

Abstract
The immature brain is especially vulnerable to lead (Pb2+) toxicity, which is considered an environmental neurotoxin. Pb2+ exposure during development compromises the cognitive and behavioral attributes which persist even later in adulthood, but the mechanisms involved in this effect are still unknown. On the other hand, the kynurenine pathway metabolites are modulators of different receptors and neurotransmitters related to cognition; specifically, high kynurenic acid levels has been involved with cognitive impairment, including deficits in spatial working memory and attention process. The aim of this study was to evaluate the relationship between the neurocognitive impairment induced by Pb2+ toxicity and the kynurenine pathway. The dams were divided in control group and Pb2+ group, which were given tap water or 500 ppm of lead acetate in drinking water ad libitum, respectively, from 0 to 23 postnatal day (PND). The poison was withdrawn, and tap water was given until 60 PND of the progeny. The locomotor activity in open field, redox environment, cellular function, kynurenic acid (KYNA) and 3-hydroxykynurenine (3-HK) levels as well as kynurenine aminotransferase (KAT) and kynurenine monooxygenase (KMO) activities were evaluated at both 23 and 60 PND. Additionally, learning and memory through buried food location test and expression of KAT and KMO, and cellular damage were evaluated at 60 PND. Pb2+ group showed redox environment alterations, cellular dysfunction and KYNA and 3-HK levels increased. No changes were observed in KAT activity. KMO activity increased at 23 PND and decreased at 60 PND. No changes in KAT and KMO expression in control and Pb2+ group were observed, however the number of positive cells expressing KMO and KAT increased in relation to control, which correlated with the loss of neuronal population. Cognitive impairment was observed in Pb2+ group which was correlated with KYNA levels. These results suggest that the increase in KYNA levels could be a mechanism by which Pb2+ induces cognitive impairment in adult mice, hence the modulation of kynurenine pathway represents a potential target to improve behavioural alterations produced by this environmental toxin.
Subject terms: Neurochemistry, Environmental impact, Risk factors
Introduction
Lead (Pb2+) is one of the main environmental neurotoxins that represents a health problem due to its multiple industrial, domestic, medical and technological applications1. Because its wide distribution, Pb2+ exposure can occur via eating, drinking or inhalation. This metal can cross placental and blood brain barrier (BBB) and also it is accumulated in several tissues, such as the lungs, liver, kidneys, spleen, brain and bones2. It has been reported that the central nervous system (CNS) is a target of lead exposure3–5. The developing brain is especially vulnerable to Pb2+ neurotoxicity since this heavy metal crosses through the placenta during pregnancy and also is able to pass into milk during the lactation period, reaching the developing brain impacting its functionality and morphology6,7. There are reports showing that even low doses of Pb2+ cause cognitive and motor functions impairment in humans and animal models8, mainly when the exposure occurs during brain development, where there are intense cellular proliferation, differentiation and synaptogenesis.
In this regard, it has been shown that Pb2+, at low micromolar concentrations, inhibits in an age-dependent manner the N-methyl-D-aspartate receptor (NMDAr) activation, being more harmful in the neonatal than in adult brain and producing long-term cognitive impairment3,9,10. During the early postnatal life, the NMDAr activation is essential for the development of neuronal networks; its inhibition in this period leads to severe neuromorphological and behavioral deficits11–15. Concerning to this, kynurenine pathway (KP) constitutes the main catabolic route of the essential amino acid tryptophan in mammals, which produces important metabolites with redox and neuroactive properties16. Specifically, kynurenic acid (KYNA) has been involved in brain development17 and cognitive process18 since it is the unique endogenous antagonist of NMDA receptor and a negative allosteric modulator of alpha7-nicotinic receptors. It has been suggested that the high KYNA levels found in fetal brain as well as its rapid decline during the postnatal period, are involved in NMDAr modulation, necessary for the normal brain development19,20. In this context, it has been shown that the fluctuation in brain KYNA levels can cause impairment in working memory, sensorimotor gating and in the attention process in adult rodents21–23.
Numerous publications have recently demonstrated that both Pb2+ exposure as well as KYNA levels fluctuation during the pre- and postnatal period, lead to cognitive impairment. However, there are no studies that have linked them, even knowing that there are several assumptions and converging findings shared: (1) kynurenine pathway is modulated by redox and inflammatory environment, which are altered by Pb2+ toxicity, (2) both Pb2+ and KYNA are able to inhibit NMDAr, (3) the developing brain is especially susceptible to lead toxicity as well as to KYNA levels fluctuations and (4) Pb2+ and high KYNA levels at early life induce behavioral alterations. Hence, to explore KP alterations as a new toxic mechanism by which Pb2+ induces cognitive impairment, represents a novel approach to modulate tryptophan catabolism as a therapeutic strategy. Therefore, in this work, we evaluated whether Pb2+ exposure, during the lactation period, induces changes in KP metabolites that correlate with long-term cognitive impairment. For this purpose, the dams received lead acetate in drinking water from 0 to 23 PND (lactation period). Immediately after weaning or one month after Pb2+ exposure, behavioral performance, KP metabolites and enzyme activity as well as morphological changes were analyzed. Our results support that KP metabolites, particularly the high levels of KYNA, are correlated with long-term cognitive impairment induced by Pb2+ during the lactation period.
Results
Brain Pb2+ levels in mice after 23 days of exposure
The concentration of Pb2+ (500 ppm) used in this study had no observable effects on mortality of the litters and had no observable adverse physical effects. Figure 1 shows the reached levels of Pb2+ in each brain region evaluated after 23 days of treatment (lactation period). The levels of Pb2+ found in hippocampus, striatum, cortex and cerebellum were 23 ± 0.8, 26.7 ± 5.6, 24.1 ± 4.4, 23.4 ± 2.8 µg Pb2+/mg of tissue, respectively in mice treated with Pb2+during lactation period. No detectable levels were found in control group.
Figure 1.
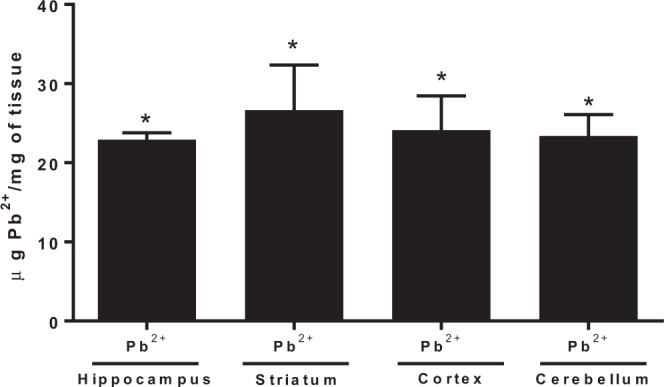
Lead concentration in different brain regions of mice at 23 PND, after Pb2+ exposure during lactation period. Determinations were made in hippocampus, striatum, cortex and cerebellum of mice. Data are mean ± S.E.M. T-test and Mann-Whitney test were used for comparisons, *P < 0.01 vs. each respective brain region control, n = 5 per group.
Locomotor activity alterations induced by lead exposure
First, we examined the immediate effect of lead treatment during lactation period (23 PND) on locomotor activity of mice (Fig. 2). We found that the total distance traveled (Fig. 2A) as well as the ambulatory time (Fig. 2B) significantly decreased in the mice of Pb2+ group compared to control group. However, when long-term effect (60 PND) was determined, no changes were found between control group and Pb2+ group (Fig. 2C,D).
Figure 2.

Locomotor activity of mice at 23 PND (A,B) and at 60 PND (C,D) in open field after Pb2+ exposure in drinking water during lactation period. Motor activity was assessed as the total distance walked (A,C) and the ambulatory time (B,D). Data are mean ± S.E.M. *P < 0.01 vs. control, T-test and Mann-Whitney test. N = 10–15 per group and per age.
Lead exposure during the lactation period induce memory impairment
Learning and memory of mice (60 PND) treated with Pb2+ during the lactation period were examined through the buried food location test (Fig. 3). Training phase is shown in panel A. Pb2+− group mice showed the same learning pattern that control group, in which they learned to reach the target. These data suggest that Pb2+ did not affect the acquisition phase. However, when long-term memory was evaluated 24 h after acquisition, both, the distance travelled as well as the time to reach the target were higher in Pb2+ group than in control group, suggesting memory consolidation impairment in the Pb2+− group.
Figure 3.

Long-term memory evaluation after Pb2+ exposure during lactation period. Memory impairment in adult mice was evaluated through the distance traveled (B) and the time to reach target (C) 24 h after acquisition phase (A). Data are mean ± S.E.M. *P < 0.01 vs. control, T-test and Mann-Whitney test. N = 9–15 per group.
Redox environment alterations and cellular dysfunction induced by Pb2+ treatment
Reactive oxygen species (ROS), lipid peroxidation and GSH levels were evaluated in Pb2+− treated mice at 23 and 60 PND (Table 1). As expected, ROS levels and lipid peroxidation were higher (50–70% and 40–110%, respectively) in Pb2+ group than in controls, in all brain regions determined at 23 PND. However, the high levels of ROS were not observed at 60 PND in the group treated with Pb2+. Lipid peroxidation was higher in hippocampus and cerebellum (~75% and 120%, respectively) of mice treated with Pb2+ during lactation period compared with control group. The GSH levels decreased around 20% in cerebellum of mice under treatment with Pb2+ compared to control at both times evaluated. Cellular function determined by MTT reduction, decreased around 40–50% in all brain regions of mice treated with Pb2+ compared to controls at both times evaluated.
Table 1.
Effects of Pb2+ exposure on brain redox environment.
| 23 PND | ||||||||
|---|---|---|---|---|---|---|---|---|
| Hippocampus | Striatum | Cortex | Cerebellum | |||||
| C | Pb2+ | C | Pb2+ | C | Pb2+ | C | Pb2+ | |
| ROS (mg of DCF/mg of protein) | 0.33 + 0.03 | 0.57 + 0.05* | 0.85 + 0.08 | 1.46 + 0.08* | 1.30 + 0.09 | 1.52 + 0.03* | 0.79 + 0.06 | 1.20 + 0.07* |
| Lipid peroxidation (µmoles of MDA/mg of protein) | 0.037 + 0.001 | 0.077 + 0.008* | 0.042 + 0.004 | 0.069 + 0.001* | 0.026 + 0.002 | 0.036 + 0.001* | 0.021 + 0.001 | 0.046 + 0.004* |
| MTT (% vs. Control) | 100 + 5.1 | 65.77 + 4.77* | 100 + 4.2 | 68.75 + 4.75* | 100 + 5.0 | 48.05 + 5.55* | 100 + 8.4 | 60.11 + 5.61* |
| GSH (nmol/g of tissue) | 2689.72 + 84.72 | 2630.15 + 89.85 | 2485.13 + 184.87 | 2531.12 + 278.88 | 2541.32 + 51.32 | 2603.96 + 88.96 | 2742.99 + 52.99 | 2548.51 + 48.51* |
| 60 PND | ||||||||
| ROS (mg of DCF/mg of protein) | 0.86 + 0.084 | 0.93 + 0.068 | 1.22 + 0.203 | 1.69 + 0.31 | 0.35 + 0.017 | 0.27 + 0.051 | 0.21 + 0.011 | 0.19 + 0.021 |
| Lipid peroxidation (µmoles of MDA/mg of protein) | 0.42 + 0.057 | 0.737 + 0.143* | 0.464 + 0.071 | 0.7 + 0.1 | 0.26 + 0.028 | 0.25 + 0.055 | 0.110 + 0.01 | 0.25 + 0.07* |
| MTT (% vs. Control) | 100 + 4.2 | 50.76 + 5.74* | 100 + 16 | 46.54 + 3.46* | 100 + 4.1 | 53.38 + 3.62* | 100 + 7.2 | 75.76 + 5.24* |
| GSH (nmol/g of tissue) | 2583.66 + 111.34 | 2573.11 + 131.89 | 2146.92 + 233.08 | 2237.48 + 137.52 | 2323.63 + 66.37 | 2246.58 + 153.42 | 2668.05 + 91.95 | 2256.63 + 83.37* |
Data were obtained in different cerebral regions: hippocampus, striatum, cortex and cerebellum at 23 and 60 PND. Data are mean ± S.E.M. T-test and Mann-Whitney test were used for comparisons, *P < 0.01 vs. each respective brain region control, n = 5–8 per group.
Kynurenine Pathway alterations by Pb2+ exposure during lactation period of mice
To evaluate the impact of Pb2+ exposition on KP during lactation period, we analyzed both arms of the pathway: by one side, kynurenine aminotransferase (KAT II) activity as well KYNA levels, and on the other side, kynurenine monooxygenase (KMO) activity as well 3-HK levels, immediately after removing drinking water with Pb2+ (23 PND) and also at 60 PND, to examine long-term effects (Fig. 4). The KAT II activity (the main enzyme to produce KYNA) did not show changes in Pb2+ group compared to control group (Fig. 4A). However, KYNA levels showed an increase in each cerebral region evaluated (Fig. 4B, 528 ± 18, 500 ± 58, 752 ± 150 and 255 ± 17% for hippocampus, striatum, cortex and cerebellum, respectively) compared with its respective brain region control (p < 0.001). KMO activity and 3-hydroxykynurenine levels at 23 PND showed a significant increase (p < 0.05) in all brain regions analyzed (Fig. 4C,D, respectively).
Figure 4.

Kynurenine pathway alterations induced by Pb2+. (A) KAT II activity, (B) KYNA levels, (C) KMO activity and (D) 3-HK levels were evaluated in hippocampus, striatum, cortex and cerebellum of mice at 23 PND, after Pb2+ exposure during lactation period. Data are mean ± S.E.M. *P < 0.01 vs. each respective brain region control, T-test and Mann-Whitney test. N = 9 per group.
When long-term effects were evaluated, we found no changes in KAT II activity (Fig. 5A); however, KYNA levels were still high (around double compared to control) in all the cerebral regions evaluated (p < 0.01; Fig. 5B). KMO activity decreased significantly in hippocampus and cortex (Fig. 5C), and 3-HK levels decreased significantly in striatum, cortex and cerebellum (Fig. 5D) of mice treated with Pb2+ during lactation period compared to control group (p < 0.01). Additionally, we found that KYNA levels determined at 60 PND, correlated with the cognitive impairment assessed by total distance traveled to reach the objective and time to reach the target (Fig. 6), suggesting that KYNA is related with the cognitive impairment induced by Pb2+ treatment during lactation period. No correlation was found between 3-HK levels and cognitive impartment parameters.
Figure 5.

Long-term alterations on kynurenine pathway generated by Pb2+ treatment during lactation period of mice. (A) KAT II activity, (B) KYNA levels, (C) KMO activity and (D) 3-HK levels were evaluated in hippocampus, striatum, cortex and cerebellum of mice at 60 PND. Data are mean ± S.E.M. *P < 0.01 vs. each respective brain region control, T-test and Mann-Whitney test. N = 8 per group.
Figure 6.

The lower triangular matrix contains the scatterplot for each pair of variables for both groups (control and Pb2+). The upper triangular matrix contains the Spearman’s rank correlation coefficient (r) and its associated p-value (p) calculated for each group.
Lead treatment decreases the number of neurons and changes the cellular proportion of KMO+ and KAT II+ brain cells
After evaluating the KP metabolites and observing that KYNA levels were higher in Pb2+ group but not KAT II activity, we decided to evaluate the expression of KAT II in the positive brain cells, both, in control group as Pb2+ group at 60 PND. Figure 7A,C show the expression of KAT II and KMO in full brain, respectively; no changes in the mean fluorescence intensity were found. However, the number of positive cells from both KAT II and KMO increased significantly, indicating that the cellular proportion that express these enzymes changed (Fig. 7B,D, respectively) with Pb2+. This would mean that Pb2+ treatment during the lactation period induces neuronal cell death and in consequence the proportion of brain cells changes. Figure 8A,B show the representative histological micrographs of the hematoxylin-eosin stained hippocampus of control and Pb2+ groups (60 PND). The majority of neurons in the hippocampal CA1 subfield of mice in the Pb2+ group appeared shrunken with eosinophilic cytoplasm, pyknotic nuclei and slight interstitial edema; while in control group there was no evidence of neuronal damage, cells exhibited round-shaped, with a lack of eosinophilic cytoplasm or evident pyknotic nuclei. The number of surviving neurons in Pb2+ group was significantly lower than in the control group (P < 0.01; Fig. 8C).
Figure 7.

Effect of Pb2+ treatment during lactation on the proportion of KAT II + and KMO + brain cells. Expression of KAT II (A) and KMO (C) was determined in the brain of mice at 60 PND. Number of positive cells to KATII (B) and KMO (D) were evaluated in the same samples. Data are mean ± S.E.M. *P < 0.05 vs. each respective brain region control, T-test and Mann-Whitney test.
Figure 8.

Representative photomicrographs of hematoxylin-eosin-stained hippocampus sections (100x) of control and Pb2+ groups. (A) Control group, normal cells exhibited round-shaped and pale-stained nuclei. (B) Pb2+ group, dead cells exhibited a shrunken cytoplasm with pyknotic nuclei (black arrows). (C) Percentage of cellular damage in the hippocampal region of mice exposed to Pb2+. Values are expressed as the percentage of damaged cells counted in ten fields per area, in three slides per mouse. *P < 0.001 vs. control, T-test and Mann-Whitney test.
Discussion
In the present work, we studied the effect of Pb2+ exposition in mice, during lactation period, in KP and its relationship with long-term cognitive impairment induced in this toxic model. Lead is a heavy metal considered one of the most widespread and harmful environmental toxin24. Many mechanisms have been involved in Pb2+ toxicity and some of them converge as oxidative stress, inflammation, energetic alterations, calcium homeostasis perturbations, among others25–28. These factors can influence the tryptophan catabolism trough KP, which produces metabolites with neuroactive and redox properties that affect brain function29–31. Alterations in KP during brain development lead to cognitive impairment in adulthood32. The KP relationship with Pb2+ toxicity and with the cognitive impairment induced by this metal, could begin to be recognized by the fact that Pb2+ generates an oxidant environment that can cause fluctuations in the KP metabolites having repercussion in the cognition33. However, this is the first study relating increased KYNA levels and Pb2+ exposure.
In this study, we used a model of Pb2+ exposure in mice, during lactation period, knowing that the brain in prenatal and early postnatal periods undergoes rapid growth and is very sensitive to environmental pollutants, including Pb2+, as well as taking in consideration that KP plays an important role in brain development19,34. First, Pb2+ concentration was determined in different brain regions founding that it was deposited in CNS and as was expected, the redox environment and cellular function were also affected after Pb2+ exposure during lactation of mice at 23 PND. These results indicated that the oxidant cellular environment may induce tryptophan catabolism, mainly through KP. According to this, high levels of KYNA and 3-HK, as well as increased KMO activity were found in the brain of Pb2+ group at 23 PND. However, no changes were found when the KAT II activity -the main enzyme responsible of KYNA production- was evaluated in these mice, indicating that the high levels of KYNA were produced by no canonical routes (30,35. In this context, new pathways of KYNA production have been described, one of them involves the interaction of reactive oxygen species with L-kynurenine (the precursor of KYNA and 3-HK) or tryptophan30,35,36. According to this background, the high brain levels of ROS that were found, under our experimental conditions, can contribute directly to KYNA production but also can promote the augment in the KP, since indoleamine-2, 3-dioxygenase (IDO, enzyme responsible of the first step of KP, via tryptophan cleavage) and KMO are modulated by superoxide anion.
Considering these premises and findings, which indicate that both, Pb2+ and KP fluctuations, during early postnatal life, impact on cognition in the adulthood, the next step was to evaluate if fluctuations in KP metabolites levels induced by Pb2+ toxicity during lactation period, correlated with long-term cognition repercussions generated by this heavy metal. Our data showed that Pb2+ exposition during lactation induced cognitive impairment at 60 PND, this parameter was evaluated by the buried food location test. In this test, the mice were able to learn the location of the food; analyzing the training curve, both control and Pb2+ group, decreased the time to reach the target according to the training trial (acquisition phase). However, when long-term memory was evaluated 24 h after of training, the mice of Pb2+ group spent more time and travelled a longer distance to reach the precise location of buried food, compared to control group, indicating impairment in memory consolidation. Several studies have shown that Pb2+ toxicity induce cognitive impairment and that this effect is stronger when the exposition occurs during the prenatal period or early postnatal life, than in older animals3. However, the particular mechanism by which this heavy metal induces motor and cognitive impairment remains elusive. This work shows the first evidence that Pb2+ exposure at early life, can impair brain tryptophan catabolism, which is correlated with the cognitive damage associated to this heavy metal. This study also demonstrates that Pb2+ induces fluctuations on KP and, considering our results, as well as previous evidence, the effect of this heavy metal on long term memory could be due to the fact that both Pb2+ and KYNA, interact with NMDAr. The NMDAr activation is essential during the development of neuronal networks; its inhibition during the early postnatal life reduces synchronous network activity both in neocortex and in hippocampus15,37,38 leading to impairment in memory functions9.
In this regard, it has been described that Pb2+ can modulate NMDAr by different routes: (1) Pb2+ blocks NMDAr in the synaptic region disrupting the calcium signaling mechanisms and (2) Pb2+ exposure enhances NR2B subunit expression of NMDA receptor in the extra synaptical area producing an increase in calcium concentration39–41. Regarding KYNA, it has been demonstrated that it is an endogenous competitive antagonist to glycine site of the NMDAr42,43, and the prenatal exposure to its immediate precursor, L-kynurenine, is associated with reduction of NR1 and NR2A expression, leading to lower NR2A/NR2B ratio44–46. It has also been suggested that high KYNA levels in the prenatal brain inhibit neurite branching and development of excitatory synapses; after birth, KYNA levels decline to allow excitatory synapse formation47,48. In our model, the excessive blocked of NMDAr in early postnatal life by KYNA and Pb2+, could produce alterations in different processes during neurodevelopment, resulting in long-term cognitive deficits as was observed under our experimental conditions. Alternatively, KYNA can impact brain development and cognitive processes since it is an endogenous negative allosteric modulator of α7 nicotinic acetylcholine receptors (α7nAChRs), which play critical roles in regulating neuronal plasticity49–51 and cellular survival in the brain49,52–54.
Coupled to possible structural alterations evoked by Pb2+ exposure and KYNA levels fluctuations during the early postnatal life, the physiopathological consequences of the variations in brain levels of KYNA found in mice at 60 PND, raise the possibility that KYNA might interact with less receptors due cell death caused by Pb2+, exposure, having influence in the neurotransmission and intensifying the alterations in long term memory. In light of the present results, it is likely that the high levels of KYNA may be due at least in part to the neuronal cell damage induced by Pb2+, which is reflected in a larger number of astrocytes in relation to neurons.
On the basis of the results showed herein, it is tempting to speculate that the cognitive impairment observed in humans exposed to Pb2+ is related with elevated levels of KYNA or some other KP metabolites. Further studies in our laboratory are focused to elucidate the impact of KP modulation during brain development as well as on cognitive impairment induced by Pb2+ toxicity.
Conclusion
In summary, this is the first study that demonstrates the positive correlation between high levels of KYNA and cognitive impairment induced by Pb2+ toxicity in early postnatal life. Therefore, manipulations of the KP may represent a potential novel treatment strategy for conditions that are linked to motor and cognitive perturbations induced by this heavy metal.
Material and Methods
Animals
Experimental protocol (Number 109/15) was approved by the Institutional Research Committee of the National Institute of Neurology and Neurosurgery and by the Institutional Committee for the care and use of laboratory animals (CICUAL-INNN). All procedures with animals were carried out according to regulations specified by the Bioethical Committee, and the Mexican Regulation for the production, care and use of laboratory animals (NOM-159 062-ZOO-1999). During the dissections, all efforts were made to minimize animal suffering. FVB mice (28–32 g) from the animal house of the National Institute of Neurology (Mexico City) were employed throughout the study. Animals were housed in acrylic cages with standard commercial mice diet and water ad libitum. The housing room was maintained under constant conditions of temperature (25 ± 3 °C), humidity (50 ± 10%) and lighting (12 h light/dark cycles
Each dam was housed with a male mouse in individual cages until vaginal plug was detected, then the male mouse was removed from the cage. At birth, litters were assigned randomly to one of two groups: 1) Control group and 2) Pb2+ group. Offspring were biochemistry and behavioral tested at 23 and 60 PND by a researcher who was blind to the treatment conditions.
Lead exposure
Dams and their offspring from control group1 received normal drinking water during the study, while dams and their offspring from Pb2+ group2 received Pb2+ in the drinking water (500 ppm of lead acetate), from the birth to 23 PND. Only males were analyzed to avoid any influence of the female hormonal system on metabolism. After weaning at 23 days of age, the male offspring from different litters were randomly housed, 2–3 per cage, for both groups and received normal drinking water to evaluate long-term effects (60 PND). For the biochemistry and behavioral evaluation, experimental subjects were the progeny from 9–11 litters per group, with no more than 3 pups from any given litter. This design was implemented in order to minimize the contribution of individual litters.
Lead determination in brain regions of mice
Pb2+ levels were evaluated in different brain regions, hippocampus, striatum, cortex and cerebellum of mice (23 PND) of both groups by graphite furnace atomic absorption spectrophotometry. Tissue samples were weighted and 65% suprapure nitric acid (Merck, Mexico) (1:10, w/v) was added, then the tissue was digested in a shaking water bath at 60 °C for 30 min. Afterwards, the samples were diluted (1:10 or 1:20, v/v) with deionized water. Samples were injected into an atomic absorption spectrophotometer (Model 3110; Perkin-Elmer, Norwalk, CT, USA) equipped with graphite furnace (HGA-600) and auto-sampler (AJS60) adjusted to a wavelength of 283.3 nm. Quality control standards and calibration curves (of known amounts of Pb2+ standard) were used for each analysis. Results were expressed as µg of lead for mg of tissue55.
Evaluation of motor activity in mice
Locomotor activity of mice from both groups at 23 and 60 PND was evaluated in an Opto-Varimex 4 system (Columbia, Ohio, USA). Briefly, the motor/kinetic activity was recorded during 5 min after the habituation period and the cage was cleaned with alcohol between each test to exclude any effect of the previous animal. The results are expressed as total distance walked (cm) and ambulatory time (sec) recorded in 5 min.
Buried food location test (BFLT)
A researcher who was unaware of the treatment condition performed all behavioral experiments.
The buried food location test (BFLT) was performed in mice at 60 PND. BFLT is an adaptation of the model described by Lehmkuhl et al. for olfactory dysfunction evaluation56.
This version of the test comprises two sessions. The training session (acquisition) consisted of 6 trials (1–6, 2 minutes inter-trial interval) and a 0 trial in which each mouse was placed in an acrylic box (1 m2), covered with a 3 cm-sawdust layer and a highly palatable food (sugary pellet, “froot loops”, to which they were previously familiarized) buried 1 cm under the sawdust, in a fixed quadrant of the box. The location of the pellet in all the trials was the same and the box exhibited spatial clues (black color geometric figures of 10 cm × 10 cm, placed in the middle of each face of the box at a height of 13 cm). In case the mice were unable to find the froot-loop within 180 s in the first trial, they were gently guided to it (the froot loop should be placed on the surface). The mice were allowed to eat the froot loop for 5 s. During this session, the animals were fasted for the previous 24 hours with ad libitum access to water. After each trial, the mice were returned to its home cage, the testing area was cleaned with a 10% ethanol solution and the sawdust removed in order to eliminate odoriferous marks. The long-term memory was evaluated 24 hours after the acquisition session, in a 3 minutes retention test where the food was removed from the sawdust and mice were allowed to freely explore the arena for 180 s; the time spent in reaching the precise location of buried food (same location as in the acquisition) was recorded and the exploration time spent in the food location quadrant was also measured. All the sessions were video recorded allowing us to analyze the videos offline with a tracking software ImageJ, USA. The results are expressed as the distance travelled to reach the objective (cm), the time to reach the target and the time spent searching for the food in the target quadrant (sec).
Tissues treatment
After treatments, offspring of both groups of mice were sacrificed by decapitation and brain regions were obtained (hippocampus, striatum, cortex and cerebellum). For reactive oxygen species (ROS), cellular function (MTT) and lipid peroxidation (LP) determination, brain regions were sonicated in 500 µl of Krebs buffer pH 7.4 (19 mM NaCl, 5 mM KCl, 2 mM CaCl2, 1.2 mM MgSO4, 5 mM glucose, 13 mM NaH2PO4 y 3 mM Na2HPO4; J.T. Baker, USA). KP metabolites were determined in the brain regions previously weighted and homogenized in deionized water (1:10, w/v). The homogenates were centrifuged at 14600 g for 10 min in a Select Bioproducts centrifuge (SB products; PO Box, Edison NJ) and the supernatant was used to inject into the HPLC.
ROS quantification
ROS were evaluated through DCF-DA oxidation29. Briefly, brain regions homogenates were incubated with DCF-DA (75 μM, cat. No. 35845, Sigma-Aldrich, USA) for 30 min at 37 °C in darkness and then centrifuged at 6000 × g (SB products; PO Box, Edison NJ) for 10 minutes. After incubation, ROS formation was quantified in supernatants through fluorescence spectrophotometry in a plate reader Flx-800 (Biotek Instruments, Winooski, VT, USA) at an excitation wavelength of 448 nm and 532 nm of emission. Results were expressed as milligram of DCF per milligram protein.
Lipid peroxidation determination
Lipid peroxidation was evaluated through production of thiobarbituric acid reactive species (TBA-RS). Briefly, 125 µl of brain regions homogenates were boiled with 250 µl of TBA (0.375 g of thiobarbituric acid (cat. No. T5500, Sigma-Aldrich, USA) + 15 g of trichloroacetic acid (TCA) + 2.54 mL of HCl in 100 ml (J.T. Baker, USA)) in a water bath during 15 min, then they were placed on ice and finally, they were centrifuged at 9800 × g (SB products; PO Box, Edison NJ) for 10 min. The optical density of supernatant was obtained in an Eon microplate reader (Biotek Instruments, Winooski, VT, USA) at a wavelength of 532 nm57. Results were expressed as micromoles of malondialdehyde per milligram protein.
MTT reduction assay
Cellular function was evaluated by MTT reduction assay57–59. Briefly, 100 µl of homogenate of brain regions were incubated with 4 µl of MTT (5 mg/mL, cat. No. M5655, Sigma Aldrich, USA) at 37 °C during 15 min. Samples were centrifuged at 12000 × g for 3 minutes and supernatant was eliminated. 250 µl of acidified isopropyl alcohol were added to dissolve the formazan. Optical density was obtained in an Eon microplate reader (Biotek Instruments, Winooski, VT, USA) at a wavelength of 570 nm. Results were expressed as the percentage of MTT reduction in relation to control values for each brain region.
GSH determination
Tissue GSH levels were determined in four brain regions using an adapted fluorescence method with o-phthalaldehyde; the assay was performed in a microplate where the GSH reacts with o-phthaldialdehyde (OPA) forming a highly fluorescent isoindole derivative60. Brain regions were homogenized (1:10 w/v) in buffer A (154 mM KCl, 5 mM diethylenetriaminepenta-acetic acid (DTPA) and 0.1 M potassium phosphate buffer pH 6.8 proteases inhibitor and PMSF). Then, it was added cold B buffer (40 mM HCl, 10 mM DTPA, 20 mM ascorbic acid and trichloroacetic acid 10%). The tissues were centrifuged at 14000 × g during 20 min. Then, 5 µl of supernatant was added with OPA to obtain the isoindole. The fluorescence was measured at 370 nm of excitation and 420 nm of emission in a microplate lector (Flx-800, BioTec). The results were expressed as nanomoles of GSH/g of tissue.
Kynurenic acid (KYNA) determination
KYNA levels were determined in brain regions of mice of 2 different ages by an HPLC method with fluorescence detection (model S200). 20 µl of sample or standard solution were injected into Eclipse XDB-C18 reverse phase column (5-μm, 4.6 × 150 mm, Agilent, Santa Clara, CA, USA) and isocratically eluted with a polar mobile phase (250 mM zinc acetate (cat. No. 2740, Meyer, Mex), 50 mM sodium acetate (cat. No. 79714, Sigma-Aldrich, USA), pH 6.2 adjusted with glacial acetic acid (J. T. Baker, USA) and 3% of acetonitrile (HPLC grade, Sigma- Aldrich, USA). Samples were eluted at a flow of 1.0 ml/min. KYNA was detected by fluorescence at emission wavelength of 398 nm and 344 nm of excitation wavelength35. Retention time was ~7 min. The results were presented as fmoles of KYNA/mg of protein. The results were presented as fmoles KYNA/mg of protein.
Kynurenine aminotransferase (KAT-II) activity
KAT-II activity was measured in brain regions of mice of 23 and 60 days old, which were homogenized (1:10, v/v) in homogenization buffer (containing 0.5 M tris-base buffer pH 8 (J. T. Baker, USA), 50 µM of pyridoxal-5-phosphate (P5P) and 10 mM 2-mercaptoethanol (cat. No. P9855, M6250 Sigma-Aldrich)). 100 µl of homogenized tissues were incubated with 100 µl of cocktail (containing 100 µM of L-kyn (cat. No. K8625, Sigma-Aldrich), 80 µM of P5P, 1 mM of sodium pyruvate (cat. No. P2256, Sigma-Aldrich) containing 150 mM tris-base buffer pH 7.4) and incubated at 37 °C for 1 h. The reaction was stopped with 20 µl of trichloroacetic acid 50% and 1 mL of HCl 0.1 M (J.T. Baker, USA). Samples were centrifuged 10 min at 14000 g. KYNA was detected by fluorescence at emission wavelength of 398 nm and 344 nm of excitation wavelength (Perkin Elmer fluorescence detector series 200a)61. The results were presented as pmoles of KYNA/h/mg of protein.
3-hydroxykynurenine (3-HK) determination
3-HK levels were determined in brain regions (cortex, hippocampus, striatum and cerebellum) by HPLC using an electrochemical method62. Briefly, 40 µl of supernatant were eluted at a constant flow rate of 0.5 ml/min with a mobile phase (950 ml deionized water, 5.9 ml phosphoric acid (J.T. Baker, USA), 20 ml acetonitrile (HPLC grade), 9 ml triethylamine, 100 mg sodium EDTA, and 1.2 g heptane sulphonic acid (cat. No. T0886, E1644 and H2766, Sigma-Aldrich, USA)) through an Adsorbosphere Catecholamine C18 reverse phase column (3-μm, 4.6 mm × 100 mm, Fisher Scientific, Hampton, Nuevo Hampshire, USA). Oxidation voltage was 0.500 V, at a range 1.0 nA and a filter of 0.10 Hz (LC-4C detector, BAS). The retention time was ~11 min. The results were presented as pmoles of 3-HK/mg of protein.
Kynurenine monooxygenase (KMO) activity
KMO activity was measured in brain regions homogenates. 100 µl of homogenized tissues were diluted in 400 µl of KMO buffer activity (containing 100 mM TRIS, 10 mM KCl and 1 mM EDTA, Sigma-Aldrich, USA). Then, 80 µl of homogenate were incubated with 100 µl of assay cocktail (1 mM of NADPH, 3 mM of G6P, 1 U/ml of G6PDH and 100 µM of L-kynurenine sulfate (cat. No. N1630, G6378 and K2750 Sigma-Aldrich respectively)) and incubated at 37 °C for 1 h. The reaction was stopped with 25 µl of perchloric acid 6%. Samples were centrifuged 10 min at 14000 g. 40 µl of supernatants were injected to HPLC and 3HK levels were detected with the above mentioned method62. The results were presented as pmoles of 3-HK/h/mg of protein.
Protein determination by Lowry
Protein quantification was evaluated through Lowry methodology, 10 µl of homogenized tissue was diluted with 190 µl of water. It was added 1 ml of C solution (A solution: Na2CO3 2%, NaOH 0.4% and 0.2% of sodium tartrate + B solution: Cu(SO4)3 0.5% Sigma-Aldrich, USA). Samples were incubated 10 min at room temperature and then Folin reactive (50%, cat. No. F9252, Sigma Aldrich, USA) was added and mixed. They were incubated for 30 min at room temperature. Absorbance was determined at 550 nm in a plate reader (Eon Biotek Instruments, Winooski, VT, USA)63.
Percentage of KAT and KMO positive cells and expression in brain by flow cytometry
Expression of the kynurenine pathway enzymes was determined in mice brains from lead treated and control groups at 60 PND. Mice brains were washed with cold PBS 1Χ and cut into small pieces, then 1 ml of accutase (cat. No. L0950, BioWest) was added. Accutase was inactivated with DMEM/F-12 (cat. No. R36/37/38, BioWest) supplemented with 10% FBS (cat. No. S1650, BioWest). Disaggregated brains were filtered in 0.7 mm pore filters, cells were centrifuged at 200 × g for 5 minutes, the supernatant was discarded, and cells were washed twice. After this, cells were resuspended in 30% Percoll (cat. No. 17-0891-01, GELife Sciences) and then centrifuged at 200 × g for 20 minutes. Following centrifugation, the myelin layer was totally discarded, cells were washed with PBS 1Χ and centrifuged. After washing, cells were resuspended with FACS permeabilizing solution (cat. No. 340973, BD FACS), and incubated during 30 minutes at room temperature. After incubation, cells were washed with PBS 1Χ and centrifuged. Then, cells were resuspended with mouse anti-KAT (0.1 µg/ml cat. No. B0408 SC-67376, Santa Cruz Biotechnology) or anti-KMO (0.1 µg/ml cat. No. OAAB05255, AVIVAS) and incubated during 30 minutes at room temperature. Following incubation with the primary antibodies, cells were washed with PBS 1Χ, centrifuged and then, resuspended with Alexa Fluor 594 anti-rabbit IgG (0.1 µg/ml cat. No. A11012, Molecular Probes), cells were incubated during 30 minutes at room temperature at darkness. After incubation, cells were washed with PBS 1Χ and fixed with 1% paraformaldehyde. Percentage of positive cells and mean fluorescence intensity (MFI) was determined by flow cytometry using a BD FACSCalibur FL4 instrument (BD Biosciences), analysis was done using CellQuest and Flowjo v.10 software.
Histology assay
The mice (60 PND) were deeply asleep with pentobarbital overdoses and perfused transcardiacally with chilled 10% w/v buffered formalin solution. The whole brains were removed immediately after perfusion and fixed for histological examination into 10% formaldehyde solution for 24 h, dehydrated and embedded in paraffin. Coronal sections of 5 µm thickness were cut with a microtome and stained with hematoxylin–eosin. Hippocampus sections were evaluated using the mouse brain atlas and stained to determine cell damage through morphology, including neurons with a normal appearance, or identifying those showing signs of damage (hyperchromatic cytoplasm, cellular shrinking, and nuclear fragmentation). The histopathology analysis was performed by independent pathologist blind to the experimental conditions. Three slides with 10 fields per mice were analyzed under light microscopy for morphometric analysis; the digitization and storage of images were performed with a light microscope Axio Lab A1 Zeiss equipped with Axicocam ICC5 camera at a 1000X magnification An unbiased counting frame was placed directly onto the screen of the PC over the reference section and the section under analysis, and counting was performed using basic principles of the optical dissector counting method reported by West (1993). In the optical dissector, the cells were counted only if a feature of interest was clearly recognized within the dissector height and located inside the counting frame or touching the inclusion line. Neurons touching the exclusion line were not counted. The percentages of damaged cells were obtained by dividing the number of damaged cells between number of total cells in the ten fields counted in each slide, and the total number of cells was expressed as the percentage of cells per field64.
Data analysis
Data were corrected by mg of protein. Results were expressed as mean values ± S.E.M. Both T-test and Mann-Whitney test were used to compare distributions of Control and Pb2+ groups using the Prism software (GraphPad, San Diego, CA, USA). Values of p < 0.01 were considered statistically significant. Additionally, the correlation between each pair of the following variables was assessed using the Spearman’s rho coefficient: hippocampus KYNA levels, time to reach the target and distance traveled to reach the objective.
Acknowledgements
This work was supported by CONACYT Grant 286885 (V.P.C.). Daniela Ramírez Ortega is scholarship holder of CONACyT-México (308054) in the Programa de Doctorado en Ciencias Bioquímicas at the Universidad Nacional Autónoma de México. S.G.-M. was supported by Recursos Fiscales para Investigación Program from the Instituto Nacional de Pediatría INP 031/2018, 038/2019; J.M.-Q. was supported by Cátedras CONACYT (2184) project number 2057 and by Recursos Fiscales para Investigación Program from the Instituto Nacional de Pediatría INP 024/2017.
Author contributions
D.R.O.: performed the biochemistry experiments and analyzed the experimental results. P.O.R. and G.R.R.: Performed the behavioral test and analyzed the results. B.P.O. and G.I.V.C. designed and performed enzyme expression by cytometry. D.G.E. and L.A.R.C. performed G.S.H. determination and enzyme activity. G.P.C. contributed to statistical analysis and data interpretation. A.D. and C.R. performed lead determination. M.M.A. performed histology assays. J.M.Q. and S.G.M. performed enzyme activities. V.P.C. and B.P. conceived and designed the present study. V.P.C. wrote the manuscript. All authors reviewed and approved the final manuscript.
Data availability
Data used to support the findings of this study are available with the corresponding author upon request.
Competing interests
The authors certify that they have no affiliations with or involvement in any organization or entity with any financial interest (such as honoraria; educational grants; participation in speakers’ bureaus; membership, employment, consultancies, stock ownership, or other equity interest; and expert testimony or patent-licensing arrangements), or non-financial interest (such as personal or professional relationships, affiliations, knowledge or beliefs) in the subject matter or materials discussed in this manuscript.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Garza A, Vega R, Soto E. Cellular mechanisms of lead neurotoxicity. Med. Sci. Monit. 2006;12:RA57–65. [PubMed] [Google Scholar]
- 2.Glass HC, et al. Antenatal and intrapartum risk factors for seizures in term newborns: a population-based study, California 1998–2002. J. Pediatr. 2009;154:24–28 e21. doi: 10.1016/j.jpeds.2008.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kuhlmann AC, McGlothan JL, Guilarte TR. Developmental lead exposure causes spatial learning deficits in adult rats. Neurosci. Lett. 1997;233:101–104. doi: 10.1016/s0304-3940(97)00633-2. [DOI] [PubMed] [Google Scholar]
- 4.Reddy GR, Zawia NH. Lead exposure alters Egr-1 DNA-binding in the neonatal rat brain. Int. J. Dev. Neurosci. 2000;18:791–795. doi: 10.1016/s0736-5748(00)00048-4. [DOI] [PubMed] [Google Scholar]
- 5.Chetty CS, et al. Perinatal lead exposure alters the expression of neuronal nitric oxide synthase in rat brain. Int. J. Toxicol. 2001;20:113–120. doi: 10.1080/109158101317097692. [DOI] [PubMed] [Google Scholar]
- 6.Zhang J, Pu L, Peng B, Gao Z. The impact of urban land expansion on soil quality in rapidly urbanizing regions in China: Kunshan as a case study. Env. Geochem. Health. 2011;33:125–135. doi: 10.1007/s10653-010-9326-6. [DOI] [PubMed] [Google Scholar]
- 7.Zhang YM, Liu XZ, Lu H, Mei L, Liu ZP. Lipid peroxidation and ultrastructural modifications in brain after perinatal exposure to lead and/or cadmium in rat pups. Biomed. Env. Sci. 2009;22:423–429. doi: 10.1016/S0895-3988(10)60021-9. [DOI] [PubMed] [Google Scholar]
- 8.Lancranjan I, Popescu HI, Olimpia GA, Klepsch I, Serbanescu M. Reproductive ability of workmen occupationally exposed to lead. Arch. Env. Health. 1975;30:396–401. doi: 10.1080/00039896.1975.10666733. [DOI] [PubMed] [Google Scholar]
- 9.Guilarte TR, Miceli RC. Age-dependent effects of lead on [3H]MK-801 binding to the NMDA receptor-gated ionophore: in vitro and in vivo studies. Neurosci. Lett. 1992;148:27–30. doi: 10.1016/0304-3940(92)90796-a. [DOI] [PubMed] [Google Scholar]
- 10.Alkondon M, Costa AC, Radhakrishnan V, Aronstam RS, Albuquerque EX. Selective blockade of NMDA-activated channel currents may be implicated in learning deficits caused by lead. FEBS Lett. 1990;261:124–130. doi: 10.1016/0014-5793(90)80652-y. [DOI] [PubMed] [Google Scholar]
- 11.Kaindl AM, et al. Brief alteration of NMDA or GABAA receptor-mediated neurotransmission has long term effects on the developing cerebral cortex. Mol. Cell Proteom. 2008;7:2293–2310. doi: 10.1074/mcp.M800030-MCP200. [DOI] [PubMed] [Google Scholar]
- 12.Elhardt M, Martinez L, Tejada-Simon MV. Neurochemical, behavioral and architectural changes after chronic inactivation of NMDA receptors in mice. Neurosci. Lett. 2010;468:166–171. doi: 10.1016/j.neulet.2009.10.091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Harris LW, Sharp T, Gartlon J, Jones DN, Harrison PJ. Long-term behavioural, molecular and morphological effects of neonatal NMDA receptor antagonism. Eur. J. Neurosci. 2003;18:1706–1710. doi: 10.1046/j.1460-9568.2003.02902.x. [DOI] [PubMed] [Google Scholar]
- 14.Cserep C, et al. NMDA receptors in GABAergic synapses during postnatal development. PLoS One. 2012;7:e37753. doi: 10.1371/journal.pone.0037753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yang JW, Hanganu-Opatz IL, Sun JJ, Luhmann HJ. Three patterns of oscillatory activity differentially synchronize developing neocortical networks in vivo. J. Neurosci. 2009;29:9011–9025. doi: 10.1523/JNEUROSCI.5646-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gonzalez Esquivel D, et al. Kynurenine pathway metabolites and enzymes involved in redox reactions. Neuropharmacol. 2017;112:331–345. doi: 10.1016/j.neuropharm.2016.03.013. [DOI] [PubMed] [Google Scholar]
- 17.Goeden N, et al. Prenatal Dynamics of Kynurenine Pathway Metabolism in Mice: Focus on Kynurenic Acid. Dev. Neurosci. 2017;39:519–528. doi: 10.1159/000481168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pocivavsek A, Elmer GI, Schwarcz R. Inhibition of kynurenine aminotransferase II attenuates hippocampus-dependent memory deficit in adult rats treated prenatally with kynurenine. Hippocampus. 2019;29:73–77. doi: 10.1002/hipo.23040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Notarangelo FM, Pocivavsek A. Elevated kynurenine pathway metabolism during neurodevelopment: Implications for brain and behavior. Neuropharmacol. 2017;112:275–285. doi: 10.1016/j.neuropharm.2016.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pershing ML, et al. Prenatal kynurenine exposure in rats: age-dependent changes in NMDA receptor expression and conditioned fear responding. Psychopharmacol. 2016;233:3725–3735. doi: 10.1007/s00213-016-4404-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Akagbosu CO, Evans GC, Gulick D, Suckow RF, Bucci DJ. Exposure to kynurenic acid during adolescence produces memory deficits in adulthood. Schizophr. Bull. 2012;38:769–778. doi: 10.1093/schbul/sbq151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chess AC, Bucci DJ. Increased concentration of cerebral kynurenic acid alters stimulus processing and conditioned responding. Behav. Brain Res. 2006;170:326–332. doi: 10.1016/j.bbr.2006.03.006. [DOI] [PubMed] [Google Scholar]
- 23.DeAngeli NE, et al. Exposure to Kynurenic Acid during Adolescence Increases Sign-Tracking and Impairs Long-Term Potentiation in Adulthood. Front. Behav. Neurosci. 2014;8:451. doi: 10.3389/fnbeh.2014.00451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mushak P, Davis JM, Crocetti AF, Grant LD. Prenatal and postnatal effects of low-level lead exposure: integrated summary of a report to the U.S. Congress on childhood lead poisoning. Env. Res. 1989;50:11–36. doi: 10.1016/s0013-9351(89)80046-5. [DOI] [PubMed] [Google Scholar]
- 25.He L, Poblenz AT, Medrano CJ, Fox DA. Lead and calcium produce rod photoreceptor cell apoptosis by opening the mitochondrial permeability transition pore. J. Biol. Chem. 2000;275:12175–12184. doi: 10.1074/jbc.275.16.12175. [DOI] [PubMed] [Google Scholar]
- 26.Flora SJ, Mittal M, Mehta A. Heavy metal induced oxidative stress & its possible reversal by chelation therapy. Indian. J. Med. Res. 2008;128:501–523. [PubMed] [Google Scholar]
- 27.Valko M, Morris H, Cronin MT. Metals, toxicity and oxidative stress. Curr. Med. Chem. 2005;12:1161–1208. doi: 10.2174/0929867053764635. [DOI] [PubMed] [Google Scholar]
- 28.Dobrakowski M, et al. Association between subchronic and chronic lead exposure and levels of antioxidants and chemokines. Int. Arch. Occup. Env. Health. 2016;89:1077–1085. doi: 10.1007/s00420-016-1144-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lugo-Huitron R, et al. On the antioxidant properties of kynurenic acid: free radical scavenging activity and inhibition of oxidative stress. Neurotoxicol Teratol. 2011;33:538–547. doi: 10.1016/j.ntt.2011.07.002. [DOI] [PubMed] [Google Scholar]
- 30.Ramos-Chavez LA, et al. Relevance of Alternative Routes of Kynurenic Acid Production in the Brain. Oxid. Med. Cell Longev. 2018;2018:5272741. doi: 10.1155/2018/5272741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schwarcz R, Stone TW. The kynurenine pathway and the brain: Challenges, controversies and promises. Neuropharmacol. 2017;112:237–247. doi: 10.1016/j.neuropharm.2016.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pocivavsek A, Thomas MA, Elmer GI, Bruno JP, Schwarcz R. Continuous kynurenine administration during the prenatal period, but not during adolescence, causes learning and memory deficits in adult rats. Psychopharmacol. 2014;231:2799–2809. doi: 10.1007/s00213-014-3452-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rahman A, Rao MS, Khan KM. Intraventricular infusion of quinolinic acid impairs spatial learning and memory in young rats: a novel mechanism of lead-induced neurotoxicity. J. Neuroinflammation. 2018;15:263. doi: 10.1186/s12974-018-1306-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kim P, et al. The plasticity of human maternal brain: longitudinal changes in brain anatomy during the early postpartum period. Behav. Neurosci. 2010;124:695–700. doi: 10.1037/a0020884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Blanco Ayala T, et al. Alternative kynurenic acid synthesis routes studied in the rat cerebellum. Front. Cell Neurosci. 2015;9:178. doi: 10.3389/fncel.2015.00178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Swartz KJ, During MJ, Freese A, Beal MF. Cerebral synthesis and release of kynurenic acid: an endogenous antagonist of excitatory amino acid receptors. J. Neurosci. 1990;10:2965–2973. doi: 10.1523/JNEUROSCI.10-09-02965.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Allene C, et al. Sequential generation of two distinct synapse-driven network patterns in developing neocortex. J. Neurosci. 2008;28:12851–12863. doi: 10.1523/JNEUROSCI.3733-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Crepel V, et al. A parturition-associated nonsynaptic coherent activity pattern in the developing hippocampus. Neuron. 2007;54:105–120. doi: 10.1016/j.neuron.2007.03.007. [DOI] [PubMed] [Google Scholar]
- 39.Jusko TA, et al. Blood lead concentrations <10 microg/dL and child intelligence at 6 years of age. Env. Health Perspect. 2008;116:243–248. doi: 10.1289/ehp.10424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang XY, Liu AP, Ruan DY, Liu J. Effect of developmental lead exposure on the expression of specific NMDA receptor subunit mRNAs in the hippocampus of neonatal rats by digoxigenin-labeled in situ hybridization histochemistry. Neurotoxicol Teratol. 2002;24:149–160. doi: 10.1016/s0892-0362(01)00210-0. [DOI] [PubMed] [Google Scholar]
- 41.Neal AP, Worley PF, Guilarte TR. Lead exposure during synaptogenesis alters NMDA receptor targeting via NMDA receptor inhibition. Neurotoxicology. 2011;32:281–289. doi: 10.1016/j.neuro.2010.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stone TW. Neuropharmacology of quinolinic and kynurenic acids. Pharmacol. Rev. 1993;45:309–379. [PubMed] [Google Scholar]
- 43.Parsons CG, Danysz W, Quack G. Glutamate in CNS disorders as a target for drug development: an update. Drug. N. Perspect. 1998;11:523–569. doi: 10.1358/dnp.1998.11.9.863689. [DOI] [PubMed] [Google Scholar]
- 44.Shouval HZ, Bear MF, Cooper LN. A unified model of NMDA receptor-dependent bidirectional synaptic plasticity. Proc. Natl Acad. Sci. USA. 2002;99:10831–10836. doi: 10.1073/pnas.152343099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yashiro K, Philpot BD. Regulation of NMDA receptor subunit expression and its implications for LTD, LTP, and metaplasticity. Neuropharmacol. 2008;55:1081–1094. doi: 10.1016/j.neuropharm.2008.07.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xu Z, et al. Metaplastic regulation of long-term potentiation/long-term depression threshold by activity-dependent changes of NR2A/NR2B ratio. J. Neurosci. 2009;29:8764–8773. doi: 10.1523/JNEUROSCI.1014-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sunyer J, et al. Association between traffic-related air pollution in schools and cognitive development in primary school children: a prospective cohort study. PLoS Med. 2015;12:e1001792. doi: 10.1371/journal.pmed.1001792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lacasana-Navarro M, Romieu I, Sanin-Aguirre LH, Palazuelos-Rendon E, Hernandez-Avila M. Calcium intake and blood lead in women in reproductive age. Rev. Invest. Clin. 1996;48:425–430. [PubMed] [Google Scholar]
- 49.Albuquerque EX, Schwarcz R. Kynurenic acid as an antagonist of alpha7 nicotinic acetylcholine receptors in the brain: facts and challenges. Biochem. Pharmacol. 2013;85:1027–1032. doi: 10.1016/j.bcp.2012.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Broide RS, Leslie FM. The alpha7 nicotinic acetylcholine receptor in neuronal plasticity. Mol. Neurobiol. 1999;20:1–16. doi: 10.1007/BF02741361. [DOI] [PubMed] [Google Scholar]
- 51.Mansvelder HD, McGehee DS. Long-term potentiation of excitatory inputs to brain reward areas by nicotine. Neuron. 2000;27:349–357. doi: 10.1016/s0896-6273(00)00042-8. [DOI] [PubMed] [Google Scholar]
- 52.Zoli M, Picciotto MR, Ferrari R, Cocchi D, Changeux JP. Increased neurodegeneration during ageing in mice lacking high-affinity nicotine receptors. EMBO J. 1999;18:1235–1244. doi: 10.1093/emboj/18.5.1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kihara T, et al. alpha 7 nicotinic receptor transduces signals to phosphatidylinositol 3-kinase to block A beta-amyloid-induced neurotoxicity. J. Biol. Chem. 2001;276:13541–13546. doi: 10.1074/jbc.M008035200. [DOI] [PubMed] [Google Scholar]
- 54.Vizi ES, Lendvai B. Modulatory role of presynaptic nicotinic receptors in synaptic and non-synaptic chemical communication in the central nervous system. Brain Res. Brain Res Rev. 1999;30:219–235. doi: 10.1016/s0165-0173(99)00016-8. [DOI] [PubMed] [Google Scholar]
- 55.Rojas-Castaneda JC, et al. Alterations induced by chronic lead exposure on the cells of circadian pacemaker of developing rats. Int. J. Exp. Pathol. 2011;92:243–250. doi: 10.1111/j.1365-2613.2011.00761.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lehmkuhl, A. M., Dirr, E. R. & Fleming, S. M. Olfactory assays for mouse models of neurodegenerative disease. J Vis Exp, e51804, 10.3791/51804 (2014). [DOI] [PMC free article] [PubMed]
- 57.Perez-De La Cruz V, et al. Time-course correlation of early toxic events in three models of striatal damage: modulation by proteases inhibition. Neurochem. Int. 2010;56:834–842. doi: 10.1016/j.neuint.2010.03.008. [DOI] [PubMed] [Google Scholar]
- 58.Reyes-Ocampo J, et al. Mitochondrial dysfunction related to cell damage induced by 3-hydroxykynurenine and 3-hydroxyanthranilic acid: Non-dependent-effect of early reactive oxygen species production. Neurotoxicology. 2015;50:81–91. doi: 10.1016/j.neuro.2015.08.003. [DOI] [PubMed] [Google Scholar]
- 59.Herrera-Mundo MN, et al. S-Allylcysteine prevents the rat from 3-nitropropionic acid-induced hyperactivity, early markers of oxidative stress and mitochondrial dysfunction. Neurosci. Res. 2006;56:39–44. doi: 10.1016/j.neures.2006.04.018. [DOI] [PubMed] [Google Scholar]
- 60.Ramos-Chavez LA, et al. Neurological effects of inorganic arsenic exposure: altered cysteine/glutamate transport, NMDA expression and spatial memory impairment. Front. Cell Neurosci. 2015;9:21. doi: 10.3389/fncel.2015.00021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Giorgini F, et al. Targeted deletion of kynurenine 3-monooxygenase in mice: a new tool for studying kynurenine pathway metabolism in periphery and brain. J. Biol. Chem. 2013;288:36554–36566. doi: 10.1074/jbc.M113.503813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Heyes MP, Quearry BJ. Quantification of 3-hydroxykynurenine in brain by high-performance liquid chromatography and electrochemical detection. J. Chromatogr. 1988;428:340–344. doi: 10.1016/s0378-4347(00)83925-0. [DOI] [PubMed] [Google Scholar]
- 63.Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- 64.West MJ. New stereological methods for counting neurons. Neurobiol. Aging. 1993;14:275–285. doi: 10.1016/0197-4580(93)90112-o. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data used to support the findings of this study are available with the corresponding author upon request.