

Abstract
Here, we sought to elucidate the role of CAR (a cyclic peptide) in the accumulation and distribution of fasudil, a drug for pulmonary arterial hypertension (PAH), in rat lungs and in producing pulmonary specific vasodilation in PAH rats. As such, we prepared liposomes of fasudil and CAR-conjugated liposomal fasudil and assessed the liposomes for CAR conjugation, physical properties, entrapment efficiencies, in vitro release profiles, and stabilities upon incubation in cell culture media, storage, and aerosolization. We also studied the cellular uptake of fasudil in different formulations, quantified heparan sulfate (HS) in pulmonary arterial smooth muscle cells (PASMCs), and investigated the distribution of the liposomes in the lungs of PAH rats. We assessed the drug accumulation in a close and recirculating isolated perfused rat lung model and studied the pharmacokinetics and pharmacological efficacy of the drug and formulations in Sugen/hypoxia-induced PAH rats. The entrapment efficiency of the liposomal fasudil was 95.5 ± 4.5%, and the cumulative release was 93.95 ± 6.22%. The uptake of CAR liposomes by pulmonary arterial cells and their distribution and accumulation in the lungs were much greater than those of no-CAR-liposomes. CAR-induced increase in the cellular uptake was associated with an increase in HS expression by rat PAH-PASMCs. CAR, when conjugated with liposomal fasudil and given via an intratracheal instillation, extended the elimination half-life of the drug by four-fold compared with fasudil-in-no-CAR-liposomes given via the same route. CAR-conjugated liposomal fasudil, as opposed to fasudil-in-no-CAR-liposomes and CAR pretreatment followed by fasudil-in-no-CAR-liposomes, reduced the mean pulmonary arterial pressure by 40–50% for 6 h, without affecting the mean systemic arterial pressure. On the whole, this study suggests that CAR aids in concentrating the drug in the lungs, increasing the cellular uptake, extending the half-life of fasudil, and eliciting a pulmonary-specific vasodilation when the peptide remains conjugated on the liposomal surface, but not when CAR is given as a pretreatment or alone as an admixture with the drug.
Keywords: liposomes, peptide as a targeting moiety, pulmonary hypertension, fasudil, isolated perfused rat lung
Graphical Abstract
1. INTRODUCTION
Targeted drug delivery systems can improve the therapeutic indices of a variety of drugs by extending the drug residence time in the blood or pathological sites, increasing the pharmacological potency, and decreasing the toxicity of the encapsulated drug molecules.1–3 In fact, for efficient targeting or extending drug circulation time in the blood, particulate carriers such as nanoparticles and liposomes are functionalized with various homing devices including antibodies, aptamers, enzymes, proteins, or peptides.4–11 Of the various targeting moieties, short peptides offer several advantages including the lack of immune reactions because of their small size and the flexibility in conjugating them with surfaces of various carriers.12 Peptides with a propensity to bind to cell surface molecules or ability to penetrate cells are often used to modify the surfaces of drug-embedded particles to extend drug’s residence time, retard drug clearance, and improve site specific accumulation. Indeed, various molecules with therapeutic potentials are packaged into nanocarriers and then targeted toward a desired site by coating the nanoparticles with various short peptides such as RGD,13 TAT,14 and CAR.15
CAR is a cyclic peptide comprising nine amino acids (CARSKNKDC). This peptide has shown a tremendous promise as a targeting moiety.16 Reported first in 2007,15 this short peptide has highly selective cell penetrating properties and binds to heparan sulfate (HS) on the target cells.17 CAR appears to recognize a unique sulfation pattern of HS proteoglycans on the target cells.18 As such, CAR and CAR-conjugated nanoparticles are reported to accumulate in various inflammatory lesions such as hypertensive pulmonary arterial lesions and injured tissues caused by trauma, surgery, and inflammation.16–18 We and others have used CAR as a targeting moiety for delivering drugs for the treatment of pulmonary arterial hypertension (PAH),19–21 a rare disease of the pulmonary vasculature, manifested as elevated mean pulmonary arterial pressure (mPAP), narrowing of pulmonary arteries, vascular remodeling, inflammation, and intimal and medial hypertrophy.22,23 CAR specifically homes to hypertensive pulmonary arteries of PAH-induced rats and accumulates in all layers of the remodeled arteries [endothelium, medial smooth muscle (SMC), and adventitia of pulmonary arteries) and vasculature and distributes in the extravascular space (interstitial space and alveolar lumen] and entire wall of hypertensive pulmonary arteries.17
In a series of studies, we have demonstrated that when administered directly into the rat lungs via intratracheal (IT) instillation, CAR-conjugated liposomes and nanoerythrosomes21,24 containing fasudil, an investigational anti-PAH drug, help accumulate the drug in the lungs and elicit pulmonary preferential vasodilation in PAH rats.25 CAR also increases the retention or residence time of liposomes in isolated perfused rat lungs (IPRL) and enhances the uptake by rat pulmonary arterial SMC cells (PASMCs).19,26 In contrast, others showed that CAR elicits its pulmonary specific vasodilation by means of a “bystander effect”, a phenomenon when drug is not required to be present in CAR-conjugated particles. Instead, administration of the drug as an admixture with CAR produces targeting or site-specific vasodilation. Indeed, a published study demonstrated that intravenously administered CAR plus fasudil increases the accumulation of the drug in PAH lungs by the bystander effect.27 However, the CAR-induced bystander effect may not be biologically feasible because CAR does not have any anti-PAH effect.
As such, here we seek to elucidate the role of CAR as a targeting moiety in enhancing the site-specific accumulation and extending pulmonary retention of liposomal formulations of fasudil. Toward this end, we prepared unmodified liposomes and CAR-conjugated liposomes containing fasudil (Figure 1), optimized them for physicochemical properties, determined whether CAR binds to liposomes, assessed for drug release profiles, evaluated the uptake of drug by various cells, investigated the distribution and clearance of the liposomes in Sugen/hypoxia induced PAH lungs, quantified HS expression in PASMCs, and monitored the pharmacokinetic profiles in both control and Sugen/hypoxia induced PAH rats. Using the same rat model, we assessed the relative vasodilatory effects of CAR-conjugated liposomal formulations of fasudil after administration via the intravenous (IV) and IT routes. In an IPRL model, we further studied whether CAR increases the retention of the drug or liposomal formulations in the lungs of PAH afflicted versus those of control animals.
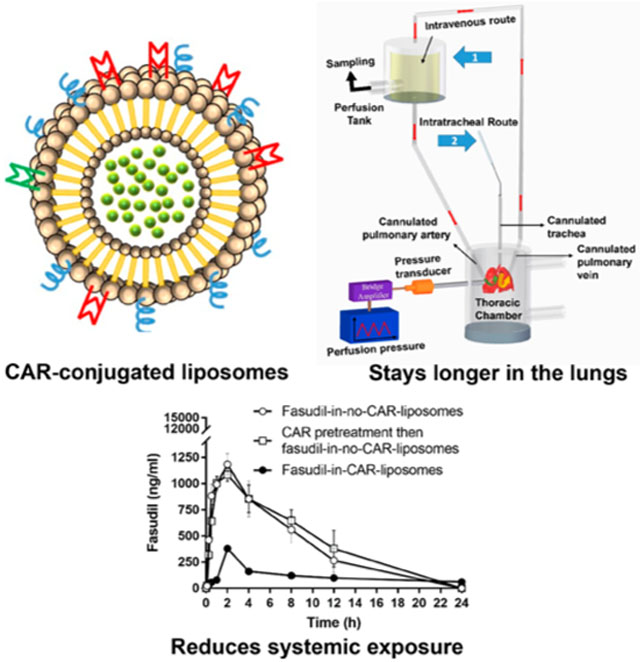
Figure 1.

Structure of targeted CAR-conjugated liposome containing fasudil.
2. EXPERIMENTAL SECTION
2.1. Materials.
Cholesterol, 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC), 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(poly ethylene glycol)-2000] (DSPE-PEG2000); and 1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine (DPPE) were purchased from Avanti Polar Lipids Inc. (Alabaster, AL). N-Succinimidyl 1,3-(2-pyridyldithio) propionate (SPDP) was obtained from Molecular Biosciences (Boulder, CO). Carboxyfluorescein (FAM)-labeled CAR peptide (currently licensed by Vascular BioSciences, Durham, NC) containing an extra cysteine for conjugation through the free sulfhydryl was custom synthesized (LifeTein, LLC, Somerset, Nj). The empirical formula for the fluorescent-labeled CAR is FAM-Cys-6-aminohexanoic acid-(CARSKNKDC)-amide. The extra cysteine was used for attaching the fluorescent probe and thus to keep the cyclic form of the peptide intact, which occurs via a disulfide bridge between Cys1 and Cys9. All other chemicals, purchased either from Fisher Scientific (Pittsburgh, PA) or Sigma-Aldrich Inc. (St Louis, MO), were of analytical grades and used without further purification. Diseased and control endothelial (ECs), SMC, and fibroblast cells (FBCs) were either isolated from the pulmonary arteries of Sugen/hypoxia-induced-PAH rats by Cell Biologic Inc. (Chicago, IL) or purchased from the same vendor. All media supplements were purchased from Invitrogen (Carlsbad, CA).
2.2. Preparation of Liposomal Fasudil.
Using DPPC, cholesterol, DSPE-PEG, and DPPE at a molar ratio of 70:30:5:5 and 50 μmol total lipid, we prepared liposomal formulations of fasudil (fasudil-in-no-CAR-liposomes), as reported in our published studies.28 In short, in a round-bottom flask, we dissolved 2 mL of chloroform and methanol at a ratio of 4:1 and dried the resulting mixture in a rotary evaporator at 45 °C to form a thin film (Buchi Rotor Evaporator R-114; BUCHI Labortechnik AG, Switzerland). Rehydrating the dried lipid film with 250 mM ammonium sulfate solution (pH 5.4) and sonicating the rehydrated film for an hour at 65 °C (formulation F-5), we prepared large multilamellar vesicles. We then extruded the resulting vesicles using LiposoFast Extruder (Avestin, Inc., Canada) at 65 °C and collected small unilamellar vesicles (SUVs) after 21 cycles of extrusion through 400 and 200 nm polycarbonate membranes (Avanti Polar Lipids, Inc., Alabaster, Alabama). We then replaced the external buffer, ammonium sulfate, with phosphate buffer saline (PBS, pH 7.4) using a PD-10 column (Sephadex-25, GE Healthcare, Piscataway, Nj). Incubating 10 mg of fasudil with liposomes (formulation F6) for 2 h at 65 °C, we entrapped the drug within liposomes and separated the unentrapped drug using a PD-10 column. By changing process parameters (speed of rotation of the rotary evaporator, sonication, and solvent evaporation time), we prepared five formulations, listed in Table 1, and stored at 4 °C for further evaluation.
Table 1.
Physical Characteristics and Drug Contents of the Liposomes Containing Fasudila
| sonication | drug | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| formulations | rotation speed | rotation time (h) | solubilization of dry films and then sonication | direct sonication without solubilization of the dry film | solution | powder | zeta potential (mV) | size (nm) | PDI | encapsulation (%) |
| F1 | 50 | 1 | × | × | −30.4 ± 11.3 | 212.1 ± 1.3 | 0.05 ± 0.01 | 46.5 ± 2.5 | ||
| F2 | 150 | 1 | × | × | −28.35 ± 5.5 | 205.2 ± 0.8 | 0.07 ± 0.06 | 77 ± 2.4 | ||
| F3 | 250 | 1 | × | × | −26.9 ± 14.8 | 209.7 ± 2.6 | 0.08 ± 0.02 | 31 ± 5.3 | ||
| F4 | 150 | 12 | × | × | 206.2 ± 2.8 | 0.03 ± 0.01 | 62.1 ± 8 | |||
| F5 | 150 | 1 | × | × | 203.6 ± 1.2 | 0.06 ± 0.01 | 95.5 ± 4.5 | |||
| F6 | 150 | 1 | × | × | 179.6 ± 8.1 | 0.07 ± 0.02 | 95.5 ± 4.5 | |||
Data represent mean ± SD, n = 3.
Following one of our published methods,29 we prepared CAR-conjugated liposomes (fasudil-in-CAR-liposomes) using DSPE, DSPE-PEG2000, DPPE, and DPPC. The thiol group in N-terminal Cys was used for conjugating the peptide with the free amino groups of liposomal lipids.19,24,29,30 For conjugating CAR with liposomes containing fasudil, we first activated the amine groups of the phospholipids of liposomes by incubating with SPDP (25 μmol) dissolved in 30 μL of dimethyl formamide for 1 h, removed the excess SPDP by ultracentrifugation at 80 000g for 1 h at 4 °C (TL-100 Ultracentrifuge, Beckman, USA), finally resuspended the purified liposomes in PBS, and incubated with 1 mg of CAR with a terminal-free thiol group for 1 h at room temperature. Finally, we removed the unconjugated peptide by ultracentrifugation and resuspended CAR-modified liposomes in PBS and stored at 4 °C for further evaluation. We calculated the amount of CAR in liposomes from the release of pyridine-2-thione, which was equivalent to 4.7 μmol CAR in each μmol of lipids. This calculation was based on the assumption that the reaction of 1 mol of CAR with SPDP-linked-liposomes releases 1 mol of pyridine-2-thione (Figure 2 step 2), which is equivalent to 0.53 mg of pyridine-2-thione, as determined by a UV spectroscopic method. As such, we determined the total amount of conjugated CAR from the amount of pyridine released using the following equation:26,29 mols of CAR = (1380.48 g CAR/1 mol CAR) × 5.3 × 10−4 g pyridine-2-thione × (1 mol pyridine-2-thione/111.2 g pyridine-2-thione) = 6.5 × 10−3 g CAR = 4.7 μmol CAR For preparation of fluorescent liposomes, we used rhodamine-labeled DPPE and followed the same procedure as above.
Figure 2.

Chemical reactions involved in the conjugation of CAR peptide with the amino groups of the lipids of liposomes.
2.3. Nuclear Magnetic Resonance.
The nuclear magnetic resonance (NMR) samples were prepared by dissolving the liposomes or SPDP in DMSO-d6. The lipid concentration in the samples was ~5 μM. Proton, nitrogen, and 2D-correlation spectroscopy (COSY) NMR spectra were recorded on a Bruker 400 MHz AVANCE III HD spectrometer at 400 and 100 MHz for 1H and 15N, respectively, with trimethylsilane as an internal standard (IS). Multiplicities are indicated by s (single), d (doublet), dd (doublet of doublets), t (triplet), q (quartet), m (multiplet), and br (broad). Data were acquired and processed using Bruker TopSpin 4.0 NMR software, chemical shifts (δ) were recorded in ppm relative to the reference signal, and coupling constant (J) values were recorded in hertz (Hz).
2.4. Physical Characterization.
We characterized the liposomal formulations for size, polydispersity index (PDI), zeta potential, and entrapment efficiency, as reported earlier.28 For measuring the size and zeta potential, we diluted 20 μL of liposomes with 1000 μL of PBS and recorded the size and zeta potential in a Nano ZS90 Zetasizer (Malvern Instruments Ltd., Worcestershire, U.K.). To determine the entrapment efficiency, after mixing with 980 μL of methanol, we first lysed 20 μL of liposomes, by sonication for 15 min, separated the drug from lipids by centrifuging at 17 000g (Legend Micro 17R, Thermo Scientific) for 15 min, and determined the amount of fasudil using a UV spectrophotometer (UV/vis 918, GBC Scientific Equipment, Hampshire, Illinois, USA) at 320 nm. Finally, we calculated the entrapment efficiency (%) using the following equation: amount of drug in liposomes/amount of drug originally added × 100.
2.5. In Vitro Release Study.
For studying drug release from the liposomes, we used dialysis cassettes (Slide-A-Lyzer, 3500 MWCO, 0.1–0.5 mL, Thermo-Scientific, Waltham, MA). In short, we first hydrated the cassettes with PBS (pH 7.4), loaded 100 μL liposomes or CAR-conjugated liposomes using a syringe, placed the cassettes containing liposomes in a beaker containing 100 mL PBS, and incubated at 37 °C under moderate stirring. We then collected 1 mL of sample at predetermined time intervals, replenished immediately with 1 mL of fresh PBS solution, determined the amount of drug released spectrophotometrically as described above, and calculated % drug release using the following equation: (the amount of fasudil at time t/the amount of fasudil when 100% released) × 100.
2.6. Storage and Aerosolization Stability of Liposomes.
For in vitro stability studies, we stored 500 μL of liposomes at 4 °C and collected samples every 7 days for 28 days and assessed the samples for vesicle size, PDI, and entrapment efficiency as described above. For studying the stability upon aerosolization, we measured the size, PDI, zeta potential, and entrapment efficiency of the formulations before and after aerosolization of the formulations with a PennCentury MicroSprayer (Model IA-1B, PennCentury, PA). For this study, we loaded a suspension of the liposomes into a Hamilton syringe attached to the microsprayer, sprayed three times, collected the droplets, and measured the above stability parameters.
2.7. Development of Sugen 5416/Hypoxia Induced PAH Model.
To develop a rat model of PAH, we administered Sugen 5416 (20 mg/kg) into male Sprague Dawley rats (200–225 g) and housed them in a hypoxia chamber (10% oxygen) (Whole Animal Hypoxia Chamber, BioSpherix, Lacona, NY) for first 3 weeks and then in normoxia for an additional week.31,32 After these four weeks, we divided the rats into various groups to perform five sets of studies: particle distribution, pharmacokinetics, pulmonary retention, and in vivo and ex vivo efficacy of fasudil and its liposomal formulations, as described below.
2.8. Determination of Pulmonary Retention of Fasudil and Liposomal Formulations.
To study the fraction of drug retained in the lungs after IT instillation and those accumulates in the lungs after systemic administration, we used an IPRL system (Hugo Sachs Elektronik, Harvard Apparatus GmbH, March-Hugstetten, Germany). We surgically removed the lungs from control and PAH rats, developed as described above, after anesthetizing with an intramuscular injection of ketamine/xylazine (90/10 mg/kg). For this surgical procedure, we made an incision from the abdomen to the neck, identified the trachea, inserted a cannula for ventilation of the lungs and keeping the lungs inflated, then opened the chest cavity, and administered 200 IU/Kg heparin into the right ventricle to prevent blood clotting. We then inserted a cannula into the pulmonary artery via a small incision in the trunk of the right ventricle and another cannula into the left atrium and maneuvered the cannula into the pulmonary vein and used those cannulas as the inlet and outlet for perfusions (Figure 3). Next, we perfused the lungs with a physiological lung solution made of CaCl2, NaCl, KC1, MgSO4, NaH2 PO4, glucose, NaHCO3, and Ficoll at pH 7.4 and 37 °C and passed a mixture of 95% O2:5% CO2 gas into the medium in the reservoir. Subsequently, we removed the lung–heart block along with the cannulas bathed with perfusion medium and placed the lungs in a humid artificial thoracic chamber under the negative pressure at 37 °C. To prevent deflation of the lungs, we maintained negative pressure within the thoracic chamber, let the lungs stabilized in the artificial thoracic chamber after 5 min of perfusion, recorded the tidal volume and ventilation frequency, and let the media perfuse at 3–9 mL, 60 cycles/min and 10 mL/min, respectively. Finally, we administered the plain fasudil or formulations containing fasudil (3 mg/kg) via the cannulated pulmonary artery or trachea to mimic the IV and pulmonary routes (IT), respectively (Figure 3). For IV administration of the formulations, we first dispersed the formulation in a 50 mL perfusion medium and then instilled the dispersion via the arterial cannula (Figure 3, arrow 1), and for IT administration, we administered the formulation directly into the lung via the tracheal cannula (Figure 3, arrow 2) using a PennCentury MicroSprayer. As such, the lungs collected from sham rats (no PAH) and PAH-afflicted rats received, via the trachea or pulmonary artery, 3 mg/kg fasudil as (i) plain fasudil, (ii) fasudil plus CAR admixture in saline, (iii) CAR pretreatment for 15 min and then administration of plain fasudil, (iv) fasudil in-no-CAR-liposomes, (v) fasudil-in-CAR-liposomes, and (vi) CAR pretreatment for 15 min and then administration of fasudil-in-no-CAR-liposome formulation. We then perfused the lungs with the above perfusion media and periodically collected an aliquot of the perfusate from 50 mL perfusate solution for 2 h and replaced the withdrawn samples with an equivalent amount of fresh perfusate.
Figure 3.

IPRL system showing the routes of administration, reservoir for sampling and thoracic chamber. Arrow 1 and 2 show IV- and inhalation-mimicking routes, respectively.
At the end of perfusion studies, we removed the lungs from the thoracic chamber and stored them at −80 °C for analyzing the drug content in lung tissues. To extract the drug from the lungs, we homogenized the lungs, centrifuged, collected the supernatant, and separated the drug from the supernatant by methanol precipitation (5:1 v/v) followed by centrifugation at 13 300g for 15 min and determined the drug using a liquid chromatography–tandem mass spectrometry (LC–MS/MS) method as described below and finally normalized the drug content to protein content of the tissue using a bicinchoninic acid assay (BCA). For all treatment groups, we determined the amount of fasudil in the IPRL circuitry by subtracting the amount of fasudil in the perfusate and lung homogenates from the dose of fasudil administered. Specifically, the total mass of fasudil was calculated by adding the amount of drug retained in the lungs with that released in the perfusate and the amount lost in the IPRL circuitry.
We measured the fasudil content in the perfusate and in lung homogenates using a validated LC–MS/MS method.33,34 In short, preparing samples in methanol (5:1 v/v) and using ranitidine as an IS, we used an isocratic elution and a Kintex XB-C18 column (1.7 μm, 100 × 2.1 mm) for separation. We let the mobile phase, comprising 65% ammonium formate (2 mM) plus 35% methanol, flow at 0.3 mL/min and at 40 °C and used a AB Sciex 5500 Qtrap tandem mass spectrometer for detection. Multiple reaction monitoring using the precursor to product ion was m/z 291.7/99.2 for fasudil and m/z 314.8/176.2 for IS. All quantifications were based on a freshly prepared standard curve of plain fasudil in plasma where the elution time was 4 min. For the calibration curve, we used plasma as the vehicle and used that curve for the drug concentration in a given sample.
2.9. Cellular Uptake Studies.
We quantified the cellular uptake of fasudil by incubating the drug or liposomal formulations of the drug with the three cells of the pulmonary arteries of PAH (PAH-PAECs, PAH-PASMCs, and PAH-PAFBCs) or healthy rats (control-PAECs, control-PASMCs, and control-PAFBCs). After growing the cells in 6-well culture plates until confluence, we removed the media and finally treated the cells with plain fasudil or fasudil in plain or CAR-liposomes for 24 h. Next, we removed the medium, washed the cells with ice-cold PBS buffer thrice, lysed the cells with 50 μL lysis buffer containing 1% (w/v) Triton X-100, 2% (v/v) protease, 2% (v/v) phosphatase, 2% (v/v) β glycine, and 2% (v/v) sodium fluoride and finally determined the fasudil concentration using the above LC–MS/MS method. By measuring the protein concentrations using a BCA protein assay kit (Bio-Rad Hercules, CA), we normalized the cellular uptake.
2.10. Stability of CAR Liposomes in Cell Culture Media and Quantification of HS.
We used an indirect method to assess whether CAR peptide undergoes any degradation in the presence of cell culture media. For this study, we first grew PASMCs from PAH and control rats in 48-well culture plates until confluence and used those cells to conduct two sets of experiments. In the first set of experiment, we treated the cells with the liposomes soon after mixing the formulation with the media. In the second set of experiment, we treated the cells with CAR-liposomes after 24 h of mixing the formulation with the media, that is, after incubating the liposomes in the cell culture medium for 24 h. We then fixed the cells with 4% paraformaldehyde, blocked with goat serum and 0.3% Triton X-100 for 40 min at room temperature, and washed thrice with PBS buffer. After incubating the fixed cells with anti-β-actin primary antibodies (Sigma-Aldrich, St. Louis, MO) overnight, we washed the cells with PBS, incubated with Alexa Fluor 594 goat anti-mouse IgG (invitrogen, Grand Island, NY), stained the cell nuclei with a mounting medium containing DAPI (4,6-diamidino-2-phenylindole), and finally imaged the cells using a fluorescent microscope (DMi8 Epifluorescence, Leica, IL). Using a commercially available ELISA kit (Immunotag, G-Biosciences, MO), we determined the levels of HS expression in PASMCs collected from both PAH and control rats.
2.11. Distribution and Clearance of Liposomes in Rat Lungs.
For studying the distribution of liposomal formulations and subsequent clearance from the lungs, we administered fluorescent-labeled no-CAR-liposomes and CAR-conjugated liposomes intratracheally into the lungs of PAH rats, developed as described above. Before the treatment, we confirmed the development of PAH by measuring mPAP as described below. We collected the lungs at 12, 24, and 48 h after IT administration of the formulations, visualized them under IVIS Lumina XR (Caliper Life Sciences, Inc.) and calculated the fluorescence intensity in the lungs using living image software (IVIS lumina series, PerkinElmer, MA). To determine whether particles reach the alveolar region, we prepared 5 μm. sections of the lungs, collected them 24 h post administration of either formulation, blocked the sections with goat serum plus Triton-X 100, and incubated them with mouse anti-prosurfactant protein C antibody (Sigma-Aldrich, St. Louis, MO). Next day, after washing the sections with PBS, we incubated them with Alexa Fluor 488 goat anti-mouse IgG (Invitrogen, Grand Island, NY), stained the nuclei with a mounting medium containing DAPI, and finally imaged the tissue sections using a fluorescent microscope (DMi8 Epifluorescence, Leica, IL).
2.12. Pharmacokinetic Studies.
We performed the pulmonary absorption studies of plain fasudil and liposomal formulations in control (no-PAH) and PAH-afflicted adult male SD rats (250–300 g) as reported earlier.35 Anesthetized control and PAH rats, that are divided into 14 groups (7 control rats plus 7 PAH rats; n = 3), received 3 mg/kg fasudil in the following forms: plain fasudil via (i) IV and (ii) IT routes; fasudil-in-no-CAR-liposomes via (iii) IV and (iv) IT; CAR pretreatment followed by fasudil-in-no-CAR-liposomes via (iv) IV and (v) IT; and fasudil-CAR-liposomes via (vi) IV and (vii) IT routes. For IV, we administered the drug or formulations via the penile vein and for IT administration, we used a PennCentury MicroSprayer for rats and sprayed the drug or formulation directly into the lungs, as described previously.19 Collecting blood samples, via tail-vein milking, in a citrated micro centrifuge tubes for 24 h, we separated the plasma by centrifuging the blood at 10 000 rpm for 10 min and quantified fasudil in the plasma using the above LC–MS/MS method. To extract the drug from plasma, we first deproteinized the plasma with methanol (5:1 v/v), vortexed, centrifuged at 13 300 rpm for 15 min at 4 °C, and then injected the supernatant into the LC–MS/MS. For all animal studies, we followed the NIH Guidelines for the Care and Use of Laboratory Animals under a protocol approved by Texas Tech University Health Sciences Center (TTUHSC) Animal Care and Use Committee (AM-10012).
2.13. Acute Hemodynamics Studies in Intact PAH Rats and Lungs Isolated from PAH Rats.
For measuring mPAP and mean systemic arterial pressure (mSAP) in intact PAH animals and mPAP in isolated lungs from PAH rats, we used Sugen/hypoxia-induced PAH rats developed above. For studies involving intact PAH rats, we first catheterized the pulmonary and carotid arteries of anesthetized PAH rats with polyvinyl (PV-1, Tygon, Lima, OH) and polyethylene tubes (PE50-BD Intramedic, Sparks, MD) to measure mPAP and mSAP, respectively.28 Then, we attached the hypertonic fluid-filled catheters to the MEMSCAP SP844 physiological pressure transducers (MEMSCAP, AD Instruments Inc., Colorado Springs, CO), connected them with the bridge amplifiers, and recorded the data using a PowerLab 16/30 system using LabChart Pro 7.0 software (AD Instruments, Inc., Colorado Springs, CO). After recording the baseline values for mPAP and mSAP for 10 min, we gave the rats (n = 3) 3 mg/kg of fasudil via IT instillation as (i) fasudil-in-no-CAR-liposomes, (ii) CAR pretreatment followed by fasudil-in-no-CAR-liposomes, and (iii) fasudil-in-CAR-liposomes and recorded mPAP and mSAP for 6 h. We presented the data in % initial of mPAP and mSAP and calculated the lung targeting index (LTI) of the formulations from the ratio of the area above the pressure/time curve (AAC) for mPAP and mSAP using the equation LTI = AAC of mPAP/AAC of mSAP.
For the ex vivo efficacy study, we used lungs from PAH rats, which we removed lung–heart en bloc from the rats and placed the block in the thoracic chamber as discussed in the IPRL system above. Before removing the heart–lung block from the rats, we confirmed the development of PAH by measuring mPAP as described above. However, to measure PAP ex vivo, we used a pressure transducer (P75, type 379; Harvard Apparatus, Germany), which we connected via the port for measuring perfusion pressure with the dc bridge amplifiers of the IPRL system (Figure 3). This transducer can measure pressure as low as 0 to ±75 mmHg. After placing the lungs in the thoracic chamber of the IPRL system, we recorded the baseline pressure for 10 min and gave six groups of lungs (n = 3) the same treatment as above.
2.14. Data Analysis.
All data are presented as mean ± SEM, and the difference among various groups were analyzed by one-way ANOVA followed by a post hoc analysis using Tukey’s comparison (GraphPad Prism, version 5.0, GraphPad Software, San Diego, CA). In statistical analysis, the p value less than 0.05 was considered statistically significant. Pharmacokinetic parameters were calculated by standard noncompartmental analysis using Phoenix WinNonlin (Sunnyvale, CA).
3. RESULTS
3.1. Drug Loading and Physical Properties of Liposomal Fasudil.
We used ammonium sulfate-mediated transmembrane gradient method, an active drug loading process, to prepare liposomal fasudil because this drug, a hydrophilic compound with a pKa of 9.727,36 shows poor entrapment in lipid-based nanocarriers.37 To increase fasudil’s entrapment efficiency, we optimized the processing variables including speed of rotation of the rotary evaporator, duration for solvent evaporation and sonication, and drug loading procedure (Table 1), which are known to influence the physical characteristics of lipid-based nanocarriers. The drug loading was the highest when the speed of the rotation and duration of evaporation was 150 rpm and 1 h, respectively and when liposomes were incubated directly with solid-state fasudil. However, when the duration of evaporation was longer than an hour, the encapsulation efficiency declined. Further, unlike direct sonication of the dry lipid film, solubilization of the lipid film and then sonication of the resulting solution has resulted in increased drug entrapment. On the other hand, unlike the previously published method, when liposomes were incubated with fasudil dissolved in PBS,29 incubation of liposomes with fasudil in the solid state increased the encapsulation efficiency from 77 ± 2.4 to 95.5 ± 4.5%.
To assess whether CAR was chemically conjugated with the liposomes, we performed proton and nitrogen NMR on the plain liposome, SPDP (cross linker), and CAR-conjugated liposomes. The 1H and 15N NMR spectra of SPDP, unmodified liposomes-SPDP, and CAR-liposomes are shown in Supporting Information Figure 1. Lack of signals at δ = 406.98 ppm in the NMR spectra of liposome-SPDP suggest binding of SPDP to plain liposomes. In fact, the NMR spectra of SPDP showed a signal for nitrogen in the succinimide ring at 406.98 ppm because of the shielding effect of two oxygen molecules in the ring. However, the spectrum for liposome-SPDP showed no peak at 406.98 ppm, which belonged to the succinimide ring, suggesting that SPDP was conjugated with plain liposome via removal of the succinimide ring from the SPDP and formation of an amide bond (Figure 2, step 1). The peaks for two CH2 units specific to the succinimide ring in 1H NMR were not identifiable because of overlapping signals of liposomes in this area of the spectrum.
The 1H NMR of the liposome-SPDP showed a doublet signal at δ = 8.46 ppm, triplet signal at δ = 7.84 ppm, and doublet signal at δ = 7.79 ppm which are assigned to hydrogen 1, 3, and 4 of the pyridine ring, respectively. Also, COSY showed a signal at 7.26 ppm representing hydrogen 2 of the pyridine ring, thus providing further evidence of linkage to the liposome after work up (Supporting Information Figure 2). In contrast, proton NMR of CAR-conjugated liposomes did not show any peaks in this chemical shift, suggesting conjugation of CAR with the liposome-SPDP via removal of the pyridine ring (Figure 2, step 2). To investigate whether fasudil was encapsulated in CAR-conjugated liposomes, we also performed proton NMR for fasudil and CAR-liposome-containing fasudil (Supporting Information Figure 3). Fasudil showed a 1H singlet at δ = 9.52 ppm and a 1H doublet at δ = 8.72 ppm, which belongs to the two hydrogens alpha and to the nitrogen of the isoquinoline ring. Similarly, proton NMR of CAR-conjugated liposomes containing fasudil showed similar peaks in these chemical shift, suggesting that fasudil was within the core of the CAR-liposome.
The average hydrodynamic diameter of the liposomes was between 174.5 ± 3 and 202.2 ±6.1 nm (Table 1) and PDIs were between 0.039 ± 0.09 and 0.063 ± 0.01, indicating a monodispersed nature of the formulation. Given that the zeta potential of the liposomes was between −26.9 ± 14.8 and 30.4 ± 11.3 mV, liposomal formulations are likely to be stable. Conjugation of CAR and subsequent purification and separation from unconjugated peptide had no appreciable impact on the physical properties of the liposomes. The final formulation that we have used in studies described below had 4.76 μmol CAR in each μmol of lipids which was indirectly determined from the release of pyridine-2-thione and entrapment efficiencies of 95.5 ± 4.5% (Table 1).
3.2. Storage and Aerosolization Stability and in Vitro Release Profiles.
We examined the stability of the optimized formulation by measuring the entrapment efficiency and vesicle size upon storing for 28 days. Neither the percentage drug encapsulation nor particle size underwent any major changes after 4 weeks of storage (Figure 4A,B). The particle size remained unaltered because of optimum zeta potential that prevents the particles from aggregating. We have also assessed the stability of the liposomes during aerosolization. Because the microsprayer exerts certain force to aerosolize the formulations, we assessed liposomal stability upon spray. Creating a spray using a PennCentury MicroSprayer did not change the size, zeta potential, and entrapment efficiency of the liposomes (Table 2), suggesting that liposomes’ physical properties would remain unaltered in case of IT instillation. Both CAR-modified and unmodified liposomes (fasudil-in-no-CAR-liposomes) released the drug in a continuous fashion, with a cumulative release of 93.95 ± 6.22 and 83.87 ± 4.58% drug, respectively (Figure 4C). Like previously published studies, dialysis cassettes played no roles in sustained drug release.28
Figure 4.
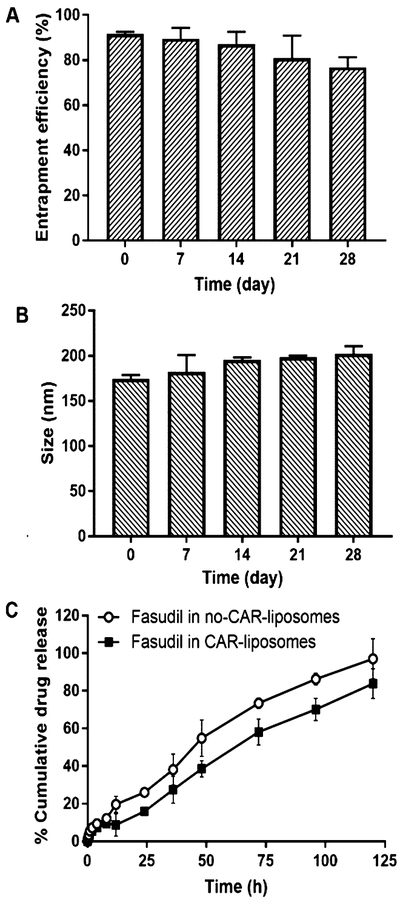
Changes in (A) size and (B) entrapment efficiency upon storage of various liposomal formulations for 28 days at 4 °C. In vitro release profiles of (C) fasudil-in-no-CAR and fasudil-in-CAR-liposomes in PBS at 37 °C (data represent mean ± SD, n = 3).
Table 2.
Changes in Liposomes upon Aerosolizationa
| nebulization | size (nm) | PDI | entrapment efficiency (%) |
|---|---|---|---|
| before | 188.4 ± 1.31 | 0.074 ± 0.02 | 87.8 ± 3.1 |
| after | 198.7 ± 10.1 | 0.098 ± 0.012 | 84.3 ± 4.5 |
Data represent mean ± SD, n = 3.
3.3. Retention of Liposomal Fasudil by Lungs from PAH and Sham Rats.
To study the influence of CAR on the pulmonary retention of plain fasudil, CAR pretreatment, and fasudil in unmodified (fasudil-in-no-CAR-liposomes) or CAR-modified liposomes (fasudil-in-CAR-liposomes), we determined the amount of fasudil retained or taken up by the lungs, collected from both PAH and control animals, using a previously established and validated IPRL model.26 Various IPRL models have been used to study the lung uptake, metabolism, and deposition of endogenous or exogenous molecules.38–40 Because this model does not involve recirculation of the blood from the peripheral compartments, this ex vivo model has been extensively used to study lung-specific effects of drugs administered via the vascular or airway route. The IPRL system that we have used here mimics the systemic routes of administration such as IV or oral routes when the drug is administered along with the perfusion media via the pulmonary artery (Figure 3). To mimic IT delivery in this IPRL model, drug is sprayed or aerosolized directly into the trachea. As such, when this model is used to mimic a systemic route such as the IV route, the concentration of a drug would decline in the perfusate and in case of pulmonary delivery, the drug concentration rises in the perfusate. In the former case, the drug concentration declines because of the uptake of the drug by the lungs, but in the latter case, the drug concentration in the perfusate increases because of the absorption of the drug from the lung into the systemic circulation (Figure 3).
Consistent with the above assumption for an IPRL system, the concentration of fasudil, when administered in various forms or after CAR pretreatment via the IV-mimicking route, in the perfusate declined in a time-dependent fashion (Figure 5). The decline in the drug concentration was very similar in both PAH and sham lungs (Figure 5A–D) and the amount of drug lost during perfusion is close to what was found in the lung homogenates (Figure 7A) and in IPRL tubes (Table 3). The concentration of the drug in the perfusate for each treatment, for example, declined from 40 to 10–20 μg/mL, and the concentration of the drug in the lung homogenate was also ~300–500 μg/g. Further, when we calculated the percentage drug remaining in the lungs by subtracting the amount of drug in the perfusate from the total amount administered, we observed an increase in the percentage remaining in the lung along with the decrease in drug concentration in the perfusate (Supporting Information Figure 4). Similar to drug concentration in the perfusate, fasudil concentration in the lung homogenates remain unaffected by the type of formulations (Figure 7).
Figure 5.

Fasudil concentration in the perfusate after the treatments with the plain drug and various liposomal formulations of the drug given via the IV-mimicking route (cannulated pulmonary artery) to (A,C) sham and (B,D) PAH lungs. The dose of fasudil was 3 mg/kg (rat body weight) or liposomes containing equivalent amounts of fasudil and the dose of CAR was 0.5 mg/kg (data represent mean ± SD, n = 3).
Figure 7.

Fasudil in the lung homogenates after treating the lungs with plain fasudil, CAR pretreatment, and liposomal formulations of fasudil given via (A) IV- and (B) IT-mimicking routes to sham and PAH lungs. The dose of fasudil was 3 mg/kg (rat body weight) or liposomes containing equivalent amounts of fasudil and the dose of CAR was 0.5 mg/kg (data represent mean ± SD, n = 3, *p < 0.05).
Table 3.
Fasudil (%) in the Perfusate, Lungs, and IPRL Tubes After 2 h of Instillations of Various Formulationsa
| recovery dose (%) | |||||||
|---|---|---|---|---|---|---|---|
| perfusate | lung | IPRL tubes | |||||
| drug/formulations | route | Sham | PAH | Sham | PAH | Sham | PAH |
| plain fasudil | IV | 56.5 ± 5.3 | 39.1 ± 3.9 | 34.7 ± 3.2 | 49.6 ± 6.2 | 8.7 ± 2.2 | 11.3 ± 3 |
| plain fasudil plus CAR | IV | 59.6 ± 8.5 | 54.7 ± 2.1 | 31 ± 5.4 | 38.4 ± 1.4 | 9.4 ± 3.1 | 6.9 ± 0.8 |
| CAR then plain fasudil | IV | 63.5 ± 6.4 | 44.1 ± 1.8 | 26.5 ± 4.2 | 37.7 ± 3.3 | 10 ± 3.9 | 18.2 ± 1.8 |
| fasudil in no-CAR-liposome | IV | 52.9 ± 6.8 | 49.4 ± 2.4 | 26.8 ± 7.3 | 46.5 ± 3.2 | 10.3 ± 3.3 | 4.1 ± 1.1 |
| CAR pretreatment then fasudil in no-CAR-liposomes | IV | 52.2 ± 9.2 | 40.3 ± 1.8 | 38.9 ± 7.5 | 49.6 ± 1.1 | 8.9 ± 2.6 | 10.1 ± 2.7 |
| fasudil in CAR-liposome | IV | 33.2 ± 2.7 | 29.1 ± 3.9 | 60.5 ± 3 | 57.5 ± 3.6 | 6.3 ± 0.3 | 13.4 ± 6.8 |
| plain fasudil | IT | 20.8 ± 2.7 | 13.3 ± 2.6 | 71.8 ± 2 | 70.1 ± 7.3 | 7.4 ± 1 | 16.6 ± 7.7 |
| plain fasudil plus CAR | IT | 30.1 ± 1.2 | 23.3 ± 5.2 | 60 ± 2.6 | 69.3 ± 3.1 | 9.9 ± 1.7 | 7.4 ± 2.4 |
| CAR then plain fasudil | IT | 27.1 ± 7.1 | 18 ± 4.7 | 60.1 ± 4.6 | 63.4 ± 2.1 | 12.8 ± 5.9 | 18.6 ± 6.3 |
| fasudil in no-CAR-liposomes | IT | 20.4 ± 0.4 | 11.6 ± 1.8 | 62.5 ±8.1 | 77 ± 1.4 | 17.1 ± 8.2 | 11.4 ± 0.7 |
| CAR pretreatment then fasudil in no-CAR-liposome | IT | 9.1 ± 0.4 | 9.7 ± 1.2 | 69.3 ± 5.6 | 73.3 ± 1.8 | 21.6 ± 5.2 | 17 ± 2.9 |
| fasudil in CAR-liposome | IT | 4.5 ± 1.9 | 2.2 ± 1.2 | 72.3 ± 2.6 | 90.6 ± 2 | 23.1 ± 0.9 | 7.2 ± 0.9 |
Data represent mean ± SD, n = 3.
However, the opposite was true for the drug concentration profiles in the perfusate, when the drug formulations were administered via the trachea or inhalation-mimicking route of the IPRL system (Figure 6). Unlike the IV-mimicking route in the IPRL system, the drug concentration in the perfusate increased in a time-dependent manner, suggesting that fasudil underwent absorption from the lung (Figure 6A–D). Echoing the reduced concentration in the perfusate, the concentration of fasudil, after administration via the IT route, in lung homogenates was greater than that administered via the IV-mimicking route (Figure 7B). Further, the fasudil concentration in the perfusate for the formulations administered to PAH lungs was lower than that in the perfusate for those administered to sham lungs, suggesting that the presence of the disease plays a role in CAR-induced retention of the drug in the lungs. When fasudil-in-no-CAR-liposomes were administered to sham and PAH lungs, the drug concentration in the perfusate was 1 μg/mL, which was about 10 μg/mL when fasudil alone was given. The lowest drug concentration in the perfusate was observed when fasudil-in-CAR-liposomes was given intratracheally. Compared with the reduction in drug concentration for sham lungs, the decline in the PAH lungs was the lowest. Consistent with the principle of IT administration, the % drug remaining in the lungs was also higher than that after IV administration (Supporting Information Figure 5). However, the effect of CAR treatment on sham animals was rather unexplainable (Supporting Information Figure 5C) because CAR pretreatment had no effect in other three major treatment groups. The amount of drug that was recovered in the lungs and perfusate was >80%, and regardless of the route of administration, the total amount of the drug in three components of the IPRL system (lung, perfusate, and IPRL circuitry) was close to 100% (Table 3), suggesting that IPRL is a valid tool for conducting ex vivo disposition of various formulations and the total amount of the drug in the system was accounted for. Further, the percentage of fasudil lost, calculated, in the tubes of IPRL system was statistically similar for all treatment groups.
Figure 6.

Fasudil concentration in the perfusate after the treatments with the plain drug and various liposomal formulations of the drug given via the inhalation-mimicking route (IT instillation) to (A,C) sham and (B,D) PAH lungs. The dose of fasudil was 3 mg/kg (rat body weight) or liposomes containing equivalent amount of fasudil and the dose of CAR was 0.5 mg/kg (data represent mean ± SD, n = 3).
A comparison of drug concentration–time curve for IT versus IV administration of various forms of fasudil suggest that the drug concentration in the perfusate for the IT route was several folds lower than that in the perfusate for the IV-mimicking route (Figures 5 and 6). Further, administration of plain fasudil 15 min after giving CAR (CAR pretreatment) appears to play no roles in the accumulation of the drug in the lungs, as evidenced from drug concentration profiles for both IT and IV routes, suggesting that CAR is unlikely to elicit a bystander effect, regardless of the route of administration and disease state. Further, a remarkably reduced concentration in the perfusate for IT administration of all three treatments (fasudil-in-no-CAR-liposomes, CAR pretreatment then fasudil-in-no-CAR liposomes, and fasudil-in-CAR-liposomes) suggests that the presence of CAR-conjugated liposomes, not CAR plus fasudil, plays an important role in retention of the drug in the lungs. Moreover, the extent of drug distributed in the lung (μg of fasudil/g of lung) was significantly greater when the drug or formulations were given via IT instillation than when the same treatment was given via IV instillation (Figure 7). This observation is consistent with the assumption that IT delivery facilitates localized delivery in the lung, whereas IV administration delivers drugs throughout the body and thus reduces total drug reaching the lungs.
3.4. Quantification of HS in PASMCs, Cellular Uptake, and Stability of CAR Peptides in Culture Media.
To assess whether fasudil in CAR-liposomes were preferentially taken up by the pulmonary arterial cells (PACs) of PAH-afflicted rats, we quantified the amount of fasudil in cells treated with two liposomal formulations. When PACs were treated with fasudil-in-CAR-liposomes versus fasudil-in-no-CAR-liposomes, drug uptake was greater from CAR-liposomes than that for no-CAR liposomes (Figure 8). The extent of uptake by PACs from PAH-afflicted rats was greater than the uptake by the PACs from control rats. Thus, the magnitude of drug uptake by the cells was influenced both by the presence or absence of CAR in the liposomes and the types of cells, that is, PACs collected from diseased versus those collected from healthy animals. These statistically significant increases in the drug uptake by PAH-afflicted PACs are consistent with the IPRL data (Figures 5–7). Similar to the quantitative cellular uptake data, fluorescent microscopic study suggests a much higher uptake of CAR-conjugated liposomes by PAH-SMCs, when compared with no-CAR-liposomes (Figure 10A). As such, both quantitative and qualitative studies suggest that CAR-conjugated liposomes are preferentially taken up by the cells of PAH-afflicted pulmonary arteries. Based on the observation that fasudil uptake by PASMCs was greater than that by PAECs and PAFBCs, we measured HS in PASMCs, the target cell for fasudil’s vasodilatory action. In agreement with published study concerning overexpression of HS in the PAH lesions,15,17,27,41,42 our study also suggests that the levels of HS in PASMCs from PAH rats were much greater than that of PASMCs from control rats (Figure 9).
Figure 8.

Fasudil concentration in the cell lysate. Plain fasudil or various liposomal formulations of fasudil were incubated with EC, SMCs, and FBCs collected from the pulmonary arteries of Sugen-hypoxia-induced PAH and control rats (data represent mean ± SD, n = 3; *p < 0.05, **p < 0.005, ***p < 0.001, ****p < 0.001).
Figure 10.

Representative fluorescence microscopy images showing the uptake of liposomes by PAH-SMCs after (A) immediate and (B) 24 h of incubation of (i) fluorescent liposomes without CAR peptide and (II) fluorescent CAR-conjugated liposomes in cell culture media. Green, beta actin; blue, DAPI; red, rhodamine B-labeled liposomes. The rightmost panel shows the overlay.
Figure 9.

Concentration of HS in the cell lysate of SMCs collected from the pulmonary arteries of Sugen-hypoxia-induced PAH and control rats, (data represent mean ± SD, n = 3; *p < 0.05, **p < 0.005, ***p < 0.001, ****p < 0.001).
To confirm that CAR peptide does not undergo any degradation in the cell culture media, we treated CAR-liposomes with culture media and determined the cellular uptake at 0 and 24 h. Incubation of CAR-liposomes with the culture media for 24 h had no effect on the cellular uptake of the formulations. Thus, the extent of the cellular uptake of CAR liposomes determined after 24 h of incubation in culture media was no different from that was determined soon after mixing the (Figure 10B), suggesting that cell culture medium had no adverse effect on the stability of the peptide.
3.5. Particle Deposition and Clearance in Rat Lungs.
For studying the distribution and subsequent clearance of the particles, we imaged the whole lungs and lung sections collected from PAH-induced rats. For assessing the particle clearance, we quantitated fluorescent intensity in the lungs after 12, 24, and 48 h post administrations of fluorescent liposomes. The IVIS Lumina XR acquired images of whole lungs suggest that both no-CAR and CAR-liposomes reach the deep lung, and the extent of the distribution of CAR liposomes was greater than that of no-CAR liposomes (Figure 11A). One day after administration, ~38% of CAR-conjugated liposomes were eliminated from the lungs, while ~50% of liposomes without CAR were cleared off. This difference in the extent of retention was significantly higher after 48 h of IT administration of liposomes (Figure 11B). Fluorescent microscopic observation of the particles in lung tissue sections suggest that similar to the images of whole lungs, CAR-conjugated liposomes distributed more extensively in the lung tissues compared with liposomes without CAR Further, microscopy data show that particles indeed reach the alveolar region of the lungs (Figure 11C).
Figure 11.

Images of the lungs treated with fluorescent liposomes: (A) whole lung: (i) no treatment, (II) liposomes without CAR, and (III) CAR-conjugated liposomes. (B) Clearance of various formulations of the liposomes from PAH-induced rat lungs after IT administration. (C) Images of lung tissue sections collected 24 h after administration of fluorescent particles: (I) liposomes without CAR, and (II) CAR-conjugated liposomes (green, anti-prosurfactant protein C; blue, DAPI; red, rhodamine B-labeled liposomes). (Data represent mean ± SD, n = 3; *p < 0.05).
3.6. Inhalational of CAR-Conjugated Liposomes Reduces Systemic Exposure of Fasudil.
Using both sham and PAH-afflicted male SD rats, we assessed IV and pulmonary absorptions of various forms of fasudil: plain fasudil, fasudil-in-no-CAR-liposomes, CAR pretreatment then administration of fasudil-in-no-CAR-liposomes, and fasudil-in-CAR-liposomes. In both sham and PAH animals, IV administration of plain fasudil or different formulations led to a quick rise (Table 4) followed by a rapid decline in drug plasma concentration (Figure 12A,B). The overall bioavailability of fasudil-in-CAR-liposomes was smaller than that for the other two formulations (fasudil-in-no-CAR-liposomes and CAR pretreatment then fasudil-in-no-CAR-liposomes), perhaps because of slower drug release and accumulation of fasudil-in-CAR-liposomes in various tissue compartments. Interestingly, the Cmax and AUC for fasudil-in-CAR-liposomes were much smaller in PAH rats than in sham rats, suggesting that fasudil-in-CAR-liposomes has possibly accumulated in PAH lesions and other tissues expressing HS. Because of the presence of CAR and accumulation of fasudil-in-CAR-liposomes in tissue compartments, the drug from the liposomes was released into tissue compartments instead of the plasma compartment and thus the AUC and Cmax for fasudil-in-CAR-liposomes, which is based on plasma concentration, in PAH rats were l/3rd of those in sham rats (Table 4). For the same reason, the elimination half-life for fasudil-in-CAR-liposomes in PAH rats is 2–5-fold longer that that in sham animals.
Table 4.
Pharmacokinetic Parameters of Plain Fasudil and Various Formulations after IT and IV Administration into PAH and Sham Ratsa
| Cmax (ng/mL) | t1/2 (h) | AUC0-inf (ng·h/mL) | |||||
|---|---|---|---|---|---|---|---|
| drug/formulations | route | Sham | PAH | Sham | PAH | Sham | PAH |
| plain fasudil | IV | 14 150 ± 1050 | 0.2 ± 0.004 | 5967.1 ± 25.9 | |||
| fasudil in no-CAR-liposome | IV | 11 700 ± 778.9 | 10 365 ± 1135 | 0.3 ± 0.05 | 0.4 ± 0.1 | 9737.9 ± 544.3 | 9537.3 ± 1204 |
| CAR pretreatment then fasudil no-CAR-liposomes | IV | 13 200 ± 1186 | 12 200 ± 1268**** | 0.5 ± 0.06 | 0.35 ± 0.1 | 11 890.8 ± 975.3*** | 6118.8 ± 520* |
| fasudil in CAR-liposome | IV | 12 466.7 ± 1302**** | 4080 ± 630* | 0.7 ± 0.3 | 1.1 ± 0.3 | 5213 ± 152.8 | 1274 ± 218 |
| plain fasudil | IT | 3035 ± 635 | 0.7 ± 0.08 | 5967.1 ± 25.9 | |||
| fasudil in no-CAR-liposomes | IT | 1433.3 ± 87 | 1183.3 ± 83 | 3.3 ± 1.3* | 4.7 ± 0.9* | 9737.9 ± 544.3 | 10 191.8 ± 2016 |
| CAR pretreatment then fasudil in no-CAR-liposome | IT | 1390 ± 150 | 1085 ± 45 | 3.9 ± 0.1 | 6.9 ± 1.8 | 11 890.8 ± 975.3* | 12 677.1 ± 2910**** |
| fasudil in CAR-liposome | IT | 614 ± 102 | 379.7 ± 11.9 | 12.9 ± 4.6*** | 16.1 ± 4.1*** | 5832 ± 1365 | 4210.6 ± 985 |
Data represent mean ± SD, n = 3;
p < 0.05,
p < 0.005,
p < 0.001,
p < 0.001.
Figure 12.

Absorption profiles of fasudil and liposomal formulations of fasudil given intravenously to (A) sham and (B) PAH rats and those given intratracheally to (C) sham and (D) PAH rats. The dose of fasudil was 3 mg/kg (rat body weight) or liposomes containing equivalent amount of fasudil and the dose of CAR was 0.5 mg/kg (data represent mean ± SD, n = 3).
However, IT instillation reduced the drug concentration in the plasma (Figure 12C,D) and extended the elimination half-lives (t1/2) of both plain fasudil and liposomal formulations of fasudil. Similarly, the Cmax for plain fasudil was much larger than that for liposomal fasudil (Table 4) because the rate of absorption of the drug from liposomal fasudil was much slower than that for the plain drug. This observation is consistent with the IPRL data (Figure 6), wherein much of the drug remained in the lungs after administration via the IT-mimicking route. Importantly, the most dramatic change in the half-life occurred when fasudil-in-CAR-liposomes was and PAH rats. The half-life of fasudil-in-CAR-liposomes was about 4-fold longer than that of fasudil-in-no-CAR-liposomes. This observation is strikingly similar to the increased accumulation of fasudil-in-CAR-liposomes in lung homogenates and reduced concentration of fasudil in the perfusate (Figures 5 and 6).
3.7. Pulmonary Hemodynamics in Rats and Isolated Lungs.
Based on the above ex vivo disposition and pharmacokinetic studies, we chose three liposome-based formulations/treatments in studying the pulmonary hemodynamics using lungs collected from PAH-afflicted rats and intact PAH animals. For both ex vivo and in vivo hemodynamic studies, we used Sugen/hypoxia-induced rat model of PAH. For ex vivo studies, we first developed PAH in male rats and confirmed the presence of PAH by measuring mPAP, as discussed above. As shown in Figure 3, using a highly sensitive pressure transducer, we measured perfusion pressure-mimicking PAP, which we will call here ex vivo PAP. In fact, for the first time here we have used an ex vivo model for studying the effect of a drug formulation on PAH-afflicted lungs (Figure 13). When three forms of the liposomal formulations were administered via the IV-mimicking route of the IPRL system, ex vivo mPAP declined in a time-dependent fashion. Although, all three formulations produced about 60% reduction in PAP, there was no statistically significant differences between the treatments. Similarly, when the PAH lungs were treated with the same formulations and given via the IT-mimicking route, the reduction in PAP was more pronounced than the reduction produced by the intravenously administered formulations. Fasudil-in-CAR-liposomes produced about 80% reduction in ex vivo PAP, which was statistically significant compared with other two formulations given via the same route. The maximum reduction induced by fasudil-in-CAR-liposomes is consistent with the increased accumulation of the same formulations in IPRL studies (Figures 5 and 6).
Figure 13.

Effect of liposomal fasudil on the ex vivo PAP of lungs collected from Sugen/hypoxia PAH rats, determined using IPRL model. The percent reduction of initial ex vivo PAP after administration of various forms of liposomal fasudil via (A) IV- and (B) IT-mimicking routes. The dose of fasudil was 3 mg/kg (rat body weight) or liposomes containing equivalent amount of fasudil and the dose of CAR was 0.5 mg/kg (data represent mean ± SD, n = 3).
Finally, we studied the effect of the formulations on mPAP and mSAP in PAH rats. First two treatments, fasudil-in-no-CAR-liposomes and CAR pretreatment and then fasudil-in-no-CAR-liposomes, reduced the mPAP to 28 and 34% of the initial mPAP, respectively, and the duration for the effect was 200 min (Figure 14A,B). However, echoing the data on drug disposition and pharmacokinetics, the CAR-conjugated liposomes containing fasudil produced much pronounced reduction in mPAP (53%) and the vasodilation persisted over 360 min (Figure 14C). Unlike their effect on mPAP, fasudil-in-no-CAR-liposomes and CAR pretreatment-then-fasudil-in-no-CAR-liposomes showed a sharp decline in mSAP, but returned to the baseline in 30–60 min. However, when fasudil-in-CAR-liposomes was given via the IT route, mSAP did not change, suggesting a pulmonary preferential vasodilation. LTI gave further credence to the lung specificity of CAR-liposomes. The LTI for fasudil-in-CAR-conjugated liposomes was about three-fold greater than those for two other treatments (Figure 14D), suggesting a greater lung accumulation and reduced peripheral vasodilation.
Figure 14.

Effect of liposomal fasudil on the pulmonary hemodynamics in Sugen/hypoxia PAH rats after administration of (A) fasudil in no-CAR-liposomes, (B) CAR pretreatment followed by fasudil in no-CAR-liposomes and (C) fasudil in CAR-liposomes. (D) Targeting indices of fasudil after IT administration in various forms. The dose of fasudil was 3 mg/kg (rat body weight) or liposomes containing equivalent amount of fasudil and the dose of CAR was 0.5 mg/kg (data represent mean ± SD, n = 3; *p < 0.05, §p < 0.005, Ƶp < 0.001).
4. DISCUSSION
In this study, we conjugated CAR onto the surface of the liposomes and performed a series of in vitro, ex vivo, and in vivo studies to delineate the effect of CAR on the targeted delivery of fasudil in PAH treatment. We demonstrated that CAR, the homing peptide, helps augment the therapeutic effect of fasudil formulated in liposomes by increasing the lung residence time, reducing particle clearance, and elevating the fraction retention of liposomal fasudil in the lungs. We further showed that CAR does not elicit its targeting effect when given alone or in combination with the drug.
We have developed a stable formulation with favorable drug encapsulation efficiency and physicochemical properties using an active loading method. In this active loading process, ammonium sulfate is first encapsulated within the liposomes and thus the hydrophilic core of the liposomes had a higher concentration of ammonium sulfate. Because of high permeability, ammonia, a neutral species with a permeability coefficient of 0.13 cm/s, diffuses out of the liposomal core into the external phase of the liposomes. As such, for every NH3 molecule that leaves liposomal core, one proton species is released, which remains trapped within the core. Consequently, a pH gradient develops, and the core of the liposomes becomes more acidic compared with the external phase.43 When incubated with the ammonium sulfate-rich liposomes, fasudil enters the liposome bilayer as unprotonated species and gets trapped in the aqueous acidic core because of ionization. Further, the PEG chains of PEG-functionalized lipids, used to prepare liposomes, form a protective sheath around the liposome and thus prevent degradation of liposomes in biological fluids and slow clearance by macrophages44 (Figure 1) and consequently increase the fasudil retention time in the lungs. Our results showed that optimizing the processing variables remarkably affect the entrapment efficiency of fasudil in the liposome. Encapsulation efficiency declined upon longer evaporation time because of low phase transition temperature of DPPC, which precipitated out of the solution. The increase in drug entrapment after sonication of the solubilized lipid film may have resulted from creation of SUVs and increased surface area due to sonication. Finally, reduced encapsulation of fasudil in solution versus increased encapsulation in solid state can be explained by the phenomenon that fasudil undergoes hydration in PBS but not when fasudil is in solid state. Hydrated molecules are less likely to enter the liposomal core, and this thus leads to reduced encapsulation, but in case of solid-state fasudil, more drug enters the liposomal core and thus increases the encapsulation.45–48
Because the vesicle size was less than 250 nm, these liposomes are unlikely to be phagocytosed by alveolar macrophages and thus they can escape lung clearance mechanism and dwell longer in the lungs.29,49 Colloidal stability for liposomal formulations is important because any instability may adversely affect the hydrodynamic diameter and thus affect the extent and mechanism of cellular uptake.50 Large negative zeta potential of our particles offers protection against aggregation due to mutual electrostatic repulsive forces, which keeps particle separated and helps prevent coalescence. Further, the cell-penetrating properties of CAR remained unaffected in the presence of cell culture media. The net negative charge has arisen from the zwitterionic phosphatidylcholine and from the amide linkage that results from the conjugation of CAR with liposomes, as evidenced by the NMR analysis.
The in vitro release profiles showed continuous release of fasudil from liposomal formulations, which may have resulted from the higher phase transition temperature of DSPE-PEG and DPPE. A continuous release of the drug from the liposome formulations and lack of burst release in the first 30 min indicates that fasudil diffuses out from the aqueous core of the liposome through the lipid bilayer instead of being released due to disruption of liposomes. The diffusion may be result of the formation of electrochemically neutral complexes of protonated fasudil and anionic sulfates, which can diffuse out from the core via the lipid membranes. All in all, in vitro release study suggests that both formulations are likely to deliver fasudil into the distal pulmonary capillaries in a controlled fashion and thus provide sustained vasodilatory effect. Further, published studies suggest that unlike micro-particles, inhaled particles of 100–500 nm size range reach the alveolar region of the lungs where gas exchange occurs. Thus, this liposomal formulation is expected to deposit chiefly in the deep lung deposition, but only slightly in the upper respiratory tract.51–54
However, the distribution of liposomal particles and their clearance from the lung merit further explanation, given the fact that the microsprayer used in this study produces 20–50 μm size droplets, which are not expected to deposit in the alveolar region of the lung. Nevertheless, based on the efficacy of liposomal formulations in reducing mPAPs in PAH rats, we assume that droplets delivered by the sprayer break into liposomal vesicles upon landing in the surfactant-rich respiratory epithelium, and the resulting liposomal vesicles distribute throughout the lungs including the alveolar region of the lungs. Thus, we studied the distribution and clearance of the liposomal formulations in PAH lungs. True to our assumption, the residence time for CAR liposomes in the lungs was longer than that for no-CAR liposomes (Figure 11A,B) and the accumulation of CAR-liposomes in the alveolar region was greater than that of no-CAR liposomes (Figure 11C).
For studying drug accumulation in and disposition from the lungs, we used a closed IPRL system, initially reported by Tronde et al.55,56 Our IPRL system (Hugo Sachs Elektronik, Harvard Apparatus GmbH, March-Hugstetten, Germany) prevents loss of perfusate due to evaporation and allows administration of drug directly into the lungs. The system allows maintaining the perfusate buffer constant at 50 mL by replenishing the withdrawn volume. Unlike IPRL circuitry proposed by Byron et al.,57–59 in which drug loss was 10–20%, drug loss in our system was minimal because our system recirculates perfusates and is completely closed. Thus, consistent with the data reported by Tronde et al., wherein drug recovery was 95%, our mass balance calculation showed that the total amount of the fasudil in the perfusate, lung, and IPRL tubes was close to 100% (Table 3). Further, lungs showed no signs of edema during the 120 min study; the tidal volume and perfusion flow were stable throughout the experiments.
Our finding based on the IPRL data demonstrated that, after IV administration of the fasudil in different formulations, the time-dependent decline in drug concentration appears to be unaffected by the type of formulations (plain fasudil, CAR pretreatment, CAR plus fasudil admixture, fasudil-in-no-CAR-liposomes vs fasudil-in-CAR-liposomes) and type of the lungs, diseased versus healthy lungs. In other words, when various formulations were administered via the IV-mimicking route of the IPRL system, CAR appears to have no effect on the pulmonary retention of the drug irrespective of the formulation or lung types, healthy or PAH afflicted. In contrast, the concentration of fasudil in the perfusate increased after IT administration. In fact, upon inhalational delivery, drug formulations undergo wetting and dissolution in the respiratory epithelium and subsequently enter the systemic circulation by traversing the respiratory epithelium.60 Because fasudil is a water-soluble drug and the liposomes contain highly soluble PEG chains, both quickly dissolved in the airway lining fluids and crossed the air–blood barrier and thus increased the drug concentration in the perfusate, which mimics the circulating blood in the IPRL system.61,62 The higher amount of fasudil in the lung after IT administration of various formulations is consistent with our previous observation that IT administration extends the residence time in the lungs, reduces its systemic exposure and clearance, and thus increases the pulmonary retention.63 The greater retention of fasudil in various formulations after IT administration in PAH lungs point to preferential accumulation of liposomes in PAH lungs, which overexpress HS in inflammatory conditions such as PAH, as we have observed in the cellular uptake study presented in Figure 8. Increased retention of fasudil in CAR-conjugated liposomes in sham rats is because normal lungs also express HS,17 which we reconfirmed here in this study by measuring HS in PASMCs from PAH rats versus control rats (Figure 9). Overall, this ex vivo disposition study suggests that the presence of CAR, when conjugated on the liposomal surface, produces pulmonary retention of the drug more than when present in the unconjugated form. Further, CAR-based pulmonary retention is greater in PAH-afflicted lungs than that in sham lungs.
This study addressed whether the CAR-liposomes preferentially accumulate on PACs afflicted by PAH versus non-PAH PACs. Previous studies suggest that CAR binds to various layers of diseased pulmonary vasculatures more extensively compared with negatively charged extracellular matrices of non-PAH pulmonary arteries.15,16 Increased uptake of fasudil by PAH-afflicted PACs compared with control PACs was because of increased expression of HS by PAH-PACs as discussed above. Compared with PAH-ECs and PAH-FBCs, a greater uptake of the drug by PAH-SMCs was perhaps because SMC is the site of action of fasudil and CAR may have a superior penetrating property for SMCs compared with other cells.
The extended half-lives of IT liposomal fasudil compared with IV liposomal fasudil in both sham and PAH rats are the result of the absorption of the drug from the lungs and lack of an absorption phase after IV administration (Table 4), suggesting that the drug largely accumulated in the lungs after IT instillation. The extended elimination half-life of CAR-liposomes may have stemmed from a number of factors including slower release of the drug from the liposomes, absorption controlled elimination, and binding of CAR to HS. These factors may have reduced the rate of arrival of the drug in the blood and that slowed the rate of elimination. On the whole, the absorption profiles of IV and IT administration of fasudil in various formulations accurately emulate the ex vivo disposition data. Both studies suggest that the presence of CAR in liposomes in conjugated forms plays an important role in the accumulation of fasudil in the lungs. Although fasudil-in-no-CAR-liposomes and CAR pretreatment appears to influence the accumulation of the drug in the lungs, the effect of CAR-liposomes in extending the half-life and reducing the overall plasma concentration is far greater than any other forms of the drug. Importantly, the effect of CAR-conjugated liposomes in reducing plasma concentration is consistent across the four major treatment groups: IV versus IT routes of administration and sham versus PAH animals. Further, because CAR pretreatment or administration of CAR plus fasudil admixture had no influence in ex vivo deposition or absorption, it is reasonable to argue that CAR per se has no anti-PAH or bystander effect. For CAR to function as a targeting peptide, it should be administered in the conjugated form with liposomes.
Ex vivo and in vivo hemodynamic studies demonstrated the prolonged pulmonary vasodilation of fasudil in CAR-conjugated liposomes after IT administration. This site-specific effect is due to the accumulation of the CAR-conjugated liposomal fasudil in the lungs and reduced availability of the drug in the systemic circulation. Overall, akin to the pharmacokinetic data, lung homogenates, and IPRL studies, the hemodynamic studies demonstrate that CAR pretreatment had no influence on pulmonary-specific retention and vasodilation.
However, although the above study provides convincing evidence that CAR-conjugated liposomes of fasudil are more efficacious in increasing pulmonary retention and producing pulmonary vasculature specific vasodilation, a number of questions remain unanswered and a few assumptions remain untested. First, here, we have not assessed the effect of the admixture of fasudil plus CAR on PAH lung or PAH animals. Although CAR pretreatment had no effect on the retention of fasudil in sham or PAH lungs, the presence of CAR as an admixture with fasudil may augment pulmonary retention in PAH lungs. Because of its amphiphilic characteristic, CAR is likely to form micelles in contact with water and encapsulate fasudil in the micellar core. Thus CAR-based micellar formulations can potentially elicit a similar effect as that elicited by CAR-conjugated liposomes. In addition to the shortcomings surrounding the IPRL and pharmacokinetic study, we have not used parallel IV control in hemodynamic studies. The use of all eight treatments, which were used in disposition studies, in pulmonary hemodynamic studies would have further strengthened data in support of CAR’s role in pulmonary retention of fasudil and subsequent fasudil-mediated vasodilation. The effect of CAR concentration on the cellular uptake also remains unelucidated. A study of the concentration-dependent effect of CAR was not feasible using the formulation reported in this manuscript because we used an excess of CAR to prepare liposomes saturated with CAR.
5. CONCLUSIONS
In this study, we have for the first time demonstrated that CAR, a cyclic peptide, increases drug accumulation in the lung and extend circulation life of the drug when the peptide is conjugated with a particulate carrier. The presence of CAR as a simple physical mixture with particulate carriers has little or no influence on endowing a drug with the property of controlled release or spatial accumulation. The cellular uptake of fasudil in various formulations demonstrated that CAR-liposomes preferentially accumulate on various PAH cells, especially proliferative SMC cells. As such, the distribution of liposomes and disposition profiles of fasudil, determined using IPRL and intact animal models, demonstrate that fasudil concentrated more in the lungs than in the blood when the drug was administered as a CAR-conjugated liposomal formulation, not when the lungs were pretreated with CAR The study also showed that liposomes, although delivered as large droplets, reach the alveolar region of the lungs. We have also demonstrated that an IPRL model can be used as a surrogate for pulmonary hemodynamics, especially for measuring mPAP. True to the disposition data, both ex vivo and in vivo pulmonary hemodynamics point to the assumption that CAR-conjugated liposomes, not CAR-pretreated liposomes, produce pulmonary preferential vasodilation.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported in part by multiple National Institutes of Health (NIH) and Department of Defense (DoD) grants: R01HL114677 (NHLBI) awarded to F.A., 1R35HL139726-01 (NHLBI) awarded to E.N.-G.; P01HL014985 (NHLBI), R01HL114887 (NHLBI) and PR140977 (DoD) awarded to Kurt Stenmark. We thank Dr. Paul Trippier for his help in interpretation of NMR data.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.molpharmaceut.9b00208.
15N NMR of SPDP conjugation on the liposome; 1H NMR of CAR conjugation on the liposome; 1H NMR of entrapped fasudil in the liposome; and retention of fasudil in the lungs after IV and IT administration (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).Iyer AK; Khaled G; Fang J; Maeda H Exploiting the enhanced permeability and retention effect for tumor targeting. Drug Discovery Today 2006, 11, 812–818. [DOI] [PubMed] [Google Scholar]
- (2).Malam Y; Loizidou M; Seifalian AM Liposomes and nanoparticles: nanosized vehicles for drug delivery in cancer. Trends Pharmacol Sci. 2009, 30, 592–599. [DOI] [PubMed] [Google Scholar]
- (3).Tacar O; Sriamornsak P; Dass C R Doxorubicin: an update on anticancer molecular action, toxicity and novel drug delivery systems. J. Pharm. Pharmacol 2013, 65, 157–170. [DOI] [PubMed] [Google Scholar]
- (4).Betancourt T; Byrne JD; Sunaryo N; Crowder SW; Kadapakkam M; Patel S; Casciato S; Brannon-Peppas L PEGylation strategies for active targeting of PLA/PLGA nanoparticles. J. Biomed. Mater. Res., Part A 2009, 91, 263–276. [DOI] [PubMed] [Google Scholar]
- (5).Brooks NA; Pouniotis DS; Tang C-K; Apostolopoulos V; Pietersz GA Cell-penetrating peptides: application in vaccine delivery. Biochim. Biophys. Acta 2010, 1805, 25–34. [DOI] [PubMed] [Google Scholar]
- (6).Chacko A-M; Li C; Nayak M; Mikitsh JL; Hu J; Hou C; Grasso L; Nicolaides NC; Muzykantov VR; Divgi CR; Coukos G Development of 124I immuno-PET targeting tumor vascular TEM1/endosialin. J. Nucl. Med 2014, 55, 500–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Jain A; Mishra SK; Vuddanda PR; Singh SK; Singh R; Singh S Targeting of diacerein loaded lipid nanoparticles to intra-articular cartilage using chondroitin sulfate as homing carrier for treatment of osteoarthritis in rats. Nanomedicine 2014, 10, e1031–e1040. [DOI] [PubMed] [Google Scholar]
- (8).Kornberger P; Skerra A Sortase-catalyzed in vitro function-alization of a HER2-specific recombinant Fab for tumor targeting of the plant cytotoxin gelonin. mAbs 2014, 6, 354–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Pradhan P; Giri J; Rieken F; Koch C; Mykhaylyk O; Döblinger M; Banerjee R; Bahadur D; Plank C Targeted temperature sensitive magnetic liposomes for thermo-chemotherapy. J. Controlled Release 2010, 142, 108–121. [DOI] [PubMed] [Google Scholar]
- (10).Ruoslahti E Peptides as targeting elements and tissue penetration devices for nanoparticles. Adv. Mater 2012, 24, 3747–3756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Zhang X; Zhang J; Ma Y; Pei X; Liu Q; Lu B; Jin L; Wang J; Liu J A Cell-based Single-stranded DNA Aptamer Specifically Targets Gastric Cancer. Int. J. Biochem. Cell Biol 2013, 46, 1–8. [PubMed] [Google Scholar]
- (12).AlDeghaither D; Smaglo BG; Weiner LM Beyond peptides and mAbs-current status and future perspectives for biotherapeutics with novel constructs. J. Clin. Pharmacol 2015, 55, S4–S20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Bellis SL Advantages of RGD peptides for directing cell association with biomaterials. Biomaterials 2011, 32, 4205–4210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Dinca A; Chien W-M; Chin M Intracellular Delivery of Proteins with Cell-Penetrating Peptides for Therapeutic Uses in Human Disease. Int. J. Mol. Sci 2016, 17, 263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Järvinen TAH; Ruoslahti E Molecular changes in the vasculature of injured tissues. Am. J. Pathol 2007, 171, 702–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Jarvinen TAH; Ruoslahti E Target-seeking antifibrotic compound enhances wound healing and suppresses scar formation in mice. Proc. Natl. Acad. Sci. U.S.A 2010, 107, 21671–21676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Urakami T; Järvinen TAH; Toba M; Sawada J; Ambalavanan N; Mann D; McMurtry I; Oka M; Ruoslahti E; Komatsu M Peptide-directed highly selective targeting of pulmonary arterial hypertension. Am. J. Pathol 2011, 178, 2489–2495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Järvinen TAH; Ruoslahti E Targeted Antiscarring Therapy for Tissue Injuries. Adv. Wound Care 2013, 2, 50–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Gupta N; Al-Saikhan FI; Patel B; Rashid J; Ahsan F Fasudil and SOD packaged in peptide-studded-liposomes: Properties, pharmacokinetics and ex-vivo targeting to isolated perfused rat lungs. Int. J. Pharm 2015, 488, 33–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Gupta N; Rashid J; Nozik-Grayck E; McMurtry IF; Stenmark KR; Ahsan F Cocktail of Superoxide Dismutase and Fasudil Encapsulated in Targeted Liposomes Slows PAH Progression at a Reduced Dosing Frequency. Mol. Pharm 2017, 14, 830–841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Rashid J; Nahar K; Raut S; Keshavarz A; Ahsan F Fasudil and DETA NONOate, Loaded in a Peptide-Modified Liposomal Carrier, Slow PAH Progression upon Pulmonary Delivery. Mol. Pharm 2018, 15, 1755–1765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Barman S; Zhu S; White RE RhoA/Rho-kinase signaling: a therapeutic target in pulmonary hypertension. Vasc. Health Risk Manage 2009, 5, 663–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Runo JR; Loyd JE Primary pulmonary hypertension. Lancet 2003, 361, 1533–1544. [DOI] [PubMed] [Google Scholar]
- (24).Gupta N; Patel B; Nahar K; Ahsan F Cell permeable peptide conjugated nanoerythrosomes of fasudil prolong pulmonary arterial vasodilation in PAH rats. Eur. J. Pharm. Biopharm 2014, 88, 1046–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Fukumoto Y; Matoba T; Ito A; Tanaka H; Kishi T; Hayashidani S; Abe K; Takeshita A; Shimokawa H Acute vasodilator effects of a Rho-kinase inhibitor, fasudil, in patients with severe pulmonary hypertension. Heart 2005, 91, 391–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Nahar K; Rashid J; Absar S; Al-Saikhan FI; Ahsan F Liposomal Aerosols of Nitric Oxide (NO) Donor as a Long-Acting Substitute for the Ultra-Short-Acting Inhaled NO in the Treatment of PAH. Pharm. Res 2016, 33, 1696–1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Toba M; Alzoubi A; O’Neill K; Abe K; Urakami T; Komatsu M; Alvarez D; Järvinen TAH; Mann D; Ruoslahti E; McMurtry IF; Oka M A novel vascular homing peptide strategy to selectively enhance pulmonary drug efficacy in pulmonary arterial hypertension. Am. J. Pathol 2014, 184, 369–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Gupta V; Gupta N; Shaik IH; Mehvar R; McMurtry IF; Oka M; Nozik-Grayck E; Komatsu M; Ahsan F Liposomal fasudil, a rho-kinase inhibitor, for prolonged pulmonary preferential vasodilation in pulmonary arterial hypertension. J. Controlled Release 2013, 167, 189–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Nahar K; Absar S; Gupta N; Kotamraju VR; McMurtry IF; Oka M; Komatsu M; Nozik-Grayck E; Ahsan F Peptide-coated liposomal fasudil enhances site specific vasodilation in pulmonary arterial hypertension. Mol. Pharm 2014, 11, 4374–4384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Absar S; Nahar K; Kwon YM; Ahsan F Thrombus-targeted nanocarrier attenuates bleeding complications associated with conventional thrombolytic therapy. Pharm. Res 2013, 30, 1663–1676. [DOI] [PubMed] [Google Scholar]
- (31).de Raaf MA; Kroeze Y; Middelman A; de Man FS; de Jong H; Vonk-Noordegraaf A; de Korte C; Voelkel NF; Homberg J; Bogaard HJ Serotonin transporter is not required for the development of severe pulmonary hypertension in the Sugen hypoxia rat model. Am. J. Physiol.: Lung Cell. Mol. Physiol 2015, 309, L1164–L1173. [DOI] [PubMed] [Google Scholar]
- (32).Taraseviciene-Stewart L; Kasahara Y; Alger L; Hirth P; Mc Mahon G; Waltenberger J; Voelkel NF; Tuder RM Inhibition of the VEGF receptor 2 combined with chronic hypoxia causes cell death-dependent pulmonary endothelial cell proliferation and severe pulmonary hypertension. FASEB J. 2001, 15, 427–438. [DOI] [PubMed] [Google Scholar]
- (33).Chen H; Lin Y; Han M; Bai S; Wen S Simultaneous quantitative analysis of fasudil and its active metabolite in human plasma by liquid chromatography electro-spray tandem mass spectrometry. J. Pharm. Biomed. Anal 2010, 52, 242–248. [DOI] [PubMed] [Google Scholar]
- (34).Hanson SFL; Terry MH; Moretta DT; Power GG; Wilson SM; Alam F; Ahsan F; Blood AB; Giri PC Inhaled Fasudil Lacks Pulmonary Selectivity in Thromboxane-Induced Acute Pulmonary Hypertension in Newborn Lambs. J. Cardiovasc. Pharmacol. Ther 2018, 23, 472–480. [DOI] [PubMed] [Google Scholar]
- (35).Gupta N; Ibrahim HM; Ahsan F Peptide-micelle hybrids containing fasudil for targeted delivery to the pulmonary arteries and arterioles to treat pulmonary arterial hypertension. J. Pharm. Sci 2014, 103, 3743–3753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Ishida T; Takanashi Y; Kiwada H Safe and efficient drug delivery system with liposomes for intrathecal application of an antivasospastic drug, fasudil. Biol. Pharm. Bull 2006, 29, 397–402. [DOI] [PubMed] [Google Scholar]
- (37).Haran G; Cohen R; Bar LK; Barenholz Y Transmembrane ammonium sulfate gradients in liposomes produce efficient and stable entrapment of amphipathic weak bases. Biochim. Biophys. Acta 1993, 1151, 201–215. [DOI] [PubMed] [Google Scholar]
- (38).Ewing P; Blomgren B; Ryrfeldt A; Gerde P Increasing exposure levels cause an abrupt change in the absorption and metabolism of acutely inhaled benzo(a)pyrene in the isolated, ventilated, and perfused lung of the rat. Toxicol Sci. 2006, 91, 332–340. [DOI] [PubMed] [Google Scholar]
- (39).Ewing P; Ryrfeldt Å; Sjöberg C-O; Andersson P; Edsbäcker S; Gerde P Vasoconstriction after inhalation of budesonide: a study in the isolated and perfused rat lung. Pulm. Pharmacol Ther 2010, 23, 9–14. [DOI] [PubMed] [Google Scholar]
- (40).Selg E; Acevedo F; Nybom R; Blomgren B; Ryrfeldt Å; Gerde P Delivering horseradish peroxidase as a respirable powder to the isolated, perfused, and ventilated lung of the rat: the pulmonary disposition of an inhaled model biopharmaceutical. J. Aerosol Med. Pulm. Drug Delivery 2010, 23, 273–284. [DOI] [PubMed] [Google Scholar]
- (41).Chang Y-T; Tseng C-N; Tannenberg P; Eriksson L; Yuan K; de Jesus Perez VA; Lundberg J; Lengquist M; Botusan IR; Catrina S-B; Tran P-K; Hedin U; Tran-Lundmark K Perlecan heparan sulfate deficiency impairs pulmonary vascular development and attenuates hypoxic pulmonary hypertension. Cardiovasc. Res 2015, 107, 20–31. [DOI] [PubMed] [Google Scholar]
- (42).Gotha L; Lim SY; Osherov AB; Wolff R; Qiang B; Erlich I; Nili N; Pillarisetti S; Chang Y-T; Tran P-K; Tryggvason K; Hedin U; Tran-Lundmark K; Advani SL; Gilbert RE; Strauss BH Heparan sulfate side chains have a critical role in the inhibitory effects of perlecan on vascular smooth muscle cell response to arterial injury. Am. J. Physiol.: Heart Circ. Physiol 2014, 307, H337–H345. [DOI] [PubMed] [Google Scholar]
- (43).Tu S; McGinnis T; Krugner-Higby L; Heath TD A mathematical relationship for hydromorphone loading into liposomes with trans-membrane ammonium sulfate gradients. J. Pharm. Sci 2010, 99, 2672–2680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Wang R; Xiao R; Zeng Z; Xu L; Wang J Application of poly(ethylene glycol)-distearoylphosphatidylethanolamine (PEG-DSPE) block copolymers and their derivatives as nanomaterials in drug delivery. Int. J. Nanomed 2012, 7, 4185–4198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Essa E Effect of formulation and processing variables on the particle size of sorbitan monopalmitate niosomes. Asian J. Pharm 2014, 4, 227. [Google Scholar]
- (46).Lapinski MM; Castro-Forero A; Greiner AJ; Ofoli RY; Blanchard GJ Comparison of liposomes formed by sonication and extrusion: rotational and translational diffusion of an embedded chromophore. Langmuir 2007, 23, 11677–11683. [DOI] [PubMed] [Google Scholar]
- (47).Liang MT; Davies NM; Toth I Encapsulation of lipopeptides within liposomes: effect of number of lipid chains, chain length and method of liposome preparation. Int. J. Pharm 2005, 301, 247–254. [DOI] [PubMed] [Google Scholar]
- (48).Petit J-C; Mea GD; Dran J-C; Magonthier M-C; Mando PA; Paccagnella A Hydrated-layer formation during dissolution of complex silicate glasses and minerals. Geochim. Cosmochim. Acta 1990, 54, 1941–1955. [Google Scholar]
- (49).Allen TM; Hansen C; Rutledge J Liposomes with prolonged circulation times: factors affecting uptake by reticuloendothelial and other tissues. Biochim. Biophys. Acta 1989, 981, 27–35. [DOI] [PubMed] [Google Scholar]
- (50).Natarajan JV; Chattopadhyay S; Ang M; Darwitan A; Foo S; Zhen M; Koo M; Wong TT; Venkatraman SS Sustained release of an anti-glaucoma drug: demonstration of efficacy of a liposomal formulation in the rabbit eye. PLoS One 2011, 6, e24513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Byron PR Prediction of drug residence times in regions of the human respiratory tract following aerosol inhalation. J. Pharm. Sci 1986, 75, 433–438. [DOI] [PubMed] [Google Scholar]
- (52).Heyder J Deposition of inhaled particles in the human respiratory tract and consequences for regional targeting in respiratory drug delivery. Proc. Am. Thorac. Soc 2004, 1, 315–320. [DOI] [PubMed] [Google Scholar]
- (53).Kleinstreuer C; Zhang Z; Donohue JF Targeted drug-aerosol delivery in the human respiratory system. Annu. Rev. Biomed. Eng 2008, 10, 195–220. [DOI] [PubMed] [Google Scholar]
- (54).Cohen BS; Li W; Xiong JQ; Lippmann M Detecting H +in Ultrafine Ambient Aerosol Using Iron Nano-Film Detectors and Scanning Probe Microscopy . Appl. Occup. Environ. Hyg 2000, 15, 80–89. [DOI] [PubMed] [Google Scholar]
- (55).Tronde A; Krondahl E; von Euler-Chelpin H; Brunmark P; Hultkvist Bengtsson U; Ekström G; Lennernäs H High airway-to-blood transport of an opioid tetrapeptide in the isolated rat lung after aerosol delivery. Peptides 2002, 23, 469–478. [DOI] [PubMed] [Google Scholar]
- (56).Tronde A; Nordén B; Jeppsson A-B; Brunmark P; Nilsson E; Lennernäs H; Bengtsson UH Drug Absorption from the Isolated Perfused Rat Lung-Correlations with Drug Physicochemical Properties and Epithelial Permeability. J. Drug Targeting 2003, 11, 61–74. [DOI] [PubMed] [Google Scholar]
- (57).Byron PR; Niven RW A novel dosing method for drug administration to the airways of the isolated perfused rat lung. J. Pharm. Sci 1988, 77, 693–695. [DOI] [PubMed] [Google Scholar]
- (58).Byron PR; Roberts SRN; Clark AR An isolated perfused rat lung preparation for the study of aerosolized drug deposition and absorption. J. Pharm. Sci 1986, 75, 168–171. [DOI] [PubMed] [Google Scholar]
- (59).Niven RW; Byron PR Solute absorption from the airways of the isolated rat lung. II. Effect of surfactants on absorption of fluorescein. Pharm. Res 1990, 7, 8–13. [DOI] [PubMed] [Google Scholar]
- (60).Sahib M; Darwis; Peh; Abdulameer, S. A.; Tan. Rehydrated sterically stabilized phospholipid nanomicelles of budesonide for nebulization: physicochemical characterization and in vitro, in vivo evaluations. Int. J. Nanomed 2011, 6, 2351–2366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Immordino ML; Dosio F; Cattel L Stealth liposomes: review of the basic science, rationale, and clinical applications, existing and potential. Int. J. Nanomed 2006, 1, 297–315. [PMC free article] [PubMed] [Google Scholar]
- (62).Shibuya M; Asano T; Sasaki Y Effect of Fasudil HCl, a protein kinase inhibitor, on cerebral vasospasm. Acta Neurochir. Suppl 2001, 77, 201–204. [DOI] [PubMed] [Google Scholar]
- (63).Sung JC; Pulliam BL; Edwards DA Nanoparticles for drug delivery to the lungs. Trends Biotechnol. 2007, 25, 563–570. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.