40]. In this model, very few BAX molecules would conceivably need to be activated (or primed) by interaction with BH3-only proteins. This amplification step may also be affected by reduced BAX concentration, especially if the recruitment of BAX by primed BAX molecules was by random chance (i.e., primed BAX essentially snared other BAX molecules as they ventured close to the MOM). While clearly more studies are warranted to examine this mechanistic effect of reduced BAX concentration further, we predict that it is the initial interaction with BH3-only proteins that is the rate-limiting step for the following reasons.
First, here we have shown that BAX depletion impairs BAX translocation to the MOM, 1 week after ONC, similar to observations made in tissue culture cells expressing titrated levels of BAX [38]. As noted above, new evidence seems to correlate the activation of BAX by BH3-only proteins with the process of BAX translocation. Second, the majority of RGCs with depleted BAX do not die. This likely rules out the possibility that low levels of BAX are being primed and translocated, but that further recruitment is impaired, leaving cells with the appearance of cytosolic BAX. Studies in tissue culture cells show that MOMP occurs early in the BAX recruitment process, even before BAX puncta are detectable by light microscopy [41]. It is notable that caspase inhibitors provide only transient protection to RGCs [42,43]. Since MOMP releases cytochrome c, which activates the caspase cascade, we would expect even just priming BAX would be lethal to RGCs.
Aside from possibilities in which BAX activation is affected by reduced concentrations of this protein, there are other factors that may be affecting RGC susceptibility in Bax+/− mice. The concentration of BAX in a cell may have a role in determining cellular health, independent of the role of BAX in intrinsic apoptosis. Emerging evidence points to a role for BAX in regulating mitochondrial fusion and metabolic activity [18,20], and lower BAX levels may be optimal for this function. Therefore, the possibility that BAX depleted cells are intrinsically more resilient to apoptotic stimuli cannot be ruled out. Lastly, the protective effect of reduced BAX levels may occur from extrinsic factors, such as modified behavior from glial cells that extends their trophic support role. Reducing the BAX level is clearly protective, but the mechanism by which this occurs remains to be investigated.
BAM deactivation and atrophy of qRGCs
Our experiments show that Bax+/− RGCs exhibit a prolonged activation of the BAM, but that around 8 weeks after optic nerve damage, this activation becomes muted and even shuts down in surviving cells. The factors precipitating this shut down are not known. One possible explanation is that the principal damaging stimulus that signals RGCs to execute the apoptotic pathway is removed. This seems unlikely, if it is assumed that this stimulus is the starvation of retrograde neurotrophic molecules like brain-derived neurotrophic factor (BDNF), since there is no evidence that damaged RGCs have re-established their synaptic connections to visual centers in the brain. With respect to this concept, it may also be possible that the physiology of surviving RGCs changes where they no longer require a spectrum of neurotrophins considered essential for functioning RGCs. It has been well-documented that Bax−/− RGCs exhibit atrophy, including nuclear shrinkage, the formation of heterochromatin, and the loss of RGC-specific gene expression [11,44–46] after optic nerve damage. The presumption is that these cells, while still alive, no longer have the capacity to function as RGCs. Interestingly, the deactivation of the BAM generally correlates with a quieting of the glial activation response, and it is reasonable to speculate that re-establishment of the normal trophic functions of the retinal glial population may play an important role in supporting surviving RGCs in their quiescent state. Further work, however, is needed to assess the interaction between qRGCs and the retinal glia and to determine if these two events are related to one another. Alternatively, BAM deactivation may be dependent on some other factor like severity of initial injury, local glial activation, proximity to the vasculature etc. Future studies could leverage Bax+/− RGCs to explore how these factors influence recovery from injury.
While a change in RGC physiology and renewal of glial support may account for the timing of deactivation the BAM, the process by which this happens may be, in some respects, absolute. This is demonstrated by the failure of anisomycin to reactivate phospho-cJUN in qRGCs. It is clear that some components of the BAM require transcriptional activation, such as the JUN-dependent expression of BH3-only genes. These genes may be epigenetically silenced, such as during the process of heterochromatin formation, a phenomenon that is driven in damaged RGCs by the activation of histone deacetylases [23,47,48]. Why surviving Bax+/− cells are able to maintain the necessary metabolic pathways for life but not the more complex signal transduction cascades associated with being a functional RGC must be explored further.
Bax deficient RGCs survive indefinitely after severe axonal injury [10] and are potential candidates for regenerative therapies. Lowering the concentration of BAX in a cell is a much more likely clinical paradigm than total knockout of the Bax gene. In spite of this, significant hurdles remain. The deactivation of the BAM and loss of RGC specific gene expression and failure of anisomycin to reactivate the JNK pathway clearly demonstrate that qRGCs atrophy even though they are no longer actively attempting to undergo apoptosis. A contributor to this quiescent state may be nuclear atrophy [45]. Together, these results suggest that significant rejuvenation will be necessary to return functionality to these cells. A recent study compared the regenerative potential of Bax−/− RGCs from immediately after to 8 weeks after ONC and found that there was no difference in regenerative potential between conditions [49]. Importantly, this study was conducted on cells right at the cusp of when we observe deactivation of the BAM, so it is not clear if they were still actively attempting to run the cell death program or had become quiescent. It remains to be seen whether or not this regenerative potential is greater, or can even be activated, in cells after they become quiescent. We anticipate that achieving such a condition is contingent upon reversing the quiescent state.
BAX as a therapeutic target for neurodegenerative disease
In this study, ONC model has been utilized to demonstrate that Bax+/− neurons are less susceptible to apoptosis after an acute axonal injury. RGCs are long projection neurons of the CNS, and therefore may represent a model that can inform a variety of other neurodegenerative conditions. This is relevant because BAX activation is implicated in numerous neurodegenerative diseases including Glaucoma, Alzheimer’s Disease, Parkinson’s Disease, Amyotrophic Lateral Sclerosis, Multiple Sclerosis, and peripheral neuropathies [50–54]. Thus, BAX activation is a conserved component of neurodegeneration and, therefore, the effect of BAX depletion on disease progression in a variety of diseases warrants further study.
Conclusion
Reducing, but not eliminating the level of BAX is effective at preventing apoptosis of RGCs following axonal injury. Damaged RGCs attempt to undergo apoptosis for several months following injury but deactivate their BAM by 8–12 weeks after ONC. Targeting BAX activation may be a viable treatment option for neurodegenerative disease.
Supplementary Material
Supplemental Figure 1: All qPCR data from Bax+/+ mice after optic nerve crush
qPCR data for wild type animals 1, 3 and 7 days after optic nerve crush (ONC). Bars represent the fold change in the retinas of experimental eyes relative to the contralateral control eyes. The data for the groups collected 1 and 3 days after ONC are the average of 2 cohorts of 3 mice each, whose eyes were pooled for analysis. The data for the group collected 7 days after ONC is the average of 4 cohorts of 3 mice each, whose eyes were pooled for analysis. Error bars represent the standard deviation from the mean. P values were calculated using a one sided t test, assuming equal variance. *P<0.05, **P<0.01, ***P<0.001.
{kind=link}
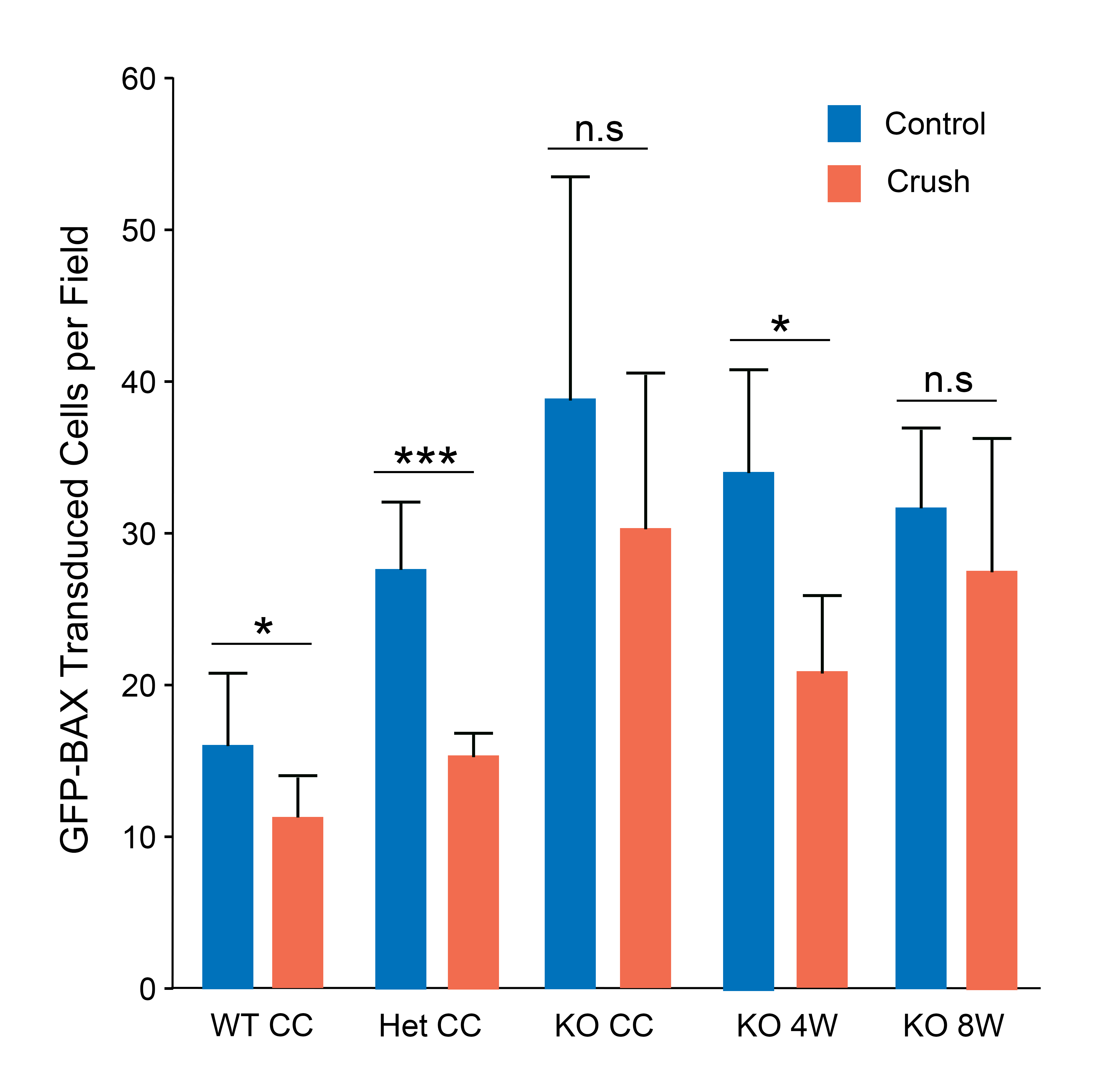
Supplemental Figure 2: The average number of cells labeled per field in each cohort of mice that was transduced with AAV2/2-Pgk-GFP-Bax
Bars represent the mean number of GFP-BAX positive cells labeled in each image for each group. Error bars represent the standard deviation from the mean. P values were calculated using a one-sided test, assuming equal variance between groups. Number of mice per group is as follows: 10 for Bax+/+ transduced before ONC, 5 for Bax+/− transduced before ONC, 9 for Bax−/− transduced before ONC, 7 for Bax−/− transduced 4 weeks after ONC and 7 for Bax−/− transduced 8 weeks after ONC. *P<0.05, **P<0.01, ***P<0.001.
{kind=link}
Acknowledgements
This work was supported by National Eye Institute grants R01 EY012223 (RWN), R01 EY030123 (RWN), T32 EY027721 (Department of Ophthalmology and Visual Sciences, University of Wisconsin-Madison), and a Vision Science Core grant P30 EY016665 (Department of Ophthalmology and Visual Sciences, University of Wisconsin-Madison), an unrestricted funding grant from Research to Prevent Blindness (Department of Ophthalmology and Visual Sciences, University of Wisconsin-Madison), the Frederick A. Davis Endowment (RWN), and the Mr. and Mrs. George Taylor Foundation (RWN). We would like to thank Satoshi Kinoshita at the Translational Research Initiative in Pathology (TRIP) lab at the University of Wisconsin – Madison for cutting all retinal sections analyzed in this manuscript.
Abbreviations
- RGC
retinal ganglion cell
- ONC
optic nerve crush
- MOM
mitochondrial outer membrane
- MOMP
mitochondrial outer membrane permeabilization
- BH3
BCL2-homology domain, 3
- BAX
BCL2 associated X, apoptosis regulator
- BAM
BAX activation mechanism
- JNK
c-JUN N-terminal kinase
- DAPI
4’,6-diamidino-2-phenylindole
- qRGC
quiescent retinal ganglion cell
Footnotes
Competing Interests
The authors declare no competing interests.
Declarations
Ethics approval
Adult mice were used for this study and handled according to the Association for Research in Vision and Ophthalmology statement for the use of animals in research and all experiments were approved by the Animal Care and Use Committee at the University of Wisconsin - Madison.
Availability of data and materials
The data collected during this study that is not present in the article are available from the corresponding author upon request.
Publisher's Disclaimer: This Author Accepted Manuscript is a PDF file of a an unedited peer-reviewed manuscript that has been accepted for publication but has not been copyedited or corrected. The official version of record that is published in the journal is kept up to date and so may therefore differ from this version.
References
- 1.Howell GR, Libby RT, Jakobs TC, Smith RS, Phalan FC, Barter JW, et al. Axons of retinal ganglion cells are insulted in the optic nerve early in DBA/2J glaucoma. J Cell Biol. 2007;179:1523–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Li Y, Schlamp CL, Nickells RW. Experimental induction of retinal ganglion cell death in adult mice. Invest Ophthalmol Vis Sci. 1999;40:1004–8. [PubMed] [Google Scholar]
- 3.Youle RJ, Strasser A. The BCL-2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol. 2008;9:47–59. [DOI] [PubMed] [Google Scholar]
- 4.Edlich F, Banerjee S, Suzuki M, Cleland MM, Arnoult D, Wang C, et al. Bcl-xL retrotranslocates Bax from the mitochondria into the cytosol. Cell. 2011;145:104–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Levin LA, Schlamp CL, Spieldoch RL, Geszvain KM, Nickells RW. Identification of the bcl-2 family of genes in the rat retina. Invest Ophthalmol Vis Sci. 1997;38:2545–53. [PubMed] [Google Scholar]
- 6.Harder JM, Ding Q, Fernandes KA, Cherry JD, Gan L, Libby RT. BCL2L1 (BCL-X) promotes survival of adult and developing retinal ganglion cells. Mol Cell Neurosci. 2012;51:53–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sun YF, Yu LY, Saarma M, Timmusk T, Arumae U. Neuron-specific Bcl-2 homology 3 domain-only splice variant of Bak is anti-apoptotic in neurons, but pro-apoptotic in non-neuronal cells. J Biol Chem. 2001;276:16240–7. [DOI] [PubMed] [Google Scholar]
- 8.Li Y, Schlamp CL, Poulsen KP, Nickells RW. Bax-dependent and independent pathways of retinal ganglion cell death induced by different damaging stimuli. Exp Eye Res. 2000;71:209–13. [DOI] [PubMed] [Google Scholar]
- 9.Libby RT, Li Y, Savinova O V., Barter J, Smith RS, Nickells RW, et al. Susceptibility to neurodegeneration in a glaucoma is modified by Bax gene dosage. PLoS Genet. 2005;1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Semaan SJ, Li Y, Nickells RW. A single nucleotide polymorphism in the Bax gene promoter affects transcription and influences retinal ganglion cell death. ASN Neuro. 2010;2:87–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Maes ME, Schlamp CL, Nickells RW. Bax to Basics: How the BCL2 gene family controls the death of retinal ganglion cells. Prog Retin Eye Res. 2017;57:1–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fernandes KA, Harder JM, Fornarola LB, Freeman RS, Clark AF, Pang I-H, et al. JNK2 and JNK3 are major regulators of axonal injury-induced retinal ganglion cell death. Neurobiol Dis. 2012;46:393–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fernandes KA, Harder JM, Kim J, Libby RT. JUN regulates early transcriptional responses to axonal injury in retinal ganglion cells. Exp Eye Res. 2013;112:106–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Putcha GV, Le S, Frank S, Besirli CG, Clark K, Chu B, et al. JNK-mediated BIM phosphorylation potentiates BAX-dependent apoptosis. Neuron. 2003;38:899–914. [DOI] [PubMed] [Google Scholar]
- 15.Welsbie DS, Yang Z, Ge Y, Mitchell KL, Zhou X, Martin SE, et al. Functional genomic screening identifies dual leucine zipper kinase as a key mediator of retinal ganglion cell death. Proc Natl Acad Sci. 2013;110:4045–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Watkins T a, Wang B, Huntwork-Rodriguez S, Yang J, Jiang Z, Eastham-Anderson J, et al. DLK initiates a transcriptional program that couples apoptotic and regenerative responses to axonal injury. Proc Natl Acad Sci. 2013;110:4039–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Syc-Mazurek SB, Fernandes KA, Wilson MP, Shrager P, Libby RT. Together JUN and DDIT3 (CHOP) control retinal ganglion cell death after axonal injury. Mol Neurodegener. 2017;12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim EM, Jung C-H, Song J-Y, Park JK, Um H-D. Pro-apoptotic Bax promotes mesenchymal-epithelial transition by binding to respiratory complex-I and antagonizing the malignant actions of pro-survival Bcl2 proteins. Cancer Lett. 2018;424:127–35. [DOI] [PubMed] [Google Scholar]
- 19.Reyna DE, Garner TP, Lopez A, Kopp F, Choudhary GS, Sridharan A, et al. Direct activation of BAX by BTSA1 overcomes apoptosis resistance in acute myeloid leukemia. Cancer Cell. 2017;32:490–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Karbowski M, Norris KL, Cleland MM, Jeong S-Y, Youle RJ. Role of Bax and Bak in mitochondrial morphogenesis. Nature. 2006;443:658–62. [DOI] [PubMed] [Google Scholar]
- 21.Mosinger Ogilvie J, Deckwerth TL, Knudson CM, Korsmeyer SJ. Supression of developmental retinal cell death but not of photoreceptor degeneration in Bax-deficient mice. Invest Ophthalmol Vis Sci. 1998;39:1713–20. [PubMed] [Google Scholar]
- 22.Knudson CM, Tung KS, Tourtellotte WG, Brown GA, Korsmeyer SJ. Bax-deficient mice with lymphoid hyperplasia and male germ cell death. Science. 1995;270:96–9. [DOI] [PubMed] [Google Scholar]
- 23.Schmitt HM, Pelzel HR, Schlamp CL, Nickells RW. Histone deacetylase 3 (HDAC3) plays an important role in retinal ganglion cell death after acute optic nerve injury. Mol Neurodegener. 2014;9:39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods. 2001;25:402–8. [DOI] [PubMed] [Google Scholar]
- 25.Schlamp CL, Montgomery AD, MacNair CE, Schuart C, Willmer DJ, Nickells RW. Evaluation of the percentage of ganglion cells in the ganglion cell layer of the rodent retina. Mol Vis. 2013;19:1387–96. [PMC free article] [PubMed] [Google Scholar]
- 26.Harder JM, Libby RT. BBC3 (PUMA) regulates developmental apoptosis but not axonal injury induced death in the retina. Mol Neurodegener. 2011;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Harder JM, Fernandes KA, Libby RT. The Bcl-2 family member BIM has multiple glaucoma-relevant functions in DBA/2J mice. Sci Rep. 2012;2:530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yang X, Luo C, Cai J, Pierce WM, Tezel G. Phosphorylation-dependent interaction with 14–3-3 in the regulation of Bad trafficking in retinal ganglion cells. Invest Ophthalmol Vis Sci. 2008;49:2483–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Happo L, Strasser A, Cory S. BH3-only proteins in apoptosis at a glance. J Cell Sci. 2012;125:1081–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mac Nair CE, Nickells RW. Neuroinflammation in glaucoma and optic nerve damage. Prog Mol Biol Transl Sci. 2015;134:343–63. [DOI] [PubMed] [Google Scholar]
- 31.Nickells RW, Schmitt HM, Maes ME, Schlamp CL. AAV2-mediated transduction of the mouse retina after optic nerve injury. Invest Opthalmology Vis Sci. 2017;58:6091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Laderoute KR, Webster KA. Hypoxia/reoxygenation stimulates Jun Kinase activity through redox signaling in cardiac myocytes. Circ Res. 1997;80:336–44. [DOI] [PubMed] [Google Scholar]
- 33.Duan X, Qiao M, Bei F, Kim I-J, He Z, Sanes JR. Subtype-specific regeneration of retinal ganglion cells following axotomy: Effects of osteopontin and mTOR signaling. Neuron. 2015;85:1244–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Daniel S, Clark A, McDowell C. Subtype-specific response of retinal ganglion cells to optic nerve crush. Cell Death Discov. 2018;5:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Youle RJ, Van Der Bliek AM. Mitochondrial fission, fusion, and stress. Science. 2012;337:1062–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Robin AY, Iyer S, Birkinshaw RW, Sandow J, Wardak A, Luo CS, et al. Ensemble properties of Bax determine its function. Structure. Cell Press; 2018;26:1346–59. [DOI] [PubMed] [Google Scholar]
- 37.Nickells RW. Variations in the rheostat model of apoptosis: What studies of retinal ganglion cell death tell us about the functions of the Bcl2 family proteins. Exp Eye Res. 2010;91:2–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Semaan SJ, Nickells RW. The apoptotic response in HCT116 cancer cells becomes rapidly saturated with increasing expression of a GFP-BAX fusion protein. BMC Cancer. 2010;10:554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Adams JM, Cory S. The Bcl-2 apoptotic switch in cancer development and therapy. Oncogene. 2007;26:1324–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Valentijn AJ, Upton J-P, Bates N, Gilmore AP. Bax targeting to mitochondria occurs via both tail anchor dependent and independent mechanisms. Cell Death Differ. 2008;15:1243–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Maes ME, Schlamp CL, Nickells RW. Live-cell imaging to measure BAX recruitment kinetics to mitochondria during apoptosis. PLoS One. 2017;12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kermer P, Klöcker N, Labes M, Thomsen S, Srinivasan A, Bähr M. Activation of caspase-3 in axotomized rat retinal ganglion cells in vivo. FEBS Lett. 1999;453:361–4. [DOI] [PubMed] [Google Scholar]
- 43.Kermer P, Klöcker N, Labes M, Bähr M. Insulin-like growth factor-I protects axotomized rat retinal ganglion cells from secondary death via PI3-K-dependent Akt phosphorylation and inhibition of caspase3 In vivo. J Neurosci. 2000;20:2–8. [PubMed] [Google Scholar]
- 44.Schlamp CL, Johnson EC, Li Y, Morrison JC, Nickells RW. Changes in Thy1 gene expression associated with damaged retinal ganglion cells. Mol Vis. 2001;7:192–201. [PubMed] [Google Scholar]
- 45.Janssen KT, Mac Nair CE, Dietz JA, Schlamp CL, Nickells RW. Nuclear atrophy of retinal ganglion cells precedes the bax-dependent stage of apoptosis. Invest Ophthalmol Vis Sci. 2013;54:1805–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pelzel HR, Schlamp CL, Waclawski M, Shaw MK, Nickells RW. Silencing of Fem1cR3 gene expression in the DBA/2J mouse precedes retinal ganglion cell death and is associated with histone deacetylase activity. Invest Ophthalmol Vis Sci. 2012;53:1428–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pelzel HR, Schlamp CL, Nickells RW. Histone H4 deacetylation plays a critical role in early gene silencing during neuronal apoptosis. BMC Neurosci. 2010;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schmitt HM, Schlamp CL, Nickells RW. Role of HDACs in optic nerve damage-induced nuclear atrophy of retinal ganglion cells. Neurosci Lett. 2016;625:11–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yungher BJ, Ribeiro M, Park KK. Regenerative responses and axon pathfinding of retinal ganglion cells in chronically injured mice. Invest Ophthalmol Vis Sci. 2017;58:1743–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ahmad K, Baig M, Gupta GK, Kamal M, Pathak N, Choi I. Identification of common therapeutic targets for selected neurodegenerative disorders: An in silico approach. J Comput Sci. 2016;17:292–306. [Google Scholar]
- 51.Kudo W, Lee H-P, Smith M a, Zhu X, Matsuyama S, Lee H-G. Inhibition of Bax protects neuronal cells from oligomeric Aβ neurotoxicity. Cell Death Dis. 2012;3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kozin MS, Kulakova OG, Favorova OO. Involvement of mitochondria in neurodegeneration in multiple sclerosis. Biochem. 2018;83:813–30. [DOI] [PubMed] [Google Scholar]
- 53.Cosker KE, Pazyra-Murphy MF, Fenstermacher SJ, Segal RA. Target-derived neurotrophins coordinate transcription and transport of Bclw to prevent axonal degeneration. J Neurosci. 2013;33:5195–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nickells RW, Semaan SJ, Schlamp CL. Involvement of the Bcl2 gene family in the signaling and control of retinal ganglion cell death. Prog Brain Res. 2008;173:423–35. [DOI] [PubMed] [Google Scholar]
- 55.Nadal-Nicolas FM, Jimenez-Lopez M, Sobrado-Calvo P, Nieto-Lopez L, Canovas-Martinez I, SalinasNavarro M, et al. Brn3a as a marker of retinal ganglion cells: Qualitative and quantitative time course studies in naïve and optic nerve–injured retinas. Invest Opthalmology Vis Sci. 2009;50:3860–8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1: All qPCR data from Bax+/+ mice after optic nerve crush
qPCR data for wild type animals 1, 3 and 7 days after optic nerve crush (ONC). Bars represent the fold change in the retinas of experimental eyes relative to the contralateral control eyes. The data for the groups collected 1 and 3 days after ONC are the average of 2 cohorts of 3 mice each, whose eyes were pooled for analysis. The data for the group collected 7 days after ONC is the average of 4 cohorts of 3 mice each, whose eyes were pooled for analysis. Error bars represent the standard deviation from the mean. P values were calculated using a one sided t test, assuming equal variance. *P<0.05, **P<0.01, ***P<0.001.
Supplemental Figure 2: The average number of cells labeled per field in each cohort of mice that was transduced with AAV2/2-Pgk-GFP-Bax
Bars represent the mean number of GFP-BAX positive cells labeled in each image for each group. Error bars represent the standard deviation from the mean. P values were calculated using a one-sided test, assuming equal variance between groups. Number of mice per group is as follows: 10 for Bax+/+ transduced before ONC, 5 for Bax+/− transduced before ONC, 9 for Bax−/− transduced before ONC, 7 for Bax−/− transduced 4 weeks after ONC and 7 for Bax−/− transduced 8 weeks after ONC. *P<0.05, **P<0.01, ***P<0.001.